

Background: The prion domain (PrD) of Sup35p can aggregate to form the [PSI+] prion.
Results: Introduction of charged lysine residues (sup35KK) in the Sup35p PrD alters prion properties.
Conclusion: Some sup35KK alleles lead to the formation of new prion variants.
Significance: Establishment of molecular interactions influencing [PSI+] prion stability and maintenance is a step toward an understanding of prion folding.
Keywords: Amyloid, Prions, Protein Misfolding, Protein Structure, Translation Release Factors, Saccharomyces cerevisiae, [PSI+], Superpleated β-Structure, Translation Termination
Abstract
Recent studies have shown that Sup35p prion fibrils probably have a parallel in-register β-structure. However, the part(s) of the N-domain critical for fibril formation and maintenance of the [PSI+] phenotype remains unclear. Here we designed a set of five SUP35 mutant alleles (sup35KK) with lysine substitutions in each of five N-domain repeats, and investigated their effect on infectivity and ability of corresponding proteins to aggregate and coaggregate with wild type Sup35p in the [PSI+] strain. Alleles sup35-M1 (Y46K/Q47K) and sup35-M2 (Q61K/Q62K) led to prion loss, whereas sup35-M3 (Q70K/Q71K), sup35-M4 (Q80K/Q81K), and sup35-M5 (Q89K/Q90K) were able to maintain the [PSI+] prion. This suggests that the critical part of the parallel in-register β-structure for the studied [PSI+] prion variant lies in the first 63–69 residues. Our study also reveals an unexpected interplay between the wild type Sup35p and proteins expressed from the sup35KK alleles during prionization. Both Sup35-M1p and Sup35-M2p coaggregated with Sup35p, but only sup35-M2 led to prion loss in a dominant manner. We suggest that in the fibrils, Sup35p can bind to Sup35-M1p in the same conformation, whereas Sup35-M2p only allowed the Sup35p conformation that leads to the non-heritable fold. Mutations sup35-M4 and sup35-M5 influence the structure of the prion forming region to a lesser extent, and can lead to the formation of new prion variants.
Introduction
The devastating symptoms of transmissible fatal spongiform encephalopathies or infectious prion diseases are caused by an aberrant conformational change of the normally innocuous cellular prion protein PrP, leading to its deposition in the form of amyloid fibrils (1). Apart from the mammalian PrP3 protein, clear evidence for prions has only been found in fungal systems (2), with over 10 prions identified in Saccharomyces cerevisiae (3, 4). One of the most studied prions to date is [PSI+], which arises as a consequence of prionization of yeast Sup35p (translation termination factor eRF3). In the non-prion state, Sup35p is essential for robust translation termination. Cells bearing [PSI+] have a reduced pool of soluble Sup35p, which leads to increased read through of stop codons during translation (5, 6).
The Sup35 protein consists of three distinct functional domains (Fig. 1A). The C-proximal region has significant homology with the translation elongation factor eEF1-A, it is required and sufficient for translation termination and cell viability (7). The N-proximal region, or prion-forming domain, is uniformly rich in Gln and Asn residues and contains six oligonucleotide repeats (8). This region is not essential for viability or termination (9), but is required for [PSI+] induction (10) and propagation (11). The charged middle (M) region is not required for viability or termination, but can influence [PSI+] induction or propagation because of interaction with the protein disaggregase Hsp104p (12).
FIGURE 1.
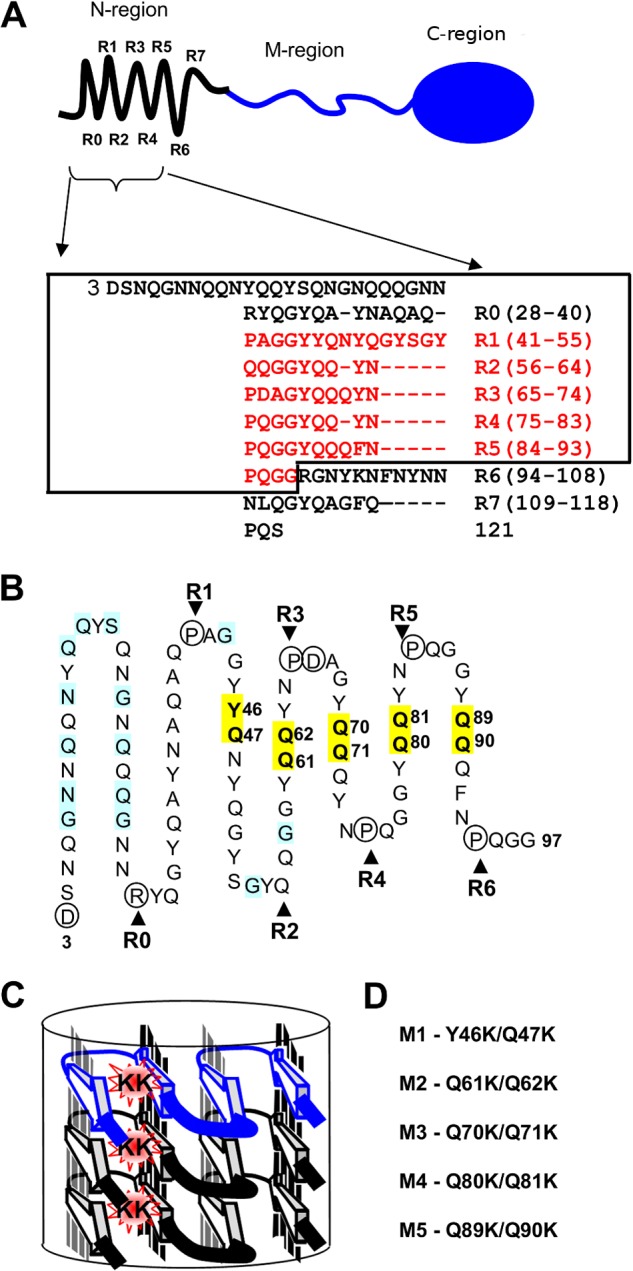
Localization of mutated Sup35 residues regarding the structural model of the N-terminal domain. A, top, schematic representation of Sup35p domain organization: the N-terminal region (1–123 amino acids) is shown with an array of eight repeats (R0–R7), discovered with the T-REKS program, the M-region (124–245 amino acids), C-terminal region (246–685 amino acids) has a globular-fold of the GTP-binding domain essential for robust translation termination. Bottom, new assignment of the Sup35p tandem repeats (T-REKS), which includes a previous one (8) colored in red. B, sequence of the N-terminal domain (the rectangle within the panel A) arranged in a serpentine with linear regions and bends, corresponding to putative β-strands and loop regions, respectively. The circled residues tend to localize to the loop regions (proline and charged residues). Positions where substitutions with charged residues lead to the PNM phenotype in previous reports (19, 36, 48) are highlighted in blue. Residues highlighted in yellow are mutated to lysines in this study. C, a model for the super-pleated β-structure that demonstrates how the introduction of charged residues in the β-strand region destabilizes the fibril by the electrostatic repulsion of same-sign charges along the fibril axis. D, sup35 mutations studied in this work (numbering of the mutations corresponds to the repeat number shown above).
Over the past decade, substantial progress has been made toward elucidating the structural arrangements of the prion fibrils of Sup35p. Their overall molecular architecture represents an approximate 8-nm wide backbone fibril of the N-region, which is surrounded by a diffuse 65-nm wide cloud of globular C-domains connected to the fibril by the extended M-region (13). Two types of structural model of the N-region fibrils have been proposed, based on experimental data: a β-helical model (14, 15) and a superpleated β-structure model (16). Current data favor the superpleated β-structure model (17, 18). In this structure, each polypeptide chain zig-zags in a planar serpentine-fold, and successive chains are stacked in register, one on top of another (Fig. 1B). This arrangement generates an array of elongated parallel β-sheets, each composed of identical strands and aligned with the fibril axis. The strands form a so-called cross-β structure that runs perpendicular to the filament axis.
Despite progress in understanding the structural arrangement of the Sup35p fibrils, it is still unclear which part(s) of the N-domain of full-length protein is critical for fibril formation. Mutational analysis of the Sup35 protein (substitutions with proline, insertions of glycine, or deletions of the repeats) has shown that only the first half of the N-domain may be required for [PSI+] phenotype and fibril formation (19–22). Region 38–56 amino acids appears to be essential for the incorporation of a polypeptide into a growing amyloid fibril (20). Similarly to mammalian PrP, Sup35p can adopt multiple conformational states of the aggregates or “strains,” which differ in phenotypic or biochemical characteristics (10). Species barrier or interference between different prion strains have been reported for both mammalian and yeast prions (23–25). For most prion strains characterized to date, the first half of the N-domain is either involved in formation of the β-structure (22, 26, 27) or critical for the infectivity of amyloid fibrils (22). On the other hand, other studies suggested that in addition to this region, a downstream region (amino acids 102–107) can also initiate amyloid formation (26, 27). Furthermore, the solid-state NMR data suggest that almost the entire N-domain and even part of the M-domain have an in-register parallel β-structure (28).
Thus, a complete understanding of the relationship between the N-domain sequence and its ability to form fibrils and maintain the [PSI+] phenotype is still lacking. To address this question, here we designed a set of alleles encoding Sup35p with substitutions in the N-domain and probed their effect on the propagation of [PSI+].
EXPERIMENTAL PROCEDURES
Strains, Media, and Growth Condition
S. cerevisiae strain 7A-D832 (Matα ade1–14 his7–1 leu2 lys2 trp1 ura3 sup35Δ::TRP1 [pYCH-U2-SUP35] [psi−] [PIN+] and its isogenic derivative bearing the strong variant of [PSI+] 10-7A-D832 (both generous gifts of A. Borchsenius) were used in this study. Both strains contain the sup35Δ::TRP1 knock-out on the chromosome, compensated by plasmids bearing the SUP35 gene.
Yeast cultures were maintained on YPD (yeast extract/peptone/dextrose medium) or on synthetic complete minimal medium (SC) (29) at 30 °C. Solid media were prepared with the addition of agar (2.5%). YPD medium containing 4 mm guanidine hydrochloride (GdnHCl) was used to eliminate [PSI+] (30). We utilized solid SC media with 5-fluoroorotic acid (1 mg/ml) for negative selection of plasmids bearing URA3 (31). Nonsense suppression induced in [PSI+] cells was detected by the ability to grow on SC medium lacking adenine (SC-Ade). Suppression was also scored by the color test on 1/4 YEPD (32) or MD 1/5Ade; this variant of MD medium (33) contained all the appropriate auxotrophic supplements and adenine at 4 mg/liters. [PSI+] cells suppressing the ade1–14 allele were white in color, whereas [psi−] cells accumulated red pigment.
Plasmids
Plasmids bearing sup35 mutations were constructed by subcloning the corresponding PCR products in centromeric plasmids pRSU1 (LEU2 SUP35) and pRSU2 (URA3 SUP35) (34), which express SUP35 from its own promoter. Point mutations were generated using the fusion PCR approach (35), primer sequences are available upon request. The final PCR product was cloned into pRSU1 or pRSU2 digested using HindIII. To obtain pRSU1-PNM2 or pRSU2-PNM2 the HindIII fragment of pSM128 (36) was subcloned in the corresponding sites of pRSU1 or pRSU2. The centromeric URA3 plasmid CEN-GAL-SUP35 contains the complete S. cerevisiae SUP35 gene under control of the GAL promoter (10). We obtained derivatives of this vector bearing mutant alleles by site-directed mutagenesis using highly processive DNA polymerase (AccuPrime Pfx, Invitrogen). Primer sequences are available upon request. Products of amplifications were digested by DpnI and transformed in Escherichia coli competent cells. All mutations were verified by sequencing. Centromeric plasmid pRS315 encoded Sup35p fused with the 3-HA tag (37) was the generous gift of M.D. Ter-Avanesyan.
Genetic and Microbiological Procedures
Yeast transformation was performed by the lithium acetate protocol (38).
Plasmid Shuffle
Direct plasmid shuffle (from wild type to mutant allele) was performed as follows: the [PSI+] sup35Δ strain with the SUP35 gene on a URA3 plasmid was transformed with LEU2 plasmids bearing the wild type or mutant SUP35 allele. Transformants, selected on a uracil- and leucine-omitted SC medium, were tested for suppression of the ade1–14 mutation to determine the presence of [PSI+]. Then transformants were streaked out on YPD media to allow for spontaneous loss of plasmids. Colonies were replica plated on SC-Leu or SC-Ura medium to identify which had lost one of the plasmids. The suppressor phenotype of strains obtained was analyzed on SC-Ade or 1/4 YEPD. Reverse shuffle (from mutant to wild type allele) was performed in a similar way.
[PSI+] Loss and Transmission
[PSI+] curing and transmission were scored according to the previously described procedure (37) with minor modifications. The [PSI+] sup35Δ [SUP35 URA3] strain was transformed with a LEU2 plasmid bearing the SUP35 or its mutant allele. To estimate [PSI+] curing, due to the presence of the mutated sup35 variant, three transformants for each combination of SUP35 and sup35KK alleles (sup35 double mutations with lysines substituted for two consecutive residues in the equivalent positions of Sup35p repeats) were passaged three times on a uracil-omitted medium, then suspended in water and plated on MD 1/5 Ade medium to obtain single colonies. The fraction of Ura+ Leu− [PSI+] colonies was estimated by a color test and replica plating to SC-Leu and SC-Ura.
To determine the efficiency of [PSI+] transmission from Sup35p to mutant Sup35 proteins, 50 transformants for each combination of WT (wild type) SUP35 and sup35KK alleles were passaged on leucine-omitted medium with uracil to enable the cells to lose plasmid coding for wild type SUP35. To estimate the efficiency of [PSI+] transmission, Ade+ colonies were scored among Ura−Leu+ isolates.
[PSI+] Induction
The plate assays for [PSI+] induction were performed as described previously (10). Yeast transformants bearing plasmid CEN-GAL-SUP35, or CEN-GAL with mutant variants of SUP35, were grown on glucose-Ura medium and replica plated onto -Ura and -Ura/Gal media. After 3 to 4 days of incubation, each plate was replica plated onto glucose -Ade medium. [PSI+] induction was detected as heterogeneous growth on -Ade medium after 7 to 10 days of incubation.
Prion Loss in Exponentially Growing Cultures
For quantitative estimation of [PSI+] elimination, the sup35Δ [SUP35 LEU2] strain was transformed by URA3 plasmids bearing sup35-M2, sup35-M3, or PNM2 mutations. Transformants were grown in SC-Leu-Ura liquid medium, which allowed selection for both plasmids. The culture was diluted at 12-h intervals with fresh medium to ensure exponential growth. Cell concentration was estimated by A600. Equivalent amounts of cells were periodically plated on 5-FOA (5-fluoroorotic acid) solid medium to select for cells that lost plasmid with the sup35KK allele. The fraction of [PSI+] colonies was estimated by a color test after replica plating on MD 1/5 Ade medium. The kinetics of [PSI+] elimination by GdnHCl treatment was estimated according to the previously published procedure (32). Each experiment was performed three times.
β-Gal Assay
The β-galactosidase reporter system was used for quantitative characterization of nonsense suppression (39). Vectors utilized in this assay were the generous gift of S. Peltz and W. Wang. The efficiency of suppression was calculated as the ratio of β-galactosidase activity in cells harboring lacZ with the premature termination codon to that in cells harboring lacZ (WT). Values for liquid β-galactosidase assay represent the mean of five independent assays. For statistical analysis the Mann-Whitney U test was performed (40).
Protein Analysis
Sup35p amounts in different strains were quantified using the Western blotting procedure with rabbit polyclonal anti-eRF3 antibodies (41). Monoclonal anti-tubulin antibodies (T6074, Sigma) were used for tubulin detection. Densitometry measurements were performed in ImageJ software. The Mann-Whitney U test (40) was performed for statistical analysis. SDD-PAGE with additional boiling (42) was performed to detect Sup35–3HAp and Sup35p in the aggregated and soluble fractions. For the analysis of Sup35p amyloid aggregates, semidenaturing detergent-agarose gel electrophoresis (SDD-AGE) (43, 44) was used.
RESULTS
Rationale Behind the Design of Mutations in the N-region of Sup35p
We used the T-REKS program for identification of tandem repeats in sequences (45) and detected eight repeats (R0–R7) between residues 28 and 118, instead of the previously identified six repeats spanning the region of amino acids 41–97 (8) (Fig. 1A). To select a set of mutations, we assumed that the Sup35p fibrils have the parallel and in-register stack of β-serpentines. Taking into consideration the repetitive sequence pattern and the fact that proline and charged side chains are preferentially located in the loops, we derived a β-serpentine spanning the entire N-domain with putative β-strands and bends (Fig. 1B). The interior of the fibril may accommodate both apolar residues and uncharged polar Asn and Gln engaged in H-bonded ladders, as well as Ser, Thr, and Cys, interacting with each other and/or with the polypeptide backbone (46, 47). In contrast, charged residues (Lys, Arg, Glu, or Asp) that are not involved in the salt bridges can destabilize a parallel in-register stacking of β-strands because of electrostatic repulsion of the juxtaposed charged residues of the same sign that are placed one over another along the fibril (Fig. 1C). This repulsion is particularly strong if such a charged axial ladder is located inside the fibril. Therefore, the substitution of uncharged residues for charged ones in the region that forms the parallel and in-register β-structure could inhibit fibril formation and may affect the [PSI+] phenotype. In the alternative β-solenoid model, the inserted charged residues will be further away from each other and the repulsive effect should be, in general, negligibly small. Importantly, the inhibitory effects of the other already tested mutations, such as deletions, insertions, or substitutions for proline residues (21, 22), are not specific to the parallel and in-register β-structures and can also be explained by the β-solenoid arrangements. Thus, the substitution of uncharged residues for charged residues appears to be the most suitable type of mutation for the establishment of boundaries of the parallel and in-register β-structures.
Previous reports have shown that many known SUP35 mutants with the PNM ([PSI+] no more) phenotype have neutral residues substituted for charged residues in the region upstream of the tandem repeats and within the first R1 and R2 repeats of Sup35p (Fig. 1B) (19, 36, 48). The parallel superpleated β-structure provides a straightforward explanation for this effect (Fig. 1C). Our goal was to test whether the effect of charged residues is also observed downstream of the R2 repeat. For this purpose, we constructed five double mutations with lysines substituted for two consecutive residues in the equivalent positions of Sup35p repeats R1–R5 (Fig. 1D). Two charged residues in each mutant were introduced to ensure the destabilization effect on the assumed parallel β-structure. Five mutant alleles (collectively termed the sup35KK alleles) were constructed as follows, with the numbering (M1–M5) corresponding to the mutated repeat (R1–R5): sup35-M1 (Y46K/Q47K), sup35-M2 (Q61K/Q62K), sup35-M3 (Q70K/Q71K), sup35-M4 (Q80K/Q81K), and sup35-M5 (Q89K/Q90K) (Fig. 1). A well characterized PNM2 mutation (G58D) (36, 49, 50) was used as a control for prion destabilization.
The Effect of sup35KK Alleles on [PSI+] Prion in the Presence of Wild Type SUP35
First, we sought to determine the effect of sup35KK alleles on the [PSI+] prion in the presence of wild type SUP35. The 10-7A-D832 (sup35Δ [PSI+]) strain that ectopically expresses wild type Sup35p (URA3 plasmid) was transformed with LEU2 plasmids containing either wild type SUP35 or one of the five sup35KK alleles. A LEU2 plasmid expressing the PNM2 allele was used as a control for prion destabilization. Leu+,Ura+ transformants were plated on media lacking adenine to test for suppression of ade1–14 (UGA), which occurs if [PSI+] is present. Eight transformants for each combination of plasmids were tested (except for PNM2, for which five transformants were studied) (Fig. 2A). Substitutions within the R2 repeat (sup35-M2) in half of the tested transformants, as well as the PNM2 mutation (in all transformants tested) abolished growth on adenine-deficient medium, indicating elimination of [PSI+] in these isolates (Fig. 2B). Thus, in some transformants the sup35-M2 allele can cause an effective [PSI+] loss in a dominant manner. In contrast, other sup35KK alleles (sup35-M1, sup35-M3, sup35-M4, and sup35-M5) failed to eliminate the nonsense suppression phenotype in the presence of wild type SUP35.
FIGURE 2.

sup35-M2 and PNM2 eliminate [PSI+] in the presence of wild type SUP35. A, transformants of yeast strain 10-7A-D832 (sup35Δ [PSI+]) contain two plasmids: pYCH-U2 (URA3) with a wild type copy of SUP35, and pRSU1 (LEU2) with wild type or mutant alleles of SUP35 (schematic representation of plasmid combination shown at bottom). Eight transformants were tested for each combination (except for PNM2 where 5 transformants were tested). In the case of sup35-M2 the [PSI+]-dependent suppression of ade1–14 was eliminated in 4 of 8 transformants. B, five serial dilutions of the indicated transformants were spotted onto SC medium selective for both plasmids with or without adenine, the tested sup35 allele combinations are shown on the left. A typical Ade− transformant of sup35-M2 is shown. Yeast strain 7A-D832 (sup35Δ [psi−]) bearing two plasmids with a wild type copy of SUP35 was used as a negative control. C, 10-7A-D832 (sup35Δ) derivatives bearing pRSU1-SUP35 or pRSU1-sup35KK plasmids were analyzed by Western blotting with an anti-eRF3 antibody. The captions below the lanes indicate the sup35KK allele. The “ratio” represents the relative abundance of Sup35p in each mutant compared with the amount of tubulin.
To determine whether sup35KK alleles themselves have a nonsense suppression phenotype, we transformed an isogenic [psi−] strain (7A-D832) with plasmids bearing mutant alleles. Transformants failed to grow on media lacking adenine (data not shown). In addition, we measured steady-state levels of Sup35p in cells ectopically expressing the sup35KK alleles. No significant variation in Sup35p levels in the [psi−] strain was observed (Fig. 2C).
Effects of sup35KK Alleles on the [PSI+] Phenotype in the Homozygous State
To check [PSI+] propagation in the presence of mutant alleles alone, we selected clones that lost the LEU2 plasmid encoding wild type Sup35p. The nonsense suppression phenotype of the selected clones was tested on medium lacking adenine. We found that PNM2, sup35-M1, and sup35-M2 have a strong inhibitory effect on [PSI+] propagation. Each of these alleles was able to eliminate growth of the [PSI+] strain on adenine-deficient medium upon loss of wild type (Fig. 3A) in all colonies tested (Fig. 3B). A strong effect of sup35-M1 and sup35-M2 mutations was also evident for clones that retained the plasmid with the wild type SUP35 gene, as some of them also lost [PSI+] (Fig. 3B). Loss of [PSI+] was irreversible for sup35-M1, sup35-M2, and PNM2 alleles, as the growth of mutant strains on adenine-deficient medium was not restored after shuffling back the plasmid encoding wild type Sup35p (Fig. 3C).
FIGURE 3.

sup35KK alleles have the opposite effect on [PSI+] phenotype in the absence of wild type SUP35. A, five serial dilutions of 10-7A-D832 (sup35Δ) derivatives containing the pRSU1 plasmid with wild type or mutant alleles of SUP35. B, amount of [PSI+] and [psi−] isolates after the loss of one of the plasmids. C, isolates shown on panel A were retransformed with pRSU2 (SUP35 (WT)) with subsequent loss of the pRSU1 plasmid, one representative clone from 16 tested in each case is shown. In both panels A and C one representative transformant was spotted onto YPD and SC-Ade. Derivatives of 7A-D832 (sup35Δ [psi−]) containing the corresponding plasmids with a wild type copy of SUP35 were used as a negative control in panels A–C. Schematic representation of the plasmid shuffling assay is shown under panels A and C. Arrows indicate where the corresponding phenotype is demonstrated. D and E, read through efficiency in [PSI+] 10-7A-D832 derivatives bearing sup35-M4 mutation after direct (C) and reverse (D) shuffle measured by in vivo β-galactosidase activity assay. The efficiency of suppression was calculated as a ratio of β-galactosidase activity in cells harboring lacZ with premature termination codon to lacZ (WT) control. The results are from at least three separate experiments. Significant differences from wild type [PSI+] are shown by one or two asterisks (p < 0.05 and p < 0.01, respectively). F, SDS-PAGE with additional boiling of [PSI+] aggregates in 10-7A-D832 (sup35Δ) derivatives containing the pRSU1 plasmid with wild type SUP35 or sup35KK. The captions below the lanes indicate the sup35 allele. A schematic representation of the plasmid combination is shown near the panel. G, SDD-AGE analysis of Sup35p aggregates in transformants shown in panel A. The arrow marks the position of thyroglobulin (Tg, 670 kDa) in gel revealed by Coomassie staining.
Interestingly, the sup35-M4 allele alone led to an increase in nonsense suppression efficiency compared with the original [PSI+] strain. This was observed by the presence of white colonies in 1/4 YEPD medium (data not shown) and better growth on adenine-deficient medium compared with wild type SUP35 (Fig. 3A). Read through efficiency for UGA and UAA stop codons in strains bearing the sup35-M4 mutation was also significantly higher (p < 0.05) than that of the [PSI+] strain with wild type SUP35, as measured with the β-galactosidase assay. No difference was observed for the UAG codon (Fig. 3D). This strong [PSI+] phenotype was maintained after reverse shuffle (from sup35-M4 to SUP35) that was confirmed by better growth on adenine-deficient medium as compared with wild type SUP35 (Fig. 3B) and statistically significant read through efficiency for UGA and UAG stop codons (Fig. 3E). Suppression of the ade1–14 mutation in the presence of sup35-M3 was indistinguishable from the original [PSI+] strain (Fig. 3A), and was not changed after reverse shuffle (Fig. 3C). We did not find any significant difference in read through efficiency between the native [PSI+] strain and the [PSI+] variants supported by the sup35-M3 and sup35-M5 alleles (data not shown). These data suggest that the [PSI+] variant supported by the sup35-M4 allele is stronger than the initial [PSI+].
The sup35-M5 allele alone decreases the nonsense suppression efficiency, as observed from weak growth on adenine-deficient medium (Fig. 3A). Replacement of sup35-M5 by wild type SUP35 led to failure of growth on adenine-deficient medium (Fig. 3C). Among 17 independent isolates of the [PSI+]M5 (prion variants maintained by sup35-M5) strain tested after reverse shuffle, none retained the nonsense suppression phenotype, indicating the loss of [PSI+] due to the inability of wild type SUP35 to support the [PSI+]M5 prion isolate (data not shown).
We next sought to characterize [PSI+] aggregates in the presence of sup35KK alleles using a modified version of SDS-PAGE with additional boiling (42) and SDD-AGE (43, 44). Using the SDD-AGE approach, differences in the size of the aggregates among prion strain variants can be detected as changes in electrophoretic mobility in agarose gels. Prion aggregates from a strong [PSI+] strain migrate further into the gel than aggregates from a weak [PSI+] strain (43). Using both techniques, we found that the presence of sup35-M1 or sup35-M2 in the homozygous state led to a complete absence of prion aggregates, with Sup35p present only in the soluble fraction (Fig. 3F and data not shown). This finding is consistent with the lack of nonsense suppression phenotype in these strains, and provides additional support for the conclusion that sup35-M1 and sup35-M2 in the homozygous state lead to the elimination of [PSI+].
Mutant Sup35-M3, Sup35-M4, and Sup35-M5 proteins from corresponding ade1–14 suppressing cells were found in aggregates (Fig. 3G). There was no significant difference in the sizes of the Sup35-M3p and wild type Sup35p aggregates (Fig. 3G), whereas both the Sup35-M4p and Sup35-M5p aggregates migrated more slowly through the gel than [PSI+] aggregates from SUP35WT cells (Fig. 3G). In the case of sup35-M5, this observation is consistent with weak growth on the adenine-deficient medium (Fig. 3A), suggesting the presence of a “weak” [PSI+] variant in this strain. However, in regard to sup35-M4, this is inconsistent with the strong suppressor phenotype of this mutation (Fig. 3, A and D).
In summary, the absence of wild type SUP35, sup35-M1, and sup35-M2 mutations led to the elimination of both the nonsense suppression phenotype and Sup35p aggregates in yeast cells, indicating a complete cure of the [PSI+] prion. We did not observe any significant influence of sup35-M3 on [PSI+]-mediated phenotypes, whereas sup35-M4 increased the strength of nonsense suppression and formed larger aggregates. [PSI+] prion isolates in the presence of sup35-M5 were characterized by decreased strength of nonsense suppression of ade1–14 and by aggregates of higher molecular weight that could not be supported by wild type SUP35 upon loss of sup35-M5.
The Effects of sup35KK Alleles on Prion Transmission from [PSI+]WT to [PSI+]mut and [PSI+]WT Loss
The inability of strains bearing sup35-M1 and sup35-M2 alleles in the homozygous state to grow on medium without adenine may reflect the inability of the mutant protein to adopt prion conformation. To study this phenomenon, we employed methods previously used in the characterization of the species barrier for transmission of [PSI+] (37). We used the cells that ectopically express wild type SUP35 and sup35KK alleles from two independent vectors (Fig. 2A). We induced spontaneous loss of one of the two vectors by growing the cells in a medium selective for only one of the two plasmids. The nonsense suppressor phenotype observed upon loss of the wild type plasmid allowed us to estimate the transfer of the prion state from the wild type Sup35p to mutant protein. Concomitantly, loss of the plasmid with the sup35KK allele and a subsequent assessment of the percentage of [PSI+] cells allowed us to estimate the effect of the mutant allele on the stability of native [PSI+].
The prion state was transmitted with 100% efficiency from the wild type to mutant proteins Sup35-M3, Sup35-M4, and Sup35-M5 (Fig. 4A). PNM2 was characterized by a low capacity for prionization, because only 7.5% of the clones maintained the [PSI+] phenotype. Transfer of the [PSI+] state for the mutant proteins Sup35-M1 and Sup35-M2 was not observed, in accordance with the data shown in Fig. 3A.
FIGURE 4.

Influence of sup35KK alleles on [PSI+] transmission, loss, induction, and protein coaggregation. A, [PSI+] transmission from the wild type to the indicated sup35 allele. A fraction of cells that retained prions after loss of the wild type allele is shown on the graph. B, [PSI+] loss induced by transient expression of the sup35KK alleles. The fraction of cells that have lost prions after loss of the sup35KK allele is shown. Significant differences from control variant (WT) in A and B are shown by asterisks (p < 0.05). C, transient overexpression of sup35-M1 or sup35-M2 leads to [PSI+] induction. The appearance of prions was detected by growth on the adenine-omitted medium. Transformants of the 7A-D832 strain (SUP35 WT [psi−]) were incubated on -Ura/Gal medium and replica plated onto glucose -Ade medium. Plates were photographed after 5 days of incubation. D, cell lysates of 10-7A-D832 derivatives containing wild type Sup35p-HA together with mutant Sup35p were separated using SDS-PAGE with additional boiling. Blotted proteins were probed with anti-eRF3 antibody, which revealed Sup35p-HA and Sup35p migrating in monomeric and aggregate fractions. Schematic representations of experiments and the plasmid combinations are shown below each panel.
The transient presence of sup35-M2 caused destabilization of the native [PSI+] (Fig. 4B), and the PNM2 allele showed similar results, although to a lesser extent. The presence of sup35-M1, sup35-M3, sup35-M4, and sup35-M5 alleles did not affect the stability of the original [PSI+].
To test the influence of sup35-M1 and sup35-M2 alleles on de novo [PSI+] formation we overproduced wild type SUP35 and its mutant variants in [psi−] background (Fig. 4C). The de novo appearance of [PSI+] was detected in all transformants except for transformants with a control empty vector, indicating the potential ability of the sup35-M1 and sup35-M2 alleles to form prions.
To determine whether the mutant Sup35-M1 and Sup35-M2 proteins are found in aggregates in the presence of wild type Sup35p, we used SDS-PAGE with additional boiling (42). For these experiments we use the derivative of the 10-7A-D832 [PSI+] strain bearing SUP35-HA on a centromeric vector (37). This construction encodes wild type hemagglutinin-tagged Sup35p (Sup35WT-HAp), which allowed us to discriminate it from the mutant Sup35p via an increase in molecular weight. The strain was transformed with plasmids coding for Sup35-M1p or Sup35-M2p. Transformants were selected and analyzed for coaggregation of Sup35KKp with wild type Sup35p. We observed coaggregation of both Sup35-M1p and Sup35-M2p with Sup35WT-HAp, whereas the amount of monomers in both alleles was higher compared with the control variant (SUP35 in the homozygous state) (Fig. 4D).
Prion Loss in the Presence of sup35-M2 but Not sup35-M1 Depends on Generation Number
To obtain further insight into the molecular mechanisms of [PSI+] loss in the presence of sup35-M1 and sup35-M2 alleles, we determined the kinetics of loss of [PSI+] during cell division. GdnHCl treatment of the SUP35 WT [PSI+] strain was used for comparison. The [PSI+] sup35Δ strain ectopically expressing wild type Sup35p was transformed with plasmids bearing either sup35-M1 or sup35-M2 alleles. Transformants were grown in liquid medium selective for both plasmids, and cultures were maintained in the logarithmic growth phase by regular dilution with fresh medium. Equivalent amounts of cells were regularly plated onto solid medium with 5-FOA to select for cells that lost the URA3 plasmid carrying the sup35KK mutant allele. The obtained colonies were screened for [PSI+] by replica plating on MD 1/5 Ade medium. White, pink, and sectored colonies were scored as [PSI+], whereas red colonies were scored as [psi−]. This experiment allowed us to study the effect of the transient expression of selected sup35KK alleles on [PSI+] stability.
The sup35-M1 allele did not lead to an increase in red colonies upon loss of mutant allele expression compared with wild type SUP35 (Fig. 5A). This result is in agreement with our data showing that this allele does not lead to loss of [PSI+] in the heterozygous condition (Fig. 2). In control experiments, [PSI+] cells containing plasmids with wild type SUP35 were white, and the isogenic [psi−] cells remained red (data not shown).
FIGURE 5.

Kinetics of [PSI+] loss in the presence of sup35-M1 and sup35-M2 alleles. A, transformants of 10-7A-D832 bearing two plasmids (with SUP35WT and sup35KK) were grown in liquid medium selective for both plasmids. At regular time points, culture aliquots were taken and plated on 5-FOA solid medium (see ”Experimental Procedures“). 5-FOA resistant colonies were tested for the presence of [PSI+] by replica plating onto MD 1/5 Ade medium. The percentages of [PSI+] cells have been plotted as a function of generation number passed from the beginning of the experiment, which was estimated from measurements of A600. [PSI+] elimination in the presence of 4 mm GdnHCl was used as control. B, SDD-AGE analysis of [PSI+] aggregates in independent Ade+ isolates that have lost sup35-M2 after more than 50 generations of growth as in panel A (top panel). WT, wild type strain 10-7A-D832. The arrow marks the position of thyroglobulin (Tg, 670 kDa) in the gel as revealed by Coomassie staining. Ade+ phenotype of the same isolates (lower panel). Ten serial dilutions of one representative clone were spotted onto SC medium without adenine. Schematic representation of the plasmid shuffling assay is shown to the right of each panel.
Transient expression of sup35-M2 led to destabilization of the [PSI+], as evidenced by the scoring of 70% of cells as [psi−] after 20 generations (Fig. 5A). However, after this observation and until the 50th generation, we did not detect significant prion loss. One of the possible explanations for this phenomenon is that sup35-M2 may convert the original strong [PSI+] strain into a weaker version, characterized by reduced mitotic stability. To test this hypothesis, we maintained plasmids encoding wild type and mutant SUP35 in [PSI+] sup35Δ cells for more than 50 generations. Cells from seven independent isolates obtained on 5-FOA to eliminate the URA3 plasmid bearing the sup35-M2 allele did not display any red [psi−] colonies when subcloned on MD 1/5 Ade and did not differ in growth in Ade− medium from the wild type strain (Fig. 5B, lower panel). SDD-AGE analysis of Sup35p aggregates in these seven isolates revealed variation in their sizes (Fig. 5B, top panel). Thus, the sup35-M2 allele led to destabilization of the [PSI+], but did not cause complete elimination of [PSI+].
DISCUSSION
We constructed five mutant alleles of the SUP35 gene, each carrying a pair of positively charged lysine residues in equivalent positions in the five repeats, to uniformly cover the entire repeat-containing N-domain up to the C-terminal end of the putative prion-forming region (Fig. 1). We demonstrated that the sup35-M1 and sup35-M2 mutant alleles (amino acid positions 46–47 and 61–62) led to prion loss in direct shuffling experiments (Fig. 3A). The other alleles were able to maintain the [PSI+] prion with 100% efficiency (Fig. 4A). Introduction of the charged residues into the parallel in-register superpleated structure would destabilize the prion fibrils, whereas the charged residues placed outside of the prion-forming region would have no effect. This suggests that the critical part of the parallel in-register β-structure for the studied [PSI+] prion variant lies within the first 63–69 residues.
It is also noteworthy that both proteins Sup35-M1 and Sup35-M2 can induce prions when overproduced (Fig. 4C). This resembles the PNM2 (G58D) allele, which can also induce the de novo appearance of [PSI+] upon overexpression (50). Thus, the observed effects of corresponding alleles on [PSI+] propagation is not associated with a global influence on prion folding but rather interferes with a specific prion variant. As the dominant-negative effect of the sup35-M2 allele is similar to that of PNM2 (Figs. 2, A and B, and 3, A–C), this property could be explained by the increased sensitivity of Sup35-M2p and Sup35p heteroaggregates to Hsp104p as shown for the PNM2 allele coexpressed with wild type SUP35 (51) (see below).
At the same time, our study reveals a more complex interplay between the Sup35p wild type and mutant proteins. Mutant proteins Sup35-M1 and Sup35-M2 both coaggregate with Sup35p but lead to a transmission barrier in accordance with a previous observation where it was shown that the ability to coaggregate does not exclude the existence of a cross-species barrier (52). Prion transmission from wild type SUP35 to both sup35-M1 and sup35-M2 was not observed (Fig. 4A). We hypothesize that the mechanisms leading to this outcome are different for the two sup35KK alleles. In the case of sup35-M1, in addition to the inability to transmit prions from wild type to sup35-M1 allele, the transient expression of Sup35-M1p does not induce prion loss (Fig. 4B). Finally, Sup35-M1p was able to coaggregate with Sup35p (Fig. 4D). To explain these findings we hypothesized that the failure of prion transmission in the case of sup35-M1 is caused by the inability of the mutant protein to form homoaggregates due to electrostatic repulsion in the parallel and in-register β-structure (Fig. 6). This hypothesis is in accordance with data that residues 47 and 48 lie within one of the three consensus sequences of amyloid stretches (53). The sup35-M1 can maintain [PSI+] in the presence of the wild type SUP35 allele because Sup35-M1p molecules are required to be interspersed with Sup35p along the axis of the prion fibril so that positively charged lysine residues of Sup35-M1p are placed further away from each other. This arrangement does not significantly change prion conformation, as the loss of sup35-M1 expression does not induce a loss of [PSI+], suggesting that the conformation of the heteropolymer fibril is still compatible with maintenance by wild type Sup35p only.
FIGURE 6.
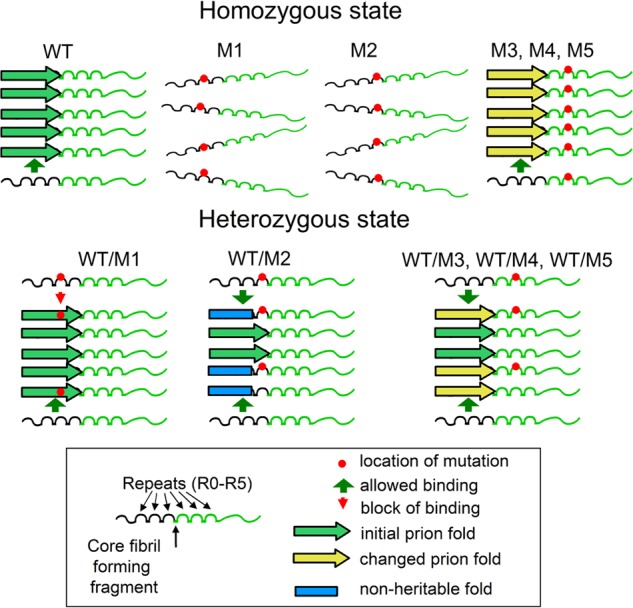
The model describing interactions of mutant Sup35p with prion filaments. Mutations sup35-M1 and sup35-M2 are located within the core region of the parallel and in-register superpleated β-structure. In the homozygous state, both Sup35-M1p and Sup35-M2p do not form fibrils. In the heterozygous state, Sup35-M1p and Sup35-M2p can coaggregate with Sup35p. Sup35-M1p does not affect the prion, whereas Sup35-M2p leads to conformational changes, formation of non-heritable fibril-folds, and hence loss of [PSI+] in the majority of cells. We suggest that in the case of Sup35-M1p, homoaggregates are not permitted. Thus in the growing fibril Sup35p and Sup35 are perfectly interspersed, one following the other along the axis of the prion filament. We speculate that regions mutated in Sup35-M3p, Sup35-M4p, and Sup35-M5p lie outside of the superpleated β-structure of Sup35p in the conformation of the prion strain utilized in this study. Hence these alleles cannot eliminate [PSI+] under the conditions tested.
The sup35-M2 mutant allele also could not maintain [PSI+] in the absence of wild type SUP35. However, in contrast to sup35-M1, it has a dominant-negative effect and eliminated [PSI+] even in the presence of wild type SUP35 in a large proportion of cells (Fig. 2). We show that prion elimination by Sup35-M2p depends on cell divisions (Fig. 5A). It is worth mentioning that prion elimination in the SUP35/sup35-M2 heterozygote was not complete, and ∼30% of cells maintained prion. [PSI+] maintained in these cells was stable in mitosis. Our data show evidence of heteroaggregation of Sup35-M2p and Sup35p (Fig. 4D). We speculate that only part of the prion-forming region of Sup35-M2p can bind aggregates of wild type protein and change β-structure arrangement (Fig. 6). The next protein molecule (mutant or wild-type) that joins the end of the fibril replicates the change made by Sup35-M2p in the β-structure. It is plausible that some aggregates (∼70%) acquired a non-heritable fold that led to prion loss. Evidence for such a conformational change comes from the observation of aggregates of varying size in cells that retain [PSI+] after the presence of sup35-M2 (Fig. 5B). Similar reasoning has been previously used to explain the mechanism of the Sup35p prion interspecies barrier (37). A minor fraction of the fibrils could preserve the prion-fold, thus maintaining the ability to persist through generations of cells.
Alternatively, prion loss could be explained by the terminal blocking of fibril growth, called “growth poisoning,” by mutant Sup35-M2p (37, 52, 53). Such a scenario implies a significant increase in the monomeric fraction of Sup35p; however, we observed only a mild increase in the soluble fraction when SUP35 and sup35-M2 were co-expressed in cells that managed to maintain [PSI+] (Fig. 4D). Another potential mechanism for prion loss could be due to Sup35-M2p and Sup35p heteroaggregates being more susceptible to Hsp104p disaggregase activity. This explanation was proposed for PNM2 mutation (51). Indeed, an optimal level of this chaperone activity is required to maintain [PSI+] (12, 54). If Hsp104p fails to act on heteroaggregates, the kinetics of prion loss should match those obtained under GdnHCl treatment (55), eventually leading to complete prion-free cultures. In the case of the SUP35/sup35-M2 heterozygote, 30% of cells contained prion stably maintained during cell divisions (Fig. 5A), thus rendering the hypothesis of reduced Hsp104p activity less plausible. Although we cannot rule out any of these scenarios, we favor our initial hypothesis, according to which Sup35-M2p induces a conformational switch in heteroaggregates (Fig. 6). Such a switch may lead, in most cases, to destabilization of prion structure and prion loss; however, some conformations can permit weak [PSI+] characterized by long aggregates that are stably inherited from one generation to the other.
The mutations sup35-M4 and sup35-M5 in the N-region downstream of sup35-M1 and sup35-M2 have some effect, albeit to a lesser extent. Alleles sup35-M4 and sup35-M5 increased and decreased the nonsense suppressor phenotype of [PSI+], respectively (Fig. 3A). Sup35p aggregates increased in size in the presence of sup35-M4 and sup35-M5 alleles (Fig. 3G). Mutant alleles also differed in the ability for reverse transmission of prion to the wild type allele. Indeed, the prion determinant was lost upon reverse shuffle of [PSI+] cells from sup35-M5 back to wild type (Fig. 3C). An analogous phenomenon was mentioned in the literature as asymmetry of cross-species prion transmission (52, 53). Hence, we conclude that the [PSI+] strain maintained with sup35-M5 differs substantially from the [PSI+] strain maintained with the wild type SUP35 allele. In our opinion, the observed asymmetry of the “cross-strain” barrier stems from the inability of the wild type Sup35p to efficiently adopt prion conformation of the Sup35-M5p fibril. Allele sup35-M4 leads to irreversible alteration of prion properties (Fig. 4, D and E). Analogous results were obtained previously during investigation of the inter-species barrier and it is noteworthy that the region responsible for changes in [PSI+] properties includes position of the sup35-M4 mutation (53).
We suggest that although sup35-M4 and sup35-M5 mutations are located outside of the core fibril-forming region and can maintain [PSI+], they may still cause rearrangement of the fibril structure. It is worth mentioning that the superpleated β-structure is more permissive to such polymorphisms compared with the other types of protein structures. This fold allows different locations and numbers of β-strands and turns within the same sequence. Structural variations of this kind offer a plausible explanation for the phenomenon of prion variants (22, 56). A similar hypothesis of the effect of amino acid substitutions in the PrP protein on prion fibril conformation was previously proposed by Chen and colleagues (57) in their in silico study. We propose that sup35-M4 and sup35-M5 lead to the formation of new prion variants. Analogous phenomena were previously described, for example, when the SUP35 WT allele was combined with one from another species or a mutant one (53, 58) and for the PrP prion when it was transmitted between species (23). Based on an analytical model of prion strain variations (59), we speculate that [PSI+]M5 has a decreased division rate. This assumption is in agreement with the increase in size of the weaker nonsense-suppression phenotype Sup35p aggregates. [PSI+]M4 (prion variants maintained by sup35-M4) possesses a stronger nonsense-suppression phenotype compared with [PSI+] in the 10-7A-D832 strain, although the prion aggregates are slightly increased in size. These data can be explained by the increased fibril growth rate.
Taken together, our results suggest that residues 46–47 (sup35-M1) and 61–62 (sup35-M2) are located within the parallel and in-register superpleated β-structure for the [PSI+] variant studied in this work. These mutants do not form homoaggregates and lead to prion loss (Fig. 6). However, they influence propagation of the Sup35p aggregates in a different manner. The sup35-M1 mutant does not affect the prion in the presence of SUP35, but sup35-M2 leads to prion loss in a dominant manner and the formation of non-heritable aggregates. To explain these data, we suggest that in the fibrils, Sup35p can bind to Sup35-M1p in the same conformation, whereas Sup35-M2p allowed the Sup35p conformation that leads to non-heritable fibrils (Fig. 6). The smaller effect of the mutations on sup35-M4 and sup35-M5 can be explained by their location within the parallel in-register β-structure but outside of its critical core region. These mutations have some influence on protein structure and can lead to the formation of new prion variants.
Acknowledgments
We are very grateful to A. Borchsenius and M. D. Ter-Avanesyan for plasmids and yeast strains, A. Masharsky for sequencing, E. Besedina and N. Trubicina for help in some experiments, A. Galkin and P. Drozdova for critical reading of the manuscript, Y. Chernoff and members of the Physiological Genetics laboratory of Saint Petersburg State University for fruitful discussions and a “Dynasty Foundation” for travel grant (to A.V.K.).
This work was supported in part by the Research Resource Center “Molecular and Cell Technologies” of Saint-Petersburg State University, Grant 10-04-00237 from the Russian Foundation of Basic Research, a grant from Russian Academy of Science program “The origin of life and the biosphere formation” (to S. A. B. and G. A. Z.), and The Ministry of Education and Science of the Russian Federation project Grant 8831 (to A. V. K.).
- PrP
- prion protein
- GdnHCl
- guanidine hydrochloride
- PNM
- [PSI+] no more mutation
- 5-FOA
- 5-fluoroorotic acid
- SDD-AGE
- semi-denaturing detergent-agarose gel electrophoresis.
REFERENCES
- 1. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wickner R. B. (1994) [URE3] as an altered URE2 protein. Evidence for a prion analog in Saccharomyces cerevisiae. Science 264, 566–569 [DOI] [PubMed] [Google Scholar]
- 3. Wickner R. B., Edskes H. K., Shewmaker F. P., Kryndushkin D., Nemecek J., McGlinchey R., Bateman D. (2010) The relationship of prions and translation. Wiley Interdiscip. Rev. RNA 1, 81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Crow E. T., Li L. (2011) Newly identified prions in budding yeast, and their possible functions. Semin. Cell Dev. Biol. 22, 452–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tuite M. F., Lindquist S. L. (1996) Maintenance and inheritance of yeast prions. Trends Genet. 12, 467–471 [DOI] [PubMed] [Google Scholar]
- 6. Ter-Avanesyan M. D., Kushnirov V. V. (1999) Prions. Infectious proteins with genetic properties. Biochemistry 64, 1382–1390 [PubMed] [Google Scholar]
- 7. Inge-Vechtomov S., Zhouravleva G., Philippe M. (2003) Eukaryotic release factors (eRFs) history. Biol. Cell 95, 195–209 [DOI] [PubMed] [Google Scholar]
- 8. Kushnirov V. V., Ter-Avanesyan M. D., Smirnov V. N. (1995) Structure and functional similarity of yeast Sup35p and Ure2p proteins to mammalian prions. Mol. Biol. (Mosk) 29, 750–755 [PubMed] [Google Scholar]
- 9. Ter-Avanesyan M. D., Kushnirov V. V., Dagkesamanskaya A. R., Didichenko S. A., Chernoff Y. O., Inge-Vechtomov S. G., Smirnov V. N. (1993) Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non-overlapping functional regions in the encoded protein. Mol. Microbiol. 7, 683–692 [DOI] [PubMed] [Google Scholar]
- 10. Derkatch I. L., Chernoff Y. O., Kushnirov V. V., Inge-Vechtomov S. G., Liebman S. W. (1996) Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 144, 1375–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ter-Avanesyan M. D., Dagkesamanskaya A. R., Kushnirov V. V., Smirnov V. N. (1994) The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics 137, 671–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Helsen C. W., Glover J. R. (2012) A new perspective on Hsp104-mediated propagation and curing of the yeast prion [PSI+]. Prion 6, 234–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baxa U., Keller P. W., Cheng N., Wall J. S., Steven A. C. (2011) In Sup35p filaments (the [PSI+] prion), the globular C-terminal domains are widely offset from the amyloid fibril backbone. Mol. Microbiol. 79, 523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kishimoto A., Hasegawa K., Suzuki H., Taguchi H., Namba K., Yoshida M. (2004) β-Helix is a likely core structure of yeast prion Sup35 amyloid fibers. Biochem. Biophys. Res. Commun. 315, 739–745 [DOI] [PubMed] [Google Scholar]
- 15. Krishnan R., Lindquist S. L. (2005) Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature 435, 765–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kajava A. V., Baxa U., Wickner R. B., Steven A. C. (2004) A model for Ure2p prion filaments and other amyloids. The parallel superpleated β-structure. Proc. Natl. Acad. Sci. U.S.A. 101, 7885–7890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baxa U., Cassese T., Kajava A. V., Steven A. C. (2006) Structure, function, and amyloidogenesis of fungal prions. Filament polymorphism and prion variants. Adv. Protein Chem. 73, 125–180 [DOI] [PubMed] [Google Scholar]
- 18. Shewmaker F., Wickner R. B., Tycko R. (2006) Amyloid of the prion domain of Sup35p has an in-register parallel β-sheet structure. Proc. Natl. Acad. Sci. U.S.A. 103, 19754–19759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. King C. Y. (2001) Supporting the structural basis of prion strains. Induction and identification of [PSI] variants. J. Mol. Biol. 307, 1247–1260 [DOI] [PubMed] [Google Scholar]
- 20. Osherovich L. Z., Cox B. S., Tuite M. F., Weissman J. S. (2004) Dissection and design of yeast prions. PLoS Biol. 2, E86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shkundina I. S., Kushnirov V. V., Tuite M. F., Ter-Avanesyan M. D. (2006) The role of the N-terminal oligopeptide repeats of the yeast Sup35 prion protein in propagation and transmission of prion variants. Genetics 172, 827–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chang H. Y., Lin J. Y., Lee H. C., Wang H. L., King C. Y. (2008) Strain-specific sequences required for yeast [PSI+] prion propagation. Proc. Natl. Acad. Sci. U.S.A. 105, 13345–13350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schutt C. R., Bartz J. C. (2008) Prion interference with multiple prion isolates. Prion 2, 61–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gonzalez Nelson A. C., Ross E. D. (2011) Interactions between non-identical prion proteins. Semin. Cell Dev. Biol. 22, 437–443 [DOI] [PubMed] [Google Scholar]
- 25. Sigurdson C. J., Bartz J. C., Nilsson K. P. (2011) Tracking protein aggregate interactions. Prion 5, 52–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pastor M. T., Esteras-Chopo A., Serrano L. (2007) Hacking the code of amyloid formation. The amyloid stretch hypothesis. Prion 1, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Toyama B. H., Kelly M. J., Gross J. D., Weissman J. S. (2007) The structural basis of yeast prion strain variants. Nature 449, 233–237 [DOI] [PubMed] [Google Scholar]
- 28. Shewmaker F., Kryndushkin D., Chen B., Tycko R., Wickner R. B. (2009) Two prion variants of Sup35p have in-register parallel β-sheet structures, independent of hydration. Biochemistry 48, 5074–5082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaiser C., Michaelis S., Mitchell A. (1994) Methods in Yeast Genetics. A Laboratory Course Manual, pp. 234, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 30. Tuite M. F., Mundy C. R., Cox B. S. (1981) Agents that cause a high frequency of genetic change from [psi+] to [psi−] in Saccharomyces cerevisiae. Genetics 98, 691–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boeke J. D., LaCroute F., Fink G. R. (1984) A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast. 5-Fluoro-orotic acid resistance. Mol. Gen. Genet. 197, 345–346 [DOI] [PubMed] [Google Scholar]
- 32. Eaglestone S. S., Ruddock L. W., Cox B. S., Tuite M. F. (2000) Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI+] of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 97, 240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Inge-Vechtomov S. G. (1974) Identification of certain linkage groups in Petergoff genetical lines of yeast. Sov. Genet. 7, 1190–1199 [PubMed] [Google Scholar]
- 34. Volkov K. V., Aksenova A. Y., Soom M. J., Osipov K. V., Svitin A. V., Kurischko C., Shkundina I. S., Ter-Avanesyan M. D., Inge-Vechtomov S. G., Mironova L. N. (2002) Novel non-mendelian determinant involved in the control of translation accuracy in Saccharomyces cerevisiae. Genetics 160, 25–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sambrook J., Fritsch E. F., Maniatis T. (1989). Molecular Cloning. A Laboratory Manual, pp. 2344, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 36. Doel S. M., McCready S. J., Nierras C. R., Cox B. S. (1994) The dominant PNM2− mutation which eliminates the psi factor of Saccharomyces cerevisiae is the result of a missense mutation in the SUP35 gene. Genetics 137, 659–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Afanasieva E. G., Kushnirov V. V., Tuite M. F., Ter-Avanesyan M. D. (2011) Molecular basis for transmission barrier and interference between closely related prion proteins in yeast. J. Biol. Chem. 286, 15773–15780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gietz R. D., Schiestl R. H., Willems A. R., Woods R. A. (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11, 355–360 [DOI] [PubMed] [Google Scholar]
- 39. Stansfield I., Akhmaloka, Tuite M. F. (1995) A mutant allele of the SUP45 (SAL4) gene of Saccharomyces cerevisiae shows temperature-dependent allosuppressor and omnipotent suppressor phenotypes. Curr. Genet. 27, 417–426 [DOI] [PubMed] [Google Scholar]
- 40. Mann H. B., Whitney D. R. (1947) On a test of whether one of two random variables is stochastically larger than the other in Annals of Mathematical Statistics, Vol. 18, 50–60 [Google Scholar]
- 41. Chabelskaya S., Kiktev D., Inge-Vechtomov S., Philippe M., Zhouravleva G. (2004) Nonsense mutations in the essential gene SUP35 of Saccharomyces cerevisiae are non-lethal. Mol. Genet. Genomics 272, 297–307 [DOI] [PubMed] [Google Scholar]
- 42. Kushnirov V. V., Alexandrov I. M., Mitkevich O. V., Shkundina I. S., Ter-Avanesyan M. D. (2006) Purification and analysis of prion and amyloid aggregates. Methods 39, 50–55 [DOI] [PubMed] [Google Scholar]
- 43. Kryndushkin D. S., Alexandrov I. M., Ter-Avanesyan M. D., Kushnirov V. V. (2003) Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 278, 49636–49643 [DOI] [PubMed] [Google Scholar]
- 44. Halfmann R., Lindquist S. (2008) Screening for amyloid aggregation by semi-denaturing detergent-agarose gel electrophoresis. J. Vis. Exp. 17, 838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jorda J., Kajava A. V. (2009) T-REKS. Identification of Tandem REpeats in sequences with a K-meanS based algorithm. Bioinformatics 25, 2632–2638 [DOI] [PubMed] [Google Scholar]
- 46. Hennetin J., Jullian B., Steven A. C., Kajava A. V. (2006) Standard conformations of β-arches in β-solenoid proteins. J. Mol. Biol. 358, 1094–1105 [DOI] [PubMed] [Google Scholar]
- 47. Kajava A. V., Steven A. C. (2006) β-Rolls, β-helices, and other β-solenoid proteins. Adv. Protein Chem. 73, 55–96 [DOI] [PubMed] [Google Scholar]
- 48. DePace A. H., Santoso A., Hillner P., Weissman J. S. (1998) A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 93, 1241–1252 [DOI] [PubMed] [Google Scholar]
- 49. Kochneva-Pervukhova N. V., Paushkin S. V., Kushnirov V. V., Cox B. S., Tuite M. F., Ter-Avanesyan M. D. (1998) Mechanism of inhibition of [PSI+] prion determinant propagation by a mutation of the N-terminus of the yeast Sup35 protein. EMBO J. 17, 5805–5810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Derkatch I. L., Bradley M. E., Zhou P., Liebman S. W. (1999) The PNM2 mutation in the prion protein domain of SUP35 has distinct effects on different variants of the [PSI+] prion in yeast. Curr. Genet. 35, 59–67 [DOI] [PubMed] [Google Scholar]
- 51. DiSalvo S., Derdowski A., Pezza J. A., Serio T. R. (2011) Dominant prion mutants induce curing through pathways that promote chaperone-mediated disaggregation. Nat. Struct. Mol. Biol. 18, 486–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen B., Newnam G. P., Chernoff Y. O. (2007) Prion species barrier between the closely related yeast proteins is detected despite coaggregation. Proc. Natl. Acad. Sci. U.S.A. 104, 2791–2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen B., Bruce K. L., Newnam G. P., Gyoneva S., Romanyuk A. V., Chernoff Y. O. (2010) Genetic and epigenetic control of the efficiency and fidelity of cross-species prion transmission. Mol. Microbiol. 76, 1483–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chernoff Y. O. (2007) Stress and prions. Lessons from the yeast model. FEBS Lett. 581, 3695–3701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ferreira P. C., Ness F., Edwards S. R., Cox B. S., Tuite M. F. (2001) The elimination of the yeast [PSI+] prion by guanidine hydrochloride is the result of Hsp104 inactivation. Mol. Microbiol. 40, 1357–1369 [DOI] [PubMed] [Google Scholar]
- 56. Kajava A. V., Baxa U., Steven A. C. (2010) Beta arcades. Recurring motifs in naturally occurring and disease-related amyloid fibrils. FASEB J. 24, 1311–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen W., van der Kamp M. W., Daggett V. (2010) Diverse effects on the native β-sheet of the human prion protein due to disease-associated mutations. Biochemistry 49, 9874–9881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lin J. Y., Liao T. Y., Lee H. C., King C. Y. (2011) Inter-allelic prion propagation reveals conformational relationships among a multitude of [PSI] strains. PLoS Genet. 7, e1002297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tanaka M., Weissman J. S. (2006) An efficient protein transformation protocol for introducing prions into yeast. Methods Enzymol. 412, 185–200 [DOI] [PubMed] [Google Scholar]