

Background: mTORC1 is a protein kinase that plays a central role in cell growth and metabolism.
Results: mTORC1 signaling activity is affected by expression levels of two apoptotic proteins, Bcl-2 and Bcl-XL.
Conclusion: Bcl-2 and Bcl-XL control mTORC1 activity by affecting the binding of mTORC1 with its inhibitor FKBP38.
Significance: The novel cross-talk between Bcl-2/XL and mTORC1 signaling is likely to play an important role in cancer development.
Keywords: Bcl-2, Mitochondria, mTOR Complex (mTORC), S6 Kinase, Signaling, Bcl-XL, FKBP38, mTORC1
Abstract
Mammalian target of rapamycin complex 1 (mTORC1) is a key regulator of cell growth and metabolism. Its activity is controlled by various types of signals, including growth factors, nutrients, and stresses. In this study, we show that changes in expression levels of two antiapoptotic proteins, Bcl-2 and Bcl-XL, also affect mTORC1 signaling activity. In cells overexpressing Bcl-XL, mTORC1 activity is increased and becomes less sensitive to growth factor or nutrient conditions. In contrast, reduction in expression levels of the two antiapoptotic proteins inhibits mTORC1 signaling activity. Our results suggest that the effect of Bcl-2 and Bcl-XL on mTORC1 is mediated by FKBP38, an inhibitor of mTORC1. The two proteins compete with mTORC1 for FKBP38 binding and hence alter mTORC1 activity. This study reveals a novel cross-talk between Bcl-2/XL and mTORC1 signaling, which is likely to contribute to cancer development.
Introduction
Evasion of cell death is one of the hallmarks in cancer development (1). Cancer cells achieve this feat by augmenting activities of various pathways that allow survival under stress conditions. One major mechanism for enhancing cell survival involves overexpression of antiapoptotic proteins of the Bcl-2 family, such as Bcl-2 and Bcl-XL. Elevated levels of these antiapoptotic proteins are often associated with cancer progression and contribute to the resistance of cancer cells to conventional cancer therapies (2, 3).
Bcl-2 and Bcl-XL exert their antiapoptotic function by binding to proapoptotic proteins, Bax and Bak, and thereby preventing their oligomerization on the mitochondrial membrane that leads to mitochondrial permeabilization (2). To perform this function, the two proteins have to be recruited to mitochondria and attached to mitochondrial outer membrane through their transmembrane domain. The recruitment is achieved by association with FKBP38, a member of FK506-binding protein family that resides on mitochondria (4). Overexpression of FKBP38 increases the levels of Bcl-2 and Bcl-XL on mitochondria and enhances resistance to apoptosis, whereas down-regulation of FKBP38 reduces the mitochondrial localized Bcl-2 and Bcl-XL, rendering cells more vulnerable to apoptosis (4, 5).
In addition to regulating Bcl-2 and Bcl-XL function, FKBP38 also acts as an inhibitor of the mammalian target of rapamycin (mTOR),4 a PI3K-related protein kinase (6). mTOR controls many growth-related processes by integrating signals of various origins, including those of growth factors, nutrients, cellular energy, and stresses. It exists in two distinct complexes, termed as mTOR complex 1 (mTORC1) and 2 (mTORC2) (7). mTORC1 promotes cell growth mainly by stimulation of protein synthesis, which is achieved through phosphorylation of ribosomal S6 protein kinase (S6K) and 4E-BP1, two key factors in translation initiation (8). mTORC2, on the other hand, acts in a distinct way. It phosphorylates Akt and enhances its prosurvival function (9). Despite the presence of mTOR in both complexes, rapamycin, in complex with another member of the FK506 protein family, FKBP12, specifically targets mTORC1 and blocks its function. FKBP38, operating in a manner analogous to that of the rapamycin-FKBP12 complex, binds to mTORC1 and prevents its activation (10).
The association of FKBP38 with mTOR as well as with Bcl-2 and Bcl-XL is regulated by the Ras-like small GTPase Rheb in response to changes in growth factor and nutrient conditions. Upon serum or amino acid stimulation, an active Rheb binds to FKBP38 and releases Bcl-2/XL and mTORC1 from its interaction. However, under serum or amino acid deprivation conditions, an inactive Rheb allows binding of FKBP38 with these proteins, which causes down-regulation of mTORC1 as well as accumulation of Bcl-2/XL on mitochondria (5, 6).
Because both mTOR and the two antiapoptotic proteins are capable of binding to FKBP38, it is possible that Bcl-2/XL and mTOR may influence each other's function by competing for FKBP38 binding. In this regard, changes in expression levels of Bcl-2/XL are expected to alter the binding of FKBP38 with mTOR and vice versa, hence affecting each other's activity. Although fluctuations in expression level do not appear to be a major mechanism for mTOR regulation, they do affect dramatically the antiapoptotic function of Bcl-2 and Bcl-XL. An increased expression of these two proteins is often associated with the resistance to apoptosis in cancer cells. Therefore, a potential regulation may occur to mTORC1 due to changes in the levels of the antiapoptotic proteins. In this study, we examined the putative cross-talk between mTORC1 and the two apoptotic proteins.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
HEK293 cells used in this study were obtained from ATCC. HEK293-Myc-FKBP38 and its vector control line (HEK293-Myc) were created by transfecting HEK293 cells with pcDNA3.1-3×Myc-FKBP38 or control vector and selecting clones stably expressing Myc-FKBP38 at a level similar to its endogenous copy. HCT116 Bax+/+ and HCT116 Bax−/− cells were gifts from Dr. L. Zhang with permission for use from Dr. B. Vogelstein (11). Cultured cells were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS). A CellTiter-Glo® luminescent cell viability assay kit from Promega was used to assay cell viability. The oxygen consumption rate of the transfected cells was determined using an XF extracellular flux analyzer (Seahorse Bioscience). For amino acid starvation and repletion, cells were incubated in DMEM medium free of amino acids for 1 h; 50× amino acid stock mixture (Sigma) or vehicle control was added to the medium to a final concentration of 1×. Cells were harvested and lysed 30 min after the addition of amino acids or vehicle control. For serum deprivation and readdition, cells were shifted to DMEM medium contain 0.2% FBS for 24 h. FBS (HyClone) was then added back to the medium to a final concentration of 10%. Cells were harvested and lysed upon incubation for 30 min after the addition of FBS. All experiments were repeated at least three times. Representative data are shown in the figures.
Antibodies and Plasmids
Antibodies for S6K, phospho-S6K(Thr-389), S6, phospho-S6(Ser-235/236), 4E-BP1, phospho-4E-BP1(Thr-37/46), Bcl-XL, Mcl-1, β-actin, and mTOR were purchased from Cell Signaling Technology Inc. (Danvers, MA). Antibody for Bcl-2 was from Dako (Carpentaria, CA), and antibody for Bax was from Santa Cruz Biotechnology. Rabbit anti-FKBP38 antibody and pcDNA3.1-HA-Bcl-XL were created in our previous study (5). pcDNA3.1-V5-Mcl-1 was kindly provided by Dr. L. Zhang. CMV-Bcl-XL-cyb5 and CMV-Bcl-XL-acta plasmids are gifts from Dr. Andrews at McMaster University (12). Plasmid transfections were performed using Invitrogen LipofectamineTM 2000 according to manufacturer's instructions.
shRNA Constructs
Bcl-2- and Bcl-XL-specific shRNAs were designed based on previously published RNAi sequences and cloned into pSIH-H1 shRNA expression lentivector (System Biosciences). The two Bcl-2-specific RNAi sequences are: 5′-GTACATCCATTATAAGCTGTT-3′ (Bcl-2 shRNA-1) (13); 5′-GCTGCACCTGACGCCCTTCTT-3′ (Bcl-2 shRNA-2) (14). The two Bcl-XL-specific RNAi sequences are: 5′-GTGCGTGGAAAGCGTAGACAA-3′ (Bcl-XL-shRNA-1) (15); 5′-CAGGGACAGCATATCAGACTT-3′ (Bcl-XL shRNA-2) (14). The two Bcl-w-specific RNAi sequences are: 5′-GAGGAAGGTGGACTTACATAA-3′ (Bcl-w shRNA-1); 5′-TGGCAGACTTTGTAGGTTATA-3′ (Bcl-w shRNA-2). The RNAi oligonucleotides were cloned into a lentiviral expression vector, pLKO.1-puro. A nontargeting shRNA control in the same vector from Addgene was used as a control. siRNA for Mcl-1 was from Santa Cruz Biotechnology. The FKBP38-specific shRNA construct (pLKO.1-FKBP38 shRNA) was purchased from Sigma (Clone ID TRCN 0000010595). This shRNA was used to create a HEK293 line stably expressing the construct, which reduces endogenous expression of FKBP38 by ∼90%. For HEK293 and HeLa cells, shRNAs were transfected with LipofectamineTM 2000 from Invitrogen. For other cell lines, shRNAs were first packed as lentiviral particles with the package system from Invitrogen and used for infecting the cells. The effects of shRNAs were analyzed 72 h after the transfection or infection.
Co-immunoprecipitation
Cultured cells at 80–90% confluence were washed with cold PBS on ice and lysed in buffer containing 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 1 mm PMSF, and 1× protease inhibitor mixture (Roche Applied Science). Lysates (5 mg) were incubated with anti-Myc antibody (9E10)-linked protein A-agarose beads for 3 h at 4 °C with agitation. Beads were washed four times with lysis buffer and once with 20 mm Tris-HCl (pH 7.4) and boiled for 5 min in 60 μl of 2× SDS sample buffer. Samples were subjected to SDS-PAGE. Western blotting was performed by standard protocols and developed using ECL reagents (BD Biosciences). Myc-tagged and endogenous FKBP38 proteins were detected with anti-FKBP38 antibody.
RESULTS
We and others have previously shown that both mTOR and antiapoptotic proteins, Bcl-2 and Bcl-XL, are able to interact directly with FKBP38 (4–6). To test whether Bcl-2 and Bcl-XL are able to affect mTORC1 activity through binding of FKBP38, we examined the effect of Bcl-XL overexpression on mTORC1 signaling activity in HEK293 cells. As shown in Fig. 1, we found that overexpression of Bcl-XL enhanced mTORC1-dependent phosphorylation of S6K, S6 ribosomal protein (S6), and 4E-BP1 in a dose-dependent manner under serum deprivation or amino acid starvation condition (Fig. 1, A and B). Similarly, the overexpression also partially blocked dephosphorylation of S6K and 4E-BP1 induced by serum deprivation (Fig. 1C). These observations thus confirmed that high level of Bcl-XL overexpression is able to enhance mTORC1 signaling activity. An attempt to test the effect of Bcl-2 overexpression was hindered by its toxic effect on cell growth, a phenomenon that was observed previously by other groups (16, 17). Although overexpression of Bcl-XL increased mTORC1 signaling activity, we found that overexpression of another antiapoptotic protein, Mcl-1, had no obvious effect on the activity (Fig. 1D).
FIGURE 1.

Overexpression of Bcl-XL activates mTORC1. HEK293 cells transfected with HA-S6K and increasing doses of HA-Bcl-XL were deprived of serum (A) or amino acids (AA) (B). The mTORC1-dependent phosphorylation (P) of HA-S6K, S6, and 4E-BP1 was determined by Western blotting. C, HEK293 cells transfected with HA-S6K and HA-Bcl-XL were shifted to serum deprivation condition. Cells were collected at the indicated time points after the shift, and mTORC1-dependent phosphorylation of HA-S6K and 4E-BP1 in the cells was determined by Western blotting. D, mTORC1-dependent phosphorylation of HA-S6K, S6, and 4E-BP1 was determined in serum-deprived HEK293 cells transfected with HA-S6K together with either HA-Bcl-XL or V5-Mcl-1.
Although Bcl-XL functions mainly on mitochondria, it also localizes to other cellular compartments, including the nucleus and ER. To determine whether the effect of Bcl-XL is mitochondria-specific, we employed mutant Bcl-XL proteins with the membrane anchor replaced either by an ER targeting sequence of cytochrome b5 (Bcl-XL-cyb5) or by a mitochondrial targeting sequence of ActA (Bcl-XL-acta). These two chimeric proteins have been shown previously to target specifically to ER and mitochondria, respectively (12). We found that the overexpressed mitochondria-targeted Bcl-XL retained the ability to stimulate mTORC1 activity under serum deprivation condition, but the ER-targeted Bcl-XL lost this ability. Likewise, a mutant Bcl-XL without its membrane anchor (Bcl-XLΔTM) was ineffective (Fig. 2). These observations suggest that the ability of Bcl-XL in mTORC1 regulation depends on its mitochondrial localization.
FIGURE 2.
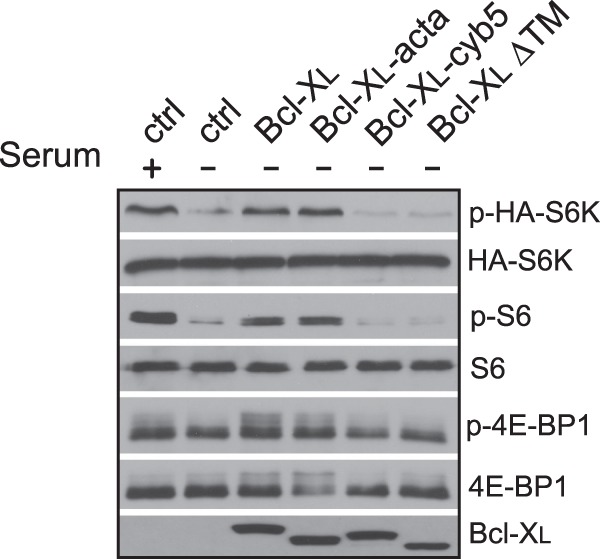
Mitochondrial localization is required for the stimulatory effect of Bcl-XL on mTORC1. HEK293 cells were transfected with HA-S6K along with empty vector (ctrl) or vectors expressing wild type (Bcl-XL), mitochondria-targeted (Bcl-XL-acta), ER-targeted (Bcl-XL-cyb5), or a mutant Bcl-XL lacking transmembrane domain (Bcl-XLΔTM). Transfected cells were grown under serum deprivation condition for 24 h. mTORC1-dependent phosphorylation (P) of HA-S6K, S6, and 4E-BP1 in the cells was determined by Western blotting.
Because overexpression of Bcl-XL enhanced mTORC1 activity, we further determined whether reduction of Bcl-2 and Bcl-XL levels had an opposite effect. Accordingly, we examined mTORC1 activity in HEK293 cells with reduced expression of Bcl-2 and Bcl-XL by specific shRNAs. As shown in Fig. 3A, we found that knockdown of Bcl-2 decreased mTORC1-dependent phosphorylation of its downstream targets. The diminished mTORC1 activity appeared to correlate with the extent of the knockdown. In cells treated with Bcl-2-shRNA-1, which reduced the level of Bcl-2 by more than 90%, we observed a drastic decrease in mTORC1 activity, whereas in cells treated with another shRNA (Bcl-2-shRNA-2) that was not as effective in the knockdown, we found that the down-regulation in mTORC1 was moderate. Similar results were obtained when HEK293 cells were treated with Bcl-XL-specific shRNAs (Fig. 3B). Again, the effectiveness of these shRNAs on mTORC1 activity correlated with their ability in knockdown of Bcl-XL. The effect of Bcl-2 and Bcl-XL knockdown was mTORC1-specific because we found that the activity of mTORC2, as measured by the mTORC2-dependent phosphorylation of Akt at position Thr-473, was not changed by the knockdown.5 To further confirm the observation from HEK293 cells, we examined the effect of Bcl-2 and Bcl-XL knockdown on mTORC1 activity in several cancer cell lines. We found that, as in HEK293 cells, knockdown of Bcl-2 or Bcl-XL reduced mTORC1 activity in lung cancer line A549, breast cancer line MCF-7, and two prostate cancer lines, LNCaP and C4-2 (Fig. 3C).5 Thus, the effect of Bcl-2 and Bcl-XL knockdown on mTORC1 activity appears to be a common response.
FIGURE 3.
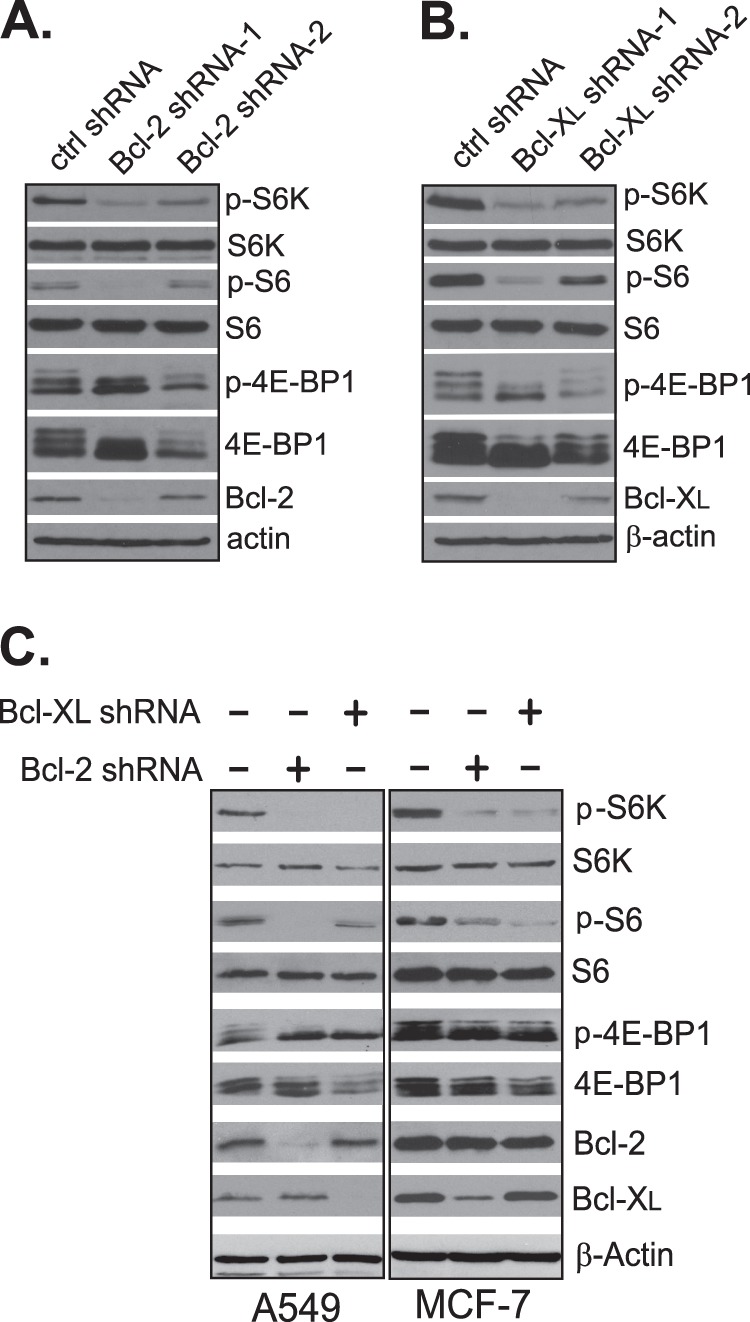
Knockdown of Bcl-2 or Bcl-XL reduces mTORC1 activity. HEK293 cells were transfected with scrambled control shRNAs (ctrl) or Bcl-2- (A) or Bcl-XL-specific shRNAs (B). Cells were harvested after 72 h, and mTORC1-dependent phosphorylation (P) was determined by Western blotting. C, cancer cells were infected with lentiviral particles expressing control shRNA, Bcl-2 shRNA-1, or Bcl-XL shRNA-1. Cells were harvested after 72 h, and protein levels and mTORC1-dependent phosphorylation in the infected cells were determined by Western blotting.
In contrast to Bcl-2 and Bcl-XL, knockdown of two antiapoptotic proteins, Mcl-1 and Bcl-w, did not have any effect on mTORC1 activity (Fig. 4, A and B), suggesting that mTORC1 activity is specifically affected by the levels of Bcl-2 and Bcl-XL. Both Bcl-2 and Bcl-XL have been shown previously to interact with FKBP38 (4, 5). However, we found that under the same condition at which Bcl-XL interacted with FKBP38, Bcl-w and Mcl-1 did not (Fig. 4C).5 In this regard, the activity of these antiapoptotic proteins in mTORC1 regulation appeared to correlate with their ability for binding with FKBP38.
FIGURE 4.
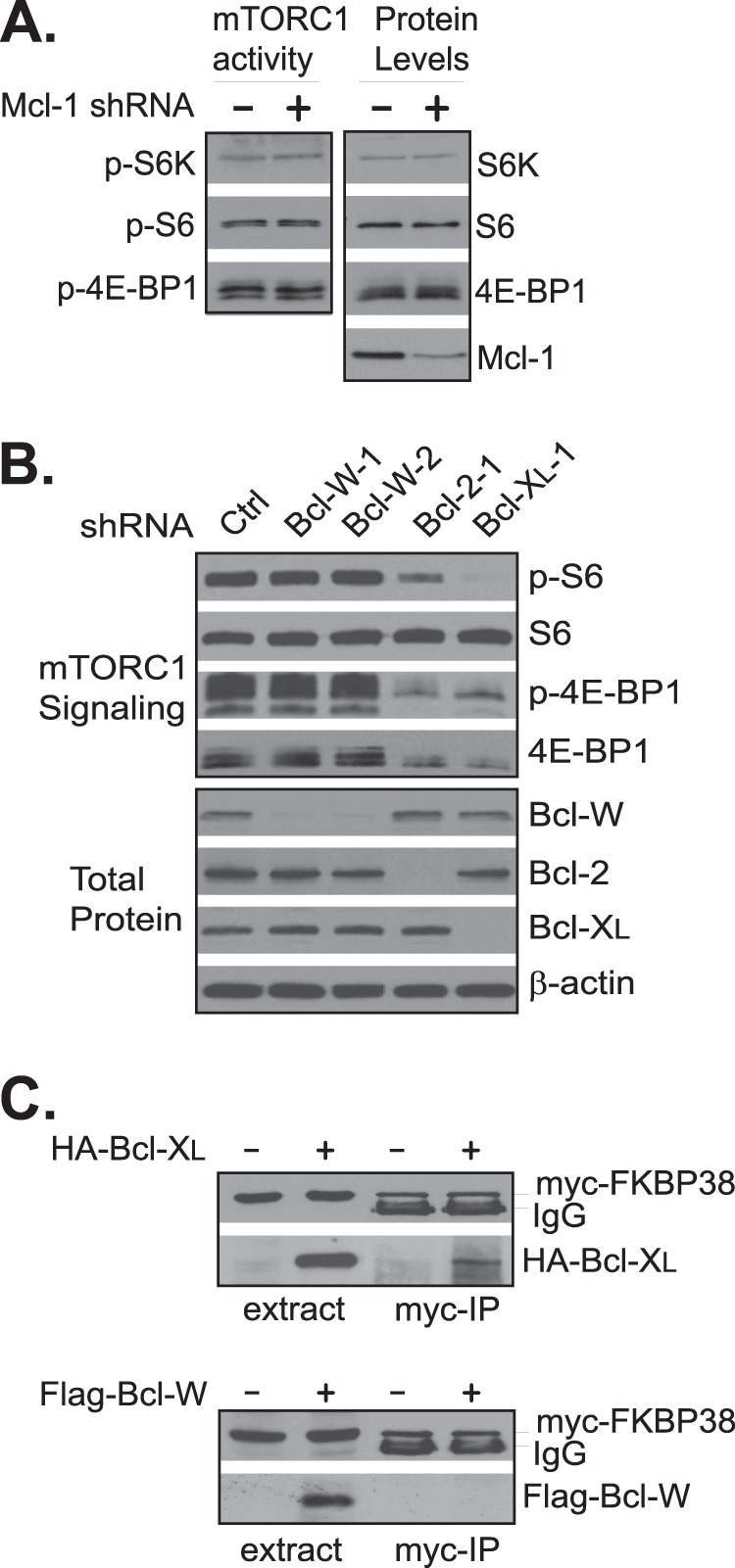
Knockdown of Mcl-1 and Bcl-w does not affect mTORC1 activity. A, HEK293 cells were transfected with control (−) or Mcl-1-specific siRNA (+). P, phosphorylation. B, HEK293 cells were transfected with control, Bcl-w-, Bcl-2-, or Bcl-XL-specific shRNAs. The corresponding protein levels and mTORC1-dependent phosphorylation in the transfected cells were determined 72 h after the transfection. C, HEK293 cells stably expressing Myc-FKBP38 were transfected with empty vector (−), HA-Bcl-XL (upper panel), or FLAG-Bcl-w (lower panel) expression plasmid (+). The interaction of Myc-FKBP38 with the expressed HA-Bcl-XL and FLAG-Bcl-w was assayed by co-immunoprecipitation with anti-Myc antibody. The levels of HA-Bcl-XL (upper panel) and FLAG-Bcl-w (lower panel) in the cell extracts and precipitates (Myc-IP) were determined by Western blotting using anti-HA and -FLAG antibodies, respectively. The levels of Myc-FKBP38 were determined using anti-Myc antibody.
Because of the role of Bcl-2 and Bcl-XL in cell survival, it was possible that the effect on mTORC1 activity associated with Bcl-2 and Bcl-XL knockdown was caused indirectly by loss of cell viability. To test this possibility, we examined the viability of cells treated with Bcl-2 and Bcl-XL shRNAs and failed to detect a significant reduction in overall cell viability under the same growth condition at which we observed a drastic down-regulation in mTORC1 activity (Fig. 5A). We also did not detect any obvious defects in mitochondrial oxygen consumption of those treated cells (Fig. 5B), ruling out mitochondria deregulation as a potential cause for the reduced mTORC1 activity. Furthermore, we found that mTORC1 activity was not affected in cells treated with Abt-263, a potent inhibitor of Bcl-2 (Fig. 5C). These findings suggest that the down-regulation in mTORC1 activity was not caused by reduction in cell viability. To further rule out the role of apoptosis in the effect induced by the Bcl-2 and Bcl-XL knockdown, we examined the mTORC1 activity in Bax null cells, which are resistant to apoptosis due to the absence of Bax, a major proapoptotic protein (11). We found that knockdown of either Bcl-2 or Bcl-XL in the Bax null cells reduced mTORC1 activity as in the wild type control (Fig. 5D). Taken together, these findings suggest that a reduced viability was not the cause for the down-regulated mTORC1 activity in cells treated with Bcl-2- and Bcl-XL-specific shRNAs.
FIGURE 5.
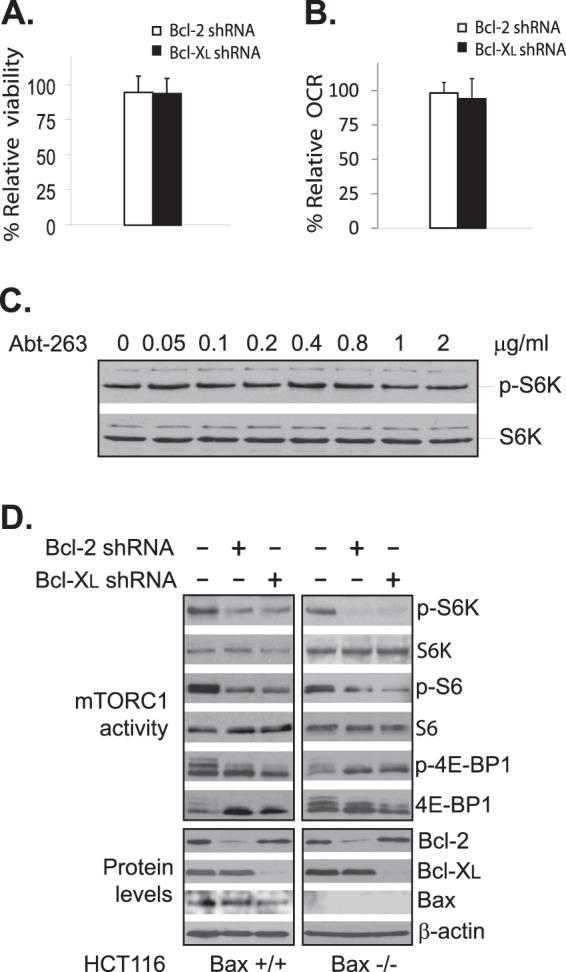
Apoptosis does not contribute to the down-regulation of mTORC1 induced by Bcl-2 or Bcl-XL knockdown. HEK293 cells were transfected with scrambled (ctrl), Bcl-2 shRNA-1, or Bcl-XL shRNA-1. Cell viability (A) and oxygen consumption rate (OCR) (B) of the transfected cells were assayed 72 h after the transfection. The viability and oxygen consumption rate of the cells transfected with Bcl-2- or Bcl-XL-specific shRNA were normalized against that of the cells transfected with scrambled shRNA. Data represent the mean ± S.D. of the values from three independent experiments. C, HEK293 cells were treated with increasing concentrations of Abt-263 for 4 h. mTORC1-dependent phosphorylation (P) of S6K (upper half) and S6K protein levels (lower half) in the treated cells were determined by Western blotting. D, HCT116 Bax null (Bax −/−) and its wild type control (Bax +/+) cells were transfected with scrambled control shRNA (−), Bcl-2 shRNA-1, or Bcl-XL shRNA-1(+). The expression levels of Bcl-2 and Bcl-XL as well as the mTORC1-dependent phosphorylation of S6K, S6, and 4E-BP1 were determined by Western blotting.
Previous studies have shown that Bcl-2, Bcl-XL, and mTOR are all able to interact with FKBP38 (5, 6), which raises the possibility that Bcl-2 and Bcl-XL may affect mTORC1 activity by competing with mTOR for FKBP38 binding. In this regard, a reduced level of Bcl-2 and Bcl-XL may allow more FKBP38 to interact with mTORC1 and hence reduce its activity. To investigate this possibility, we examined the effect of Bcl-2 or Bcl-XL knockdown on mTORC1 activity in cells with FKBP38 levels reduced by a stably expressed shRNA. As shown in Fig. 6, we found that although knockdown of Bcl-2 or Bcl-XL reduced mTORC1 activity in cells expressing a scrambled control shRNA, it failed to do so in cells expressing FKBP38 shRNA. This finding indicates that FKBP38 is involved in the Bcl-2/XL-dependent regulation of mTORC1.
FIGURE 6.
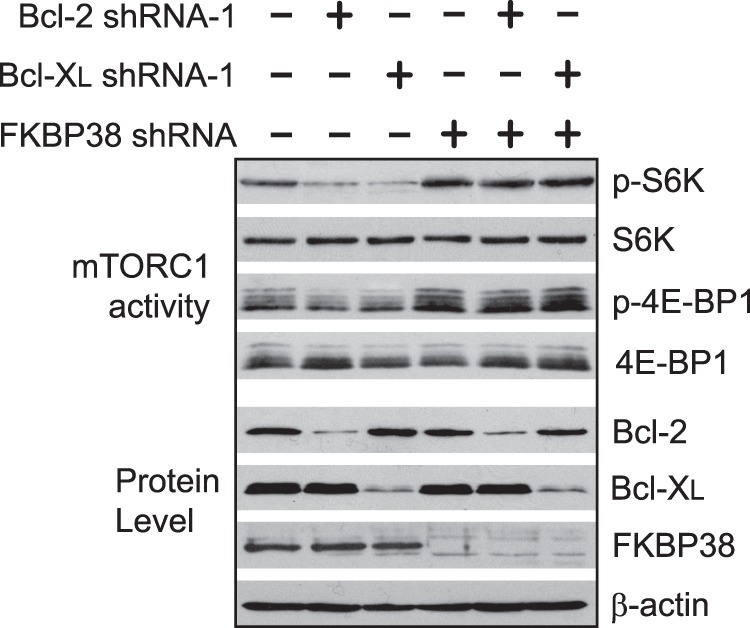
FKBP38 down-regulation abolishes the effect of Bcl-2 and Bcl-XL knockdown on mTORC1 activity. HEK293 cells stably expressing an FKBP38-specific shRNA (+) or a scrambled shRNA (−) were transfected with scrambled control shRNA (−) or Bcl-2- or Bcl-XL-specific shRNA (+). mTORC1-dependent phosphorylation (P) and expression levels of Bcl-2, Bcl-XL, FKBP38, and actin in transfected cells were determined by Western blotting.
We next determined whether the ability for binding with FKBP38 was essential for the effect of Bcl-XL. A previous study has shown that Bcl-2 interacts with FKBP38 through a region between its BH4 and BH3 domains (18). Accordingly, we created a mutant Bcl-XL incapable of FKBP38 binding by deleting the corresponding region (amino acids 45–81) in the protein (Fig. 7). We found that although a mitochondria-targeted wild type Bcl-XL (Bcl-XL-acta) stimulated mTORC1 activity under serum starvation condition, the mutant Bcl-XL (Bcl-XLΔ(45–81)-acta) failed to do so (Fig. 7B). This result suggests that the ability for FKBP38 binding is required for the effect of Bcl-XL on mTORC1. To further examine the role of FKBP38 in the process, we created a mutant protein containing the first 90 amino acids of Bcl-XL fused with GFP. This mutant protein preserved the FKBP38 binding domain of Bcl-XL but lacked the BH3, BH1, and BH2 functional domains (Fig. 7A). When expressed in cultured cells, the mutant protein (Bcl-XL-GFP-acta) was targeted to mitochondria6 and was able to bind with FKBP38 (Fig. 7C). Correlating with the ability for FKBP38 binding, this mitochondria-targeted fusion protein retained the capability to stimulate mTORC1 as its wild type counterpart (Fig. 7B). These observations demonstrate that the ability for interacting with FKBP38 but not the antiapoptotic function is required for Bcl-XL to affect mTORC1 activity.
FIGURE 7.
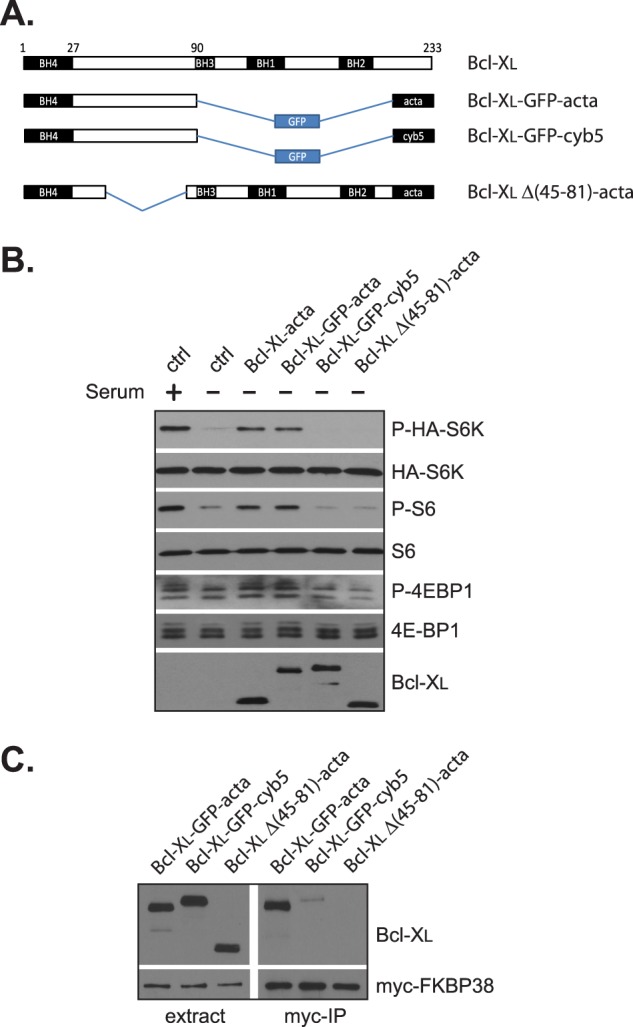
The ability for interaction with FKBP38 is required for the effect of Bcl-XL on mTORC1. A, schematic presentation of the mutant Bcl-XL constructs used in B and C. B, HEK293 cells transfected with HA-S6K and indicated Bcl-XL constructs were serum-deprived for 24 h. The expression levels of Bcl-XL as well as the mTORC1-dependent phosphorylation (P) of S6K, S6, and 4E-BP1 were determined by Western blotting. ctrl, empty vector. C, HEK293 cells stably expressing Myc-FKBP38 were transfected with Bcl-XL mutant constructs. The interaction of the mutant Bcl-XL proteins with Myc-FKBP38 was determined by immunoprecipitation with Myc antibody (Myc-IP).
To further ascertain the function of FKBP38 in the Bcl-XL-mediated effect, we tested whether the differences in mTORC1 activity induced by changes in expression levels of Bcl-XL correlated with its association with FKBP38. In our previous study, we showed that amino acid starvation induced a strong association between mTOR and FKBP38, which led to mTORC1 down-regulation (6). We hence analyzed the effect of Bcl-XL overexpression on the starvation-induced association by co-immunoprecipitation. We found that amino acid starvation increased the association in cells expressing a control vector but the effect was largely eliminated in cells overexpressing Bcl-XL (Fig. 8A). Consistent with what we observed previously (6), the changes in the association of mTOR with FKBP38 correlated inversely with the mTORC1 signaling activity. In contrast, we observed an increase in the association of mTOR with FKBP38 in cells expressing a Bcl-XL shRNA in comparison with the control cells expressing a scrambled shRNA. Again, the increase was associated with a decrease in mTORC1 signaling activity (Fig. 8B). These findings suggest that the differences in mTORC1 activity in response to changes in Bcl-2 and Bcl-XL expression levels are likely to be mediated by FKBP38.
FIGURE 8.
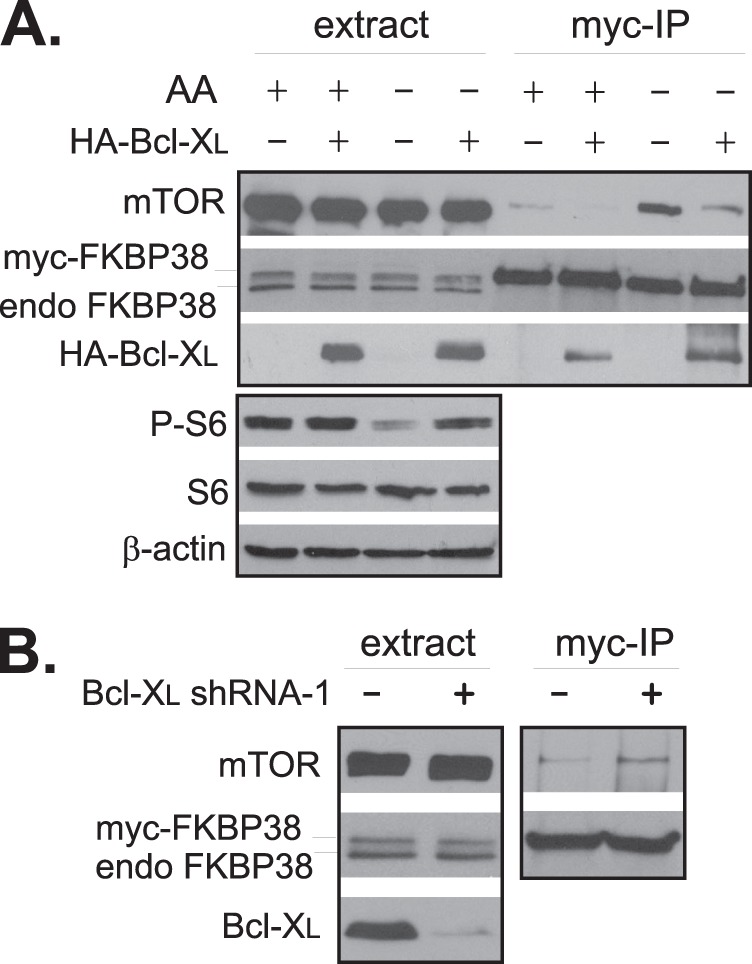
Changes in Bcl-XL levels affect the binding of FKBP38 with mTORC1. A, HEK293 cells stably expressing Myc-FKBP38 were transfected with HA-Bcl-XL expression plasmid (+) or empty vector (−). Transfected cells were starved for amino acids followed by repletion of amino acids (AA+) or vehicle control (AA−). The interaction of Myc-FKBP38 with endogenous mTOR was assayed by co-immunoprecipitation with anti-Myc antibody. The protein levels in the cell extracts and precipitates were determined by Western blotting with corresponding antibodies. endo FKBP38, endogenous expression of FKBP38; P, phosphorylation. B, HEK293 cells stably expressing Myc-FKBP38 were transfected with Bcl-XL shRNA-1 (+) or scrambled shRNA (−), and the association of Myc-FKBP38 with mTOR was determined by co-immunoprecipitation with anti-Myc antibody (Myc-IP).
DISCUSSION
Antiapoptotic proteins, Bcl-2 and Bcl-XL, and mTORC1 are important factors acting synergistically at opposite sides in controlling cell proliferation. These factors are believed to be regulated independently through different mechanisms. In the present study, we find that changes in Bcl-2 and Bcl-XL expression levels affect mTORC1 signaling activity. This finding reveals the existence of a previously unknown cross-talk that renders mTORC1 signaling activity susceptible to alterations in Bcl-2 and Bcl-XL levels. Given the fact that many cancer cells possess high levels of Bcl-2 and/or Bcl-XL, this Bcl-2/XL-dependent regulation of mTORC1 is likely to bear important implications in tumor cell survival and proliferation.
We have previously shown that Bcl-2 and Bcl-XL interact with FKBP38, an inhibitor of mTOR, which indicates that these antiapoptotic proteins are able to compete with mTOR for FKBP38 binding (5, 6). This competition may hence affect the amount of FKBP38 available for mTOR binding, allowing Bcl-2 and Bcl-XL to control mTORC1 activity. Several lines of evidence support this view. First, we find that the ability of Bcl-2 and Bcl-XL to affect mTORC1 signaling correlates with their ability to interact with FKBP38. Both Bcl-2 and Bcl-XL are able to bind to FKBP38 and regulate mTORC1 activity. However, two other antiapoptotic proteins, Bcl-w and Mcl-1, which do not bind to FKBP38 (Fig. 4C),6 are unable to control mTORC1 (Fig. 4, A and B). In addition, we show that a mutant Bcl-XL containing its FKBP38 binding domain but lacking three BH domains remains active for mTORC1 stimulation (Fig. 7), suggesting that the ability for FKBP38 binding but not the antiapoptotic function of Bcl-XL is essential for its effect on mTORC1. Second, we find that the inhibitory effect of Bcl-2 and Bcl-XL on mTORC1 is largely abolished in cells with a down-regulated FKBP38 level (Fig. 6). Third, we demonstrate that changes in Bcl-XL levels affect the association of mTOR with FKBP38, which correlates inversely with mTORC1 signaling activity (Fig. 8). It is hence likely that the cross-talk between the apoptotic proteins and mTORC1 is mediated by FKBP38.
The binding of Bcl-2 and Bcl-XL with FKBP38 is negatively regulated by nutrient and growth factor availability (5). It is thus expected that their competition with mTOR is at its strongest when growth factor or nutrient is limited. In this regard, it is surprising that under normal growth condition, when the competition is weak, down-regulation of Bcl-2 and Bcl-XL still has a remarkable impact on mTORC1 activity (Fig. 3). This phenomenon prompted us to examine other FKBP38-independent mechanisms. Because of the role of Bcl-2 and Bcl-XL in cell survival, it is possible that the inhibitory effect of their knockdown on mTORC1 is caused indirectly by reduced cell viability. However, the finding that the Bcl-2 and Bcl-XL knockdown remained effective in Bax null cells (Fig. 5D), which are largely resistant to apoptosis, argues against this possibility. In addition, we found that inhibition of Bcl-2 and Bcl-XL apoptotic function by Abt263 inhibitor, which blocks the association of these antiapoptotic proteins with proapoptotic proteins (19), had no effect on mTORC1 activity (Fig. 5C). These observations suggest that a reduced antiapoptotic function does not contribute to the effect of Bcl-2 and Bcl-XL knockdown on mTORC1. Furthermore, we did not detect any obvious effect of the knockdown on mitochondrial function (Fig. 5B), indicating that a compromised mitochondrial function is not a cause. Collectively, our data suggest that the effect of Bcl-2 and Bcl-XL on mTORC1 signaling activity is most likely to be mediated by FKBP38.
Overexpression of Bcl-2 and Bcl-XL is a common mechanism for cancer cells to elude apoptosis. The positive effect of the overexpression on mTORC1 activity indicates that up-regulation of these antiapoptotic proteins not only enhances cell survival but also stimulates mTOR-dependent proliferation in cancer cells. In support of this notion, it was found that up-regulation of Bcl-2 increased tumor cell proliferation and vascularization in prostate cancer xenografts (20), two events under control of mTORC1. Because translation of Bcl-2 and Bcl-XL is regulated by mTORC1-dependent activation of eIF4E (8), an enhanced mTORC1 activity is expected to increase the production of the two antiapoptotic proteins, which in turn is anticipated to further stimulate mTORC1 activity through the cross-talk. As such, the cross-talk may set forth a positive feed-forward cycle that promotes survival and cell proliferation and hence contributes to cancer progression.
Acknowledgments
We thank Drs. Zhang and Andrews for kindly providing expression plasmids and appreciate other laboratory members for comments and discussion.
This work was supported, in whole or in part, by National Institutes of Health Grants (CA129821 and CA169186 (to Yu Jiang)). This work was also supported by National Key Basic Research (973) Program of China 2010CB529704, NSFC Grant 81030055 and PCSIRT Grant IRT0731 (to Yong Jiang) and NSFC Grant 81272269 (to Yu Jiang).
Y. Lai and Y. Jiang, unpublished observations.
X. Zhao and Y. Jiang, unpublished observations.
- mTOR
- mammalian target of rapamycin
- mTORC
- mTOR complex
- S6K
- S6 protein kinase
- FKBP
- FK506-binding protein
- ER
- endoplasmic reticulum
- BH
- Bcl-2 homology.
REFERENCES
- 1. Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 2. Youle R. J., Strasser A. (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59 [DOI] [PubMed] [Google Scholar]
- 3. Kelly P. N., Strasser A. (2011) The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell Death Differ. 18, 1414–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shirane M., Nakayama K. I. (2003) Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat. Cell Biol. 5, 28–37 [DOI] [PubMed] [Google Scholar]
- 5. Ma D., Bai X., Zou H., Lai Y., Jiang Y. (2010) Rheb GTPase controls apoptosis by regulating interaction of FKBP38 with Bcl-2 and Bcl-XL. J. Biol. Chem. 285, 8621–8627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bai X., Ma D., Liu A., Shen X., Wang Q. J., Liu Y., Jiang Y. (2007) Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science 318, 977–980 [DOI] [PubMed] [Google Scholar]
- 7. Laplante M., Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ma X. M., Blenis J. (2009) Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318 [DOI] [PubMed] [Google Scholar]
- 9. Oh W. J., Jacinto E. (2011) mTOR complex 2 signaling and functions. Cell Cycle 10, 2305–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bai X., Jiang Y. (2010) Key factors in mTOR regulation. Cell. Mol. Life Sci. 67, 239–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang L., Yu J., Park B. H., Kinzler K. W., Vogelstein B. (2000) Role of BAX in the apoptotic response to anticancer agents. Science 290, 989–992 [DOI] [PubMed] [Google Scholar]
- 12. Fiebig A. A., Zhu W., Hollerbach C., Leber B., Andrews D. W. (2006) Bcl-XL is qualitatively different from and ten times more effective than Bcl-2 when expressed in a breast cancer cell line. BMC Cancer 6, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dai Y., Rahmani M., Corey S. J., Dent P., Grant S. (2004) A Bcr/Abl-independent, Lyn-dependent form of imatinib mesylate (STI-571) resistance is associated with altered expression of Bcl-2. J. Biol. Chem. 279, 34227–34239 [DOI] [PubMed] [Google Scholar]
- 14. Jiang M., Milner J. (2003) Bcl-2 constitutively suppresses p53-dependent apoptosis in colorectal cancer cells. Genes Dev. 17, 832–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tran N. L., McDonough W. S., Savitch B. A., Sawyer T. F., Winkles J. A., Berens M. E. (2005) The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling system regulates glioma cell survival via NFκB pathway activation and BCL-XL/BCL-W expression. J. Biol. Chem. 280, 3483–3492 [DOI] [PubMed] [Google Scholar]
- 16. Uhlmann E. J., Subramanian T., Vater C. A., Lutz R., Chinnadurai G. (1998) A potent cell death activity associated with transient high level expression of BCL-2. J. Biol. Chem. 273, 17926–17932 [DOI] [PubMed] [Google Scholar]
- 17. Wang N. S., Unkila M. T., Reineks E. Z., Distelhorst C. W. (2001) Transient expression of wild-type or mitochondrially targeted Bcl-2 induces apoptosis, whereas transient expression of endoplasmic reticulum-targeted Bcl-2 is protective against Bax-induced cell death. J. Biol. Chem. 276, 44117–44128 [DOI] [PubMed] [Google Scholar]
- 18. Kang C. B., Tai J., Chia J., Yoon H. S. (2005) The flexible loop of Bcl-2 is required for molecular interaction with immunosuppressant FK-506 binding protein 38 (FKBP38). FEBS Lett. 579, 1469–1476 [DOI] [PubMed] [Google Scholar]
- 19. Tse C., Shoemaker A. R., Adickes J., Anderson M. G., Chen J., Jin S., Johnson E. F., Marsh K. C., Mitten M. J., Nimmer P., Roberts L., Tahir S. K., Xiao Y., Yang X., Zhang H., Fesik S., Rosenberg S. H., Elmore S. W. (2008) ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 68, 3421–3428 [DOI] [PubMed] [Google Scholar]
- 20. Sakai Y., Goodison S., Kusmartsev S., Fletcher B., Eruslanov E., Cao W., Porvasnik S., Namiki K., Anai S., Rosser C. J. (2009) Bcl-2 mediated modulation of vascularization in prostate cancer xenografts. Prostate 69, 459–470 [DOI] [PubMed] [Google Scholar]