

Background: IL-4 can directly inhibit growth of several tumor cell types, but the molecular mechanism is not known.
Results: IL-4 induces senescence by increasing p21WAF1/CIP1 expression through STAT6 and p38 MAPK.
Conclusion: STAT6 and p38 MAPK play important roles in senescence induction by IL-4.
Significance: This is the first report of cellular senescence induction by IL-4 and the responsible mechanism.
Keywords: Interleukin, p38 MAPK, Renal Physiology, Senescence, STAT Transcription Factor, IL-4, Renal Cell Carcinoma, STAT6, p21WAF1/CIP1
Abstract
Interleukin (IL)-4, originally identified as a lymphocyte growth factor, can directly inhibit growth of certain tumor cell types. We reported previously that IL-4 induced cell cycle arrest in G1 phase through an increase in p21WAF1/CIP1 expression in human renal cell carcinoma (RCC) cell lines. In the present study, we investigated the underlying mechanism of IL-4-induced growth inhibition. In four of six human RCC cell lines, including Caki-1, A498, SNU482, and SNU228, IL-4 induced cellular senescence as demonstrated by enlarged and flattened morphology, increased granularity, and senescence-associated-β-galactosidase (SA-β-gal) staining. Signal tranducer and activator of transcription 6 (STAT6) and p38 MAPK were found to mediate IL-4-induced growth inhibition and cellular senescence. Both of these molecules were activated by 10 min after IL-4 treatment, and inhibition of their activity or expression prevented growth suppression and cellular senescence induced by IL-4. Inhibiting or silencing either STAT6 or p38 MAPK alone partially reduced the effect of IL-4, whereas inhibiting or silencing both molecules exerted an additive effect and almost completely abrogated the effect of IL-4. Thus STAT6 and p38 MAPK appeared to independently mediate IL-4-induced growth inhibition and cellular senescence. The p21WAF1/CIP1 up-regulation that accompanied growth inhibition and cellular senescence by IL-4 was also attenuated additively when p38 MAPK and STAT6 were silenced. Taken together, these results show that IL-4 induces cellular senescence through independent signaling pathways involving STAT6 and p38 MAPK in some human RCC cell lines.
Introduction
Cellular senescence is a complex phenotype that is characterized by irreversible cell cycle arrest, enlarged and flattened cellular morphology, up-regulation of senescence-associated-β-galactosidase (SA-β-gal)3 activity, and enhanced expression of cyclin-dependent kinase inhibitors such as p21WAF1/CIP1 and p16INK4A (1). Although cellular senescence contributes to the loss of tissue homeostasis in mammalian aging, it serves as a defense mechanism against cancer through irreversible cell cycle arrest (2, 3). Senescence can be induced by telomere erosion and other cellular stress such as telomere-independent DNA damage, oncogene activation, and oxidative stress. In addition, sustained signaling by certain anti-proliferative cytokines, such as interferon-β and transforming growth factor-β, may also induce cellular senescence (4, 5). However, IL-4 has not been shown to induce cellular senescence.
IL-4 is a pleiotropic cytokine with roles in proliferation, differentiation, and apoptosis in T and B lymphocytes and other cell types including non-hematopoietic cells (6, 7). Cellular responses to IL-4 are mediated by two different types of IL-4 receptor complex (8–10). The type I receptor, composed of IL-4Rα and the common γ (γc) chain, is expressed in hematopoietic cells, whereas the type II receptor, comprising the IL-4Rα and IL-13Rα1 chains, is expressed in non-hematopoietic cells. IL-4 binds to the IL-4Rα chain in both type I and type II receptors, and this binding induces IL-4Rα to dimerize with either the γc or the IL-13Rα1 chain to initiate activation of the Janus tyrosine kinase (JAK) family members. Activated Janus kinases phosphorylate tyrosine residues in the IL-4Rα chain, and these phosphotyrosine residues act as docking sites for downstream signaling molecules. IL-4 binding to IL-4R triggers at least two distinct signaling pathways, involving STAT6 and the insulin receptor substrate, respectively (8, 11, 12). In the first pathway, the STAT6 protein recruited to tyrosine-phosphorylated IL-4Rα is itself phosphorylated on tyrosine residues by Janus kinases. Phosphorylated STAT6 proteins dimerize and translocate to the nucleus where they bind to specific DNA elements on IL-4-responsive genes. In the second pathway, insulin receptor substrate-1 and insulin receptor substrate-2 proteins are recruited to phosphorylated IL-4Rα to activate the PI3K-AKT pathway, leading to cell proliferation and resistance to apoptosis.
The biological effect of IL-4 varies depending on the cell type and differentiation status of the cell. In cancer cells, IL-4 often promotes proliferation, metastasis, and expression of anti-apoptotic genes (13–16). On the other hand, we and others have shown that IL-4 directly inhibits growth of several human cancer cell lines including those derived from breast, gastric, colon, and renal cancers (17–22). In addition, apoptosis can be induced or suppressed by IL-4: IL-4 induced apoptosis in certain cell types, including breast and liver cancer cells, hepatocytes, endothelial cells, monocytes, and developing mast cells (23–26), whereas it increased resistance to apoptosis in mouse fibrosarcoma cell lines and STAT6-high human cancer cell lines such as HT-29 and ZR-75-1 (16, 27). Molecular mechanisms for the diverse and sometimes opposing effects of IL-4 are not yet understood. Both STAT1 and STAT6 have been proposed to mediate the growth inhibitory effect of IL-4 (28), and STAT6 to mediate apoptosis (23) as well as resistance to apoptosis by IL-4 (16, 27). In the present study, we present evidence that IL-4 induces cellular senescence in some human RCC cell lines and identify STAT6 and p38 MAPK as signaling mediators of IL-4-induced cellular senescence.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
The human RCC cell lines Caki-1, A498, 786-O, SNU482, SNU228, and A704 were obtained from the American Type Culture Collection (ATCC, Manassas, VA) or the Korean Cell Line Bank (Seoul, Korea). The cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) (Invitrogen), penicillin (100 units/ml), and streptomycin (100 μg/ml) (Invitrogen).
Chemical inhibitors of p38 MAPK (SB203580), JNK (SP600125), and MEK (PD98059) were purchased from Calbiochem (La Jolla, CA), and the SB203580 analog, SB202474, and human recombinant IL-4 were obtained from BioSource (Camarillo, CA). For electrophoretic mobility shift assays (EMSA), STAT3 and STAT6 probes were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and NF-κB and AP-1 probes were purchased from Promega (Madison, WI).
Flow Cytometric Analysis
Flow cytometry was performed using the FACScan flow cytometer (BD Biosciences). To measure cell size and granularity, cells were harvested and washed twice with 1× PBS and then suspended in 1× PBS. Cell size and granularity were analyzed in samples of 1 × 105 cells by forward scatter and side scatter measurements, respectively.
Proliferation Assay
Cell proliferation was assessed from [3H]thymidine or BrdU incorporation. For a [3H]thymidine incorporation assay, cells were seeded in a 96-well plate and cultured in the presence of varying concentrations of IL-4 for 24 h. The cells were labeled with [3H]thymidine (1 μCi/well) for the last 6 h of IL-4 exposure, and [3H]thymidine incorporation into DNA was measured using a β-scintillation counter. Results were expressed as percent growth inhibition.
A BrdU assay was performed using the BrdU Cell Proliferation Assay Kit (Cell Signaling, Boston, MA) according to the manufacturer's instructions. Briefly, cells were pulsed with 0.1 mg/ml of BrdU for 1 h and fixed. After denaturation of the genomic DNA, cells were incubated with an anti-mouse BrdU antibody and subsequently with a HRP-conjugated anti-mouse IgG antibody. Then, the HRP substrate 3,3′,5,5′-tetramethylbenzidine was added, and the colored reaction product was quantified by spectrophotometry.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and Real Time-PCR
Total cellular RNA was extracted using the acid guanidinium isothiocyanate method. The cDNAs were synthesized from total cellular RNA using oligo(dT) primers (Promega) with Moloney murine leukemia virus-reverse transcriptase (Invitrogen) at 37 °C for 1 h. The cDNA served as template for PCR amplification of the IL-4R (the IL-4Rα gene), IL-13RA1 (the IL-13Rα1 gene), and ACTB (the β-actin gene) genes. The PCR primers were as follows: IL-4R, 5′-ATG GAT GAC GTG GTC AGT-3′ and 5′-TCA GGC GAT GCA CAG AAG-3′; IL-13RA1, 5′-AGG ATG ACA AAC TCT GGA G-3′ and 5′-CTC AAG GTC ACA GTG AAG G-3′; and ACTB, 5′-CAG AGC AAG AGA GGC ATC-3′ and 5′-CGT AGA TGG GCA CAG TGT-3′. Thirty PCR cycles were conducted, and each cycle consisted of denaturation for 30 s at 95 °C, annealing for 30 s at 58 °C, and extension for 1.5 min at 72 °C.
Relative quantification of gene expression was measured by real-time PCR of the cDNA synthesized as above, using the LightCycler 480 (Roche Molecular Biochemicals). PCR amplification was performed in a 10-μl reaction volume containing 1 μl of cDNA, 0.3 μm primers, and the LightCycler-DNA Master SYBR Green I mixture (Roche Molecular Biochemicals). The reaction was performed under the following conditions: 10 s denaturation at 95 °C, 10 s annealing of primers at 60 °C, and 20 s elongation at 72 °C, for 55 cycles. Primers used for real time-PCR analysis were as follows: IL-4R, 5′-GCT ATG TCA GCA TCA CCA AGT TAA-3′ and 5′-CCC CTG AGC ATC CTG GAT TAT-3′; IL-13RA1, 5′-ATC TCA CCC CCA GAA GGT GAT-3′ and 5′-CGG GAC TGG TAT TCC TTC-3′; and ACTB, 5′-GTA CCA CTG GCA TCG TGA TGG ACT-3′ and 5′-CCG CTC ATT GCC AAT GGT GAT-3′.
Immunoblot Analysis
Cells were lysed on ice in an extraction buffer (31.25 mm Tris-HCl, pH 6.8, 150 mm NaCl, 1% SDS, 10% glycerol, and 2.5% β-mercaptoethanol). The protein concentration of the whole cell lysate was measured by the Bradford assay (Bio-Rad). Whole cell lysates were subjected to an immunoblot analysis using an antibody specific for each protein. Antibodies against STAT1, p-STAT1, STAT3, p-STAT3, p-STAT6, p-p38, p-JNK, and FLAG were purchased from Cell Signaling Technology, and those against p21WAF1/CIP1, p16INK4A, STAT6, p38, JNK1, MEK3, p-MEK3/6, ERK, p-ERK, poly(ADP-ribose) polymerase, caspase-3, and β-actin were from Santa Cruz Biotechnology. Anti-tubulin and p53 antibodies were obtained from Calbiochem.
siRNA and Plasmid Transfection
The siRNAs specific for STAT6 (STAT6-1) and p38 MAPK (1092108) or control scrambled siRNA (SN-1013) were purchased from Bioneer (Daejeon, Korea), and another STAT6-specific siRNA (STAT6-2) was purchased from Santa Cruz Biotechnology (sc-29497). Sequences of two STAT3-specific siRNAs are as follows: STAT3-1, 5′-CCC GUC AAC AAA UUA AGA-3′ and STAT3-2, 5′-AGC AGA UAU UGU CAA GUU-3′. pcDNA3-FLAG-STAT6 and pcDNA3-p38 MAPK plasmids were kind gifts from Dr. Mark H. Kaplan (Indiana University, IN) and Dr. Roger J. Davis (University of Massachusetts Medical School), respectively. siRNA and plasmid transfection was carried out as previously described (29).
Electrophoretic Mobility Shift Assay (EMSA)
Cells were washed twice with 1× PBS and resuspended in the ice-cold solution A (10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 0.5 mm DTT, and 1 mm Na3VO4) containing protease inhibitors (0.5 mm PMSF, 1 μg/ml of aprotinin, and 1 μg/ml of leupeptin). After incubating for 5 min on ice, 10% Nonidet P-40 was added. Nuclei were harvested by centrifugation and resuspended in the cold solution C (20 mm HEPES, pH 7.9, 1.5 mm MgCl2, 0.42 m NaCl, 0.2 mm EDTA, and 25% glycerol) containing protease inhibitors. Nuclear proteins were extracted by rotating the tube at 4 °C for 1 h and collected by centrifugation at 13,000 × g. Double-stranded oligonucleotide probes were radiolabeled using T4 polynucleotide kinase and [γ-32P]ATP (5,000 Ci/mmol). The probe sequences were 5′-GTG CAT TTC CCG TAA ATC TTG TCT ACA-3′ for STAT1, 5′-GAT CCT TCT GGG AAT TCC TAG ATC-3′ for STAT3, 5′-GTA TTT CCC AGA AAA GGA AC-3′ for STAT6, and 5′-CGC TTG ATG AGT CAG CCG GAA-3′ for AP-1. The binding reaction was conducted at 37 °C for 10 min in a 15-μl reaction mixture containing 4 μg of nuclear extract, 1.2 ng of 32P-labeled probe, 0.1 μg of poly(dI-dC), and 0.3 mm dithiothreitol in 1× binding buffer (12 mm Tris-HCl, pH 8.0, 2 mm MgCl2, 60 mm KCl, 0.12 mm EDTA, and 12% glycerol). The reaction mixture was loaded onto a 6% non-denaturing polyacrylamide gel prepared in 0.5× TBE buffer and subjected to electrophoresis. For a supershift experiment, the nuclear extract was incubated with an antibody against STAT1, STAT3, or STAT6 at 4 °C for 30 min prior to adding a probe. For cold competition, a 30–50-fold molar excess of an unlabeled STAT6 or AP-1 oligonucleotide was added to the binding reaction.
Immunofluorescence and SA-β-Gal Staining
Cells were cultured in an 8-well chamber slide. Following exposure to IL-4, cells were fixed with 100% acetone for 10 min at 4 °C and permeabilized with 0.1% Triton X-100 for 10 min at room temperature. Fixed cells were pretreated with 1% BSA in 1× PBS for 1 h and incubated with a rabbit anti-STAT6 antibody for 1 h. After three washes in 1× PBS, the cells were incubated with a FITC-conjugated goat anti-rabbit antibody for 1 h. To examine the senescence-associated heterochromatin foci, cells were stained with DAPI. Images were captured using a fluorescence microscope (Carl Zeiss MicroImaging, Thornwood, NY). SA-β-gal staining was performed as previously described (30).
Luciferase Reporter Assay
The STAT6 reporter plasmid, p4xSTAT6-Luc2P, was purchased from Addgene (Cambridge, MA). p4xSTAT6-Luc2P plasmid or its vector, pGL4, was transfected into Cack-1 cells. After 48 h, cells were split, transfected with control, p38 MAPK, and/or STAT6 siRNAs for 24 h. Cells were then serum-starved for 24 h and treated with IL-4 (20 ng/ml). Luciferase assays were performed 6 h after IL-4 treatment using the luciferase assay reagent (Promega) and Luminoskan Ascent luminometer (Thermo Scientific, Waltham, MA). Luciferase activity of each sample was normalized to that of the corresponding sample transfected with pGL4.
Microarray
The microarray experiments were conducted at the DNALINK (Seoul, Korea), and gene expression levels of the samples were analyzed using the Affymetrix Expression Console software (version 1.1) and R program (2.11.1). Gene Ontology analysis was conducted using the DAVID online tool. All data presented in Table 1 show the up-regulated fold-change in gene expression in Caki-1 cells treated with 10 ng/ml of IL-4 for 24 h compared with untreated control cells. All genes presented were significantly changed, and the minimum fold-change chosen for presentation was 1.8.
TABLE 1.
Up-regulated genes in Caki-1 cell treated with IL-4
The gene expression profile of Caki-1 cells treated with IL-4 (10 ng/ml) for 24 h was assessed by microarray analysis using the Affymetrix GeneChip Human Gene 2.0 ST Array, which contains more than 48,000 human genes. This table shows some of the genes up-regulated more than 1.8-fold in IL-4-treated cells compared to untreated control cells.
| Category | Accession number | Gene name | Description | -Fold |
|---|---|---|---|---|
| Adhesion molecules | NM_001078 | VCAM1 | Vascular cell adhesion molecule 1 | 1.94 |
| NM_003637 | ITGA10 | Integrin, α10 | 1.82 | |
| Growth factors and receptors | NM_001552 | IGFBP4 | Insulin-like growth factor binding protein 4 | 2.82 |
| ENST00000230990 | HBEGF | Heparin-binding EGF-like growth factor | 2.35 | |
| Cytokines, chemokines, andreceptors | ENST00000394905 | CCL26 | Chemokine (C-C motif) ligand 26 | 7.91 |
| ENST00000371936 | IL-13RA2 | Interleukin 13 receptor, α2 | 3.85 | |
| ENST00000303115 | IL-7R | Interleukin 7 receptor | 2.89 | |
| ENST00000293272 | CCL5 | Chemokine (C-C motif) ligand 5 | 2.64 | |
| ENST00000296861 | TNFRSF21 | Tumor necrosis factor receptor superfamily, member 21 | 2.34 | |
| NM_152456 | IL-34 | Interleukin 34 | 2.28 | |
| ENST00000404625 | IL-6 | Interleukin6 (interferon, β2) | 1.84 | |
| Cell cycle | NM_001220778 | CDKN1A | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | 1.81 |
| Transcription | NM_001206616 | EHF | Ets homologous factor | 4.39 |
| NM_001717 | BNC1 | Basonuclin 1 | 3.44 | |
| ENST00000407775 | ZFPM2 | Zinc finger protein, multitype 2 | 3.36 | |
| Extracellular matrix | ENST00000404816 | LTBP1 | Latent transforming growth factor β-binding protein 1 | 8.41 |
| NM_198391 | FLRT3 | Fibronectin leucine rich transmembrane protein 3 | 6.54 | |
| ENST00000380548 | ADAMTSL1 | ADAMTS-like 1 | 3.09 | |
| NM_001039348 | EFEMP1 | EGF containing fibulin-like extracellular matrix protein 1 | 3.00 | |
| NM_002317 | LOX | Lysyl oxidase | 2.27 | |
| NM_001848 | COL6A1 | Collagen, type VI, α1 | 2.19 | |
| ENST00000261037 | COL8A1 | Collagen, type VIII, α1 | 2.03 | |
| NM_001849 | COL6A2 | Collagen, type VI, α2 | 1.92 | |
| Others | NM_006919 | SERPINB3 | Serpin peptidase inhibitor, clade B (ovalbumin), member 3 | 34.85 |
| NM_013324 | CISH | Cytokine inducible SH2-containing protein | 5.92 | |
| NM_033256 | PPP1R14A | Protein phosphatase 1, regulatory (inhibitor) subunit 14A | 3.97 | |
| NM_003745 | SOCS1 | Suppressor of cytokine signaling 1 | 3.31 | |
| NM_000927 | ABCB1 | ATP-binding cassette, sub-family B (MDR/TAP), member 1 | 3.21 | |
| NM_001004343 | MAP1LC3C | Microtubule-associated protein 1 light chain 3 gamma | 3.10 | |
| NM_001102445 | RGS4 | Regulator of G-protein signaling 4 | 2.59 | |
| ENST00000296861 | TNFRSF21 | Tumor necrosis factor receptor superfamily, member 21 | 2.34 |
RESULTS
IL-4 Inhibits Growth of Human RCC Cells
IL-4 inhibits cell growth in several human cancer cell lines with or without inducing apoptosis (17–22, 28). To evaluate the effects of IL-4 on the growth of human RCC cells, Caki-1, A498, and 786-O cells were exposed to 1–10 ng/ml of IL-4 for 24 h, and cell proliferation was measured by a [3H]thymidine incorporation assay. As shown in Fig. 1A, IL-4 inhibited proliferation of Caki-1 and A498 cells. After a 24-h exposure to IL-4 (3 ng/ml), proliferation of Caki-1 and A498 cells was reduced by 51 and 23%, respectively, as compared with that of untreated control cells. Cell death was not observed in IL-4-treated cells under microscopic examination (data not shown) or by immunoblot analyses of procaspase-3 and poly(ADP-ribose) polymerase cleavage (Fig. 1B). However, proliferation of 786-O cells was not affected by IL-4.
FIGURE 1.

IL-4 suppresses proliferation of Caki-1 and A498 but not 786-O cells. A, the three RCC cell lines were exposed to IL-4 for 24 h. Proliferation was measured by a [3H]thymidine incorporation assay as described under “Experimental Procedures.” The data shown here represent three independent experiments. B, Caki-1 cells were exposed to different concentrations of IL-4 or irradiated with ultraviolet C (UVC). After 24 h, cell lysates were subjected to an immunoblot analysis. UV-irradiated cells were used as positive controls for apoptosis. C, Caki-1 and A498 cells were exposed to IL-4 at 1 ng/ml, and the expression of IL-4R (IL-4Rα) and IL-13RA1 (IL-13Rα1) mRNA was analyzed by RT-PCR. *, the number represents a signal intensity normalized to β-actin (ACTB). D, Caki-1 cells were treated with 10 ng/ml of IL-4 for 0, 1, 8, or 24 h. Relative mRNA expression levels of IL-13RA1 and IL-4R were assessed by real time RT-PCR in triplicate. The mRNA level of IL-4R or IL-13RA1 was normalized against that of β-actin, and the relative mRNA expression in IL-4-treated cells compared with that in control cells was plotted. Data were expressed as mean ± S.D. PARP, poly(ADP-ribose) polymerase.
Human RCC cells express the high-affinity IL-4 receptor (IL-4R), composed of IL-4Rα and IL-13Rα1 chains (31–33). Therefore, the expression of IL-4R components was assessed in Caki-1 and A498 cells before and after the exposure to IL-4. In contrast to Caki-1, which expressed IL-4R and IL-13RA1 mRNA in the resting state, A498 cells did not express detectable levels of IL-4R or IL-13RA1 mRNA (Fig. 1C). Consistent with the mRNA expression, the basal level of the IL-4Rα protein was much higher in Caki-1 than A498 and 786-O cells (data not shown). IL-4 increased IL-4R mRNA levels after 1 h in Caki-1 cells, and IL-4R as well as IL-13RA1 mRNA levels after 4–8 h in A498 cells (Fig. 1, C and D). These results suggested that IL-4 stimulation induced expression of the IL-4R components in RCC cells, and that a high level of basal expression and/or rapid induction of IL-4R might be at least partly responsible for the sensitivity of Caki-1 cells to IL-4.
IL-4 Induces Cellular Senescence in Human RCC Cell Lines
Because IL-4-induced growth inhibition did not accompany cell death, it was investigated whether cellular senescence was induced by IL-4 in Caki-1 and A498 cells. Indeed, after exposure to IL-4 for 3–7 days, the cells underwent a profound morphological change characteristic of cellular senescence such as flattening and enlargement (Fig. 2A). Flow cytometric analysis showed that Caki-1 cells exposed to IL-4 for 7 days increased in size and granularity (Fig. 2B). Up-regulation of SA-β-gal activity (Fig. 2C) increased protein expression of p21WAF1/CIP1 and p16INK4A (Fig. 2D, upper panel) and formation of senescence-associated heterochromatin foci (Fig. 2D, lower panel) were also shown. IL-4 also induced cellular senescence in other human RCC cell lines, SNU482 and SNU228, but not in a third RCC cell line, A704 (Fig. 2E). These results reassure that IL-4 induces cellular senescence in some, but not all, human RCC cell lines. Because serum IL-6 concentration increased with age (34), and IL-6 was known to contribute to the progression of senescence (35, 36) we investigated the capacity of IL-6 to induce cellular senescence. However, IL-6 did not induce senescence in Caki-1 cells, as assessed by SA-β-gal staining, cell proliferation, and p21WAF1/CIP1 protein expression (Fig. 2, F–H).
FIGURE 2.

IL-4 induces senescence in Caki-1 cells. A, Caki-1, A498, and 786-O cells were exposed to 10 ng/ml of IL-4 for 7 days. Cellular morphology was observed by light microscopy. B, Caki-1 cells were exposed to 10 ng/ml of IL-4 for 3 days, and the cellular morphology was observed by light microscopy. Senescent cells with an enlarged and flattened morphology are indicated by white arrows (upper panel). Flow cytometric analysis of control and IL-4-treated Caki-1 cells are indicated in the lower panel. C, SA-β-gal staining was performed in Caki-1 cells after 7 days of exposure to IL-4 (10 ng/ml) (upper panel). More than 200 cells were counted in each of three randomly selected fields, and the percentage of SA-β-gal positive cells was plotted (lower panel). *, p < 0.05 (Student's t test). D, Caki-1 cells were exposed to 10 ng/ml of IL-4. Cell lysates were immunoblotted with specific antibodies (upper panel). Caki-1 cells were incubated with 10 ng/ml of IL-4 for up to 5 days. The cells were stained with DAPI, and heterochromatic foci were visualized by fluorescence microscopy (lower panel). E, SNU482, SNU228, and A704 cells were exposed to 10 ng/ml of IL-4 for 7 days and stained for SA-β-gal activity. F and G, Caki-1 cells were exposed to 10 ng/ml of IL-4 or IL-6 for 3 days. Cells were stained for SA-β-gal activity (F) or subjected to a BrdU incorporation assay (G). H, Caki-1 cells were exposed to different concentrations of IL-4 or IL-6 for 3 days. Cell lysates were prepared and an immunoblot analysis was performed with the indicated antibodies.
IL-4 Induces Strong and Persistent STAT6 Activation in Caki-1 Cells
The heterodimerization of IL-4R subunits by IL-4 is known to activate JAK kinases, which then stimulate at least two different signaling pathways involving either STAT6 or insulin receptor substrate-1/2 (6, 37). Thus, phosphorylation of STAT6 as well as STAT1 and STAT3 was analyzed in Caki-1 cells after exposure to IL-4. As shown in Fig. 3A, STAT6 phosphorylation was predominant by 10 min after IL-4 treatment, although STAT1 and STAT3 were also phosphorylated (Fig. 3A). Although STAT6 phosphorylation in Caki-1 cells was strong and persistent, it was weak and transient in A498 and 786-O cells, consistent with the higher IL-4 sensitivity of Caki-1 as compared with the other cell lines (Fig. 3B). Phosphorylation activates STAT6 for nuclear translocation. Indeed, nuclear translocation of STAT6 was obvious by 30 min after IL-4 treatment in immunofluorescence staining (Fig. 3C).
FIGURE 3.

IL-4 activates STAT1, STAT3, and STAT6. A, Caki-1 cells were exposed to 10 ng/ml of IL-4 and analyzed for STAT phosphorylation by immunoblotting. B, Caki-1, A498, and 786-O cells were exposed to 1 ng/ml of IL-4, and STAT6 phosphorylation was analyzed by immunoblotting. C, Caki-1 cells were incubated with 1 ng/ml of IL-4, fixed, and immunostained with an anti-STAT6 antibody. D, Caki-1 cells were exposed to 10 ng/ml of IL-4 for 10 min, nuclear extracts were prepared, and EMSA was performed using a 32P-labeled STAT6 probe. Where indicated, anti-STAT1, -STAT3, or -STAT6 antibody was added for a supershift assay. The sequence specificity of the STAT6 binding was demonstrated by competition with a 50-fold excess of the cold STAT6 or AP-1 probe.
We then conducted an EMSA to examine whether IL-4 promoted DNA binding activity of STAT6. Nuclear extracts prepared from control or IL-4-treated Caki-1 cells were incubated with the radiolabeled oligonucleotide probe containing the consensus STAT6-binding motif. IL-4 treatment caused a mobility shift of the probe band compared with the untreated control (Fig. 3D, first and second lanes). In an antibody supershift assay, a supershifted band was observed with an anti-STAT6 antibody indicating STAT6 binding to the probe (Fig. 3D, fifth lane). Anti-STAT1 or anti-STAT3 antibody did not cause a supershift (Fig. 3D, third and fourth lanes). The mobility shift of the probe caused by IL-4 was prevented by competition with an excess of the non-radiolabled STAT6, but not the unrelated AP-1 probe (Fig. 3D, sixth and seventh lanes). These findings show that IL-4 activates STAT6 and induces its nuclear translocation and DNA binding activity. The nuclear extract from IL-4-treated Caki-1 cells also caused a mobility shift of STAT1 and STAT3 probes (data not shown). However, anti-STAT6 antibody supershifted both bands indicating that STAT6 could also bind to the consensus target sequences of STAT1 and STAT3.
STAT6 Mediates IL-4-induced Growth Inhibition in RCC Cells
To explore the role of STAT6 in IL-4-induced growth inhibition and cellular senescence, STAT6 expression was knocked down by transfection with specific siRNAs, and proliferation was measured in IL-4-treated cells by [3H]thymidine or BrdU incorporation assays. STAT6 silencing was confirmed by an immunoblot analysis, and it reversed IL-4-induced inhibition of proliferation greater than 50% (Fig. 4). Although STAT1 was known to suppress cell proliferation, STAT1 silencing did not affect IL-4-induced inhibition of proliferation (data not shown). These results suggest that STAT6 mediates IL-4-induced inhibition of proliferation and cellular senescence.
FIGURE 4.
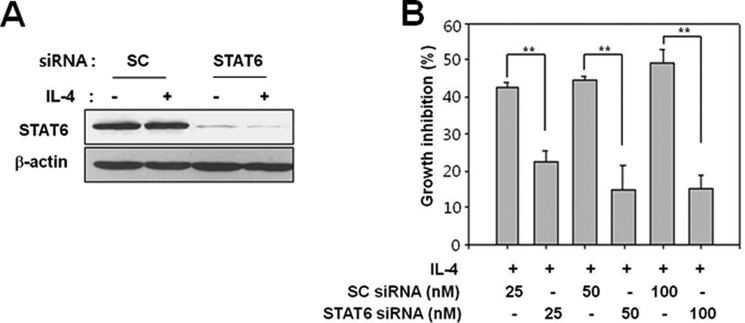
Silencing STAT6 abrogates IL-4-induced growth inhibition. Caki-1 cells were transfected with control (scrambled, SC) or STAT6 (STAT6-1) siRNA. After 48 h, cells were exposed to 10 ng/ml of IL-4 for 16 h. A, STAT6 expression was analyzed by immunoblotting 48 h after siRNA transfection. B, incorporation of [3H]thymidine was measured using a liquid scintillation counter. **, p < 0.01 (Student's t test). The data shown are representative of three independent experiments.
p38 MAPK Is Also Required for IL-4-induced Growth Inhibition
p38 MAPK is known to play an important role in the induction of cellular senescence under various stress conditions such as telomere shortening, Ras-Raf activation, oxidative stress, or inappropriate culture conditions (38–40). Besides stress, growth factors, and mitogens, several hematopoietic cytokines activate p38 MAPK. However, its activation by IL-4 is known to be cell type-specific (41). Therefore, activation of p38 MAPK by IL-4 was examined in Caki-1 cells. p38 MAPK as well as its upstream kinases, MEK3/6, were phosphorylated following IL-4 treatment (Fig. 5A). Phosphorylated levels of ERK and JNK were not altered. In A498 cells, activation of p38 MAPK by IL-4 was weaker than that in Caki-1 cells, and in 786-O cells, p38 MAPK was not activated at all (Fig. 5B). Therefore, p38 MAPK activation in these cell lines appeared to correlate with their sensitivity to IL-4. In Caki-1 cells pretreated with the p38 MAPK inhibitor, SB203580, IL-4-induced growth inhibition decreased by 12–16% (Fig. 5C), whereas neither the JNK inhibitor, SP600125, nor the ERK inhibitor, PD98059, had any effect (Fig. 5, D and E). Similarly, knock-down of p38 MAPK expression by transfection of Caki-1 cells with either one of the two specific siRNAs (814 and 940) reduced IL-4-mediated growth inhibition by ∼30% (Fig. 5F). In addition, ectopic expression of a dominant-negative mutant of p38 MAPK (p38-AGF) partially prevented an IL-4-induced increase in p21WAF1/CIP1 protein expression (Fig. 5G). Taken together, these results indicate that p38 MAPK, but not ERK or JNK, is involved in IL-4-induced growth inhibition in Caki-1 cells.
FIGURE 5.

p38 MAPK is involved in IL-4-induced growth inhibition. A, Caki-1 cells were serum-starved for 24 h and exposed to 10 ng/ml of IL-4, and cell lysates were prepared for immunoblotting. B, Caki-1, A498, and 786-O cells were serum-starved for 24 h and exposed to 10 ng/ml of IL-4 for the indicated time periods. Activation of the kinases was analyzed by immunoblotting with phospho-specific antibodies. C–E, Caki-1 cells were preincubated with varying concentrations of SB203580 (C), SP600125 (D), or PD98059 (E) for 1 h and then exposed to 10 ng/ml of IL-4. After 24 h, a [3H]thymidine incorporation assay was performed. F, Caki-1 cells were transfected with control (scrambled, SC) or p38 siRNA (814 or 940). After 48 h, p38 MAPK protein expression was analyzed by immunoblotting (left panel). Cells were exposed to 10 ng/ml of IL-4 for 24 h, and a [3H]thymidine incorporation assay was performed (right panel). *, p < 0.01 (Student's t test). G, Caki-1 cells were transfected with an empty vector (Vector) and pcDNA3-FLAG-p38AGF (p38-AGF). After 2 days, cells were exposed 10 ng/ml of IL-4 for 24 h. Cell lysates were subjected to an immunoblot analysis using the indicated antibodies.
STAT6 and p38 MAPK Independently Mediate IL-4-induced Cellular Senescence
Having demonstrated that STAT6 and p38 MAPK both contribute to IL-4-induced growth inhibition, it was investigated whether they acted through the same or different signaling pathway(s). Either transfection with STAT6 siRNA or pretreatment with SB203580 alone attenuated IL-4-induced growth inhibition as measured by [3H]thymidine incorporation (Fig. 6A). However, SB203580 pretreatment of cells transfected with STAT6 siRNA almost completely abolished IL-4-induced growth inhibition (Fig. 6A). Furthermore, an SA-β-gal assay demonstrated that IL-4-induced cellular senescence was partially prevented by transfection with either p38 MAPK or STAT6 siRNA alone, but almost completely inhibited by co-transfection with both siRNAs (Fig. 6B). These results showing an additive effect of STAT6 and p38 MAPK suggested that these molecules mediate IL-4-induced growth inhibition and cellular senescence through independent pathways.
FIGURE 6.

STAT6 and p38 MAPK independently mediate IL-4-induced growth inhibition and cellular senescence. A–C, Caki-1 cells were transfected with scrambled (SC), p38 MAPK or STAT6 (STAT6–1) siRNA. A, after 48 h, cells were serum-starved for 24 h and pretreated with SB203580 (20 μm) for 1 h. Cells were the exposed to 10 ng/ml of IL-4 for 24 h, and a [3H]thymidine incorporation assay was performed. B, 5 days after IL-4 exposure, SA-β-gal staining was performed. More than 200 cells were counted in each of three randomly selected fields, and the percentage of SA-β-gal-positive cells was plotted. C, 16 h after IL-4 exposure (10 ng/ml), cells were harvested for an immunoblot analysis. D, the control pGL4 and p4xSTAT6-Luc2P plasmids were transfected into Caki-1 cells. After 48 h, cells were split and transfected again with scrambled (SC), p38, and/or STAT6 siRNAs for 24 h. Cells were serum-starved for 24 h and incubated with 20 ng/ml of IL-4 for 6 h. Then a luciferase assay was performed with the cell lysates. The data shown are mean ± S.D. of three independent experiments. RLU, relative luciferase units. E, Caki-1 cells were pretreated with 20 μm SB203580 for 1 h and subsequently exposed to 1 ng/ml of IL-4 for 1 h. Total cell lysates were subjected to an immunoblot analysis with anti-STAT6, anti-p-STAT6, and anti-tubulin antibodies. F, Caki-1 cells were transfected with an empty vector, pcDNA3-FLAG-STAT6 (STAT6 or FL-STAT6), and/or pcDNA3-p38 MAPK (p38) as indicated. After 3 days, SA-β-gal staining was performed (left panel). Protein expression of FLAG-STAT6 and p38 MAPK was assessed by an immunoblot analysis (right panel).
Increased p21WAF1/CIP1 protein expression is one of the hallmarks of cellular senescence. Because exposure of Caki-1 cells to IL-4 induced p21WAF1/CIP1 protein expression (22), we investigated whether STAT6 and/or p38 MAPK played a key role in IL-4-induced expression of p21WAF1/CIP1. Silencing either STAT6 or p38 MAPK partially prevented an increase in p21WAF1/CIP1 protein expression by IL-4, whereas silencing both genes diminished it further (Fig. 6C). To further show that p38 MAPK mediates IL-4-triggered signaling independently of STAT6, the effect of p38 MAPK silencing on the transcriptional activity of STAT6 induced by IL-4 was investigated by a reporter assay using a STAT6 reporter plasmid. As shown in Fig. 6D, the transcriptional activity of STAT6 was not affected by p38 MAPK silencing. Likewise, phosphorylation of STAT6 by IL-4 stimulation was not affected by pretreatment with SB203580 (Fig. 6E). All these results indicate that STAT6 and p38 MAPK independently mediate growth inhibition and cellular senescence by IL-4.
Overexpression of STAT6 and/or p38 MAPK Is Sufficient to Induce Cellular Senescence
To confirm that STAT6 and p38 MAPK mediate cellular senescence induction by IL-4, these proteins were overexpressed in Caki-1 cells. As judged by SA-β-gal staining and a morphological change, ectopic expression of STAT6 or p38 MAPK induced cellular senescence even without IL-4 treatment (Fig. 6F). Moreover, overexpression of these two molecules together further accelerated senescence. These results show that activation of STAT6 and/or p38 MAPK is sufficient to induce senescence in Caki-1 cells.
DISCUSSION
The present study demonstrates for the first time that IL-4 induces cellular senescence in human RCC cell lines. Morisaki et al. (20) observed that IL-4 treatment in gastric carcinoma cells not only inhibited cell growth but also altered cellular morphology to a flattened shape. It appears that IL-4 induced cellular senescence in gastric cancer cells, although no further evidence for cellular senescence was provided. In this study, cellular senescence was induced by IL-4 in four of six RCC cell lines, including Caki-1, A498, SNU482, and SNU228 (Fig. 2, A and E). In Caki-1 cells, IL-4 inhibited proliferation in a concentration-dependent manner. At concentrations greater than 1 ng/ml, IL-4 inhibited proliferation and induced senescence, whereas at a much lower concentration (5 pg/ml), IL-4 moderately stimulated proliferation (data not shown). Caki-1 cells were more sensitive to IL-4-induced growth inhibition and cellular senescence than A498 cells. This may be due to, at least in part, a difference in the basal level of IL-4R expression. In the resting state, A498 cells did not express mRNAs of both IL-4R and IL-13RA1 at detectable levels, but their expression was induced after IL-4 treatment. In contrast, Caki-1 cells in the resting state expressed mRNAs of both IL-4R chains (Fig. 1, C and D). The basal protein level of IL-4Rα was higher in Caki-1 cells compared with A498 and 786-O cells (data not shown). Various types of tumors including RCC express significantly higher levels of IL-4R than do the non-transformed cells such as B cells, endothelial cells, and monocytes (17). In lung and ovarian cancer, 60–80% of cancer specimens display higher levels of IL-4R expression as compared with normal tissue (42, 43). It may be that IL-4 treatment is selectively beneficial in patients with cancers expressing high levels of IL-4R.
In various non-transformed and transformed cells of different tissue origins, IL-4 is reported to induce apoptosis (17, 23–26). However, there are also reports that IL-4 reduces apoptosis (17, 41). Apoptosis was not observed in Caki-1, A498, SNU482, and SNU228 cells exposed to IL-4. Rather, the cells up-regulated SA-β-gal activity and acquired the morphology characteristic of cellular senescence. In contrast to IL-4, IL-6, which is one of the cytokines secreted from senescent cells and believed to contribute to senescence induction, did not inhibit proliferation or induce cellular senescence. Although IL-4 is known to directly inhibit growth of various human non-hematopoietic cancer cell lines, the molecular mechanisms of this action have not been elucidated. STAT6 is the primary STAT protein activated by IL-4, but STAT1 and STAT3 were also activated in Caki-1 cells to a lesser extent and for a shorter duration than STAT6 (Fig. 3A). Silencing STAT6 by RNA interference partially prevented IL-4-induced growth suppression and cellular senescence (Figs. 4B and 6, A-C), indicating that STAT6 played a role in mediating these effects. Overexpression of STAT6 induced senescence even without IL-4 stimulation in Caki-1 cells, emphasizing the role of STAT6 in senescence induction (Fig. 6F). Even though STAT1 is crucial for the growth inhibitory effect of interferon (44) and IL-4 in colon carcinoma cell lines, HT29 and WiDr (28), silencing STAT1 did not interfere with IL-4-induced growth suppression in Caki-1 (data not shown). The functional consequences of STAT6 activation are likely to be dependent on cellular context. STAT6 was shown to be required for apoptosis induction by IL-4 (26, 27). However, in this study, STAT6 mediated senescence rather than apoptosis (Fig. 4B). The molecular mechanisms that determine the outcome of IL-4 signaling, i.e. apoptosis, senescence, or even proliferation, are not yet known.
Because silencing STAT6 only partially reversed IL-4-induced growth inhibition, we attempted to identify other signaling molecules involved in the growth inhibitory effect of IL-4. One plausible candidate, p38 MAPK, plays an important role in replicative and Ras-induced cellular senescence (38–40, 45, 46). Activation of p38 MAPK by IL-4 is thought to be highly dependent on cell type (41). IL-4 induces p38 MAPK activation in T and pro-B cell lines, but not in monocytes/macrophages, mast cells, or a human B cell line, Ramos 2G6 (47). We found that IL-4 activated p38 MAPK, but not ERK or JNK, in Caki-1 cells (Fig. 5A). Although p38 MAPK was activated in Caki-1 and A498 cells, it was not activated in 786-O cells. Thus, activation of p38 MAPK was correlated with sensitivity to IL-4-induced growth inhibition and senescence. In fact, p38 MAPK was required for these effects of IL-4 because interfering with the activity or expression of p38 MAPK prevented IL-4-induced growth inhibition and senescence, albeit partially (Figs. 5, C, F, and G, and 6, A–C). Moreover, overexpression of p38 MAPK induced senescence in Caki-1 cells without IL-4 stimulation (Fig. 6F). Although we did not examine the signaling pathway by which p38 MAPK is activated in these RCC cell lines, IL-4 was shown to activate p38 MAPK via the Cdc42-Rac-PAK-MKK3/6 pathway in A431 cells (48). It has been reported that p38 MAPK can directly regulate the activity of the transactivation domain of STAT6 (47). In this study, however, p38 MAPK and STAT6 appeared to function independently to induce growth inhibition and cellular senescence. Although inhibition of either one of these two molecules partially blocked the effect of IL-4, simultaneously knocking down both molecules exerted an additive effect, almost completely abrogating the effect of IL-4 (Fig. 6, A and B). Furthermore, siRNA-mediated knockdown of p38 MAPK did not significantly decrease the IL-4-induced transcriptional activity of STAT6 in a STAT6-driven reporter assay (Fig. 6D). Also, the p38 MAPK inhibitor, SB203580, did not affect STAT6 phosphorylation by IL-4 (Fig. 6E). Altogether, these data support the notion that p38 MAPK and STAT6 function independently to mediate IL-4-induced senescence in Caki-1 cells.
Our previous work revealed that IL-4-induced growth inhibition was associated with an increase in the expression of p21WAF1/CIP1 (22). IL-4 increased the mRNA and protein expression of p21WAF1/CIP1 leading to G1 cell cycle arrest through inhibition of cyclin-dependent kinase 2 activity. We supposed that the p21WAF1/CIP1 mediated IL-4-induced cellular senescence downstream of STAT6 and p38 MAPK. p21WAF1/CIP1 is highly expressed in senescent cells and may increase intracellular levels of reactive oxygen species to cause senescence (49). Moreover, IL-4 was recently reported to inhibit macrophage proliferation through STAT6-regulated p21WAF1/CIP1 expression, and STAT6 was shown to directly bind to the p21WAF1/CIP1 promoter (50). Indeed, in this study, STAT6 silencing not only prevented senescence induction but also inhibited an increase of p21WAF1/CIP1 protein. In addition to STAT6, p38 MAPK contributed to the induction of p21WAF1/CIP1 protein expression by IL-4, because IL-4-induced p21WAF1/CIP1 expression was partially prevented by silencing p38 MAPK expression (Fig. 6C). These findings suggest that STAT6 and p38 MAPK mediate IL-4-induced growth inhibition through an increase in p21WAF1/CIP1 protein expression.
IL-4-induced cellular senescence may also be relevant in clinical conditions other than RCC. Renal cellular senescence can compromise renal function in glomerulosclerosis, tubular atrophy, and interstitial fibrosis. In these conditions, renal cells show a senescence phenotype such as increased p16INK4A expression (51). Recently, many reports have suggested that IL-4 signaling is involved in renal dysfunction associated with diabetes, lupus, and renal allograft. Although IL-4 and IL-4R mRNA are weakly expressed in normal glomeruli, both mRNAs are strongly expressed in diseased glomeruli (52). In lupus-prone mice, STAT6 deficiency or anti-IL-4 Ab treatment ameliorates kidney disease, particularly glomerulosclerosis (53). Additionally, unilateral urethral obstruction induced tubulointerstitial fibrosis, which was ameliorated by knock-out of IL-4 and IL-13 (54). In our microarray data, expression levels of genes implicated in renal diseases, such as collagen VI, IL-6, LTBP1, and SOCS1, were markedly induced by IL-4 (Table 1), suggesting that IL-4 might play a crucial role in the progression of renal disease. Therefore, it is supposed that IL-4-induced senescence may be involved in the pathogenesis of renal diseases resulting in loss of kidney function.
Previous clinical trials using IL-4 as a cancer therapeutic agent for RCC were not successful. Our present data showing that IL-4 directly induces senescence in some but not all RCC cell lines suggest that IL-4 may prove effective in cancer therapy if RCC patients are selected for tumor sensitivity to IL-4. Because the growth inhibitory and senescence-inducing effects of IL-4 appear to be dependent on levels of basal IL-4R expression as well as activation of STAT6 and p38 MAPK, these may serve as markers for the sensitivity to IL-4. In addition, further understanding the molecular basis of IL-4 sensitivity may also contribute to development of a novel target for RCC therapy. For example, whereas STAT6 expression is often increased in cancer cells, possibly promoting tumor growth by several mechanisms (27, 55, 56), increased expression of STAT6 may be exploited to induce senescence in cancer cells.
This work was supported by National Research Foundation of Korea Grant 2010-0024981 from the Korean government and Korea University Grant K1132201.
- SA-β-gal
- senescence-associated-β-galactosidase
- RCC
- renal cell carcinoma.
REFERENCES
- 1. Kuilman T., Michaloglou C., Mooi W. J., Peeper D. S. (2010) The essence of senescence. Genes Dev. 24, 2463–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Prieur A., Peeper D. S. (2008) Cellular senescence in vivo. A barrier to tumorigenesis. Curr. Opin. Cell Biol. 20, 150–155 [DOI] [PubMed] [Google Scholar]
- 3. Campisi J. (2005) Senescent cells, tumor suppression, and organismal aging. Good citizens, bad neighbors. Cell 120, 513–522 [DOI] [PubMed] [Google Scholar]
- 4. Moiseeva O., Mallette F. A., Mukhopadhyay U. K., Moores A., Ferbeyre G. (2006) DNA damage signaling and p53-dependent senescence after prolonged β-interferon stimulation. Mol. Biol. Cell 17, 1583–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vijayachandra K., Lee J., Glick A. B. (2003) Smad3 regulates senescence and malignant conversion in a mouse multistage skin carcinogenesis model. Cancer Res. 63, 3447–3452 [PubMed] [Google Scholar]
- 6. Nelms K., Keegan A. D., Zamorano J., Ryan J. J., Paul W. E. (1999) The IL-4 receptor. Signaling mechanisms and biologic functions. Annu. Rev. Immunol. 17, 701–738 [DOI] [PubMed] [Google Scholar]
- 7. Horvath C. M. (2000) STAT proteins and transcriptional responses to extracellular signals. Trends Biochem. Sci. 25, 496–502 [DOI] [PubMed] [Google Scholar]
- 8. Keegan A. D., Nelms K., White M., Wang L. M., Pierce J. H., Paul W. E. (1994) An IL-4 receptor region containing an insulin receptor motif is important for IL-4-mediated IRS-1 phosphorylation and cell growth. Cell 76, 811–820 [DOI] [PubMed] [Google Scholar]
- 9. Wills-Karp M., Finkelman F. D. (2008) Untangling the complex web of IL-4- and IL-13-mediated signaling pathways. Sci. Signal. 1, pe55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Andrews A. L., Nordgren I. K., Kirby I., Holloway J. W., Holgate S. T., Davies D. E., Tavassoli A. (2009) Cytoplasmic tail of IL-13Rα2 regulates IL-4 signal transduction. Biochem. Soc. Trans. 37, 873–876 [DOI] [PubMed] [Google Scholar]
- 11. Hou J., Schindler U., Henzel W. J., Ho T. C., Brasseur M., McKnight S. L. (1994) An interleukin-4-induced transcription factor. IL-4 Stat. Science 265, 1701–1706 [DOI] [PubMed] [Google Scholar]
- 12. Pernis A., Witthuhn B., Keegan A. D., Nelms K., Garfein E., Ihle J. N., Paul W. E., Pierce J. H., Rothman P. (1995) Interleukin 4 signals through two related pathways. Proc. Natl. Acad. Sci. U.S.A. 92, 7971–7975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prokopchuk O., Liu Y., Henne-Bruns D., Kornmann M. (2005) Interleukin-4 enhances proliferation of human pancreatic cancer cells. Evidence for autocrine and paracrine actions. Br. J. Cancer 92, 921–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Taylor C. W., Grogan T. M., Salmon S. E. (1990) Effects of interleukin-4 on the in vitro growth of human lymphoid and plasma cell neoplasms. Blood 75, 1114–1118 [PubMed] [Google Scholar]
- 15. Myers J. N., Yasumura S., Suminami Y., Hirabayashi H., Lin W. C., Johnson J. T., Lotze M. T., Whiteside T. L. (1996) Growth stimulation of human head and neck squamous cell carcinoma cell lines by interleukin 4. Clin. Cancer Res. 2, 127–135 [PubMed] [Google Scholar]
- 16. Li B. H., Yang X. Z., Li P. D., Yuan Q., Liu X. H., Yuan J., Zhang W. J. (2008) IL-4/Stat6 activities correlate with apoptosis and metastasis in colon cancer cells. Biochem. Biophys. Res. Commun. 369, 554–560 [DOI] [PubMed] [Google Scholar]
- 17. Gooch J. L., Christy B., Yee D. (2002) STAT6 mediates interleukin-4 growth inhibition in human breast cancer cells. Neoplasia 4, 324–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lahm H., Schnyder B., Wyniger J., Borbenyi Z., Yilmaz A., Car B. D., Fischer J. R., Givel J. C., Ryffel B. (1994) Growth inhibition of human colorectal-carcinoma cells by interleukin-4 and expression of functional interleukin-4 receptors. Int. J. Cancer 59, 440–447 [DOI] [PubMed] [Google Scholar]
- 19. Morisaki T., Yuzuki D. H., Lin R. T., Foshag L. J., Morton D. L., Hoon D. S. (1992) Interleukin 4 receptor expression and growth inhibition of gastric carcinoma cells by interleukin 4. Cancer Res. 52, 6059–6065 [PubMed] [Google Scholar]
- 20. Morisaki T., Uchiyama A., Yuzuki D., Essner R., Morton D. L., Hoon D. S. (1994) Interleukin 4 regulates G1 cell cycle progression in gastric carcinoma cells. Cancer Res. 54, 1113–1118 [PubMed] [Google Scholar]
- 21. Cheon J., Chung D. J., Kim J. J., Koh S. K., Sohn J. (1996) Inhibitory effects of interleukin-4 on human renal cell carcinoma cells in vitro. In combination with interferon-α, tumor necrosis factor-α or interleukin-2. Int. J. Urol. 3, 196–201 [DOI] [PubMed] [Google Scholar]
- 22. Yu S. J., Kim H. S., Cho S. W., Sohn J. (2004) IL-4 inhibits proliferation of renal carcinoma cells by increasing the expression of p21WAF1 and IRF-1. Exp. Mol. Med. 36, 372–379 [DOI] [PubMed] [Google Scholar]
- 23. Aoudjehane L., Podevin P., Scatton O., Jaffray P., Dusanter-Fourt I., Feldmann G., Massault P. P., Grira L., Bringuier A., Dousset B., Chouzenoux S., Soubrane O., Calmus Y., Conti F. (2007) Interleukin-4 induces human hepatocyte apoptosis through a Fas-independent pathway. FASEB J. 21, 1433–1444 [DOI] [PubMed] [Google Scholar]
- 24. Mangan D. F., Robertson B., Wahl S. M. (1992) IL-4 enhances programmed cell death (apoptosis) in stimulated human monocytes. J. Immunol. 148, 1812–1816 [PubMed] [Google Scholar]
- 25. Lee Y. W., Kühn H., Hennig B., Toborek M. (2000) IL-4 induces apoptosis of endothelial cells through the caspase-3-dependent pathway. FEBS Lett. 485, 122–126 [DOI] [PubMed] [Google Scholar]
- 26. Bailey D. P., Kashyap M., Mirmonsef P., Bouton L. A., Domen J., Zhu J., Dessypris E. N., Ryan J. J. (2004) Interleukin-4 elicits apoptosis of developing mast cells via a Stat6-dependent mitochondrial pathway. Exp. Hematol. 32, 52–59 [DOI] [PubMed] [Google Scholar]
- 27. Zhang W. J., Li B. H., Yang X. Z., Li P. D., Yuan Q., Liu X. H., Xu S. B., Zhang Y., Yuan J., Gerhard G. S., Masker K. K., Dong C., Koltun W. A., Chorney M. J. (2008) IL-4-induced Stat6 activities affect apoptosis and gene expression in breast cancer cells. Cytokine 42, 39–47 [DOI] [PubMed] [Google Scholar]
- 28. Chang T. L., Peng X., Fu X. Y. (2000) Interleukin-4 mediates cell growth inhibition through activation of Stat1. J. Biol. Chem. 275, 10212–10217 [DOI] [PubMed] [Google Scholar]
- 29. Lee J. Y., Yu S. J., Park Y. G., Kim J., Sohn J. (2007) Glycogen synthase kinase 3β phosphorylates p21WAF1/CIP1 for proteasomal degradation after UV irradiation. Mol. Cell. Biol. 27, 3187–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim H. D., Jang C. Y., Choe J. M., Sohn J., Kim J. (2012) Phenylbutyric acid induces the cellular senescence through an Akt/p21WAF1 signaling pathway. Biochem. Biophys. Res. Commun. 422, 213–218 [DOI] [PubMed] [Google Scholar]
- 31. Kawakami K., Joshi B. H., Puri R. K. (2000) Sensitization of cancer cells to interleukin 13-pseudomonas exotoxin-induced cell death by gene transfer of interleukin 13 receptor α chain. Hum. Gene Ther. 11, 1829–1835 [DOI] [PubMed] [Google Scholar]
- 32. Obiri N. I., Hillman G. G., Haas G. P., Sud S., Puri R. K. (1993) Expression of high affinity interleukin-4 receptors on human renal cell carcinoma cells and inhibition of tumor cell growth in vitro by interleukin-4. J. Clin. Invest. 91, 88–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bernard J., Treton D., Vermot-Desroches C., Boden C., Horellou P., Angevin E., Galanaud P., Wijdenes J., Richard Y. (2001) Expression of interleukin 13 receptor in glioma and renal cell carcinoma. IL13Rα2 as a decoy receptor for IL13. Lab. Invest. 81, 1223–1231 [DOI] [PubMed] [Google Scholar]
- 34. Walston J., Arking D. E., Fallin D., Li T., Beamer B., Xue Q., Ferrucci L., Fried L. P., Chakravarti A. (2005) IL-6 gene variation is not associated with increased serum levels of IL-6, muscle, weakness, or frailty in older women. Exp. Gerontol. 40, 344–352 [DOI] [PubMed] [Google Scholar]
- 35. Kuilman T., Michaloglou C., Vredeveld L. C., Douma S., van Doorn R., Desmet C. J., Aarden L. A., Mooi W. J., Peeper D. S. (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031 [DOI] [PubMed] [Google Scholar]
- 36. Acosta J. C., O'Loghlen A., Banito A., Guijarro M. V., Augert A., Raguz S., Fumagalli M., Da Costa M., Brown C., Popov N., Takatsu Y., Melamed J., d'Adda di Fagagna F., Bernard D., Hernando E., Gil J. (2008) Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018 [DOI] [PubMed] [Google Scholar]
- 37. Keegan A., Nelms K., Paul W. E. (1994) The IL-4 receptor-signaling mechanisms. Adv. Exp. Med. Biol. 365, 211–215 [DOI] [PubMed] [Google Scholar]
- 38. Wang W., Chen J. X., Liao R., Deng Q., Zhou J. J., Huang S., Sun P. (2002) Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol. Cell. Biol. 22, 3389–3403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Iwasa H., Han J., Ishikawa F. (2003) Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells 8, 131–144 [DOI] [PubMed] [Google Scholar]
- 40. Haq R., Brenton J. D., Takahashi M., Finan D., Finkielsztein A., Damaraju S., Rottapel R., Zanke B. (2002) Constitutive p38HOG mitogen-activated protein kinase activation induces permanent cell cycle arrest and senescence. Cancer Res. 62, 5076–5082 [PubMed] [Google Scholar]
- 41. Hunt A. E., Williams L. M., Lali F. V., Foxwell B. M. (2002) IL-4 regulation of p38 MAPK signalling is dependent on cell type. Cytokine 18, 295–303 [DOI] [PubMed] [Google Scholar]
- 42. Kawakami K., Kawakami M., Puri R. K. (2001) Overexpressed cell surface interleukin-4 receptor molecules can be successfully targeted for antitumor cytotoxin therapy. Crit. Rev. Immunol. 21, 299–310 [PubMed] [Google Scholar]
- 43. Kioi M., Takahashi S., Kawakami M., Kawakami K., Kreitman R. J., Puri R. K. (2005) Expression and targeting of interleukin-4 receptor for primary and advanced ovarian cancer therapy. Cancer Res. 65, 8388–8396 [DOI] [PubMed] [Google Scholar]
- 44. Bromberg J. F., Horvath C. M., Wen Z., Schreiber R. D., Darnell J. E., Jr. (1996) Transcriptionally active Stat1 is required for the antiproliferative effects of both interferon α and interferon γ. Proc. Natl. Acad. Sci. U.S.A. 93, 7673–7678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kwong J., Hong L., Liao R., Deng Q., Han J., Sun P. (2009) p38α and p38γ mediate oncogenic ras-induced senescence through differential mechanisms. J. Biol. Chem. 284, 11237–11246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Courtois-Cox S., Jones S. L., Cichowski K. (2008) Many roads lead to oncogene-induced senescence. Oncogene 27, 2801–2809 [DOI] [PubMed] [Google Scholar]
- 47. Pesu M., Aittomäki S., Takaluoma K., Lagerstedt A., Silvennoinen O. (2002) p38 Mitogen-activated protein kinase regulates interleukin-4-induced gene expression by stimulating STAT6-mediated transcription. J. Biol. Chem. 277, 38254–38261 [DOI] [PubMed] [Google Scholar]
- 48. Wery-Zennaro S., Zugaza J. L., Letourneur M., Bertoglio J., Pierre J. (2000) IL-4 regulation of IL-6 production involves Rac/Cdc42- and p38 MAPK-dependent pathways in keratinocytes. Oncogene 19, 1596–1604 [DOI] [PubMed] [Google Scholar]
- 49. Macip S., Igarashi M., Fang L., Chen A., Pan Z. Q., Lee S. W., Aaronson S. A. (2002) Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 21, 2180–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Arpa L., Valledor A. F., Lloberas J., Celada A. (2009) IL-4 blocks M-CSF-dependent macrophage proliferation by inducing p21Waf1 in a STAT6-dependent way. Eur. J. Immunol. 39, 514–526 [DOI] [PubMed] [Google Scholar]
- 51. Melk A. (2003) Senescence of renal cells. Molecular basis and clinical implications. Nephrol. Dial. transplant. 18, 2474–2478 [DOI] [PubMed] [Google Scholar]
- 52. Furusu A., Miyazaki M., Koji T., Abe K., Ozono Y., Harada T., Nakane P. K., Hara K., Kohno S. (1997) Involvement of IL-4 in human glomerulonephritis. An in situ hybridization study of IL-4 mRNA and IL-4 receptor mRNA. J. Am. Soc. Nephrol. 8, 730–741 [DOI] [PubMed] [Google Scholar]
- 53. Singh R. R., Saxena V., Zang S., Li L., Finkelman F. D., Witte D. P., Jacob C. O. (2003) Differential contribution of IL-4 and STAT6 vs STAT4 to the development of lupus nephritis. J. Immunol. 170, 4818–4825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ito H., Yan X., Nagata N., Aritake K., Katsumata Y., Matsuhashi T., Nakamura M., Hirai H., Urade Y., Asano K., Kubo M., Utsunomiya Y., Hosoya T., Fukuda K., Sano M. (2012) PGD2-CRTH2 pathway promotes tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 23, 1797–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ni Z., Lou W., Lee S. O., Dhir R., DeMiguel F., Grandis J. R., Gao A. C. (2002) Selective activation of members of the signal transducers and activators of transcription family in prostate carcinoma. J. Urol. 167, 1859–1862 [PubMed] [Google Scholar]
- 56. Guiter C., Dusanter-Fourt I., Copie-Bergman C., Boulland M. L., Le Gouvello S., Gaulard P., Leroy K., Castellano F. (2004) Constitutive STAT6 activation in primary mediastinal large B-cell lymphoma. Blood 104, 543–549 [DOI] [PubMed] [Google Scholar]