

Background: Type III polyketide synthases (PKSs) synthesize various polyketide and alkaloid scaffolds.
Results: QNS synthesizes quinolone as the single product, whereas ACS produces acridone as the major product.
Conclusion: QNS and ACS are novel quinolone- and acridone-producing type III PKSs, respectively.
Significance: Structure-function analyses of QNS and ACS provide insights into molecular bases for alkaloid biosyntheses.
Keywords: Biosynthesis, Cloning, Enzymes, Polyketides, Structural Biology
Abstract
Two novel type III polyketide synthases, quinolone synthase (QNS) and acridone synthase (ACS), were cloned from Citrus microcarpa (Rutaceae). The deduced amino acid sequence of C. microcarpa QNS is unique, and it shared only 56–60% identities with C. microcarpa ACS, Medicago sativa chalcone synthase (CHS), and the previously reported Aegle marmelos QNS. In contrast to the quinolone- and acridone-producing A. marmelos QNS, C. microcarpa QNS produces 4-hydroxy-N-methylquinolone as the “single product” by the one-step condensation of N-methylanthraniloyl-CoA and malonyl-CoA. However, C. microcarpa ACS shows broad substrate specificities and produces not only acridone and quinolone but also chalcone, benzophenone, and phloroglucinol from 4-coumaroyl-CoA, benzoyl-CoA, and hexanoyl-CoA, respectively. Furthermore, the x-ray crystal structures of C. microcarpa QNS and ACS, solved at 2.47- and 2.35-Å resolutions, respectively, revealed wide active site entrances in both enzymes. The wide active site entrances thus provide sufficient space to facilitate the binding of the bulky N-methylanthraniloyl-CoA within the catalytic centers. However, the active site cavity volume of C. microcarpa ACS (760 Å3) is almost as large as that of M. sativa CHS (750 Å3), and ACS produces acridone by employing an active site cavity and catalytic machinery similar to those of CHS. In contrast, the cavity of C. microcarpa QNS (290 Å3) is significantly smaller, which makes this enzyme produce the diketide quinolone. These results as well as mutagenesis analyses provided the first structural bases for the anthranilate-derived production of the quinolone and acridone alkaloid by type III polyketide synthases.
Introduction
The members of the chalcone synthase (CHS)5 superfamily of type III polyketide synthases (PKSs) are distributed in diverse organisms, including plants, fungi, and bacteria, and are responsible for the syntheses of various biologically and pharmaceutically important natural products (1, 2). They are structurally simple 40–45-kDa homodimeric enzymes that utilize a conserved Cys-His-Asn catalytic triad in an active site to catalyze the iterative condensations of C2 units derived from malonyl-CoA to a CoA-linked starter molecule. The functional diversity of the type III PKSs is basically derived from small modifications within the active site architectures of the enzymes, which influence the starter substrate selection, the number of chain extensions, and the cyclization reaction mechanisms (1, 2). As one of the characteristic features of the type III PKSs, the enzymes show broad substrate promiscuity and catalytic versatility in in vitro reactions and convert structurally and chemically distinct CoA starters into various molecular scaffolds (2–6).
The quinolone and acridone alkaloids have been investigated as N-methyl-d-aspartate (NMDA) and serotonin 5-hydroxytryptamine 3 receptor antagonists and as potential antimalarial drugs, respectively (7–9). The greatest abundance of these alkaloids is found in the Rutaceae plants, and N-methylanthranilic acid is thought to be a key intermediate in their biosynthesis (10–12). Among the growing number of reported type III PKSs, two kinds of type III PKSs specifically involved in the biosynthesis of the anthranilate-derived alkaloids have been obtained from Rutaceae plants. One is acridone synthase (ACS) from Ruta graveolens, which catalyzes the iterative condensations of N-methylanthraniloyl-CoA with three molecules of malonyl-CoA to produce the tetraketide 1,3-dihydroxy-N-methylacridone (Fig. 1A) (10, 11). Although the chalcone-forming CHS does not accept the bulky N-methylanthraniloyl-CoA starter (4, 11, 13–15), R. graveolens ACS can accept 4-coumaroyl-CoA as a starter substrate to produce naringenin chalcone after three condensations with malonyl-CoA (Fig. 1B) (13, 15). The other type III PKS is the recently reported quinolone synthase (QNS) from Aegle marmelos, which produces the diketide 4-hydroxy-N-methylquinolone by the one-step condensation of N-methylanthraniloyl-CoA with one molecule of malonyl-CoA (Fig. 1A) (12). The two type III PKS enzymes thus catalyze the C–N bond-forming reactions, in addition to the C–C bond formation, to generate the anthranilate-derived alkaloid scaffolds. Notably, A. marmelos QNS does not produce the diketide quinolone specifically but also generates the tetraketide acridone from N-methylanthraniloyl-CoA in an in vitro enzyme reaction (Fig. 1A). Thus, A. marmelos QNS could be regarded as another acridone-producing ACS, which simply yields the diketide quinolone as a by-product. This is analogous to the tetraketide chalcone-forming CHS, which also produces triketide and tetraketide lactone by-products in vitro (Fig. 1B). However, we previously demonstrated that benzalacetone synthase (BAS) from Rheum palmatum, which normally produces the diketide benzalacetone by the one-step condensation of 4-coumaroyl-CoA with malonyl-CoA, also accepts N-methylanthraniloyl-CoA as a starter substrate to produce the quinolone by condensation with malonyl-CoA (Fig. 1B) (16).
FIGURE 1.

Proposed mechanism for the formation of alkaloids and polyketides by type III PKSs. Enzyme reaction products from malonyl-CoA and the following: N-methylanthraniloyl-CoA (A), 4-coumaroyl-CoA (B), benzoyl-CoA (C), and hexanoyl-CoA (D). C. microcarpa QNS produces 4-hydroxy-N-methylquinolone as the sole product from N-methylanthraniloyl-CoA and triketide lactones from benzoyl-CoA and hexanoyl-CoA, whereas C. microcarpa ACS produces all of the products.
We now report the first QNS that produces 4-hydroxy-N-methylquinolone from N-methylanthraniloyl-CoA, as well as another acridone-producing ACS, from the leaves of the natural anti-inflammatory Rutaceae plant Citrus microcarpa. Remarkably, in contrast to the previously reported quinolone- and acridone-forming A. marmelos QNS, C. microcarpa QNS produces the diketide quinolone as the “single product” by the one-step condensation of N-methylanthraniloyl-CoA with malonyl-CoA (Fig. 1A). However, the newly obtained C. microcarpa ACS shows quite promiscuous substrate specificities and produces not only the anthranilate-derived acridone and quinolone but also accepts various starter substrates to generate distinct molecular scaffolds, including chalcone, benzophenone, and phloroglucinol (Fig. 1) (1, 2, 17). To clarify the mechanistic details of the enzyme reactions, we also solved the x-ray crystal structures of C. microcarpa QNS and ACS, at 2.47- and 2.35-Å resolutions, respectively. A comparison of the crystal structures revealed the unique active site architectures of these two enzymes and provided the first structural basis for the production of the anthranilate-derived quinolone and acridone alkaloids by type III PKSs.
EXPERIMENTAL PROCEDURES
Materials
The [2-14C]malonyl-CoA (55 mCi/mmol) and [1-14C]acetyl-CoA (52.5 mCi/mmol) were purchased from Moravek Biochemicals (Brea, CA). The 4-coumaroyl-CoA and N-methylanthraniloyl-CoA were chemically synthesized as described previously (4, 18). Malonyl-CoA, hexanoyl-CoA, and benzoyl-CoA were purchased from Sigma. Authentic samples of the enzyme reaction products were obtained in our previous work (4, 18).
Gene Cloning of QNS and ACS
C. microcarpa Bunge leaves were harvested from the Matakichi farm in Okinawa, Japan, and immediately frozen in liquid nitrogen. Total RNA was extracted with an RNeasy plant mini kit (Qiagen) and was reverse-transcribed using Superscript II RT (Invitrogen) and the oligo(dT) primer (RACE 32 = 5′-GGCCACGCGTCGACTAGTACTTTTTTTTTTTTTTTTT-3′), according to the manufacturer's protocol. The single strand cDNA thus obtained was used as the template for the PCRs with inosine-containing degenerate oligonucleotide primers, designed on the basis of the conserved sequences of known CHSs, as described previously (4, 19–21): 112S = 5′-(A/G)A(A/G)GCIITI(A/C)A(A/G)GA(A/G)TGGGGICA-3′; 174S = 5′-GCIAA(A/G)GA(T/C)ITIGCIGA(A/G)AA(T/C)AA-3′; 368A = 5′-CCC(C/A)(A/T)ITCIA(A/G)ICCITCICCIGTIGT-3′; and 380A =5′-TCIA(T/C)IGTIA(A/G)ICCIGGICC(A/G)AA-3′(theprimer's number indicates the amino acid number within M. sativa CHS). Following the conditions described previously (4, 19–21), nested PCR was performed with the primer sets 112S and 380A and then with 174S and 368A to amplify the respective 550-bp core fragments of C. microcarpa QNS and ACS. The 3′-RACE, using two gene-specific primers (209S, 5′-ACATCTTGGTTGGGCAAGCTGTGTTTG-3′, and 252S, 5′-ACCAACAAGTTTATCAGGGCACACGTG-3′ for QNS, and 207S, 5′-GATTCTCTAGTGGGTCAGGCTCTTT-3′, and 245S, 5′-GCTTCTGCCTGATTCTGATGGTGCAA-3′ for ACS), and the oligo(dT) primer RACE32, was used to amplify the QNS's 472-bp cDNA and the ACS's 642-bp fragment. The 5′-RACE was performed using the 5′-RACE system (Invitrogen) and two gene-specific primers (261A, 5′-TTTCACCTGTGTGATAAACTTGTTGGT-3′, and 218A, 5′-CCAAACACAGCTTGCCCAACCAAGATG-3′ for QNS, and 253A, 5′-ATTGCACCATCAGAATCAGGCAGAAGC-3′, and 215A, 5′-AAAGAGCCTGACCCACTAGAGAATC-3′ for ACS) to amplify QNS's 643-bp and ACS's 681-bp DNA fragments. The full-length cDNA encoding M. microcarpa QNS was obtained using N- and C-terminal primers as follows: 5′-GCGAGATCTATGGAATCAATGGCGAAAGTGAAAAATTTTCTTAATGCCAAG-3′ (sense, the BglII site is underlined) and 5′-CGCCTCGAGTCAACAGGCGGAATCAATAGGGACGCT-3′ (antisense, the XhoI site is underlined). The full-length cDNA encoding C. microcarpa ACS was finally obtained using N- and C-terminal-specific primers as follows: 5′-GCGAGATCTATGGTAACCATGGAGGAGATTAGAAGGGCT-3′ (sense, the BglII site is underlined) and 5′-CGCCTCGAGTCATGCTTCTATAGGGAGACTGTGCAACA-3′(antisense, the XhoI site is underlined). The amplified full-length C. microcarpa QNS and ACS cDNA fragments were digested with BglII/XhoI and cloned into the BamHI/XhoI sites of pQE80L (Novagen), respectively. Both recombinant enzymes thus contain an additional N-terminal hexahistidine tag. The nucleotide sequence was determined by using an ABI Prism 3100 Genetic Analyzer (Applied Biosystems).
Phylogenetic Tree
A total of 52 type III PKS amino acid sequences were aligned, and a phylogenetic tree was developed with the ClustalW (1.8) program (DNA Data Bank of Japan), as reported previously (4).
Expression and Purification of C. microcarpa QNS and ACS
The plasmids containing the full-length cDNAs encoding C. microcarpa QNS and ACS were individually transformed into Escherichia coli M15. The cells harboring the plasmids were cultured to an A600 of 0.6 in LB medium containing 100 μg ml−1 ampicillin at 23 °C, and 1.0 mm isopropyl 1-thio-β-d-galactopyranoside was then added to induce protein expression. The culture was incubated further at 23 °C for 16 h. All of the following procedures were performed at 4 °C. The E. coli cells were harvested by centrifugation at 5,000 × g and resuspended in 50 mm Tris-HCl buffer (pH 7.5), containing 0.2 m NaCl, 5% (v/v) glycerol, and 5 mm imidazole (buffer A). The cells were disrupted by sonication, and the lysate was centrifuged at 12,000 × g for 30 min. The supernatant was loaded onto a nickel-Sepharose 6 Fast Flow column (GE Healthcare) equilibrated with buffer A. After washing the resin with 50 mm Tris-HCl buffer (pH 7.5), containing 0.2 m NaCl, 5% (v/v) glycerol, and 10 mm imidazole (buffer B), the recombinant protein was subsequently eluted with buffer B containing 300 mm imidazole. The protein solution was then diluted 5-fold with 50 mm Tris-HCl buffer (pH 7.5), containing 5% (v/v) glycerol and 2 mm DTT (buffer C), and applied to a Resource-Q column (GE Healthcare). The column was washed with buffer C containing 50 mm NaCl, and the protein was subsequently eluted using a linear gradient of 50–300 mm NaCl. The protein solution was concentrated to 5 ml, purified to homogeneity by gel filtration chromatography on a Superdex 200 HR column (16/60 GL; GE Healthcare), and concentrated to 10 mg/ml in 20 mm Tris-HCl buffer (pH 7.5), containing 100 mm NaCl and 2 mm DTT.
Enzymatic Reactions of C. microcarpa QNS and ACS
The standard reaction mixture contained 54 μmol of starter CoA, 108 μmol of malonyl-CoA, and 20 μg of the purified recombinant enzyme, in a final volume of 500 μl of 100 mm potassium phosphate buffer (pH 7.0). The reactions were incubated at 30 °C for 60 min and stopped by adding 50 μl of 20% HCl. The products were then extracted three times with 500 μl of ethyl acetate, concentrated by evaporation, and analyzed by RP-HPLC on a COSMOSIL® 5C18-MS-II column (4.6 × 250 mm) (Nacalai Tesque) at a flow rate of 0.8 ml/min, as described previously (4–6). Gradient elution was performed with H2O and MeOH, both containing 0.1% trifluoroacetic acid: 0–5 min, 30% MeOH; 5–17 min, 30–60% MeOH; 17–25 min, 60% MeOH; 25–27 min, 60–70% MeOH; 27–35 min, 70% MeOH; and 35–40 min, 70–100% MeOH. On-line LC-ESI-MS spectra were measured with an Agilent Technologies (Santa Clara, CA) HPLC 1100 series coupled to a Bruker Daltonics (Bremen, Germany) Esquire 4000 ion trap mass spectrometer fitted with an ESI source, as described previously (4–6). The enzyme reaction products were identified by direct comparisons to authentic compounds.
Enzyme Kinetics
Steady-state kinetic parameters were determined using [2-14C]malonyl-CoA (1.8 mCi/mmol) as the substrate. The experiments were performed in triplicate, using five concentrations (51.2, 25.6, 12.8, 6.4, and 3.2 μm) of starter-CoA and 4 μg of purified enzyme, in a final volume of 100 μl of 100 mm Tris-HCl (pH 8.0). The reactions were incubated at 30 °C for 30 min. The reaction products were extracted twice with 100 μl of ethyl acetate and separated by TLC (Merck 1.11798 silica gel 60 F254; ethyl acetate/hexane/AcOH = 63:27:5, v/v/v). Radioactivities were quantified by autoradiography, using a BAS-2000II bioimaging analyzer (Fujifilm, Tokyo, Japan). Lineweaver-Burk plots of data were employed to derive the apparent Km and kcat values (average of triplicates ± S.D.) using the EnzFitter software (BIOSOFT, Cambridge, UK).
Crystallization and Structure Refinement of QNS and ACS
Initial crystallization attempts and optimization of the crystallization conditions for both enzymes were performed at 20 °C, using the sitting-drop vapor diffusion method. First, 0.5 μl of either the purified recombinant QNS or ACS protein and an equal volume of reservoir solution were mixed and equilibrated against 50 μl of reservoir solution, using a 96-condition crystallization screen originally designed by Mitsubishi Chemical Corp. Crystals of QNS appeared the next day in a crystallization solution consisting of 100 mm Tris-HCl (pH 7.0) and 18% PEG3350. Crystals of ACS were observed a few days later in a crystallization solution consisting of 100 mm HEPES-NaOH (pH 7.5), 1,400 mm ammonium sulfate, and 2 mm CoASH. Further attempts to crystallize C. microcarpa QNS and ACS were performed, using Additive Screen (Hampton Research), at various pH values, 18% PEG3350, 100 mm HEPES-NaOH (pH 7.5), and 1,400 mm ammonium sulfate as a precipitant, respectively. Diffraction quality crystals of C. microcarpa QNS were finally obtained in 50 mm Tris-HCl (pH 7.0), 18% PEG3350, and 4% (v/v) 1-propanol, and those of C. microcarpa ACS were finally obtained in 100 mm HEPES-NaOH (pH 7.5), 1,400 mm ammonium sulfate, 2 mm NiCl2, and 2 mm CoASH using the sitting-drop vapor diffusion method at 20 °C.
The crystals of C. microcarpa QNS were transferred into a cryoprotectant solution, consisting of 50 mm Tris-HCl (pH 7.0), 18% PEG3350, 4% (v/v) 1-propanol, and 18% glycerol. After a few seconds, the crystals were picked up in a nylon loop and then flash-cooled at −173 °C in a nitrogen gas stream. X-ray diffraction data sets were collected on beamline BL17A at the Photon Factory (wavelength 0.9800 Å) using an ADSC Quantum 315 CCD detector, with a distance of 250 mm between the crystal and the detector. A total of 180 frames was recorded, with a 1° oscillation angle and 1-s exposure time.
The crystals of C. microcarpa ACS were transferred into a cryoprotectant solution, consisting of 100 mm HEPES-NaOH (pH 7.5), 1,400 mm ammonium sulfate, 2 mm NiCl2, and 20% glycerol. After a few seconds, the crystals were picked up in a nylon loop and then flash-cooled at −173 °C in a nitrogen gas stream. X-ray diffraction data sets were collected on beamline NW-12 of the Photon Factory-AR (wavelength 1.00000 Å) using an ADSC Quantum 210 CCD detector, with a distance of 150 mm between the crystal and the detector. A total of 180 frames were recorded, with a 1° oscillation angle and 1-s exposure time. The data were indexed, integrated, and scaled with the HKL-2000 program package (22).
Structure Refinement
The initial phases of the C. microcarpa QNS and ACS structures were determined by molecular replacement, using the M. sativa CHS structure (Protein Data Bank code 1CGK) as the search model. Molecular replacement was performed with MOLREP in the CCP4 suite (23, 24). The structure was modified manually with Coot (25) and refined with PHENIX (26). The final model of C. microcarpa QNS consisted of residues 1–389 of monomers A–D, two molecules of glycerol, and 198 molecules of water. The final model of C. microcarpa ACS consisted of residues 1–389 with 11 artificial residues, including an artificial Met as the initiation codon and the N-terminal His6 tags of monomers A and B, two molecules of CoA-SH, nine molecules of nickel, three molecules of SO4, and 331 molecules of water. The qualities of the final models were assessed with MolProbity (27). A total of 97.1% of the residues in the C. microcarpa QNS structure are in the favored regions of the Ramachandran plot, and 2.9% are in the allowed regions, although a total of 97.5% of the residues in the C. microcarpa ACS structure are in the most favored regions of the Ramachandran plot, and 2.5% are in the allowed regions. The coordinates and structure factors have been deposited in the Protein Data Bank (Protein Data Bank code 3WD8 for the C. microcarpa QNS apo structure and code 3WD7 for the C. microcarpa ACS CoA-SH complexed structure).
A structural similarity search was performed, using the Dali program (28). The cavity volume and the active site entrance area were calculated with the program CASTP. All crystallographic figures were prepared with PyMOL (DeLano Scientific).
Site-directed Mutagenesis
The plasmids expressing the mutants of C. microcarpa QNS (Y197A) and C. microcarpa ACS (S132M, T194M, and T197Y) were constructed with a QuikChange site-directed mutagenesis kit (Stratagene), according to the manufacturer's protocol, using the following pairs of primers (mutated codons are underlined): Y197A (5′-GATATCATGAACATGGCTTTTCATGAGCCG-3′ and 5′-CGGCTCATGAAAAGCCATGTTCATGATATC-3′) for C. microcarpa QNS, and S132M (5′-CATCTCATTTTCTGCACAATGGCAGGCGTCGACATGCC-3′ and 5′-GGCATGTCGACGCCTGCCATTGTGCAGAAAATGAGATG-3′), T194M (5′-GTTTGCTCTGAGAACATGATCCCCACTTTCCGTG-3′ and 5′-CACGGAAAGTGGGGATCATGTTCTCAGAGCAAAC-3′), and T197Y (5′-GAGAACACAATCCCCTATTTCCGTGGGCCG-3′ and 5′-CGGCCCACGGAAATAGGGGATTGTGTTCTC-3′) for C. microcarpa ACS. The mutant enzymes were expressed and purified with the same procedures as described for the wild-type enzymes and used for the enzyme reaction.
RESULTS
Sequence Analyses of QNS and ACS
Two full-length cDNAs encoding novel type III PKSs, QNS and ACS, were cloned and sequenced from the leaves of C. microcarpa by the RT-PCR method (the details of the method are described under “Experimental Procedures”). The full-length QNS and ACS cDNAs contained 1,191- and 1,176-bp open reading frames encoding Mr 43,331 and 42,830 proteins with 396 and 391 amino acids, respectively (GenBankTM accession numbers AB823730 and AB823699). Notably, the deduced amino acid sequence of C. microcarpa QNS is quite unique and shared 60% identity to that of the acridone-producing C. microcarpa ACS, 56% identity to R. graveolens ACS (10), 59% identity to the quinolone-producing A. marmelos QNS (12), 60% identity to the chalcone-producing Medicago sativa CHS (29), and 58% identity to the benzalacetone-producing R. palmatum BAS (Fig. 2) (19). No additional cDNAs encoding other type III PKS isomers were obtained in this study.
FIGURE 2.

Sequence alignment of C. microcarpa QNS/ACS with other plant type III PKSs. CmQNS, C. microcarpa QNS; CmACS, C. microcarpa ACS; AmQNS, A. marmelos QNS; RgACS, R. graveolens ACS; MsCHS, M. sativa chalcone synthase; HsPKS1, H. serrata PKS1; RpBAS, R. palmatum BAS. The catalytic triad (Cys-164, His-303, and Asn-336) is colored red, and the active site residues, 132, 133, 137, 194, 197, 211, 215, 256, 265, 338, 375, are highlighted in blue (numbering in M. sativa CHS).
The sequence analysis revealed that the C. microcarpa QNS retains the CHS's active site residues, such as Gly-211, Pro-375, and the “gatekeeper” Phe-215, as well as the conserved catalytic residues, Cys-164, His-303, and Asn-336. However, half of the conserved active site residues are uniquely altered in C. microcarpa QNS. Thus, Thr-132, Ser-133, and the gatekeeper Phe-265 in M. sativa CHS are simultaneously substituted with Met, Ala, and Leu, respectively (Fig. 2). In addition, Met-137, Thr-194, Thr-197, Gly-256, and Ser-338 of M. sativa CHS are characteristically altered to Ile, Met, Tyr, Ala, and Gly, respectively. The G256A/S338G substitutions are also found in Sorbus aucuparia biphenyl synthase (30) and Hypericum androsaemum benzophenone synthase (17). The conserved active site residues Thr-197/Gly-256/Ser-338 are altered in a number of functionally different type III PKSs and are thought to be crucial for governing the substrate and product specificities of the enzyme reactions (1, 2). However, in C. microcarpa ACS, Thr-132, Ser-133, and Phe-265 are substituted with Ser, Ala, and Val, respectively, as in the case of R. graveolens ACS and A. marmelos QNS (Fig. 2). These three residues are reportedly crucial for the substrate and product specificities of the enzyme reactions in A. marmelos QNS and R. graveolens ACS (12, 15). A phylogenetic tree analysis grouped C. microcarpa QNS and ACS with the non-chalcone-producing enzymes and the closely related biphenyl- and alkaloid-producing type III PKSs, respectively (Fig. 3).
FIGURE 3.

Phylogenetic tree analysis of plant and bacterial type III PKSs. Multiple sequence alignment, performed with ClustalW (1.8). The scale represents 0.1 amino acid substitutions per site. Abbreviations used are as follows: ALS, aloesone synthase; BBS, bibenzyl synthase; BIS, biphenyl synthase; BPS, benzophenone synthase; CTAS, 4-coumaroyltriacetic acid synthase; CUS, curcuminoid synthase; CURS, curcumin synthase; DCS, diketide-CoA synthase; HKS, hexaketide synthase; OKS, octaketide synthase; ORAS, 3′-oxoresorcinolic acid synthase; PCS, pentaketide chromone synthase; 2PS, 2-pyrone synthase; STS, stilbene synthase; THNS, tetrahydroxynaphthalene synthase; VPS, valerophenone synthase. The β-ketoacyl carrier protein synthase III (KAS III and FABH) enzymes of E. coli were employed as out groups.
In Vitro Analysis of C. microcarpa QNS Activity
The sequence analyses suggested that both of the newly obtained C. microcarpa type III PKSs are functionally distinct from the regular CHS and could possess interesting catalytic activities. The recombinant C. microcarpa QNS was functionally expressed in E. coli as an N-terminally His6-tagged protein and was purified and subjected to enzyme reactions using N-methylanthraniloyl-CoA, 4-coumaroyl-CoA, benzoyl-CoA, hexanoyl-CoA, and malonyl-CoA as substrates. Although the phylogenetic tree analyses predicted a close relationship between C. microcarpa QNS and the biphenyl-producing biphenyl synthase (Fig. 3), the LC-ESI-MS analyses of the enzyme reaction products demonstrated that it does not produce biphenyl from benzoyl-CoA (Fig. 4C). Instead, C. microcarpa QNS efficiently accepts N-methylanthraniloyl-CoA as a starter substrate to produce the diketide 4-hydroxy-N-methylquinolone as the single product by the condensation of one molecule of malonyl-CoA (Fig. 4A). Notably, C. microcarpa QNS did not accept 4-coumaroyl-CoA as a substrate (Fig. 4B) and produced only triketide lactones from benzoyl-CoA and hexanoyl-CoA as starters (Fig. 4, C and D). No additional products were detected in all of the enzyme reactions tested, even with changes in the reaction pH, temperature, and time. The steady-state kinetics values for quinolone formation by C. microcarpa QNS were Km = 37.8 μm, kcat = 15.7 min−1, and kcat/Km = 416 min−1mm−1 for N-methylanthraniloyl-CoA, with a pH optimum at 8.0 within a range of 6.5–8.5. The steady-state kinetics values for quinolone formation were better than those of R. palmatum BAS (Km = 23.7 μm, kcat = 1.5 min−1, kcat/Km = 62.4 min−1mm−1) (16) but not as good as those of A. marmelos QNS (Km = 2.9 μm, kcat = 3.8 min−1, kcat/Km = 1290 min−1mm−1) (Table 1) (12).
FIGURE 4.

HPLC elution profiles of the enzyme reaction products of C. microcarpa QNS and ACS. A–D, enzyme reaction products of C. microcarpa (Cm) ACS and QNS from N-methylanthraniloyl-CoA (A), 4-coumaroyl-CoA (B), benzoyl-CoA (C), and hexanoyl-CoA, and malonyl-CoA (D). Note that by acid treatment naringenin chalcone is converted to racemic naringenin (5,7,4′-trihydroxyflavanone) through a nonstereospecific ring-C closure.
TABLE 1.
Steady-state kinetic parameters of C. microcarpa QNS and ACS
| Product | Km | kcat | kcat/Km | Ref. | |
|---|---|---|---|---|---|
| μm | min−1 | min−1 mm−1 | |||
| N-methylanthraniloyl-CoA | |||||
| C. microcarpa QNS | Quinolone | 37.8 ± 3.2 | 15.7 ± 2.0 | 416 | This paper |
| A. marmelos QNS | Quinolone | 2.9 | 3.8 | 1290 | 12 |
| R. palmatum BAS | Quinolone | 23.7 | 1.5 | 62.4 | 16 |
| C. microcarpa ACS | Quinolone | 37.4 ± 2.4 | 4.0 ± 0.6 | 117 | This paper |
| C. microcarpa ACS | Acridone | 4.3 ± 1.7 | 1.4 ± 0.0 | 324 | This paper |
| 4-Coumaroyl-CoA | |||||
| C. microcarpa ACS | Naringenin | 13.6 ± 3.1 | 6.8 ± 2.2 | 500 | This paper |
| M. sativa CHS | Naringenin | 5.1 | 6.1 | 843 | 13 |
| C. microcarpa QNS Y197A | Benzalacetone | 28.3 ± 5.7 | 1.3 ± 0.2 | 45.5 | This paper |
| R. palmatum BAS | Benzalacetone | 10.0 | 1.8 | 179 | 19 |
In Vitro Analysis of C. microcarpa ACS Activity
The phylogenetic tree analyses suggested that C. microcarpa ACS is closely related to the acridone-producing R. graveolens ACS and A. marmelos QNS (Fig. 3). Indeed, C. microcarpa ACS efficiently accepted N-methylanthraniloyl-CoA as the starter substrate to produce the tetraketide 1,3-dihydroxy-N-methylacridone after sequential condensations with three molecules of malonyl-CoA, along with 4-hydroxy-N-methylquinolone and N-methylanthraniloyltriacetic acid lactone (Fig. 4A). The steady-state kinetics values for acridone formation by C. microcarpa ACS were Km = 4.3 μm, kcat = 1.4 min−1, and kcat/Km = 324 min−1mm−1 for N-methylanthraniloyl-CoA, with a pH optimum at 8.0 within a range of 6.5–8.5. However, with respect to the quinolone forming activity, the kinetics values were Km = 34.4 μm, kcat = 4.0 min−1, and kcat/Km = 117 min−1mm−1 for N-methylanthraniloyl-CoA, with a pH optimum at 8.0 within a range of 6.5–8.5 (Table 1). Thus, the catalytic efficiency for the formation of the acridone is 2.8-fold higher than that of the quinolone. In addition, C. microcarpa ACS also accepted 4-coumaroyl-CoA as a starter substrate to yield naringenin chalcone, along with triketide and tetraketide lactone derailment by-products (Fig. 4B). When benzoyl-CoA or hexanoyl-CoA was tested as the starter substrate, C. microcarpa ACS afforded the aromatic tetraketides 2,4,6-trihydroxybenzophenone and 2-(1-keto-hexyl)phloroglucinol, but much less efficiently (Fig. 4, C and D). With respect to the chalcone forming activity, C. microcarpa ACS showed Km = 13.6 μm, kcat = 6.8 min−1, and kcat/Km = 500 min−1mm−1 for 4-coumaroyl-CoA, and the latter value is 1.7-fold lower than that of M. sativa CHS (Km = 5.1 μm, kcat = 6.1 min−1, and kcat/Km = 843 min−1mm−1) (13) (Table 1).
Overall Structures of C. microcarpa QNS and ACS
The apo-crystal structure of the recombinant C. microcarpa QNS and the crystal structure of the recombinant C. microcarpa ACS complexed with CoA-SH were solved at 2.47 and 2.35 Å resolution, respectively. The crystallographic data and refinement statistics are summarized in Table 2. The asymmetric units of C. microcarpa QNS and ACS contained four and two nearly identical monomers, respectively, and significant backbone changes were not observed between the monomers in these structures. The structures of C. microcarpa QNS and ACS, sharing 56% amino acid sequence identity, are nearly identical, with root-mean-square deviations (r.m.s.d.) of 0.9 Å. The overall structures of C. microcarpa QNS and ACS revealed the conservation of the αβαβα-fold, observed in all structurally characterized type III PKSs (Fig. 5). The catalytic triad consisting of Cys-164, His-303, and Asn-336 is buried deep within each monomer, at the intersection of a characteristic 16-Å-long CoA binding tunnel and a large cavity, in a location and an orientation very similar to those of the other plant type III PKSs (6, 31–35). Ile-137 in C. microcarpa QNS and Met-137 in C. microcarpa ACS, corresponding to Met-137 of M. sativa CHS, protrude into the other monomer and form part of the active site wall, as in the case of M. sativa CHS. The overall structures of C. microcarpa QNS and ACS are highly homologous to those of the structurally characterized plant type III PKSs (r.m.s.d. 0.7–1.3 Å and 0.5–1.2 Å for C. microcarpa QNS and ACS, respectively). A structure-based similarity search using the Dali program revealed that the overall structures of M. sativa CHS, Huperzia serrata PKS1, and R. palmatum BAS, which are functionally related to C. microcarpa QNS and ACS, exhibited r.m.s.d. of 0.9, 0.9, and 0.7 Å to that of C. microcarpa QNS, and 0.5, 0.7, and 0.6 Å to that of C. microcarpa ACS, respectively.
TABLE 2.
Data collection and refinement statistics
| Data collection | C. microcarpa QNS | C. microcarpa ACS |
|---|---|---|
| Unit cell parameter | ||
| Space group | P21 | P6522 |
| a, b, c | 51.7, 135.9, 107.6 Å | 106.0, 106.0, 346.5 Å |
| Resolution range | 50 to 2.47 Å (2.51 to 2.47 Å) | 50 to 2.35 Å (2.39 to 2.35 Å) |
| Completeness | 99.4% (98.7%) | 99.9% (100%) |
| 〈I/σI〉 | 10.7% (2.10%) | 60.9% (10.0%) |
| Rmerge | 10.3% (58.0%) | 7.4% (36.1%) |
| Redundancy | 3.85 (3.85) | 21.0 (17.2) |
| No. of observed reflections | 188,726 | 1,031,666 |
| No. of unique reflections | 49,068 | 49,243 |
| Refinement | ||
| Resolution | 20.0 to 2.47 Å | 34.5 to 2.35 Å |
| Overall Rwork | 19.5% | 18.3% |
| Overall Rfree | 24.1% | 21.8% |
| Total atoms | 12238 | 6624 |
| No. of protein atoms | 12028 | 6173 |
| No. of waters | 198 | 331 |
| No. of ligand | 12 | 120 |
| Average B-factors | ||
| Protein atoms | 39.1 Å2 | 33.2 Å2 |
| Waters | 33.3 Å2 | 33.3 Å2 |
| Ligands | 49.6 Å2 | 56.2 Å2 |
| r.m.s.d. from ideal | ||
| Bond length | 0.01 Å | 0.009 Å |
| Bond angles | 1.301° | 1.208° |
FIGURE 5.
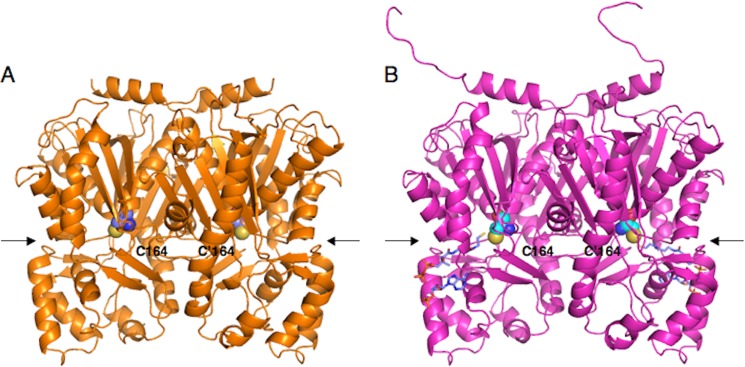
Overall structures of C. microcarpa QNS and ACS. A, C. microcarpa QNS; B, C. microcarpa ACS. The structures are represented as schematics. The catalytic Cys-164 and the substrate entrance are represented by a CPK model and an arrow, respectively. The CoASH molecules in C. microcarpa ACS are depicted as blue stick models.
Active Site Structure of C. microcarpa QNS
Similar to C. microcarpa ACS, the CHS's conserved gatekeeper Phe-265 is substituted with Leu in C. microcarpa QNS. The aromatic side chain of the highly conserved Phe-265 in the plant type III PKSs usually blocks the access of the residues behind it to the active site cavity. In addition, Thr-132, Ser-133, Met-137, Thr-194, Thr-197, Gly-256, and Ser-338 in M. sativa CHS are characteristically replaced by Met, Ala, Ile, Met, Tyr, Ala, and Gly, respectively. No such simultaneous substitutions were observed in the primary structures of the other known type III PKSs, suggesting that these changes control the unique substrate and product specificities of C. microcarpa QNS. These residues are spatial analogs among the structures of C. microcarpa QNS and M. sativa CHS. The F265L substitution in C. microcarpa QNS not only grossly changes the shape of the active site cavity, but also permits Leu-267, behind Leu-265, to form part of the active site entrance (Fig. 6A). Furthermore, because of the absence of the aromatic moiety, the side chain of Phe-215 protrudes more toward Leu-265, as compared with that of Phe-215 in M. sativa CHS, thus widening part of the active site entrance.
FIGURE 6.

Comparison of the active site entrances of C. microcarpa QNS/ACS and other type III PKSs. A, C. microcarpa QNS; Bl C. microcarpa ACS, and C, M. sativa CHS. Arrows indicate the substrate entrance in each structure.
The structural comparison also revealed the significant conformational differences of residues 159–164 (r.m.s.d. of 1.1 Å for the Cα atoms) and the slight displacement of an α-helix consisting of residues 164–179 in C. microcarpa QNS. These conformational changes are presumably caused by various neighboring amino acid replacements in C. microcarpa QNS, such as the large-to-small M159I and small-to-large A166I substitutions in these regions. As a result, the Cα atom of the catalytic residue Cys-164 in C. microcarpa QNS is displaced by 1.7 Å toward the outside of the CHS's active site cavity. This alters the shape and size of the active site cavity and also widens the active site entrance. The estimated total area of the active site entrance of QNS is 47 Å2, which is 2.8 and 1.5 times larger than those of M. sativa CHS (17 Å2) and C. microcarpa ACS (33 Å2) (Fig. 6, A–C), respectively.
However, the side chain of Met-132 of C. microcarpa QNS, corresponding to Thr-132 in M. sativa CHS, occupies the so-called coumaroyl-binding pocket that accommodates the aromatic moiety of the coumaroyl starter in M. sativa CHS (Fig. 7, A and C). The side chain of Met-194 in C. microcarpa QNS protrudes toward Gly-338, as compared with CHS's Thr-194. The coumaroyl-binding pocket in M. sativa CHS is thus absent from the active site cavity of C. microcarpa QNS, along with slight differences in the various surrounding amino acids.
FIGURE 7.

Comparison of the active site structures of C. microcarpa QNS and ACS and other type III PKSs, and their schematic representations. A, C. microcarpa QNS; B, C. microcarpa ACS; C, M. sativa CHS, and D, R. palmatum BAS. The naringenin in M. sativa CHS and the bound monoketide intermediate in R. palmatum BAS are shown as magenta and pink stick models, respectively. Arrows indicate the substrate entrance in each structure.
Interestingly, the side chain of Tyr-197 in C. microcarpa QNS, corresponding to Thr-197 lining the bottom of the active site cavity of M. sativa CHS, is inserted in front of the side chain of Met-263, corresponding to Leu-263 in M. sativa CHS, and its terminal hydroxyl group forms a hydrogen bond with the sulfur atom of Met-132 (Fig. 7, A and C). The substitution of Thr-197 with Tyr in C. microcarpa QNS thereby drastically narrows the active site cavity of C. microcarpa QNS, especially at the bottom. Additionally, the side chain of Ala-256, which is the spatial analog of Gly-256 of M. sativa CHS in C. microcarpa QNS, protrudes toward Tyr-197 and constricts the bottom part of the active site cavity of C. microcarpa QNS, along with the side chain of Tyr-197. In contrast, the large-to-small S338G and M137I substitutions in C. microcarpa QNS slightly expand its active site wall near the active site entrance, as compared with M. sativa CHS. The total cavity volume (290 Å3) of the active site of C. microcarpa QNS is about 2.6 times smaller than those of M. sativa CHS (750 Å3) and C. microcarpa ACS (760 Å3) (Fig. 7, A–C).
Active Site Structure of C. microcarpa ACS
One of the characteristic features of the C. microcarpa ACS sequence is the characteristic substitution of the CHS's conserved active site residues Thr-132, Ser-133, and Phe-265 with Ser, Ala, and Val, respectively. As in the case of C. microcarpa QNS, the location of Val-265 in the active site cavity of C. microcarpa ACS is similar to that of Phe-265 in M. sativa CHS. The loss of the aromatic moiety at this position obviously changes the shape of the active site cavity of C. microcarpa ACS. Furthermore, the side chain Cδ1 of Leu-267, corresponding to Leu-267 behind the aromatic side chain of Phe-265 in M. sativa CHS, participates in the formation of the active site entrance, with a slight displacement of its Cα atom and a significant shift of its torsion angle toward Val-265 (Fig. 6B). In addition, presumably because of the loss of the aromatic side chain at this position and of several slight conformational differences between both enzymes, such as the G216A substitution in C. microcarpa ACS, the backbone torsion angle of Phe-215 (−53, 136), as compared with that of Phe-215 (−79, 138) in M. sativa CHS, is shifted by a φ angle of −26° and a ψ angle of +2°. This is accompanied by a slight displacement of its Cα atom, and the side chain of Phe-215 protrudes toward Val-265 (Fig. 6B). These conformational differences expand the entrance to the active site cavity of C. microcarpa ACS, as compared with that of M. sativa CHS. As a result, the estimated total area of the C. microcarpa ACS's entrance is 33 Å2, which is twice as large as that of M. sativa CHS (17 Å2) (Fig. 6, B and C).
In addition to these significant conformational changes, the side chains of Val-265 and Ser-132, as compared with those of Phe-265 and Thr-132 in M. sativa CHS, slightly protrude toward the inside and the outside of the active site cavity of C. microcarpa ACS, respectively (Fig. 7, B and C). With the slight displacements of their Cα atoms and the backbone torsion angles, these conformational changes contribute toward altering the shape of the active site cavity of C. microcarpa ACS. However, Ala-133 occupies almost the same position as the active site residue Ser-133 of M. sativa CHS, with an orientation very similar to that of Ser-133. The only difference is the steric bulk between the side chains of Ala and Ser at this position. No other significant conformational differences were observed between both structures. The estimated total cavity volume of C. microcarpa ACS is 760 Å3, which is almost the same as that of M. sativa CHS (750 Å3) (Fig. 7, B and C).
Structure-based Mutagenesis of C. microcarpa QNS and ACS
To further clarify the structure-function relationship of C. microcarpa QNS and ACS, we performed site-directed mutagenesis and investigated the mechanistic consequences of the point mutations. First, to test the hypothesis that the bulky Tyr-197 plays a crucial role in the active site architecture of C. microcarpa QNS, we constructed the Y197A mutant of C. microcarpa QNS, in which the bulky Tyr-197 was substituted with a small Ala. As a result, the Y197A mutant produced the diketide quinolone, from N-methylanthraniloyl-CoA, almost as efficiently as the wild-type enzyme; however, interestingly, the QNS mutant now accepted 4-coumaroyl-CoA as the starter substrate to produce the diketide benzalacetone and triketide lactone (Fig. 8A). The kinetics values for benzalacetone formation by the Y197A mutant were Km = 28.3 μm, kcat = 1.3 min−1, and kcat/Km = 45.5 min−1mm−1. Thus, the catalytic efficiency for the formation of the benzalacetone is 3.9-fold lower than that of the native R. palmatum BAS (Km = 10.0 μm, kcat = 1.8 min−1, kcat/Km = 179 min−1mm−1) (Table 1).
FIGURE 8.

HPLC elution profiles of the enzyme reaction products of C. microcarpa QNS and ACS mutants. A, enzyme reaction products of C. microcarpa QNS, wild-type, and Y197A mutant, from 4-coumaroyl-CoA. B and C, enzyme reaction products of C. microcarpa ACS, wild-type, and mutants (S132M, T194M, and T197Y), from N-methylanthraniloyl-CoA (B), and 4-coumaroyl-CoA (C). Note that by acid treatment naringenin chalcone is converted to racemic naringenin (5,7,4′-trihydroxyflavanone) through a nonstereospecific ring-C closure.
However, the crucial active site residues, Ser-132, Thr-194, and Thr-197 in C. microcarpa ACS, were replaced with Met, Met, and Tyr, respectively, as in the case of C. microcarpa QNS. Notably, consistent with our hypothesis, all the point mutants efficiently produced quinolone as the single product and lost the tetraketide acridone and lactone producing activities from the N-methylanthraniloyl-CoA starter (Fig. 8B). Furthermore, as in the case of C. microcarpa QNS, all the ACS mutants no longer accepted 4-coumaroyl-CoA as the starter substrate to produce chalcone, tetraketide, and triketide lactones (Fig. 8C).
DISCUSSION
In vitro assays of the enzymes clearly demonstrated that C. microcarpa QNS is a novel type III PKS that produces the diketide 4-hydroxy-N-methylquinolone as the single product by the one-step condensation of N-methylanthraniloyl-CoA with malonyl-CoA (Fig. 1A). Notably, C. microcarpa QNS exhibits quite unique substrate and product specificities. The enzyme did not accept 4-coumaroyl-CoA as a substrate and produced only triketide lactones from benzoyl-CoA and hexanoyl-CoA as starter substrates (Fig. 1, B–D). These results suggested that C. microcarpa QNS can be regarded as a dedicated quinolone-producing enzyme. In contrast, the previously reported A. marmelos QNS did not produce the quinolone specifically, because it also generated the tetraketide 1,3-dihydroxy-N-methylacridone from N-methylanthraniloyl-CoA (12). Thus, A. marmelos QNS could be regarded as another acridone-producing ACS, which simply yields the diketide quinolone as a by-product. This is analogous to the tetraketide chalcone-forming CHS, which also produced triketide and tetraketide lactone by-products in the in vitro enzyme reactions. Furthermore, another major difference is that the quinolone- and acridone-forming A. marmelos QNS also accepted 4-coumaroyl-CoA to produce the diketide benzalacetone (12). However, we previously reported that the diketide-forming R. palmatum BAS also accepted both 4-coumaroyl-CoA and N-methylanthraniloyl-CoA as starter substrates to produce the benzalacetone and quinolone scaffolds, respectively (16, 19). These observations indicated that the substrate preferences of C. microcarpa QNS, A. marmelos QNS, and R. palmatum BAS are significantly different, due to the modifications of their active site architectures, as discussed below.
Interestingly, the crystal structure revealed that C. microcarpa QNS has an unusually wide active site entrance (Fig. 6A). Similar expansions of the active site entrances have also been reported for the structures of the R. palmatum BAS (34) and acridone-producing M. sativa F215S CHS mutant (13), which both accept the bulky substrate N-methylanthraniloyl-CoA. The active site entrance of C. microcarpa QNS (47 Å2) is larger than that of R. palmatum BAS (34 Å2) and is almost as large as that of the M. sativa F215S CHS mutant (41 Å2). In the structure of C. microcarpa QNS, the broadening of the active site entrance is caused by the F265L substitution and the deviation of the catalytic residue Cys-164 toward the outside of the active site cavity, as compared with the other type III PKSs. However, in R. palmatum BAS, the F208L substitution and its conformational changes expand the active site entrance. The wide active site entrance thus provides enough space to facilitate the access of the bulky N-methylanthraniloyl-CoA starter to the catalytic center of the enzymes.
In addition, the active site cavity volume of C. microcarpa QNS is quite small (290 Å3) and is even smaller than that of the diketide-forming R. palmatum BAS (350 Å3) (Fig. 7, A and D) (34). Furthermore, the crystal structure revealed the absence of the CHS's coumaroyl-binding pocket in the active site of C. microcarpa QNS, due to the unique substitutions of CHS's conserved Thr-132, Thr-194, and Thr-197 with Met, Met, and Tyr, respectively. This is the reason why C. microcarpa QNS does not accept 4-coumaroyl-CoA as a substrate. In contrast, although R. palmatum BAS also lacks the coumaroyl-binding pocket, this enzyme utilizes novel alternative pockets to bind the coumaroyl starter and produce the diketide benzalacetone (Fig. 7D) (34). Thus, the shape and the cavity volume of C. microcarpa QNS restrict the binding of the coumaroyl starter and the malonyl-CoA extender. Indeed, the mutagenesis analyses revealed that the bulky Tyr-197 drastically narrows down the active site cavity of C. microcarpa QNS and thereby controls the product chain length and specificity of the enzyme reaction. It is remarkable that the large-to-small Y197A mutant of C. microcarpa QNS gained the function of benzalacetone and triketide lactone-producing activities by the single amino acid substitution. Notably, similar steric contraction of the active site cavity was also observed for Gerbera hybrida 2-methylpyrone synthase, which utilizes the small acetyl-CoA as a starter substrate to produce triacetic acid lactone (31). The active site cavity of G. hybrida 2-pyrone synthase is even smaller (250 Å3) and also lacks the coumaroyl-binding pocket (36, 37). These observations support the unique catalytic activity of C. microcarpa QNS, which accepts the N-methylanthraniloyl-CoA starter through its wide active site entrance and catalyzes the condensation with malonyl-CoA. The chain elongation reaction is terminated at the diketide stage due to the steric contraction of the active site cavity, and this is followed by the N/C1 intramolecular lactamization of the Cys-bound linear diketide intermediate and concomitant thioester bond cleavage to produce the quinolone scaffold.
The crystal structure of C. microcarpa ACS also revealed a wide active site entrance (37 Å2), as in the case of C. microcarpa QNS (47 Å2) (Fig. 6B). However, unlike C. microcarpa QNS, the broadening of the active site entrance of C. microcarpa ACS is caused by the F265V substitution and the conformational changes of Phe-215 and Leu-267 (Fig. 6B). Furthermore, the cavity volume of C. microcarpa ACS (760 Å3) is 2.6 times larger than that of C. microcarpa QNS (290 Å3) and almost as large as that of M. sativa CHS (750 Å3). The wide active site entrance and the large active site cavity clearly affect the starter substrate preference and the product specificity of C. microcarpa ACS. As a result, C. microcarpa ACS accepts the bulky N-methylanthraniloyl-CoA as the starter substrate and catalyzes the iterative condensations with three molecules of malonyl-CoA to produce the acridone scaffold, by employing an active site cavity and catalytic machinery similar to those of CHS. Furthermore, as in the cases of the other type III PKSs, C. microcarpa ACS also exhibits promiscuous substrate specificity and accepts various starter substrates to produce distinct molecular scaffolds, including acridone, quinolone, chalcone, benzophenone, and phloroglucinol (Fig. 1, A–D). The site-directed mutagenesis studies suggested that three active site residues Ser-132, Thr-194, and Thr-197 play a crucial role for the enzyme activity of C. microcarpa ACS. The S132M, T194M, and T197Y point mutations resulted in the loss of the tetraketide acridone and chalcone forming activities and production of the diketide quinolone as the single product. Thus, C. microcarpa ACS was functionally converted into QNS. These results clearly supported our hypothesis that the volume and shape of the active site cavity control the product specificity of these enzyme reactions.
In the generation of the acridone tricyclic ring system, one of the intriguing points is the timing of the C–N bond formation, and the C-ring-forming reaction involving Claisen-type C–C bond formation and concomitant thioester bond cleavage of the linear tetraketide intermediate from the active site Cys. Interestingly, previously reported experiments for the formation of rutacridone with [1-C13]-, [2-C13]-, and [1,2-C13]acetate revealed two different labeling patterns within the C-ring (38). This result indicated that the C-ring is indeed derived from three acetate units and suggested that the different labeling patterns were caused by the rotation of a bicyclic N-methylaminobenzophenone intermediate (or a nonaromatized anthraniloyl-trione intermediate) prior to C–N bond formation. In fact, many bicyclic N-methylaminobenzophenones have been isolated from various Rutaceae plants (39–42). Our docking simulations based on the crystal structure of C. microcarpa ACS predicted that the active site cavity is large enough to accommodate the linear or cyclized tetraketide intermediate; however, there is not enough space for the rotation. The docking simulation with the linear tetraketide intermediate also predicted that the C1 carbon resides near the C6 carbon in the active site cavity. Therefore, we propose that the formation of the acridone tricyclic ring system proceeds by the initial cyclization of the linear tetraketide intermediate by Claisen-type condensation to produce the bicyclic N-methylaminobenzophenone (or a nonaromatized anthraniloyltrione), which is followed by the C–N bond formation to generate the acridone scaffold.
Finally, despite the high sequence identity with C. microcarpa ACS and R. graveolens ACS, both accepting 4-coumaroyl-CoA as the starter substrate to produce chalcone, the previously reported acridone/quinolone-producing A. marmelos QNS does not produce chalcone from 4-coumaroyl-CoA, although these three enzymes share the simultaneous substitutions of the CHS's conserved active site residues Thr-132, Ser-133, and Phe-265 with Ser, Ala, and Val, respectively. Site-directed mutagenesis studies of R. graveolens ACS and A. marmelos QNS indicated that the three residues play important roles in the substrate and product specificities of the two enzymes (12, 15). For example, the S132T/A133S double mutant of A. marmelos QNS produced chalcone from 4-coumaroyl-CoA, whereas the S132T/A133S/V265F triple mutant lost the enzyme activity, and no longer accepted the coumaroyl starter to yield any products (12). However, the S132T/A133S/V265F triple mutation of R. graveolens ACS caused full conversion into a functionally distinct chalcone-forming enzyme (15). These observations suggested that subtle structural differences exist in the active site architectures of these three acridone-producing enzymes.
In conclusion, C. microcarpa QNS is a novel type III PKS that produces the diketide quinolone by the one-step condensation of N-methylanthraniloyl-CoA and malonyl-CoA, whereas C. microcarpa ACS is a multifunctional PKS that produces not only acridone, but also chalcone, benzophenone, and phloroglucinol. The x-ray crystal structures of C. microcarpa QNS and ACS revealed the wide active site entrances of both enzymes, which facilitate the binding of the bulky N-methylanthraniloyl-CoA starter. The active site cavity of C. microcarpa QNS is significantly smaller than that of ACS, which leads to the specific production of the quinolone scaffold, whereas C. microcarpa ACS utilizes a large cavity to yield the tetraketide acridone, by employing an active site cavity and catalytic machinery similar to those of CHS. These results have provided the first structural bases for the production of the anthranilate-derived quinolone and acridone alkaloids by the type III PKSs. These findings will enable further engineering of the enzymes to create novel, structurally distinct, and biologically active molecular scaffolds for drug discovery.
This work was supported in part by grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology, Japan, CREST, Japan Science and Technology Agency, and a grant from Suntory for Bioorganic Research.
The atomic coordinates and structure factors (codes 3WD7 and 3WD8) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ACS
- acridone synthase
- BAS
- benzalacetone synthase
- CHS
- chalcone synthase
- PKS
- polyketide synthase
- QNS
- quinolone synthase
- r.m.s.d.
- root-mean-square deviation
- RACE
- rapid amplification of cDNA ends.
REFERENCES
- 1. Austin M. B., Noel J. P. (2003) The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep. 20, 79–110 [DOI] [PubMed] [Google Scholar]
- 2. Abe I., Morita H. (2010) Structure and function of the chalcone synthase superfamily of plant type III polyketide syntheses. Nat. Prod. Rep. 27, 809–838 [DOI] [PubMed] [Google Scholar]
- 3. Abe I., Watanabe T., Noguchi H. (2004) Enzymatic formation of long-chain polyketide pyrones by plant type III polyketide synthases. Phytochemistry 65, 2447–2453 [DOI] [PubMed] [Google Scholar]
- 4. Wanibuchi K., Zhang P., Abe T., Morita H., Kohno T., Chen G., Noguchi H., Abe I. (2007) An acridone-producing novel multifunctional type III polyketide synthase from Huperzia serrata. FEBS J. 274, 1073–1082 [DOI] [PubMed] [Google Scholar]
- 5. Wakimoto T., Mori T., Morita H., Abe I. (2011) Cytotoxic tetramic acid derivative produced by a plant type-III polyketide synthase. J. Am. Chem. Soc. 133, 4746–4749 [DOI] [PubMed] [Google Scholar]
- 6. Morita H., Yamashita M., Shi S. P., Wakimoto T., Kondo S., Kato R., Sugio S., Kohno T., Abe I. (2011) Synthesis of unnatural novel alkaloid scaffolds by exploiting plant polyketide synthase. Proc. Natl. Acad. Sci. U.S.A. 108, 13504–13509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hayashi H., Miwa Y., Ichikawa S., Yoda N., Miki I., Ishii A., Kono M., Yasuzawa T., Suzuki F. (1993) 5-HT3 receptor antagonists. 2,4-Hydroxy-3-quinolinecarboxylic acid derivatives. J. Med. Chem. 36, 617–626 [DOI] [PubMed] [Google Scholar]
- 8. Dittmer D. C., Li Q., Avilov D. V. (2005) Synthesis of coumarins, 4-hydroxycoumarins, and 4-hydroxyquinolinones by tellurium-triggered cyclizations. J. Org. Chem. 70, 4682–4686 [DOI] [PubMed] [Google Scholar]
- 9. Kelly J. X., Smilkstein M. J., Brun R., Wittlin S., Cooper R. A., Lane K. D., Janowsky A., Johnson R. A., Dodean R. A., Winter R., Hinrichs D. J., Riscoe M. K. (2009) Discovery of dual function acridones as a new antimalarial chemotype. Nature 459, 270–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lukacin R., Springob K., Urbanke C., Ernwein C., Schröder G., Schröder J., Matern U. (1999) Native acridone synthases I and II from Ruta graveolens L. form homodimers. FEBS Lett. 448, 135–140 [DOI] [PubMed] [Google Scholar]
- 11. Springob K., Lukacin R., Ernwein C., Gröning I., Matern U. (2000) Specificities of functionally expressed chalcone and acridone synthases from Ruta graveolens. Eur. J. Biochem. 267, 6552–6559 [DOI] [PubMed] [Google Scholar]
- 12. Resmi M. S., Verma P., Gokhale R. S., Soniya E. V. (2013) Identification and characterization of a type III polyketide synthase involved in quinolone alkaloid biosynthesis from Aegle marmelos correa. J. Biol. Chem. 288, 7271–7281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jez J. M., Bowman M. E., Noel J. P. (2002) Expanding the biosynthetic repertoire of plant type III polyketide synthases by altering starter molecule specificity. Proc. Natl. Acad. Sci. U.S.A. 99, 5323–5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lukacin R., Schreiner S., Silber K., Matern U. (2005) Starter substrate specificities of wild-type and mutant polyketide synthases from Rutaceae. Phytochemistry 66, 277–284 [DOI] [PubMed] [Google Scholar]
- 15. Lukacin R., Schreiner S., Matern U. (2001) Transformation of acridone synthase to chalcone synthase. FEBS Lett. 508, 413–417 [DOI] [PubMed] [Google Scholar]
- 16. Abe I., Abe T., Wanibuchi K., Noguchi H. (2006) Enzymatic formation of quinolone alkaloids by a plant type III polyketide synthase. Org. Lett. 8, 6063–6065 [DOI] [PubMed] [Google Scholar]
- 17. Liu B., Falkenstein-Paul H., Schmidt W., Beerhues L. (2003) Benzophenone synthase and chalcone synthase from Hypericum androsaemum cell cultures: cDNA cloning, functional expression, and site-directed mutagenesis of two polyketide synthases. Plant J. 34, 847–855 [DOI] [PubMed] [Google Scholar]
- 18. Abe I., Morita H., Nomura A., Noguchi H. (2000) Substrate specificity of chalcone synthase: enzymatic formation of unnatural polyketides from synthetic cinnamoyl-CoA analogues. J. Am. Chem. Soc. 122, 11242–11243 [Google Scholar]
- 19. Abe I., Takahashi Y., Morita H., Noguchi H. (2001) Benzalacetone synthase. A novel polyketide synthase that plays a crucial role in the biosynthesis of phenyl-butanones in Rheum palmatum. Eur. J. Biochem. 268, 3354–3359 [DOI] [PubMed] [Google Scholar]
- 20. Abe I., Utsumi Y., Oguro S., Morita H., Sano Y., Noguchi H. (2005) A plant type III polyketide synthase that produces pentaketide chromone. J. Am. Chem. Soc. 127, 1362–1363 [DOI] [PubMed] [Google Scholar]
- 21. Abe I., Oguro S., Utsumi Y., Sano Y., Noguchi H. (2005) Engineered biosynthesis of plant polyketides: chain length control in an octaketide-producing plant type III polyketide synthase. J. Am. Chem. Soc. 127, 12709–12716 [DOI] [PubMed] [Google Scholar]
- 22. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 23. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 24. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 26. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Holm L., Sander C. (1995) Dali: A network tool for protein structure comparison. Trends Biochem. Sci. 20, 478–480 [DOI] [PubMed] [Google Scholar]
- 29. Ferrer J. L., Jez J. M., Bowman M. E., Dixon R. A., Noel J. P. (1999) Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nat. Struct. Biol. 6, 775–784 [DOI] [PubMed] [Google Scholar]
- 30. Liu B., Raeth T., Beuerle T., Beerhues L. (2007) Biphenyl synthase, a novel type III polyketide synthase. Planta 225, 1495–1503 [DOI] [PubMed] [Google Scholar]
- 31. Jez J. M., Austin M. B., Ferrer J., Bowman M. E., Schröder J., Noel J. P. (2000) Structural control of polyketide formation in plant-specific polyketide synthases. Chem. Biol. 7, 919–930 [DOI] [PubMed] [Google Scholar]
- 32. Austin M. B., Bowman M. E., Ferrer J. L., Schröder J., Noel J. P. (2004) An aldol switch discovered in stilbene synthases mediates cyclization specificity of type III polyketide synthases. Chem. Biol. 11, 1179–1194 [DOI] [PubMed] [Google Scholar]
- 33. Morita H., Kondo S., Oguro S., Noguchi H., Sugio S., Abe I., Kohno T. (2007) Structural insight into chain-length control and product specificity of pentaketide chromone synthase from Aloe arborescens. Chem. Biol. 14, 359–369 [DOI] [PubMed] [Google Scholar]
- 34. Morita H., Shimokawa Y., Tanio M., Kato R., Noguchi H., Sugio S., Kohno T., Abe I. (2010) A structure-based mechanism for benzalacetone synthase from Rheum palmatum. Proc. Natl. Acad. Sci. U.S.A. 107, 669–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morita H., Wanibuchi K., Nii H., Kato R., Sugio S., Abe I. (2010) Structural basis for the one-pot formation of the diarylheptanoid scaffold by curcuminoid synthase from Oryza sativa. Proc. Natl. Acad. Sci. U.S.A. 107, 19778–19983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Helariutta Y., Elomaa P., Kotilainen M., Griesbach R. J., Schröder J., Teeri T. H. (1995) Chalcone synthase-like genes active during corolla development are differentially expressed and encode enzymes with different catalytic properties in Gerbera hybrida (Asteraceae). Plant Mol. Biol. 28, 47–60 [DOI] [PubMed] [Google Scholar]
- 37. Eckermann S., Schröder G., Schmidt J., Strack D., Edrada R. A., Helariutta Y., Elomaa P., Kotilainen M., Kilpeläinen I., Proksch P., Teeri T. H., Schröder J. (1998) New pathway to polyketides in plants. Nature 396, 387–390 [Google Scholar]
- 38. Zschunke A., Baumert A., Gröger D. (1982) Biosynthesis of rutacridone in cell cultures of Ruta graveolens: incorporation studies with [13C]acetate. J. Chem. Soc. Chem. Commun. 1263–1265 [Google Scholar]
- 39. Casey A. C., Malhotra A. (1975) TECLEANONE: a new alkaloid from Teclea grandifolia. Tetrahedron Lett. 16, 401–404 [Google Scholar]
- 40. Waterman P. G. (1975) Tecleanone from Diphasia klaineana and Teclea verdoorniana. Phytochemistry 14, 2092–2093 [Google Scholar]
- 41. Fish F., Meshal I. A., Waterman P. G. (1978) Alkaloid of Oricia suaveolens. Planta Med. 33, 228–231 [Google Scholar]
- 42. Waterman P. G., Meshal I. A., Hall J. B., Swaine M. D. (1978) Biochemical systematics and ecology of the Toddalioideae in the central part of the West African Forest Zone. Biochem. Syst. Ecol. 6, 239–245 [Google Scholar]