

Background: Leishmaniasis is a tropical disease prevalent in developing countries.
Results: Leishmania major type I nitroreductase (LmNTR) metabolizes nitroaromatic/quinone substrates, an activity essential for parasite viability.
Conclusion: LmNTR exhibits biochemical characteristics of its bacterial counterparts, and its activity is essential for parasite growth.
Significance: LmNTR can be exploited as a prodrug activation enzyme or as a target for inhibitor design in drug development.
Keywords: Drug Discovery, Drug Screening, Oxidation-Reduction, Parasite, Trypanosoma brucei, Trypanosome
Abstract
Nitroaromatic prodrugs are used to treat a range of microbial infections with selectivity achieved by specific activation reactions. For trypanosomatid parasites, this is mediated by type I nitroreductases. Here, we demonstrate that the causative agent of leishmaniasis, Leishmania major, expresses an FMN-containing nitroreductase (LmNTR) that metabolizes a wide range of substrates, and based on electron donor and acceptor preferences, it may function as an NADH:quinone oxidoreductase. Using gene deletion approaches, we demonstrate that this activity is essential to L. major promastigotes, the parasite forms found in the insect vector. Intriguingly, LmNTR+/− heterozygote promastigote parasites could readily differentiate into infectious metacyclic cells but these were unable to establish infections in cultured mammalian cells and caused delayed pathology in mice. Furthermore, we exploit the LmNTR activity evaluating a library of nitrobenzylphosphoramide mustards using biochemical and phenotypic screens. We identify a subset of compounds that display significant growth inhibitory properties against the intracellular parasite form found in the mammalian hosts. The leishmanicidal activity was shown to be LmNTR-specific as the LmNTR+/− heterozygote promastigotes displayed resistance to the most potent mustards. We conclude that LmNTR can be targeted for drug development by exploiting its prodrug activating property or by designing specific inhibitors to block its endogenous function.
Introduction
Insect-transmitted protozoan parasites belonging to the genus Leishmania are responsible for a spectrum of diseases known as leishmaniasis. These infections are endemic in 88 countries around the world with ∼350 million people living in “at risk” areas (1). Recently, as a result of military activity, population migration, modern medical practices, intravenous drug usage, and global warming, leishmaniasis has emerged as a problem in nonendemic areas (2–4).
A vaccine for visceral leishmaniasis is in phase I trials, but currently drugs are the only option available to treat leishmanial infections. Use of the frontline antimonial-based therapies is problematic as they are toxic, clinical resistance is on the rise, and they often require medical supervision to administer (5). Recent progress has been made in developing new leishmanicidal agents with several compounds such as amphotericin B, paromomycin, and miltefosine coming to market (1). However, there are issues associated with the use of these as they are expensive and require medical administration with some having teratogenic and other unwanted toxicity problems (6). Against this backdrop, the development of new cost-effective treatments is a priority, but given that leishmaniasis mainly affects people living in developing countries, these infections are not deemed commercially attractive by pharmaceutical companies. As a result, leishmaniasis is largely neglected in terms of drug development (7).
Nitroaromatic compounds encompass a wide range of compounds characterized by at least one nitro group attached to an aromatic ring (8). They have been used in medicine predominantly as antimicrobial agents, but concerns over their mutagenicity have led to many being withdrawn in Europe and the United States (9–11). However, it is now apparent that several nitro-based compounds are not as toxic as initially thought (12–14) with retrospective analysis of nitrofurantoin clinical trial data coupled with cost evaluation resulting in calls for the reinstatement of this prodrug as a treatment for uncomplicated urinary tract infections (15). This resurgence of interest has led to several nitroaromatic compounds undergoing evaluation for treatments of infectious organisms, including PA-824 and OPC-67683 against Mycobacterium tuberculosis, nitazoxanide against Giardia, Cryptosporidium, and hepatitis C (16–18), and fexinidazole against Trypanosoma brucei and Leishmania donovani (19, 20), whereas others such as SN23862, CB1954, and nifurtimox have emerged as possible anti-cancer therapies (21–23).
Most antimicrobial nitroaromatic compounds function as prodrugs and must undergo activation before producing their cytotoxic activity, a process mediated by nitroreductases (NTRs).5 Based on oxygen sensitivity and flavin cofactors, NTRs can be broadly divided into two groups (24). Type I NTRs utilize NAD(P)H as an electron donor, transferring reducing equivalents via an FMN cofactor to the substrate in a series of sequential two-electron reduction events. This nitroreduction does not involve oxygen and does not result in the production of reactive oxygen species, an activity said to be “oxygen-insensitive.” In contrast, the type II NTRs contain FAD or FMN as cofactor and catalyze the one-electron reduction of the substrate-conserved nitro group to generate a nitro radical. This radical reacts with oxygen to produce superoxide anions with the subsequent regeneration of the original nitro compound; type II NTRs is said to be “oxygen-sensitive” (25). The difference in NTR distribution is believed to underlie the specificity of most antimicrobial nitroaromatic prodrugs with type I NTRs found mainly in bacteria and absent from most eukarytotes, with a subset of fungi and protozoan parasites being the exceptions (26–28). Although some mammalian enzymes, such as NAD(P)H quinone oxidoreductase 1 and nitric-oxide synthase, can mediate two-electron reduction reactions under aerobic conditions, type II NTR activities predominate in most cell types (29).
The aim of this study was to characterize the type I NTR expressed by Leishmania major, the pathogen responsible for cutaneous leishmaniasis, and to analyze the role this enzyme plays in the parasite. Utilizing the activity, we then conducted biochemical and phenotypic screens using a library of nitrobenzylphosphoramide mustard (NBPM) compounds to identify potential leishmanicidal agents targeting the intracellular mammalian form of the pathogen.
EXPERIMENTAL PROCEDURES
Compounds
Nitrofurazone, nitrofurantoin, CB1954, coenzyme Q1, and duroquinone were purchased from Sigma. Benznidazole and nifurtimox were provided by Simon Croft (London School of Hygiene and Tropical Medicine), and metronidazole was a gift from Nubia Boechat (Far Manguinhos, Rio de Janeiro). The NBPM structures (Tables 1 and 2) were fully characterized using NMR and MS, and their purity was judged to be >90%; most were >95% based on LC-MS analysis (30–33).
TABLE 1.
Structure of cyclic nitrobenzylphosphoramide mustards
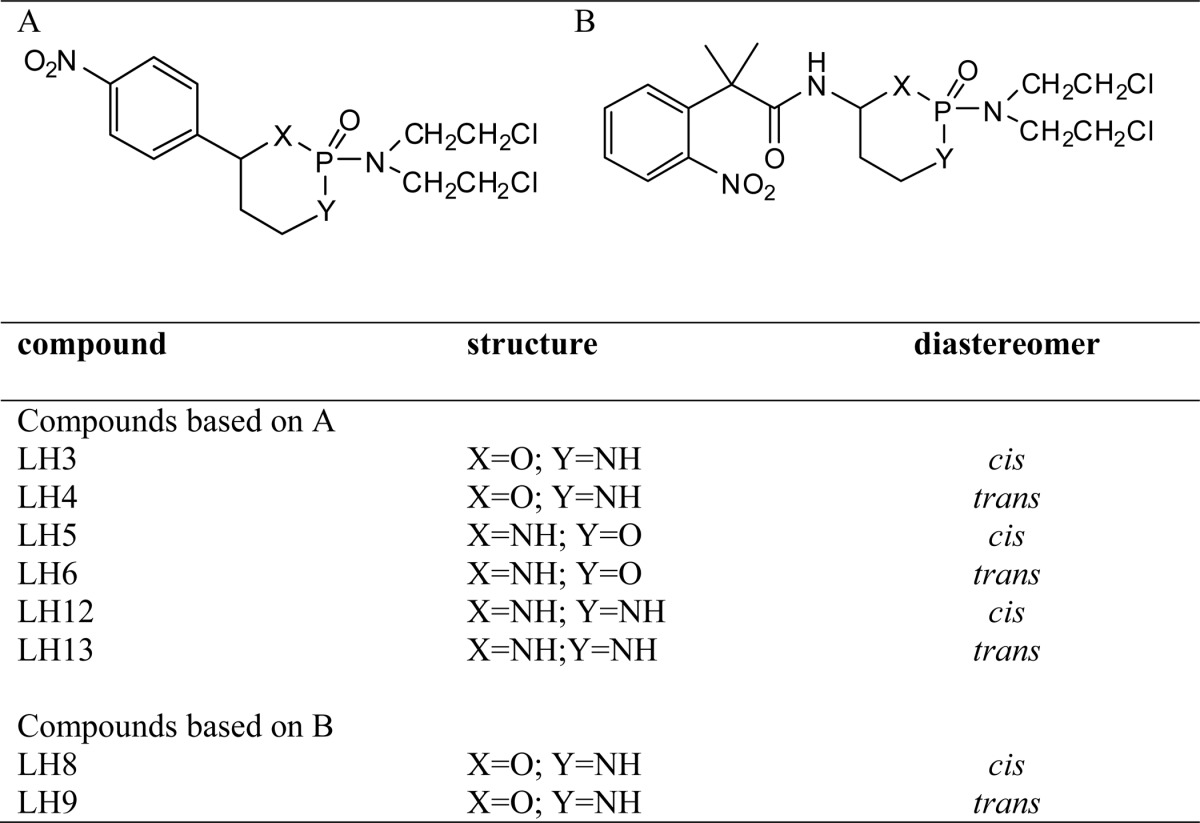
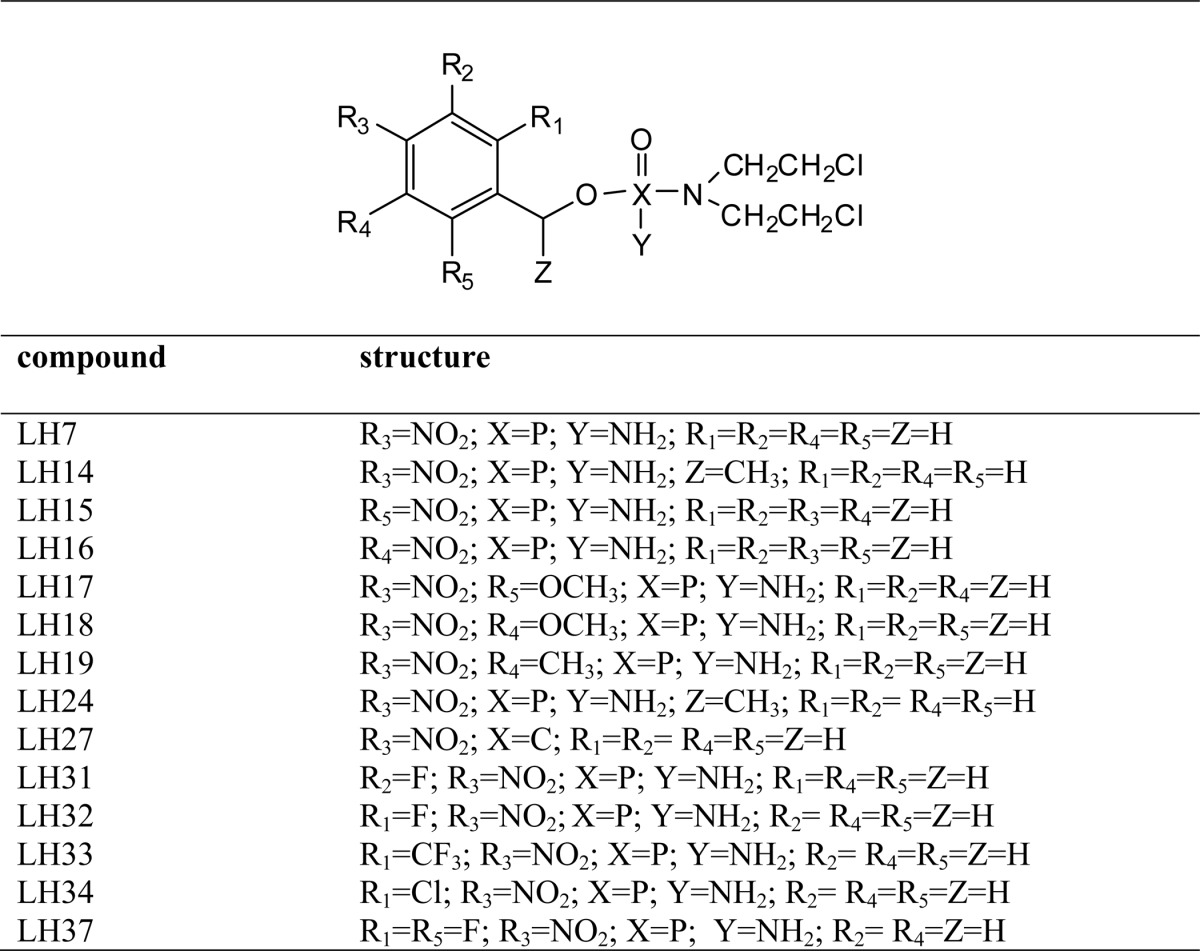
TABLE 2.
Structure of acyclic nitrobenzylphosphoramide mustards
Parasite Culture and Genetic Manipulation
L. major (MHOM/IL/80/Friedlin) promastigote form parasites were grown at 27 °C in M199 medium (Invitrogen) supplemented with 4 mm sodium bicarbonate, 40 mm HEPES, pH 7.4, 0.1 mm adenine, 0.005% (w/v) hemin, 2.5 units ml−1 penicillin, 25 μg ml−1 streptomycin, and 20% (v/v) fetal calf serum. Transformed parasites were grown in this medium containing G418 (20 μg ml−1 on agar plates, 40 μg ml−1 in broth), blasticidin (10 μg ml−1), or puromycin (20 μg ml−1). L. major metacyclic form parasites were harvested from promastigote cultures in the stationary phase of growth (7–10-day-old cultures) following agglutination of promastigote parasites with peanut lectin (Sigma) (34). The metacyclics were used to infect differentiated human acute monocytic leukemia (THP-1) cells at a ratio of 20 parasites per mammalian cell. The L. major infected monolayers were incubated overnight at 37 °C under a 5% (v/v) CO2 atmosphere in mammalian growth medium and then washed with RPMI 1640 to remove residual parasites. L. major amastigote parasites were maintained in differentiated THP-1 cells at 37 °C under a 5% (v/v) CO2 atmosphere in mammalian growth medium.
For DNA transfection, L. major promastigotes (2 × 107) in the logarithmic phase of growth were pelleted, resuspended in 100 μl of human T-cell Nucleofector® solution (Lonza AG) containing 5 μg of purified DNA, and electroporated using program U-033 on the NucleofectorTM (Lonza AG). Cells were then transferred to M199 growth medium and allowed to recover for 20–24 h at 27 °C. Parasites were pelleted, resuspended in 100 μl of promastigote growth medium, and then spread onto promastigote growth medium solidified with 1% (w/v) agar containing 1.2 μg ml−1 biopterin (Sigma) and the appropriate selective agent. The plates were incubated at 27 °C until colonies appeared. Colonies were then transferred into promastigote growth medium containing the appropriate selective agent and grown as described above.
Mammalian Cell Culturing
The human acute monocytic leukemia cell line (THP-1) was grown at 37 °C under a 5% (v/v) CO2 atmosphere in RPMI 1640 medium (PAA Laboratories Ltd.) supplemented with 2 mm pyruvate, 2 mm sodium glutamate, 2.5 units ml−1 penicillin, and 2.5 μg ml−1 streptomycin, 20 mm HEPES, pH 7.4, and 10% (v/v) fetal calf serum. Differentiation of THP-1 toward macrophage-like cells was carried out using phorbol 12-myristate 13-acetate (20 ng ml−1) (Sigma) (35).
In Vivo Studies
All animal experiments were conducted under license in accordance with regulations from United Kingdom Home Office. Female BALB/c mice were infected by subcutaneous injection of 2 × 107 purified metacyclic parasites in 100 μl of RPMI 1640 medium without serum into their shaved rump. Mice were monitored for lesion pathology, and when observed, the diameter of each lesion was measured in two dimensions at a rectangular angle with digital calipers (Jencons Scientific Ltd.), and the mean diameter was calculated. Three mice were examined for each treatment.
Antiproliferative Assays
All assays were performed in a 96-well plate format. L. major promastigote parasites (5 × 105 ml−1) or differentiated THP-1 cells (2.5 × 104 ml−1) were seeded in 200 μl of growth medium containing different concentrations of nitroaromatic agent. After incubation at 27 °C for 5 days (L. major) or at 37 °C for 3 days (THP-1), 2.5 μg of resazurin (20 μl of 0.125 μg ml−1 stock in phosphate-buffered saline) was added to each well, and the plates were incubated for a further 8–16 h. Cell densities were determined by monitoring the fluorescence of each culture using a Gemini Fluorescent Plate Reader (Molecular Devices Ltd., Wokingham, UK) at an excitation wavelength of 530 nm, emission wavelength of 585 nm, and a filter cutoff at 550 nm, and the drug concentration that inhibited cell growth by 50% (IC50) was established.
Growth inhibition of luciferase expressing L. major amastigotes was monitored as follows. THP-1 cells seeded at 2.5 × 104 ml−1 in 200 μl in growth medium containing phorbol 12-myristate 13-acetate (20 ng ml−1) were incubated at 37 °C in a 5% (v/v) CO2 atmosphere for 3 days. Macrophage monolayers were copiously washed with mammalian growth medium and then infected with purified L. major metacyclic cells (5 × 105 cells ml−1) resuspended in 200 μl of mammalian growth medium. Following incubation overnight at 37 °C in a 5% (v/v) CO2 atmosphere, the cultures were washed twice in growth medium to remove noninternalized parasites, and the supernatant was replaced with fresh growth medium containing drug. Drug-treated infections were incubated for a further 3 days at 37 °C under 5% (v/v) CO2. The growth medium was then removed, and the cells were lysed in 50 μl of cell culture lysis reagent (Promega). Activity was then measured using the luciferase assay system (Promega), and light emission was measured on a β-plate counter (PerkinElmer Life Sciences). The luminescence was proportional to the number of live cells. The IC50 value for each compound was then established.
Plasmids
The integrative L. major luciferase vector pLmRIX-luc was generated by sequentially cloning the L. major 5′-spacer/promoter rRNA and 3′-spacer rRNA regions on either side of an expression cassette containing the luciferase reporter and neomycin resistance genes. The TcMPX polypyrimidine tract/splice leader addition site sequence was inserted between the L. major 5′-spacer/promoter rRNA and the luciferase gene to provide the genetic elements required for processing the reporter mRNA transcript. The resultant construct, pLmRIX-Luc, was linearized prior to electroporation into L. major promastigotes.
The vectors used to delete LmNTR, pKO-LmNTR-PAC, and pKO-LmNTR-BLA were generated by sequentially cloning LmNTR flanking sequences on either side of a puromycin or blasticidin resistance cassette (see Fig. 3). The constructs were linearized and then electroporated into L. major promastigotes.
FIGURE 3.
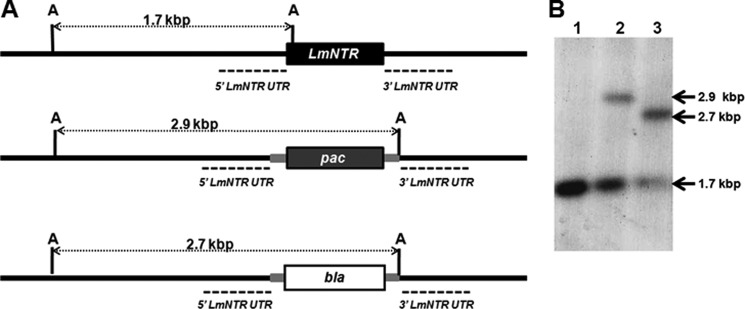
Disruption of LmNTR in L. major. A, diagram of the LmNTR alleles and the effects of gene disruption. 5′- and 3′-untranslated regions (dashed line) immediately upstream and downstream of LmNTR (black box) were amplified and cloned sequentially on either side of a puromycin (pac; dark gray box) or blasticidin (bla; white box) cassette containing T. brucei tubulin intergenic elements required for processing of mRNA (light gray boxes). The position of the predicted ApaI (A) sites plus the band sizes (in kbp) obtained after hybridization are shown. B, autoradiographs of ApaI-digested genomic DNA from L. major (lane 1), LmNTR+/−PAC and LmNTR+/−BLA heterozygous clones (lanes 2 and 3, respectively). Blots were hybridized with labeled 5′-untranslated region of LmNTR. Sizes given are in kbp.
A DNA fragment encoding for the catalytic domain of L. major NTR (LmNTR) was amplified from genomic DNA with the primers ggatccCTCGACGCCGTCGAGGCCGTCG and aagcttCTAGAACTTGTTCCACCGCAC, with lowercase italics indicating restriction sites incorporated into the primers to facilitate cloning. The fragment was digested with BamHI/HindIII and then cloned into the corresponding sites of the vector pTrcHisC (Invitrogen) to form the plasmid pTrcHisC-LmNTR. Site-directed mutagenesis was carried out using a Stratagene QuikChange mutagenesis kit (Agilent Technologies), with pTrcHisC-LmNTR as template. Amplifications were performed in accordance with the manufacturer's instructions using primers (Eurofins) to generate the desired mutation. The forward primer sequence was R96A (GCCGTCGTGCGCGACGCGTGGACGTGCCGGCAG). The relevant substitution site, incorporating the required base change, is underlined.
Protein Purification
An overnight culture of Escherichia coli BL21(+) containing the expression plasmid was diluted 1:50 in NZCYM (Sigma) medium containing 100 μg ml−1 ampicillin and grown for 2–4 h at 37 °C. The culture was transferred to 16 °C for 30 min, and the protein expression was induced by addition of isopropyl β-d-thiogalactopyranoside to a final concentration of 1 mm. The culture was then incubated overnight at 16 °C. The cells were harvested then lysed in Buffer A (50 mm NaH2PO4, pH 7.8, 500 mm NaCl) containing 1 mg ml−1 lysozyme, 10 μg ml−1 DNase, 10 μg ml−1 RNase, 1% (w/v) CHAPS, and a mixture of protease inhibitors (Roche Applied Science). The clarified supernatant was then applied to a pre-packed nickel-nitrilotriacetic acid (Qiagen) column, and the column was copiously washed with Buffer A and with Buffer A containing 50 and 100 mm imidazole. Recombinant protein was eluted off the column using Buffer A supplemented with 500 mm imidazole and 1% (w/v) CHAPS. Glycerol was then added to a final volume of 20% (v/v), and aliquots were stored at −80 °C. Protein concentration was determined by the BCA assay system (Thermo Fisher Scientific, Cramlington, UK), and the purity level was confirmed by SDS-PAGE.
Enzyme Activity
LmNTR activity was measured spectrophotometrically using several electron acceptors as substrate. A standard reaction (1 ml) containing 50 mm Tris-Cl, pH 7.5, 100 μm NADH, and 100 μm electron acceptor was incubated at room temperature for 5 min. The background reaction rate was determined, and the assay was initiated by addition of the LmNTR (35 μg). For reactions containing nitroimidazoles, nitrobenzylphosphoramides, or most quinones, activity was measured by following the change in absorbance at 340 nm corresponding to NADH oxidation (ϵ = 6,200 m−1 cm−1), although for assays involving nitrofurans, the direct reduction of the substrate itself was monitored at 435 nm for nifurtimox (ϵ = 18,000 m−1 cm−1) or at 400 nm for nitrofurazone and nitrofurantoin (ϵ = 12,000 and 15,000 m−1 cm−1, respectively (36)). For CB1954, activity was monitored by following the change in absorbance at 420 nm, corresponding to production of the hydroxylamine (ϵ = 1200 m−1 cm−1 (37)). Data were evaluated by nonlinear regression analysis using GraphPad Prism 5 (GraphPad Software).
Flavin Characterization
The flavin cofactor bound to LmNTR was established by determining the fluorescence spectrum in acidic and neutral buffers (36, 38). Purified protein was desalted and boiled for 5 min. In a total volume of 100 μl, clarified supernatants (90 μl) containing 0.5 mg of NTR were mixed with 10 μl of 50 mm NaH2PO4, pH 7.6, or 1 m HCl (final pH 2.2). The fluorescence profile for each treatment was then determined using a Gemini Fluorescent Plate Reader (Molecular Devices Ltd) with an excitation λ = 450 nm and an emission λ = 480–600 nm. The resultant patterns were compared with profiles obtained using FMN and FAD standards.
RESULTS
L. major Expresses a Functional Type I Nitroreductase
Analysis of the L. major genome database identified a 972-bp open reading frame located on chromosome 5 with the potential to encode for a 34.7-kDa enzyme related to the type I nitroreductase family of proteins (LmNTR; GenBankTM accession number XP_001687543). Full-length LmNTR is 90% identical to homologues present in Leishmania mexicana (CBZ23423) and Leishmania infantum (CAM65376) NTRs and 75% to Leishmania braziliensis NTR (CAM36899) and has 50% identity to the trypanosomal enzymes TcNTR (XP_810645) and T. brucei type I nitroreductase (TbNTR) (XP_846343). When compared with bacterial counterparts such as E. coli nfsB (NP_415110) and Bacillus cereus NAD(P)H nitroreductase (NP_832770), the highest level of sequence identity was around 20%. Based on sequence, LmNTR can be divided into two regions. The amino-terminal portion (residues 1–86), absent from prokaryotic sequences, is predicted by SignalP, PSORT II, and iPSORT algorithms to contain a mitochondrial targeting signal (data not shown), although the remainder of the enzyme (residues 87–323) constitutes the catalytic domain.
To evaluate whether LmNTR could function as a nitroreductase, the DNA sequence encoding for the putative catalytic domain (residues 87–323) was expressed as a His-tagged protein in E. coli; attempts to express full-length recombinant LmNTR failed to generate soluble protein. The recombinant enzyme could be readily purified after one round of affinity chromatography with elutions containing the purified His-tagged LmNTR having a yellow coloration, indicative of a flavoprotein (Fig. 1A). To identify the nature of the flavin and investigate NTR/cofactor interaction, recombinant LmNTR was boiled, and the fluorescence profile of clarified supernatants under neutral and acidic pH values was analyzed (Fig. 1B) (36, 38). Denaturation resulted in release of the flavin from the enzyme, indicating that the cofactor had a noncovalent association with the protein backbone (39–41), whereas the fluorescence profiles under neutral and acidic pH values identified FMN as the flavin moiety (Fig. 1B); at neutral pH, and excitation at 450 nm, flavin derived from the parasite enzyme had a peak emission around 560 nm, a signal quenched in acidic buffers, typical of FMN and distinct from FAD. In the trypanosomal enzyme TcNTR, an arginine located in the RX(S/T/A)X(R/K) motif has been shown to play a key role in cofactor binding with this region and the residue conserved in LmNTR (42). To evaluate the importance of this arginine to LmNTR activity, it was converted to alanine (R96A). Expression of the His-tagged mutant protein in E. coli resulted in recombinant protein but at levels considerably lower than that obtained with the wild type enzyme. The mutated enzyme remained in insoluble fractions despite several attempts to optimize expression and purification conditions (data not shown).
FIGURE 1.
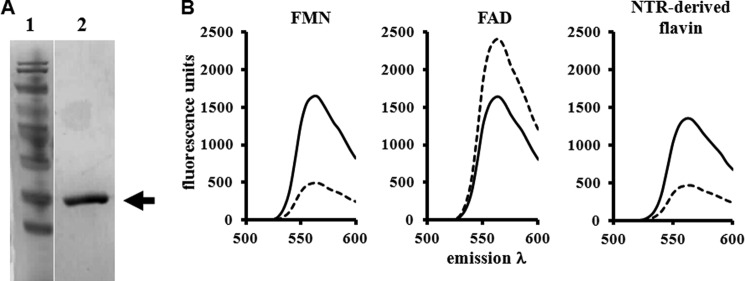
Leishmanial NTRs contain FMN as a cofactor. A, Coomassie-stained SDS-polyacrylamide gel (10%) containing purified, recombinant LmNTR (lane 2). Lane 1, size standards. B, fluorescence spectra of FMN and FAD (both 50 μm) and of supernatant from boiled and purified recombinant LmNTR (0.5 mg) at pH 7.6 (solid line) and pH 2.2 (dashed line) with excitation at 450 nm and emission between 480 and 600 nm. All the fluorescence analyses were carried out in triplicate; the profiles are derived from the mean values.
To investigate its substrate specificity, His-tagged LmNTR activity was monitored under aerobic conditions by following NADH or NADPH oxidation at 340 nm or, when a compound's absorbance spectra precluded this, by following reduction of the substrate itself (Fig. 2A). This demonstrated that the recombinant leishmanial enzyme functioned as typical type I NTR, able to catalyze the reduction of a wide range of nitroaromatics and quinones in an oxygen-insensitive fashion (Table 3). When the specificity constants (kcat/Km) generated by these substrates were compared, LmNTR showed a preference for quinone compounds; the kcat/Km values exhibited by His-tagged LmNTR toward the ubiquinone 5 (2.0 × 104) was an order of magnitude greater than for CB1954 (4.6 × 103) and benznidazole (2.5 × 103). Interestingly, recombinant LmNTR could use both NADPH and NADH as electron donors, although when using fixed concentrations of benznidazole (100 μm) and NADPH or NADH (100 μm), activity values indicated a preference for NADH. Under these conditions, LmNTR had a specific activity of 110 nmol of NADH oxidized per min−1 mg−1 as compared with 50 nmol of NADPH oxidized per min−1 mg−1. Using benznidazole (100 μm) as substrate, His-tagged LmNTR had an apparent Km for NADH of 76 μm, similar to the values reported for T. brucei type I nitroreductase (TbNTR) and TcNTR (71 and 86 μm, respectively) (42).
FIGURE 2.
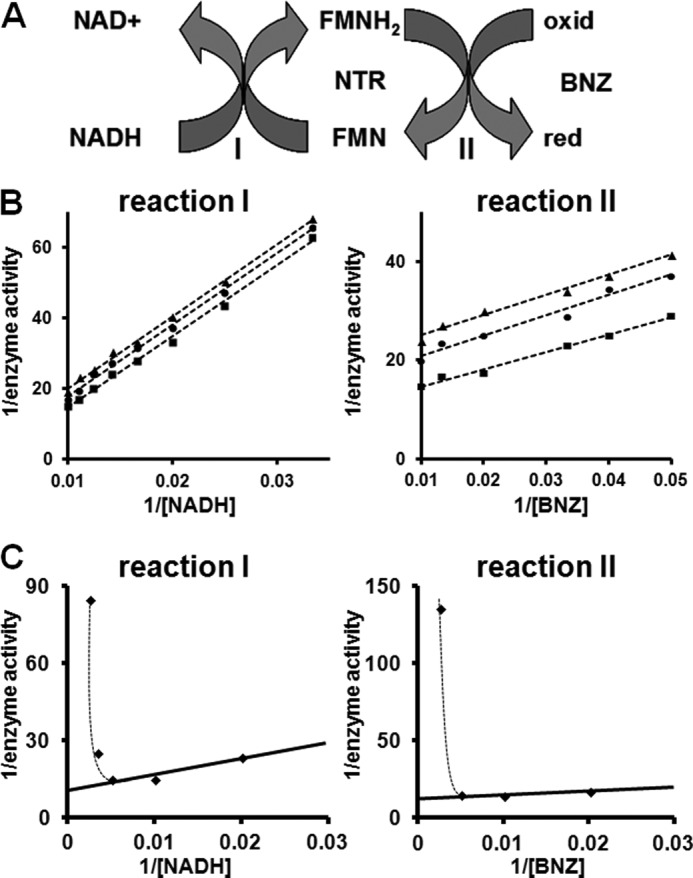
Investigation of the kinetic properties of leishmanial type I nitroreductase toward benznidazole. A, postulated scheme for the L. major type I nitroreductase-mediated reduction of benznidazole (BNZ) by using NADH as an electron donor. Red represents the reduced form, and oxid represents the oxidized form of benznidazole. The oxidized (FMN) and reduced (FMNH2) forms of the flavin cofactor are indicated. B, left panel, interaction of LmNTR with NADH (reaction I). Activity was assayed by monitoring the oxidation of NADH (30–100 μm) in the presence of benznidazole (50 μm (▴), 75 μm (●), and 100 μm (■)) and His-tagged LmNTR (35 μg). Right panel, interaction of LmNTR with benznidazole (reaction II). Activity was assayed by monitoring the oxidation of NADH (70 μm (▴), 80 μm (●), and 100 μm (■)) in the presence of benznidazole (20–100 μm) and His-tagged LmNTR (35 μg). C, inhibition of LmNTR activity by high concentrations of NADH or benznidazole. In reaction I, a fixed concentration of benznidazole (100 μm) was reduced by His-tagged LmNTR (35 μg) using various concentrations of NADH (50–400 μm). At high NADH levels (>200 μm), substrate inhibition was observed. In reaction II, a fixed concentration of NADH (100 μm) was oxidized by His-tagged LmNTR (35 μg) using various concentrations of benznidazole (50–400 μm). At benznidazole concentrations above 200 μm, substrate inhibition was observed. All assays were initiated by the addition of the parasite enzyme. LmNTR activity is expressed as micromoles of NADH oxidized per min−1 mg of protein−1, whereas [NADH] and [benznidazole] are expressed in micromolar.
TABLE 3.
Substrate specificity of LmNTR
The apparent Vmax and Km values of His-tagged LmNTR toward various nitroaromatic and quinone-based substrates were determined in the presence of NADH (“Experimental Procedures”). The kcat/Km value (the “specificity constant”) was then determined providing a useful ratio for comparing the relative rates of LmNTR activity on various substrates.
| Compound | Apparent Km | Apparent Vmax | kcat/Km |
|---|---|---|---|
| μm | nmol min−1 mg−1 | m−1 s−1 | |
| Nitroimidazole | |||
| Benznidazole | 22.0 ± 0.4 | 78.7 ± 1.0 | 2.5 × 103 |
| Metronidazole | 2.4 ± 0.0 | 17.9 ± 0.0 | 5.2 × 103 |
| Nitrofuran | |||
| Nifurtimox | 9.7 ± 1.1 | 50.9 ± 7.0 | 3.7 × 103 |
| Nitrofurazone | 4.7 ± 0.6 | 68.3 ± 10.0 | 1.0 × 104 |
| Nitrofurantoin | 3.9 ± 0.5 | 56.7 ± 8.0 | 1.0 × 104 |
| Nitrobenzyl | |||
| CB1954 | 13.1 ± 0.1 | 85.9 ± 1.0 | 4.6 × 103 |
| LH32 | 28.8 ± 2.7 | 100.7 ± 10.0 | 2.5 × 103 |
| LH33 | 8.2 ± 1.0 | 114.2 ± 15.0 | 9.8 × 103 |
| LH37 | 7.1 ± 0.4 | 71.7 ± 5.0 | 7.1 × 103 |
| Quinone | |||
| Duroquinone | 9.8 ± 0.3 | 140.6 ± 5.0 | 1.0 × 104 |
| Coenzyme Q1 | 5.1 ± 0.4 | 145.2 ± 11.0 | 2.0 × 104 |
To examine how recombinant LmNTR interacts with NADH, assays were carried out using various concentrations of reductant against a fixed concentration of benznidazole (Fig. 2B). For this substrate, double-reciprocal plots were linear at all concentrations of electron acceptor, with the slopes remaining parallel. When the converse experiments were performed using a fixed concentration of NADH and various amounts of benznidazole, a similar pattern of parallel slopes was observed (Fig. 2B). These patterns are characteristic of a ping-pong mechanism of kinetics, typical for oxidoreductase cascades. However, when benznidazole or NADH levels were above 200 μm, noncompetitive substrate inhibition was observed indicating that this mechanism of kinetics only occurs over a limited electron donor/acceptor concentration range (Fig. 2C).
Functional Analysis of LmNTR in L. major
To investigate whether the leishmanial type I NTR can activate nitroaromatic drugs in the parasite itself, we developed L. major LmNTR heterozygotes and then examined the susceptibility of the cells to nifurtimox. DNA fragments corresponding to the regions immediately upstream and downstream of LmNTR were amplified from genomic DNA and cloned sequentially on either side of cassettes containing puromycin or blasticidin resistance markers. The constructs were linearized, and the purified fragments were transformed into L. major promastigotes with clones selected on agar plates containing puromycin or blasticidin. Southern hybridization of genomic DNA showed that one LmNTR allele could be disrupted generating heterozygous parasites (LmNTR+/−PAC or LmNTR+/−BLA) (Fig. 3), with no obvious effect on parasite growth or ability to differentiate into infective metacyclic forms (Fig. 4A). To evaluate whether reduction of the LmNTR copy number affected resistance to nitroheterocyclic drugs, LmNTR+/−BLA promastigote parasites were grown in the presence of nifurtimox (Fig. 4B). From the resultant dose-response curves, the IC50 value of the heterozygote line was shown to be ∼2-fold higher than wild type parasites; L. major wild type cells exhibited an IC50 value of 6.28 ± 0.04 μm as compared with 11.50 ± 0.60 μm for the LmNTR+/− line.
FIGURE 4.
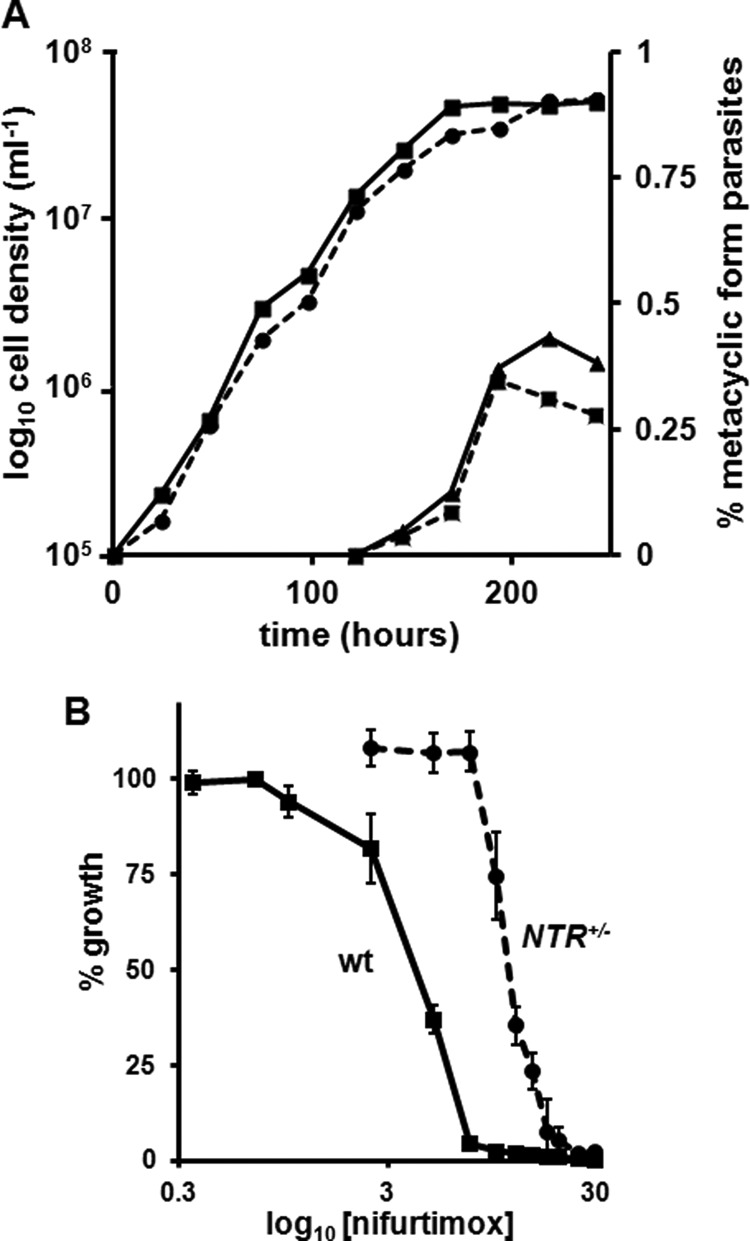
Promastigote growth and metacyclogenesis of the L. major LmNTR heterozygous line. A, L. major wild type (square; solid line) and LmNTR+/−BLA heterozygote (circle; dotted line) promastigote parasite growth was monitored until cultures were in the stationary phase of growth (time = 240 h). At 120 h onward, the number of metacyclic form parasites in wild type (triangle; solid line) and LmNTR+/−BLA heterozygote (cross; dotted line) promastigote culture was determined following purification by agglutination. The data are expressed as % metacyclic load in the total L. major population. All curves shown are derived from a single data set and are representative of experiments performed in triplicate. B, dose-response curves of nifurtimox on L. major wild type (wt) and LmNTR+/−BLA heterozygote (NTR+/−) promastigotes. Data are means from four experiments ± S.D., and the differences in susceptibility were statistically significant (p < 0.01), as assessed by Student's t test.
Attempts (16 independent transformations) to generate LmNTR null mutant lines failed, leading us to speculate that the protein is essential to promastigote form parasites. To confirm this, we constructed a range of pTEX- and pIR-SAT1-based episomal and integrative complementation vectors designed to facilitate expression of LmNTR or variants tagged at the carboxyl terminus with an epitope derived from the human c-MYC protein. Attempts (20 independent transformations) to generate parasites lines containing an ectopic copy of LmNTR failed, leading us to postulate that expression of elevated NTR levels was also deleterious to L. major promastigotes. Even when using “amastigote-specific” integrative vectors, where LmNTR expression was placed under the control of the well characterized Leishmania regulatory genetic element (the amastin 3′-UTR (43)), no recombinant parasites were obtained; the “amastigote-specific” expression system was validated using constructs expressing luciferase where the reporter activity under the control of the amastin 3′-UTR was only 2–10-fold above background, and in the absence of this regulatory genetic element reporter activity was >1000-fold higher (data not shown). This toxicity may be in part due to the LmNTR amino terminus. To facilitate localization, attempts (six independent transformations) to generate L. major promastigotes expressing the amino-terminal extension (residues 1–85) of LmNTR fused at its carboxyl terminus with GFP or red fluorescent protein failed despite using episomal or integrative expression vectors (data not shown).
To aid the characterization of L. major LmNTR heterozygotes, LmNTR+/−BLA cells were transformed with a DNA fragment that integrates into the L. major rRNA promoter/spacer region and facilitates expression of luciferase. Following selection, the luminescence activity of clonal parasite extracts was shown to be >106-fold above background, comparable with lysates generated from luciferase expressing wild type cells. These parasites also exhibit promastigote growth and metacyclogenesis rates equivalent to wild type/non-luciferase-expressing LmNTR heterozygotes (data not shown). Metacyclic form parasites from luciferase-tagged LmNTR+/− heterozygote and wild type promastigote cultures in the stationary phase of growth were then purified and used to infect differentiated THP-1 macrophage cells. Over the following 4 days, total cell extracts were generated from parasite/mammalian cultures, and the luciferase activity was determined (Fig. 5A). After correcting for background luminescence (lysed, uninfected macrophages), the reporter signal generated from tagged wild type amastigotes increased, reaching a plateau by day 4 post-infection. In contrast, LmNTR+/− heterozygote cultures exhibited luminescence values equivalent to background. This indicates that disruption of one LmNTR allele resulted in haploid insufficiency such that L. major LmNTR heterozygote amastigotes are unable to establish themselves in tissue-cultured mammalian macrophages.
FIGURE 5.
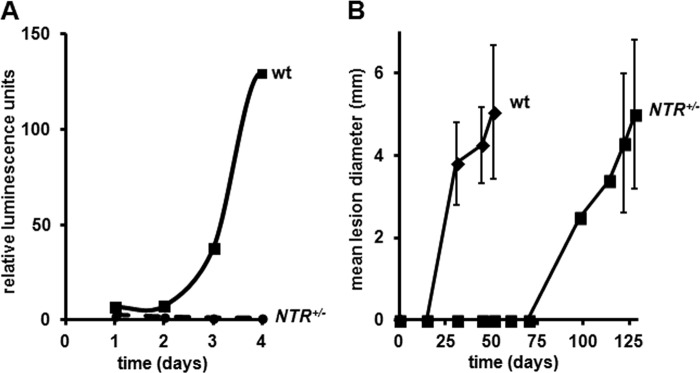
Disruption of a single LmNTR allele affects infectivity. A, purified L. major wild type (wt) and LmNTR+/−BLA heterozygote (NTR+/−) metacyclic form parasites engineered to express luciferase were used to infect differentiated THP-1 cells. Over a 4-day post-infection period, extracts were generated from each cell line, and the luciferase activity was determined. Following background correction, the luciferase activity was plotted against time. All curves shown are derived from a single data set and are representative of experiments performed in triplicate. B, purified L. major wild type (wt) and LmNTR+/−BLA heterozygote (NTR+/−) metacyclic form parasites were inoculated into the rump of BALB/c mice. Periodically, the diameter of the lesion was measured. For each cell line, three mice were infected, and the data are expressed as the mean lesion size (in mm) ± S.D. For the heterozygote infections, the lesion sizes on days 98 and 114 are derived from a single mouse as the wounds present on other animals could not be accurately determined.
To determine whether the in vitro amastigote proliferation defect translates to problems with in vivo growth, BALB/c mice were infected with purified wild type or LmNTR+/− heterozygote L. major metacyclic cells. Over a 97-day period, the presence and size of any lesion were monitored (Fig. 5B). All three mice infected with wild type parasites developed lesions by day 31 with the lesions gradually increasing in size; all mice were euthanized at day 51. Interestingly, all mice infected with LmNTR+/− heterozygote cells presented no signs of lesions until day 98, when a single mouse developed a lesion, with the remaining mice developing pathologies 24 days later (day 122). In all cases, the lesion size gradually increased until day 128, when all mice were euthanized. The altered kinetics in pathology development strongly indicate that LmNTR function is important in establishing an L. major infection in vivo with the heterozygote line struggling to grow as amastigote form parasites in BALB/c mice.
Exploiting the Prodrug Activating Properties of the Leishmania Type I NTR
We have previously shown that NBPM compounds are effective NTR-activated trypanocidal agents (36). As an initial screening strategy to determine the potential for exploiting LmNTR activity in prodrug activation, we determined whether His-tagged LmNTR displayed activity toward a library of NBPMs by monitoring the change in absorbance at 340 nm, corresponding to NADH oxidation (Fig. 6A). In total, 22 compounds were screened as follows: eight structures where either a p-nitrobenzyl group or an o-nitrophenylacetamido group was attached to cyclophosphamide (Table 1) and 14 structures where phosphoramide mustard is linked to a nitrobenzyl group with various substituents (Table 2). Based on the preliminary biochemical screen, the compounds could be divided into those that were not LmNTR substrates (activity <20 nmol of NADH oxidized per min−1 mg−1), those that stimulated a moderate activity (between 20 and 50 nmol of NADH oxidized per min−1 mg−1), and those that were readily metabolized by the parasite enzyme (>50 nmol of NADH oxidized per min−1 mg−1). Using this as a crude measure, three cyclic NBPMs (LH5, -6, and -12) were evaluated as moderate substrates, whereas the remaining five showed little/no activity. In contrast, most of the acyclic structures were metabolized by recombinant LmNTR with six (LH27, 31–34, and 37) showing high levels of activity. Of those substrates generating the higher activities, five contained at least one halogen linked to the nitro-substituted benzene ring.
FIGURE 6.

Evaluating nitrobenzylphosphoramide mustards as LmNTR substrates. A, activity of purified His-tagged LmNTR was assessed by using various NBPMs (100 μm) as substrate at a fixed concentration of NADH (100 μm). The values shown are the means of data from three experiments ± S.D. LmNTR activity was deemed to be high if it was >50 nmol of NADH oxidized per min−1 mg−1 (dotted line). The activity obtained when using nifurtimox (NFX) as a substrate is also shown. B, susceptibility of L. major wild type (wt) and LmNTR+/−BLA heterozygote (NTR+/−) promastigotes to LH33 and LH34. Data are means from four experiments ± S.D.
To determine whether there was a correlation between biochemical activity and parasite killing, all NBPMs were initially screened for leishmanicidal activity against L. major promastigote and amastigote forms. Out of the 22 compounds, 15 were shown not to affect growth of the insect stage pathogen at 30 μm with all except one (LH19) also having no effect on amastigote growth at a concentration of 10 μm (Table 4). For the remaining leishmanicidal compounds, growth inhibition assays were performed to determine their IC50 values (Table 4). Many of these displayed appreciable leishmanicidal properties, with five NBPMs (LH31–34 and -37) having significant activity (IC50 values of <10 μm) against both parasite forms. These five compounds correspond to structures previously designated “good” LmNTR substrates (Fig. 6).
TABLE 4.
Susceptibility of L. major and differentiated THP-1 cells to nitrobenzyl phosphoramide mustards
| Compounds |
L. major IC50 |
Differentiated THP-1 IC50b | Selective toxicityc | |
|---|---|---|---|---|
| Promastigotesa | Amastigotesb | |||
| μm | μm | |||
| Nifurtimox | 6.28 ± 0.04 | 2.15 ± 0.01 | >100.00 | >47 |
| LH3–9, LH12–15, LH17–18, LH24 | >30.00 | >10.00 | NDd | ND |
| LH16 | 15.60 ± 1.13 | 2.75 ± 0.40 | >100.00 | >36 |
| LH19 | >30.00 | 4.72 ± 0.55 | >100.00 | >21 |
| LH27 | 16.23 ± 0.29 | 4.65 ± 0.15 | >100.00 | >22 |
| LH31 | 9.05 ± 0.56 | 7.00 ± 0.40 | >100.00 | >14 |
| LH32 | 4.72 ± 0.23 | 1.09 ± 0.21 | >100.00 | >92 |
| LH33 | 5.88 ± 0.28 | 0.21 ± 0.07 | >100.00 | >476 |
| LH34 | 3.10 ± 0.28 | 0.77 ± 0.12 | >100.00 | >143 |
| LH37 | 1.29 ± 0.08 | 2.17 ± 0.27 | >100.00 | >46 |
a Data are means from four experiments ± S.D.
b Data are means from three experiments ± S.D.
c The therapeutic index of a compound was calculated as a ratio of the IC50 value against differentiated THP-1 cells to the IC50 value against amastigote parasites.
d ND means not determined.
The eight compounds identified as having appreciable leishmanial activity were assayed for cytotoxicity against differentiated THP-1 cells from which the selective index (IC50 against the mammalian line/IC50 against the amastigote parasite) was then determined (Table 4). In all cases, no toxicity to the mammalian line was observed at concentrations up to 100 μm. For three of the halogenated compounds identified as being preferred LmNTR substrates and having anti-parasitic activity against both L. major forms (LH32–34), selective index values against amastigote parasites of >100 were observed.
To demonstrate that NTR plays a role in NBPM prodrug activation within the parasite itself, the susceptibility of L. major LmNTR+/−BLA heterozygote promastigotes to LH33 and LH34 was investigated (Fig. 6B). For both compounds, cells with reduced levels of the nitroreductase were up to 3-fold more resistant to the agent than wild type controls; L. major wild type cells exhibited IC50 values of 4.73 ± 0.29 and 3.70 ± 0.40 μm toward LH33 and LH34, respectively, and LmNTR+/− heterozygote cells displayed values of 7.66 ± 0.23 and 12.14 ± 0.84 μm against these two compounds.
Based on our findings, NBPMs that contain the phosphoramide mustard as part of an acyclic structure and have halogen substituents on the nitrobenzyl ring are the most readily metabolized by the LmNTR enzyme and represent the most potent agents against both L. major promastigotes and amastigotes. Some (LH32, -33, and -34) have IC50 values of 1 μm or less against the intracellular stage without inducing mammalian cell toxicity.
DISCUSSION
Activation of most antimicrobial nitroaromatic prodrugs occurs through reactions catalyzed by type I NTRs (24, 26). This group of enzymes was believed to be restricted to bacteria, but it is now apparent that they are also expressed by several “lower” eukaryotes, including fungi and protozoan parasites (20, 26–28). Interestingly, type I NTRs appear to be absent from “higher” eukaryotes, including humans, and it is believed that this difference in the enzymes' distribution underlies the antimicrobial selectivity of many nitroaromatic prodrugs. Here, we demonstrate that L. major expresses a type I NTR that possesses many biochemical characteristics displayed by its bacterial counterparts. Functional studies revealed that this activity is essential for the growth of parasite forms found in the insect vector and extremely important to the intracellular forms found in mammalian cells, indicating that this prodrug-activating enzyme is a drug target in its own right.
The activity of all type I NTRs, irrespective of their origin, is dependent on FMN with the enzyme/flavin interaction occurring via a noncovalent linkage, features shared by LmNTR (Fig. 1). His-tagged LmNTR activity was associated with yellow-colored fractions, and boiling released the associated cofactor from the protein backbone with clarified supernatants exhibiting fluorescence profiles under acidic and neutral pH values identical to that observed with FMN (Fig. 1). For the leishmanial enzyme, the cofactor acts as an intermediary, accepting reducing equivalents derived preferentially from NADH and then donating these to a range of nitroaromatic and quinone-based substrates (Table 3). While catalyzing this transfer, His-tagged LmNTR displays a ping-pong type of kinetics, typical of oxidoreductases (Fig. 2). In TcNTR, cofactor binding is mediated by a specific arginine residue at position 90 present in a conserved motif. This amino acid (Arg-96 in LmNTR) plus the adjacent region is present in NTR from L. major. Attempts to express a mutated version of the leishmanial enzyme (R96A) failed to generate soluble protein suggesting that the noncovalent FMN binding to the protein backbone may be important in the correct folding of LmNTR.
Based on its substrate preferences (Table 3) and subcellular localization, as inferred from its sequence, LmNTR may function as a mitochondrial NADH:quinone oxidoreductase. In the mitochondria of most eukaryotes, oxidation of NADH to NAD+ is normally mediated by complex I of the electron transport chain. This activity serves to translocate protons across the organelle's inner membrane with the concomitant reduction of ubiquinone to ubiquinol. Ubiquinol then drives the cytochrome-dependent respiratory chains that help to form the proton motive force, which ultimately leads to ATP synthesis. In contrast to bloodstream-dwelling trypanosomes, all replicative Leishmania forms express functional cytochrome-dependent electron transport chains suggesting that energy production occurs via the well documented route within these parasites (44–46). However, the role played by the Leishmania complex I in driving these cascades is unclear. Inhibitor studies indicate that these protozoa possess an atypical complex I with bioinformatic studies revealing a subunit composition distinct from that found in other eukaryotic organisms (44, 47). As LmNTR appears to fulfill an analogous role to the leishmanial complex I, with both functioning as NADH:quinone oxidoreductases, these may act in concert to help maintain the NADH/NAD+ balance within the parasite mitochondrion.
The endogenous function of LmNTR is essential to replicating L. major: LmNTR could not be deleted from noninfectious promastigote parasites with the heterozygotes unable to establish an infection in cultured macrophages and struggled to generate lesion pathology in mice (Fig. 5). This is similar to the situation in L. donovani but distinct from that observed in trypanosomes where the essential nature of NTR is only apparent in the replicating forms present in the mammalian host (26, 48). As we were unable to express elevated levels of LmNTR in L. major heterozygotes and wild type promastigotes, the appropriate complementation experiments could not be performed. Intriguingly, complementation studies could be conducted using L. donovani promastigotes (48); L. donovani NTR null mutants could be generated only in the presence of an ectopic copy of L. major NTR. The deleterious effect of expressing LmNTR at elevated levels may not be due to the enzymatic activity of the protein itself but could reflect an inherent problem associated with the amino-terminal extension. Therefore, when expressed at levels higher than that found in wild type (and heterozygote), the LmNTR extension may interfere with mitochondrial transporter function or could interact with other essential cellular components thereby inhibiting their activities.
Nitrobenzylphosphoramide mustards are a novel class of compounds incorporating chemical motifs present in several DNA alkylating anti-cancer agents (30–33). They contain a nitrogen mustard moiety coupled to a nitrobenzyl ring via a phosphoramide linker and are designed to function as prodrugs specifically tailored to undergo activation in reactions catalyzed by type I NTRs. Here, we performed a structure-activity relationship on a library of NBPMs employing biochemical and leishmanicidal screens (Fig. 6 and Table 4). When using a group of NBPMs where the phosphoramide linker was part of a cyclic structure (Table 1), recombinant LmNTR protein was shown to metabolize some (3 out of 8) at reasonable rates, but this failed to translate into an anti-parasitic activity. Screens involving NBPMs, where the phosphoramide was part of a linearized linker (Table 2), yielded similar results for 6 out of 14 compounds with the remaining NBPMs being effective LmNTR substrates and displaying leishmanicidal effects. When comparing the anti-parasitic properties exhibited by the eight most potent NBPMs against the two replicative L. major life cycles stages, intracellular amastigote parasites were generally more susceptible to the agent under study than the promastigote form; LH33 had IC50 values of 5.88 ± 0.28 and 0.21 ± 0.07 μm against promastigote and amastigote form parasites, respectively, whereas LH34 exhibited values of 3.10 ± 0.28 and 0.77 ± 0.12 μm. To conclusively demonstrate that the LmNTR plays a role in NBPM activation in the parasite itself, the susceptibility of L. major LmNTR+/− heterozygous cells to LH33 and LH34 was evaluated. In both cases, reduction of the LmNTR gene copy number resulted in a resistance phenotype in agreement with observations made using recombinant trypanosomes and L. donovani toward other nitroaromatics (Fig. 6) (26, 48). As mammalian cells lack a type I NTR activity, they should be less susceptible to prodrugs that rely on this mechanism of activation. When the eight most potent leishmanicidal NBPMs were screened against the macrophage line in which the intracellular parasites were cultured, no cytotoxicity was observed at values up to 100 μm (Table 4). In terms of relative toxicity values, the two agents showing the highest potency against amastigote cells were >476 (LH33) and >143 (LH34) more toxic to the parasite than this particular mammalian line.
We have now shown that L. major expresses a type I NTR that in the related trypanosomal parasites activates the clinically used prodrugs nifurtimox and benznidazole (26, 49, 50). The leishmanial enzyme displays characteristics typical of this group of oxidoreductases being able to noncovalently bind with FMN and, through a ping-pong mechanism, utilizes NADH to reduce a wide range of nitroaromatics and quinones. We exploited this activity and identified several leishmanicidal NBPM compounds that have little/no cytotoxicity in mammalian cells. Based on substrate preference, the L. major type I NTRs can be regarded as NADH:quinone oxidoreductase, with this activity being important to the replicative parasite forms found in the insect and mammalian host. Therefore, our data indicate that the leishmanial enzyme can be used in drug development in two ways: either as an activator of new nitroaromatic/quinone-based prodrugs and/or through the use of inhibitors targeting this essential activity. Additionally, these two approaches could be complementary; an L. major strain showing resistance to drugs developed to inhibit LmNTR could theoretically be sensitive or even hypersensitive to prodrugs activated by this leishmanial enzyme and vice versa.
Footnotes
- NTR
- type I nitroreductase
- BLA
- blasticidin resistance gene cassette
- LmNTR
- L. major type I nitroreductase
- NBPM
- nitrobenzylphosphoramide mustard
- PAC
- puromycin resistance gene cassette
- TcNTR
- T. cruzi type I nitroreductase.
REFERENCES
- 1. Stuart K., Brun R., Croft S., Fairlamb A., Gürtler R. E., McKerrow J., Reed S., Tarleton R. (2008) Kinetoplastids: related protozoan pathogens, different diseases. J. Clin. Invest. 118, 1301–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cruz I., Morales M. A., Noguer I., Rodríguez A., Alvar J. (2002) Leishmania in discarded syringes from intravenous drug users. Lancet 359, 1124–1125 [DOI] [PubMed] [Google Scholar]
- 3. González C., Wang O., Strutz S. E., González-Salazar C., Sánchez-Cordero V., Sarkar S. (2010) Climate change and risk of leishmaniasis in North America: predictions from ecological niche models of vector and reservoir species. PLoS Negl. Trop. Dis. 4, e585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pavli A., Maltezou H. C. (2010) Leishmaniasis, an emerging infection in travelers. Int. J. Infect. Dis. 14, e1032–1039 [DOI] [PubMed] [Google Scholar]
- 5. Croft S. L., Sundar S., Fairlamb A. H. (2006) Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 19, 111–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Croft S. L., Olliaro P. (2011) Leishmaniasis chemotherapy–challenges and opportunities. Clin. Microbiol. Infect. 17, 1478–1483 [DOI] [PubMed] [Google Scholar]
- 7. Balasegaram M., Balasegaram S., Malvy D., Millet P. (2008) Neglected diseases in the news: a content analysis of recent international media coverage focussing on leishmaniasis and trypanosomiasis. PLoS Negl. Trop. Dis. 2, e234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grunberg E., Titsworth E. H. (1973) Chemotherapeutic properties of heterocyclic compounds: monocyclic compounds with five-membered rings. Annu. Rev. Microbiol. 27, 317–346 [DOI] [PubMed] [Google Scholar]
- 9. McCalla D. R. (1983) Mutagenicity of nitrofuran derivatives: review. Environ. Mutagen. 5, 745–765 [DOI] [PubMed] [Google Scholar]
- 10. Takahashi M., Iizuka S., Watanabe T., Yoshida M., Ando J., Wakabayashi K., Maekawa A. (2000) Possible mechanisms underlying mammary carcinogenesis in female Wistar rats by nitrofurazone. Cancer Lett. 156, 177–184 [DOI] [PubMed] [Google Scholar]
- 11. Hiraku Y., Sekine A., Nabeshi H., Midorikawa K., Murata M., Kumagai Y., Kawanishi S. (2004) Mechanism of carcinogenesis induced by a veterinary antimicrobial drug, nitrofurazone, via oxidative DNA damage and cell proliferation. Cancer Lett. 215, 141–150 [DOI] [PubMed] [Google Scholar]
- 12. Trunz B. B., Jędrysiak R., Tweats D., Brun R., Kaiser M., Suwiński J., Torreele E. (2011) 1-Aryl-4-nitro-1H-imidazoles, a new promising series for the treatment of human African trypanosomiasis. Eur. J. Med. Chem. 46, 1524–1535 [DOI] [PubMed] [Google Scholar]
- 13. Tweats D., Bourdin Trunz B., Torreele E. (2012) Genotoxicity profile of fexinidazole–a drug candidate in clinical development for human African trypanomiasis (sleeping sickness). Mutagenesis 27, 523–532 [DOI] [PubMed] [Google Scholar]
- 14. Matsumoto M., Hashizume H., Tomishige T., Kawasaki M., Tsubouchi H., Sasaki H., Shimokawa Y., Komatsu M. (2006) OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 3, e466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arya S. C., Agarwal N. (2009) Nitrofurantoin: the return of an old friend in the wake of growing resistance. BJU Int. 103, 994–995 [DOI] [PubMed] [Google Scholar]
- 16. Stover C. K., Warrener P., VanDevanter D. R., Sherman D. R., Arain T. M., Langhorne M. H., Anderson S. W., Towell J. A., Yuan Y., McMurray D. N., Kreiswirth B. N., Barry C. E., Baker W. R. (2000) A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405, 962–966 [DOI] [PubMed] [Google Scholar]
- 17. Adagu I. S., Nolder D., Warhurst D. C., Rossignol J.-F. (2002) In vitro activity of nitazoxanide and related compounds against isolates of Giardia intestinalis, Entamoeba histolytica, and Trichomonas vaginalis. J. Antimicrob. Chemother. 49, 103–111 [DOI] [PubMed] [Google Scholar]
- 18. Korba B. E., Montero A. B., Farrar K., Gaye K., Mukerjee S., Ayers M. S., Rossignol J. F. (2008) Nitazoxanide, tizoxanide, and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antiviral Res. 77, 56–63 [DOI] [PubMed] [Google Scholar]
- 19. Kaiser M., Bray M. A., Cal M., Bourdin Trunz B., Torreele E., Brun R. (2011) Antitrypanosomal activity of fexinidazole, a new oral nitroimidazole drug candidate for treatment of sleeping sickness. Antimicrob. Agents Chemother. 55, 5602–5608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wyllie S., Patterson S., Stojanovski L., Simeons F. R., Norval S., Kime R., Read K. D., Fairlamb A. H. (2012) The anti-trypanosome drug fexinidazole shows potential for treating visceral leishmaniasis. Sci. Transl. Med. 4, 119re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Denny W. A. (2003) Prodrugs for gene-directed enzyme-prodrug therapy (Suicide Gene Therapy). J. Biomed. Biotechnol. 2003, 48–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Saulnier Sholler G. L., Kalkunte S., Greenlaw C., McCarten K., Forman E. (2006) Antitumor activity of nifurtimox observed in a patient with neuroblastoma. J. Pediatr. Hematol. Oncol. 28, 693–695 [DOI] [PubMed] [Google Scholar]
- 23. Chen Y., Hu L. (2009) Design of anticancer prodrugs for reductive activation. Med. Res. Rev. 29, 29–64 [DOI] [PubMed] [Google Scholar]
- 24. Peterson F. J., Mason R. P., Hovsepian J., Holtzman J. L. (1979) Oxygen-sensitive and -insensitive nitroreduction by Escherichia coli and rat hepatic microsomes. J. Biol. Chem. 254, 4009–4014 [PubMed] [Google Scholar]
- 25. Moreno S. N., Mason R. P., Docampo R. (1984) Reduction of nifurtimox and nitrofurantoin to free radical metabolites by rat liver mitochondria. Evidence of an outer membrane-located nitroreductase. J. Biol. Chem. 259, 6298–6305 [PubMed] [Google Scholar]
- 26. Wilkinson S. R., Taylor M. C., Horn D., Kelly J. M., Cheeseman I. (2008) A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc. Natl. Acad. Sci. U.S.A. 105, 5022–5027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pal D., Banerjee S., Cui J., Schwartz A., Ghosh S. K., Samuelson J. (2009) Giardia, Entamoeba, and Trichomonas enzymes activate metronidazole (nitroreductases) and inactivate metronidazole (nitroimidazole reductases). Antimicrob. Agents Chemother. 53, 458–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. de Oliveira I. M., Henriques J. A., Bonatto D. (2007) In silico identification of a new group of specific bacterial and fungal nitroreductases-like proteins. Biochem. Biophys. Res. Commun. 355, 919–925 [DOI] [PubMed] [Google Scholar]
- 29. Chen S., Knox R., Wu K., Deng P. S., Zhou D., Bianchet M. A., Amzel L. M. (1997) Molecular basis of the catalytic differences among DT-diaphorase of human, rat, and mouse. J. Biol. Chem. 272, 1437–1439 [DOI] [PubMed] [Google Scholar]
- 30. Hu L., Yu C., Jiang Y., Han J., Li Z., Browne P., Race P. R., Knox R. J., Searle P. F., Hyde E. I. (2003) Nitroaryl phosphoramides as novel prodrugs for E. coli nitroreductase activation in enzyme prodrug therapy. J. Med. Chem. 46, 4818–4821 [DOI] [PubMed] [Google Scholar]
- 31. Jiang Y., Han J., Yu C., Vass S. O., Searle P. F., Browne P., Knox R. J., Hu L. (2006) Design, synthesis, and biological evaluation of cyclic and acyclic nitrobenzylphosphoramide mustards for E. coli nitroreductase activation. J. Med. Chem. 49, 4333–4343 [DOI] [PubMed] [Google Scholar]
- 32. Jiang Y., Hu L. (2008) N-(2,2-Dimethyl-2-(2-nitrophenyl)acetyl)-4-aminocyclophosphamide as a potential bioreductively activated prodrug of phosphoramide mustard. Bioorg. Med. Chem. Lett. 18, 4059–4063 [DOI] [PubMed] [Google Scholar]
- 33. Hu L., Wu X., Han J., Chen L., Vass S. O., Browne P., Hall B. S., Bot C., Gobalakrishnapillai V., Searle P. F., Knox R. J., Wilkinson S. R. (2011) Synthesis and structure-activity relationships of nitrobenzyl phosphoramide mustards as nitroreductase-activated prodrugs. Bioorg. Med. Chem. Lett. 21, 3986–3991 [DOI] [PubMed] [Google Scholar]
- 34. da Silva R., Sacks D. L. (1987) Metacyclogenesis is a major determinant of Leishmania promastigote virulence and attenuation. Infect. Immun. 55, 2802–2806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rovera G., O'Brien T. G., Diamond L. (1979) Induction of differentiation in human promyelocytic leukemia cells by tumor promoters. Science 204, 868–870 [DOI] [PubMed] [Google Scholar]
- 36. Hall B. S., Wu X., Hu L., Wilkinson S. R. (2010) Exploiting the drug-activating properties of a novel trypanosomal nitroreductase. Antimicrob. Agents Chemother. 54, 1193–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Race P. R., Lovering A. L., White S. A., Grove J. I., Searle P. F., Wrighton C. W., Hyde E. I. (2007) Kinetic and structural characterisation of Escherichia coli nitroreductase mutants showing improved efficacy for the prodrug substrate CB1954. J. Mol. Biol. 368, 481–492 [DOI] [PubMed] [Google Scholar]
- 38. Faeder E. J., Siegel L. M. (1973) A rapid micromethod for determination of FMN and FAD in mixtures. Anal. Biochem. 53, 332–336 [DOI] [PubMed] [Google Scholar]
- 39. Bryant C., DeLuca M. (1991) Purification and characterization of an oxygen-insensitive NAD(P)H nitroreductase from Enterobacter cloacae. J. Biol. Chem. 266, 4119–4125 [PubMed] [Google Scholar]
- 40. Zenno S., Koike H., Tanokura M., Saigo K. (1996) Gene cloning, purification, and characterization of NfsB, a minor oxygen-insensitive nitroreductase from Escherichia coli, similar in biochemical properties to FRase I, the major flavin reductase in Vibrio fischeri. J. Biochem. 120, 736–744 [DOI] [PubMed] [Google Scholar]
- 41. Watanabe M., Nishino T., Takio K., Sofuni T., Nohmi T. (1998) Purification and characterization of wild-type and mutant “classical” nitroreductases of Salmonella typhimurium. L33R mutation greatly diminishes binding of FMN to the nitroreductase of S. typhimurium. J. Biol. Chem. 273, 23922–23928 [DOI] [PubMed] [Google Scholar]
- 42. Hall B. S., Meredith E. L., Wilkinson S. R. (2012) Targeting the substrate preference of a type I nitroreductase to develop antitrypanosomal quinone-based prodrugs. Antimicrob. Agents Chemother. 56, 5821–5830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Charest H., Zhang W. W., Matlashewski G. (1996) The developmental expression of Leishmania donovani A2 amastigote-specific genes is post-transcriptionally mediated and involves elements located in the 3′-untranslated region. J. Biol. Chem. 271, 17081–17090 [DOI] [PubMed] [Google Scholar]
- 44. Hart D. T., Vickerman K., Coombs G. H. (1981) Respiration of Leishmania mexicana amastigotes and promastigotes. Mol. Biochem. Parasitol. 4, 39–51 [DOI] [PubMed] [Google Scholar]
- 45. Santhamma K. R., Bhaduri A. (1995) Characterization of the respiratory chain of Leishmania donovani promastigotes. Mol. Biochem. Parasitol. 75, 43–53 [DOI] [PubMed] [Google Scholar]
- 46. Van Hellemond J. J., Tielens A. G. (1997) Inhibition of the respiratory chain results in a reversible metabolic arrest in Leishmania promastigote. Mol. Biochem. Parasitol. 85, 135–138 [DOI] [PubMed] [Google Scholar]
- 47. Opperdoes F. R., Michels P. A. (2008) Complex I of Trypanosomatidae: does it exist? Trends Parasitol. 24, 310–317 [DOI] [PubMed] [Google Scholar]
- 48. Wyllie S., Patterson S., Fairlamb A. H. (2013) Assessing the essentiality of Leishmania donovani nitroreductase and its role in nitro drug activation. Antimicrob. Agents Chemother. 57, 901–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hall B. S., Bot C., Wilkinson S. R. (2011) Nifurtimox activation by trypanosomal type I nitroreductases generates cytotoxic nitrile metabolites. J. Biol. Chem. 286, 13088–13095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hall B. S., Wilkinson S. R. (2012) Activation of benznidazole by trypanosomal type I nitroreductases results in glyoxal formation. Antimicrob. Agents Chemother. 56, 115–123 [DOI] [PMC free article] [PubMed] [Google Scholar]