

Background: The function of MAGP2 was studied by inactivating its gene (Mfap5−/−) in mice.
Results: Mfap5−/− mice have a neutrophil deficiency and other phenotypes that are different from MAGP1- and fibrillin-deficient animals.
Conclusion: MAGP2 has functional roles in hematopoiesis and in vessel wall maintenance.
Significance: Characterization of MAGP2 function is crucial to the identification and treatment of microfibril-related disease.
Keywords: Bone Morphogenetic Protein (BMP), Elastin, Extracellular Matrix, Hematopoiesis, Transforming Growth Factor β (TGFβ), MAGP, Microfibril
Abstract
Microfibril-associated glycoprotein (MAGP) 1 and 2 are evolutionarily related but structurally divergent proteins that are components of microfibrils of the extracellular matrix. Using mice with a targeted inactivation of Mfap5, the gene for MAGP2 protein, we demonstrate that MAGPs have shared as well as unique functions in vivo. Mfap5−/− mice appear grossly normal, are fertile, and have no reduction in life span. Cardiopulmonary development is typical. The animals are normotensive and have vascular compliance comparable with age-matched wild-type mice, which is indicative of normal, functional elastic fibers. Loss of MAGP2 alone does not significantly alter bone mass or architecture, and loss of MAGP2 in tandem with loss of MAGP1 does not exacerbate MAGP1-dependent osteopenia. MAGP2-deficient mice are neutropenic, which contrasts with monocytopenia described in MAGP1-deficient animals. This suggests that MAGP1 and MAGP2 have discrete functions in hematopoiesis. In the cardiovascular system, MAGP1;MAGP2 double knockout mice (Mfap2−/−;Mfap5−/−) show age-dependent aortic dilation. These findings indicate that MAGPs have shared primary functions in maintaining large vessel integrity. In solid phase binding assays, MAGP2 binds active TGFβ1, TGFβ2, and BMP2. Together, these data demonstrate that loss of MAGP2 expression in vivo has pleiotropic effects potentially related to the ability of MAGP2 to regulate growth factors or participate in cell signaling.
Introduction
The ECM2 is a complex biopolymer that provides strength to tissues and plays instructive roles in organogenesis and tissue homeostasis (1). An abundant component of the ECM is the microfibril, which imparts limited elasticity to tissues, acts as a template for elastin deposition, and is a regulator of signaling events. Microfibrils are 10- to 12-nm fibers built upon a backbone of fibrillin and decorated with function-modifying accessory proteins that include growth factor complexes, fibulins, emilins, and MAGPs (2, 3). Mutations in the genes for microfibril components have varied systemic effects that are not always apparent with in vitro experimentation (3–5). The complexity of microfibril structure necessitates in vivo manipulation of proteins for an accurate understanding of microfibril function and identification of microfibril-related disease.
MAGP1 (the protein product of the microfibrillar-associated protein 2 (Mfap2) gene) and MAGP2 (the protein product of the Mfap5 gene and also known as MP-25) are a two-member family of small microfibril-associated proteins ∼31 kDa and 25 kDa in size, respectively (6, 7). Both proteins are found only in vertebrates, and phylogenetic studies suggest that MAGP2 arose from MAGP1 through gene duplication early in vertebrate evolution (8). The two MAGPs share a functional C-terminal matrix-binding domain that is characterized by conserved cysteine residues (9, 10). MAGP2 has a conserved proprotein convertase cleavage site within this domain that makes MAGP2 a substrate for multiple proprotein convertase family members (11). Also unique to MAGP2 is an RGD integrin-binding motif located at the N terminus.
Amino acid sequences in the N-terminal regions of MAGP1 and MAGP2 are dissimilar, but both are enriched in acidic amino acids. MAGP1 contains two consensus sequences for O- but not N-glycosylation (12), whereas MAGP2 contains five conserved, predicted O-linked glycosylation sequences. Functional studies with MAGP1 show that the acidic N terminus contains a growth factor interaction motif capable of binding active TGFβ and bone morphogenic protein (BMP) (13).
MAGP1 is associated with all microfibrils except those directly adjacent to the plasma membrane of aortic endothelial cells and those at the junction of the zonule of the eye (14–16). Although MAGP2 has been localized to both elastin-associated microfibrils and elastin-free microfibrils in a number of tissues, the protein exhibits patterns of tissue localization and developmental expression that are more restricted than those of MAGP1 (7, 17). The association of MAGP1 and MAGP2 with other microfibrillar proteins is covalent, requiring reducing agents for their extraction (18).
Genetic deletion of extracellular MAGP1 in mice results in an array of phenotypes, including a bleeding diathesis, obesity, and osteopenia (13, 19, 20). MAGP1-deficient animals also have a pronounced deficiency in circulating and tissue monocytes. Here we report the generation and initial characterization of mice with null alleles of the MAGP2 gene (Mfap5) and both MAGP1 and MAGP2 (Mfap2−/−;Mfap5−/−) (DKO) genes. We find that Mfap5−/− (2KO) phenotypes are non-overlapping with mice lacking MAGP1 (1KO). However, the absence of both MAGPs causes changes in large vessel architecture. Biochemical studies show that MAGP2 protein binds active TGFβ1, TGFβ2, and BMP2. Taken together, these data show that MAGP2 has unique, MAGP1-independent functions in hematopoiesis and that MAGPs have redundant functions in large vessels.
EXPERIMENTAL PROCEDURES
Generation and Breeding of MAGP2-deficient and MAGP1-MAGP2-deficient Mice
Homologous recombination was used to insert a gene-targeting cassette containing the coding sequence for neomycin resistance into the exon 9 region of the Mfap5 gene in murine ES cells. G418-resistant ES cell colonies were screened by Southern blot analysis, and ES cell clones, positive for recombination, were injected into C57Bl/6 blastocysts. Chimeric mice were bred for germ line transmission. Genotype analysis was performed on genomic DNA isolated from tail tissue using primers listed in supplemental Table 1. Generation of mice deficient in both MAGP1 and MAGP2 was achieved by breeding MAGP2 mutant mice with previously generated animals deficient in MAGP1 (13). Mice used for these studies were maintained on a mixed-strain background, and wild-type controls were generated from the same parental stock as MAGP-deficient animals. Animals were housed in a pathogen-free facility, and the Washington University Animal Studies Committee approved all procedures.
Genomic DNA Isolation and Sequencing
Genomic DNA isolated from adult mouse tail tissue was used with primers listed in supplemental Table 1 to confirm the location and orientation of the targeting construct within the Mfap5 gene locus. For all sequencing reactions, bands were extracted from agarose gel, and PCR products were isolated using a QIAquick gel extraction kit according to the protocol of the manufacturer (Qiagen Inc., Valencia, CA). Amplicons were then ligated into the pGEM-T vector using the rapid DNA ligation kit according to the protocol of the manufacturer (Roche Diagnostics, Mannheim, Germany) and introduced into DH5α Escherichia coli (Promega Corp., Madison, WI). The growth medium was inoculated with ampicillin-resistant, color-selected colonies, and plasmid products were isolated using QIAprep Spin Miniprep kits according to the instructions of the manufacturer (Qiagen). The Protein and Nucleic Acid Chemistry Laboratory (Washington University, Saint Louis, MO) performed DNA sequencing. All DNA alignments were performed using DNASTAR Lasergene 9 software.
Total RNA Isolation and in Situ Hybridization
Total RNA was isolated from adult mouse lung, kidney, and heart using TRIzol reagent, and reverse transcription cDNA amplification was performed using 1 μg of RNA and SuperScript III first-strand synthesis system with oligo(dT) according to the specifications of the manufacturer (Invitrogen). Complement DNA was amplified using primers listed in supplemental Table 1. Ethidium bromide gels for nonsense-mediated decay studies were imaged with the ChemiDoc MP system and Image Lab version 4.0 software using identical exposure and filter settings (Bio-Rad). For sequencing, PCR band excision and sequencing were performed according to methods detailed previously. RNA probe generation and in situ hybridization for Mfap5, Mfap2, and collagen1α1 (Col1a1) was performed as described previously using mRNA isolated from adult mouse heart and primers listed in supplemental Table 1 (21–23). RNA probes were 806 bp in length for Mfap5, 802 bp for Mfap2, and 846 bp for Col1a1.
Antibodies, Western Blotting, and Immunohistochemistry
Rabbit polyclonal antibodies against mouse MAGP2 (24) and MAGP1 (13) have been described previously. For Western blot analysis of serially extracted mouse tissue lysates, adult mouse lung was homogenized in 990 μl of PBS (pH7.4) and 10 μl of protease inhibitor mixture (catalog no. P8340, Sigma Aldrich, Saint Louis, MO) and extracted 24 h at 4 °C. The samples were pelleted by centrifugation at 16,000 × g or 20 min at 4 °C. The liquid portion of the sample was removed for Western blotting, and the pellet was resuspended in 1 m NaCl and protease inhibitors and extracted for 24 h. The process was repeated using 8 m urea, PBS, and 20 mm DTT (seen in Fig. 5C,D), and 8 m urea + DTT. Western blotting was performed using standard procedures. Samples were run on 7.5% or 12% SDS-PAGE gels and transferred to nitrocellulose membranes. Ponceau S stain was applied for 5 min and removed using 0.1 m NaOH. All blocking steps were performed with blocking buffer containing 5% casein, 0.1% coldwater fish skin gelatin, and 0.01% Tween 20 in TBS. Primary antibodies were applied overnight at 4 °C at a dilution of 1:10,000 for MAGP2 (in 5% BSA) or 1:1000 for MAGP1 (in 5% BSA). Secondary antibodies were applied at 1:4000 in casein block and incubated for 1 h at room temperature. ECL reagents were used for detection of horseradish peroxidase, and chemiluminescence was read using the ChemiDoc MP system and Image Lab version 4.0 software (GE Healthcare). Immunohistochemistry for paraffin sections was performed using the ultrasensitive ABC peroxidase rabbit IgG staining kit (Fisher Scientific, Pittsburgh, PA) according to the instructions of the manufacturer, with the following modifications for MAGP2 detection. After peroxidase quenching, the sections were washed in 0.1% BSA in TBS. Hyaluronidase digestion solution (1 mg/ml hyaluronidase in 0.1 m sodium acetate buffer (pH5.5) containing 0.85% NaCl) was applied, and the sections were incubated in a humid chamber for 30 min at 37 °C. The sections were washed in BSA/TBS three times for 5 min. A guanidine/DTT solution was applied to the sections for 15 min. The sections were washed with BSA/TBS three times for 5 min before incubation with iodoacetamide solution for 15 min. The sections underwent a final BSA/TBS wash before application of ultra-sensitive staining kit block solution. Primary anti-MAGP2 antibody was diluted 1:8000 in blocking solution and applied to the sections overnight at 4 °C. Secondary antibody solution was made using 2.25 μl/ml of secondary antibody. Horseradish peroxidase reactivity was visualized using the metal enhanced DAB substrate kit (Fisher). Imaging was performed on a Zeiss Axioskop using identical exposure and filter settings. Image processing was performed using Adobe Photoshop CS5 software.
FIGURE 5.

2KO and DKO mice lack MAGP2 protein. A, Western blot analyses showing that anti-MAGP2 and anti-MAGP1 antibodies are specific for their respective recombinant murine proteins (rMAGP2 and rMAGP1). B, immunohistochemistry on WT and 2KO sections demonstrates MAGP2 reactivity at the intima (arrow) and adventitia (arrowhead) of WT mouse aorta. No MAGP2 reactivity is seen in the 2KO aorta section. Scale bar = 100 μm. C, Western blotting of sequentially extracted (20 mm DTT fraction) mouse whole lung lysates with anti-MAGP2 antibody demonstrating the absence of MAGP2 protein in 2KO and DKO animal tissue. Two animals of each genotype were used, and an arrow indicates the MAGP2 protein band. Mammalian MAGP2 from tissue lysate runs as a larger protein than bacterially expressed rMAGP2 because of posttranslational modifications. D, Western blotting of sequentially extracted mouse whole lung lysates with anti-MAGP1 antibody demonstrating the absence of MAGP1 protein in DKO animals only. Two animals of each genotype were used, and arrows indicate MAGP1 protein bands. The major rMAGP1 band runs as a larger protein because of the His6 tag. Tissue lysates were run with bacterially expressed rMAGP2 or His6-tagged rMAGP1 and shown on the same blot. However, the recombinant protein section of the blot is shown at a shorter exposure interval, as indicated by a black line. Bands above the indicated MAGP proteins represent nonspecific antibody reactivity (NS). I = intima; M = media; A = adventitia.
Flow Cytometry
Monoclonal antibodies used for flow cytometry were as follows: phycoerythrin-conjugated hamster anti-mouse CD3e (BD Biosciences), FITC rat anti-mouse CD335 (NKp46, eBioscience, San Diego, CA), FITC rat anti-mouse Ly-6G (eBioscience), phycoerythrin rat anti-mouse CD115 (eBioscience), allophycocyanin rat F4/80 (Abcam, Cambridge, MA). Phycoerythrin hamster IgG1, FITC rat IgG2a, FITC rat IgG2b, phycoerythrin rat IgG2a control, and allophycocyanin rat IgG2b isotype controls were utilized according to the suggestions of the manufacturer. Mice were killed by CO2 narcosis, and the chest and abdominal cavity were exposed to reveal the heart, dorsal aorta, and spleen. Whole blood was collected from the dorsal aorta in a heparinized syringe. Red cells were lysed with NH4Cl buffer for 10 min at room temperature and washed twice with FACs buffer (1× PBS and 2% FBS). After blocking with mouse Fc Block (BD Biosciences) for 20 min, 2 × 105 cells were incubated with antigen-specific or isotype control antibodies for 30 min at 4 °C. Cells were washed twice with FACs buffer and fixed with 4% paraformaldehyde for 15 min, washed twice, and stored overnight. Spleens were removed, and splenocytes were isolated after passage through a 70-μm nylon mesh filter. Lysis, blocking, incubation, and fixation were identical to the white blood cell treatment above. Cells were sorted on a FACSCalibur machine (BD Biosciences) with Cell-Quest software (BD Biosciences). The percentage of positive cells for each cell type was determined by gating on isotype control, nonspecific labeling (percentage of positive cells within gate < 5% for control), followed by subtraction of background percentage from antibody-specific results. A minimum of 105 live cell events were recorded for each analysis. Experiments were performed with at least five animals of each genotype (n ≥ 5), and experiments were repeated at least two times with mice 2 and 6 months of age.
Microcomputed Tomography (μCT)
Microcomputed tomography analysis of left tibias from 6-month-old mice was performed as described previously (19), with the exception that the tibias were not frozen. The tibias were embedded in 2% agarose and scanned using a Scanco μCT 40 (Scanco Medical AG, Zurich, Switzerland). For trabecular bone measurements, the growth plate was excluded and 30 × 16-μm sections were contoured. For cortical bone, the sections were analyzed just above the tibiofibular junction, and three sections per tibia were analyzed. Contouring and analysis were performed by the Washington University Musculoskeletal Research Facility using Scanco MicroCT software. Identical contouring and analysis parameters were used for all tibias. The tibias from at least five male mice of each genotype (n ≥ 5) were used for analysis.
Blood Pressure and Mechanical Properties of Ascending Aorta and Left Carotid Artery
Six-month-old male mice were weighed and then anesthetized using 1.5% isoflurane, keeping the body temperature constant. A solid-state catheter was inserted into the right common carotid artery, isoflurane was reduced to 0.5%, and blood pressure measurements were recorded (25, 26). The hearts were excised and weighed, and the aortas and left carotid arteries were removed for mechanical testing as reported previously (25). At least 12 male mice of each genotype (n ≥ 12) were used for analysis.
Recombinant MAGP2 and MAGP1 Proteins and Biolayer Interferometry (BLI)
The coding sequence for mouse MAGP2 protein, excluding the N-terminal signal sequence, was cloned into pET23b as a tagless construct. This plasmid was transformed into Rosetta 2(DE3) E. coli cells for protein expression. MAGP2 proteins were purified from inclusion bodies, followed by oxidative refolding as described previously (27). Recombinant His6-tagged MAGP1, used for Western blotting, was expressed and isolated as described previously for this protein (13).
Native MAGP2 protein was expressed in mammalian cells and purified as follows. Full-length mature-form MAGP2, without the endogenous signal sequence, was cloned into the pHL-Avitag3 vector (28) containing an optimized signal sequence at the N terminus as well as a BirA biotin ligase recognition sequence and hexahistidine (His6) tag at the C terminus. Protein was expressed by large-scale transient transfection of FreeStyle 293F cells cultured in serum-free Freestyle 293 medium (Life Tech.). Culture supernatants were collected 72 h post-transfection, and protein was purified to homogeneity using nickel affinity chromatography. Purity was assessed by SDS-PAGE. Site-specific biotinylation of the C-terminal tags were carried out using E. coli BirA biotin ligase.
All surface plasmon resonance binding was performed on a Reichert SR7000 surface plasmon resonance system. To bind MAGP2 protein to the carboxymethyl dextran (CM-5) chip, the chip was preactivated with a mixture of 200 mm 1-ethyl-3(3-dimethylaminopropyl)carbodiimide hydrochloride and 50 mm N-hydroxysuccinimide, and then 100 μl MAGP2 was diluted in 5 mm acetate buffer (pH 4.5) and injected with a flow rate of 10 μl/min at a concentration of 20 μm. This resulted in ∼1300 resonance units of MAGP2 protein coupled to the chip. Excess reactive sites on the chip were then quenched with a 70-μl injection of 0.5 m ethanolamine (pH 8). To measure MAGP2 binding to active TGFβ1, carrier-free active human TGFβ1 (R&D Systems, Minneapolis, MN) was diluted in surface plasmon resonance running buffer (10 mm HEPES, 150 mm NaCl, 0.05% Triton X-100 (pH 7.4)) and injected for 100 s at a flow rate of 50 μl/min. The dissociation was monitored for 300 s. The chip was recycled with 0.2 m glycine (pH 2.3) before subsequent injections. The concentrations of TGFβ1 injected were 100, 50, 50, 25, 25, and 12.5 nm. Background subtraction and calculation of the dissociation constant for individual curves was calculated using Scrubber2 software (Center for Biolmolecular Interaction Analysis, University of Utah).
The Octet system for Biolayer interferometry (Pall Life Sciences, Ann Arbor, MI) was also used to assess MAGP2 binding to active TGFβ1 as well as TGFβ2 and BMP2. Streptavidin-coated biosensors from ForteBio were used to capture biotinylated MAGP2 onto the surface of the sensor. After reaching base line, sensors were moved to the association step containing 1000, 500, 250, 125, 62.5, 31.3, 15.6, and 7.8 nm active mouse TGFβ1, TGFβ2, or BMP2 for 300 s and then dissociated for 300 s. A buffer-only reference was subtracted from all curves. The running buffer consisted of 10 mm HEPES (pH 7.4), 150 mm NaCl, 3.4 mm EDTA, 1% BSA, 0.01% azide, 0.05% Tween, and 0.005% Triton X-100. Affinities were estimated from global kinetic analysis.
Statistical Analysis
Statistical significance was determined by Student's t test. Data are reported as a mean ± S.D.
RESULTS
MAGP2 Gene Targeting
Homologous recombination was used to disrupt the MAGP2 gene (Mfap5) by replacing exon 9 with a neomycin resistance cassette (Neo), causing a downstream frameshift. Our attempts to target exon 1 and the upstream promoter sequence were unsuccessful. Exon 9 encodes the matrix-binding domain. If expressed, the mutant protein lacking this domain would be incapable of binding to fibrillin and, hence, would be a functional null.
PCR primers flanking each exon from 2–10, as well as within the Neo cassette itself, were used to walk along the targeted Mfap5 gene and confirm localization of the Neo cassette to exon 9. Genomic DNA (gDNA) isolated from WT and Mfap5−/− mice yielded identical amplicons using primers flanking exons 2–8 of the Mfap5 gene (Fig. 1, A and B, and supplemental Table 1). An exon 9 amplicon was generated from WT but not 2KO genomic DNA, and a forward primer to the Neo sequence and a reverse primer at exon 9 produced an amplicon from 2KO and not WT gDNA (Fig. 1, A and C). These PCR bands were gel-extracted and sequenced, confirming that the Neo sequence was introduced at exon 9 of the Mfap5 gene (supplemental Fig. S1).
FIGURE 1.
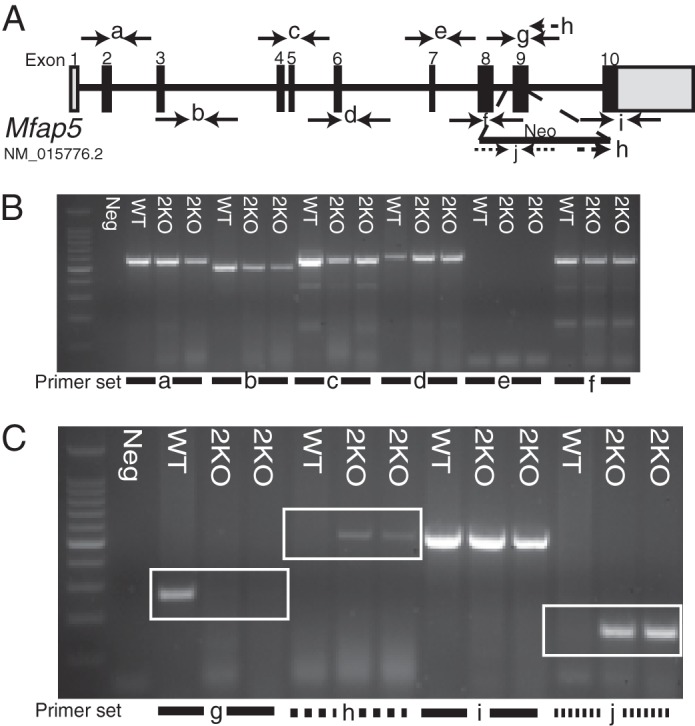
Schematic of Mfap5 gene targeting. A, schematic of the Mfap5 gene with numbered bars representing exons (gray, non-coding; black, coding). A gene-targeting cassette containing the coding sequence for neomycin resistance was introduced by homologous recombination into the murine Mfap5 gene. Lettered arrows represent primer pairs used for PCR. B and C, gDNA from one WT and two 2KO mice was subjected to PCR. Lettered lines refer to primer pairs indicated in A. A PCR amplicon is produced from the WT and not the 2KO template because of the presence of the targeting construct (boxed region, C, g). Primers to the targeting construct and Mfap5 sequence produce a PCR product from the 2KO and not the WT gDNA template (boxed region, C, h). PCR primers to Neo produce a product only from 2KO gDNA (boxed region, C, j).
To generate DKO animals, 2KO mice were bred with 1KO mice generated in a previous study (13). Fig. 4B confirms the genotype of the DKO animals. The exon 9 forward primer, Neo primer, and reverse exon 9 primer described above were used for Mfap5 genotyping reactions (Figs. 1 and 4B).
FIGURE 4.
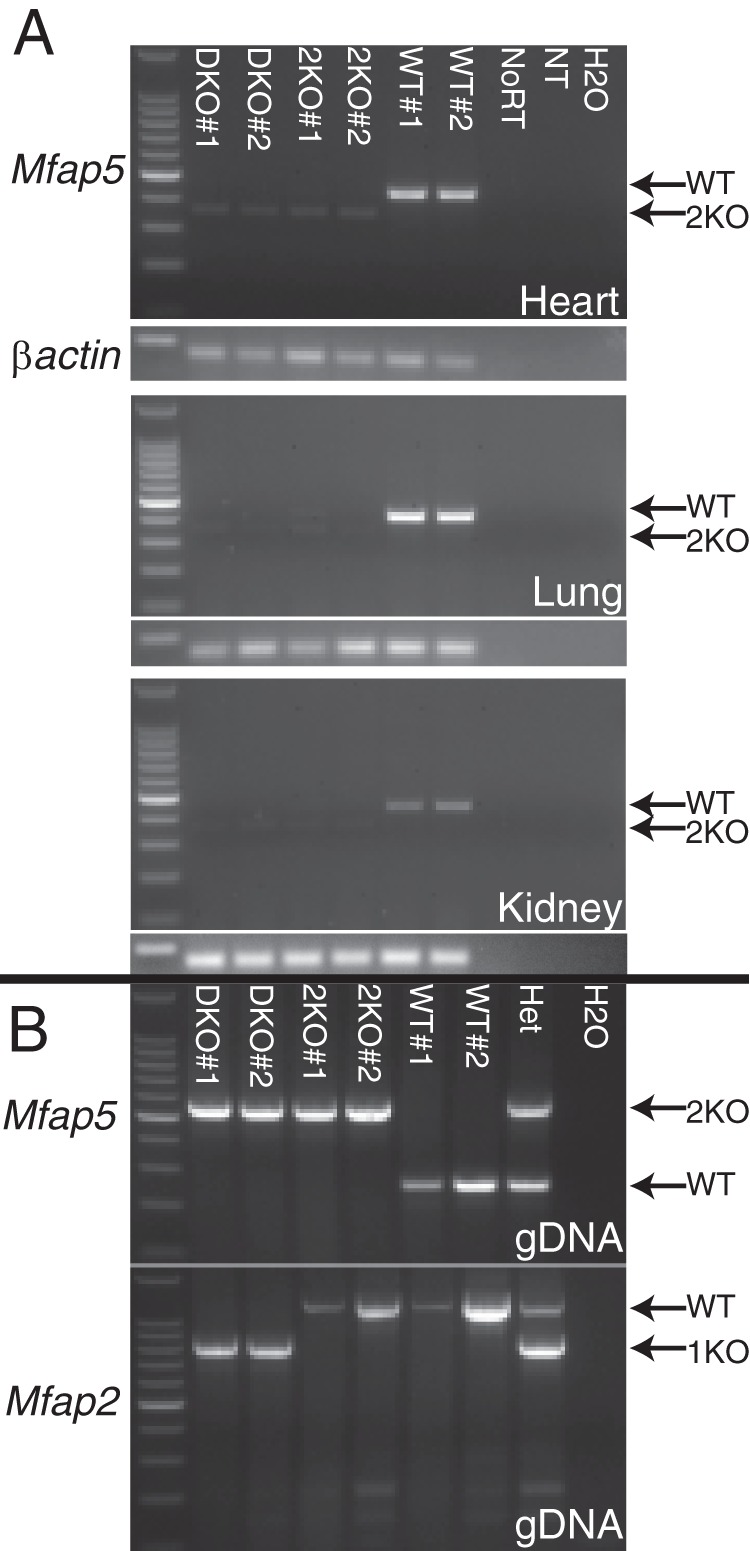
Absence of MAGP2Δ9 mRNA confirms functional knockout of MAGP2. A, polyadenylated mRNA from the heart, lung, and kidney of two DKO, 2KO, and WT animals each was reverse-transcribed and subjected to 27 rounds of PCR amplification with primers specific for the Mfap5 message. Arrows indicate WT and 2Δ9 amplicons (2KO). Reactions with primers to β-actin were run in tandem as a loading control. B, PCR genotyping of genomic DNA from animals used in the assay. WT, 2KO, and 1KO amplicons are indicated by arrows. NoRT, no reverse transcriptase control; NT, no template for reverse transcription control; H2O, no template for PCR control.
The Mfap5-targeted Allele (2Δ9) Is Transcribed, but the Mutant mRNA Is Degraded
RT-PCR amplification was used to determine whether mRNA is transcribed from the mutated Mfap5 gene in 2KO mice. PCR products representing the Mfap5 exon 2–10 sequence were amplified from both WT and 2KO cDNA. However, the 2KO amplicon is 74 bp smaller than the WT product (Fig. 2, A and B). DNA sequencing revealed that the 2KO product is a transcript with exon 9 and the Neo cassette spliced out (2Δ9) (supplemental Fig. S2 and Fig. 2C). This sequence does not occur as a natural splice variant, although we did identify a naturally occurring WT transcript lacking exon 6 in one of four WT animals tested (supplemental Fig. S3 and Fig. 2C).
FIGURE 2.
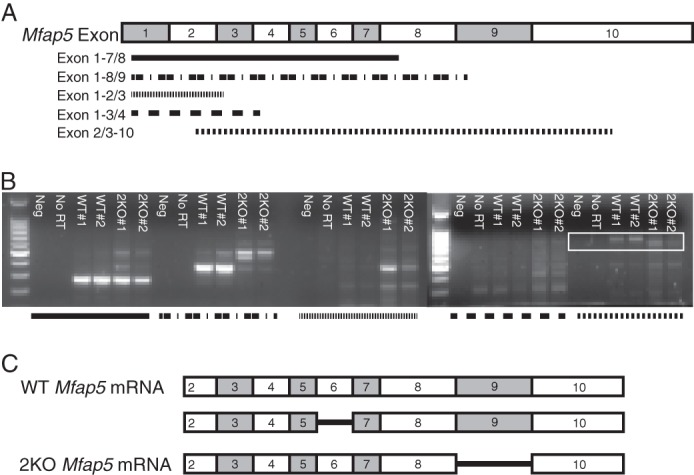
Exon 9 is spliced out of the targeted Mfap5 transcript. A naturally occurring Mfap5 splice variant lacking exon 6 also occurs. A, illustration of the Mfap5 mRNA transcript, with shaded rectangles representing exons. PCR primer pairs directed against an exon and an exon/intron junction are labeled, and the amplicon is depicted by shaded lines. B, RT-PCR was performed using the mRNA template from two WT and two 2KO mice. No reverse transcriptase (No RT) and no cDNA template (Neg) controls were included in the PCR reactions. Shaded lines refer to primer pairs illustrated in A. PCR products using exon 2/3–10 primers (boxed region) were larger for the WT template than for the 2KO template. C, schematic representation of Mfap5 mRNA transcripts from WT and 2KO mice. Shown are schematics demonstrating a full-length transcript as well as a naturally occurring splice variant lacking exon 6 from WT mice. All Mfap5 mRNA transcripts amplified from 2KO mice lack exon 9.
Because of a frameshift at the atypical exon 8–10 splice junction, there is a serine-to-arginine substitution and then a premature stop codon after domain 8 of the 2Δ9 transcript when translated (Fig. 3, A and B). Premature termination codons that occur upstream of an exon-exon junction frequently elicit nonsense-mediated decay, a RNA surveillance mechanism that degrades mutant transcripts (29). To determine whether nonsense-mediated decay affects the stability of the 2Δ9 transcript, RT-PCR was used to amplify 2Δ9 or WT mRNA isolated from the heart, lung, and kidney. After 27 cycles of PCR, there were only trace amounts of 2Δ9 PCR product compared with an abundant WT message in all three tissues (Fig. 4). These results indicate that little 2Δ9 mRNA escapes nonsense-mediated decay in 2KO animals. Similar results were found for MAGP1-MAGP2 double knockout animals (Fig. 4).
FIGURE 3.

Comparison of MAGP1 and MAGP2 sequences and predicted protein transcripts from WT and MAGP2-KO genes. The amino acid sequences of MAGP1 and MAGP2 are listed, with shaded rectangles representing protein domains. Known functional regions are labeled. Amino acids are represented by dots to denote their position in the protein sequence. Black, acidic; gray, basic; black, cysteine; black, glutamine. Threonine residues predicted to undergo O-linked glycosylation are marked (★). Black, conserved from opossum to human; white, not conserved among species. A, MAGP2 protein translated from the WT mRNA sequence. B, predicted MAGP2 protein translated from the 2KO mRNA sequence. Splicing out exon 9 creates a frameshift that introduces an arginine followed by a premature stop (asterisk) codon after exon 8. C, illustration of mature MAGP1 protein. D, comparison of mature MAGP1 and MAGP2 protein sequences. Amino acids conserved between proteins are shaded black. The MAGP homology/matrix binding domain is represented by a black line over the protein sequences.
An antibody to MAGP2 was used to confirm loss of MAGP2 protein expression in 2KO and DKO animals. This antibody reacts specifically with MAGP2 protein and does not recognize MAGP1 (Fig. 5A). Immunohistochemistry on mouse aorta sections shows MAGP2 labeling in the intimal layer as well as throughout the adventitia (Fig. 5B). This expression pattern agrees with previous reports for MAGP2 expression in fetal calf aortic tissue and supports the specificity of the MAGP2 antibody (7). MAGP2-reactive bands are present in Western blot analyses using sequentially extracted lung tissue from WT mice, whereas 2KO and DKO lung tissue lacks MAGP2 protein (Fig. 5C). Also, WT and 2KO mouse lungs are positive for MAGP1, whereas DKO mice lack this protein (Fig. 5D). Taken together, these data demonstrate that the gene targeting strategy was successful in preventing MAGP2 protein expression in 2KO and DKO animals and that DKO animals lack both MAGP2 and MAGP1.
MAGP2 Deficiency Results in Decreased Levels of Circulating Neutrophils but Normal Monocyte Counts
2KO and DKO mice are viable and fertile with no gross abnormalities in appearance or behavior. This contrasts with 1KO mice, which have complex phenotypes, including hematopoietic defects, obesity, and abnormal bone homeostasis (13, 19, 20). To investigate whether 2KO mice have defects in hematopoiesis, antibody labeling and flow cytometry were used to assess the complement of hematopoietic cells in the peripheral blood circulation or in spleen tissue. An average of 3330 ± 672 white cells/ml of blood were collected. There was no statistical difference in total white blood cells per mouse among genotypes. Using GR1 positivity as an antibody marker, our results show that WT mice have neutrophil counts of 12.5% and 8.8% of total peripheral blood and spleen cells, respectively. Similarly, 1KO mice have neutrophil counts of 13.4% of total peripheral blood (data not shown). In contrast, 2KO mice have significantly decreased neutrophils (5.6% blood and 5.1% spleen) compared with age-matched WT controls (Fig. 6). Likewise, DKO mice also exhibit neutropenia, with 6.4% of total cells in peripheral blood and 5.7% in the spleen. These findings indicate that loss of MAGP2 causes a neutrophil deficiency in 2KO mice that is not seen in 1KO animals and is not altered by loss of MAGP1 in DKO animals.
FIGURE 6.
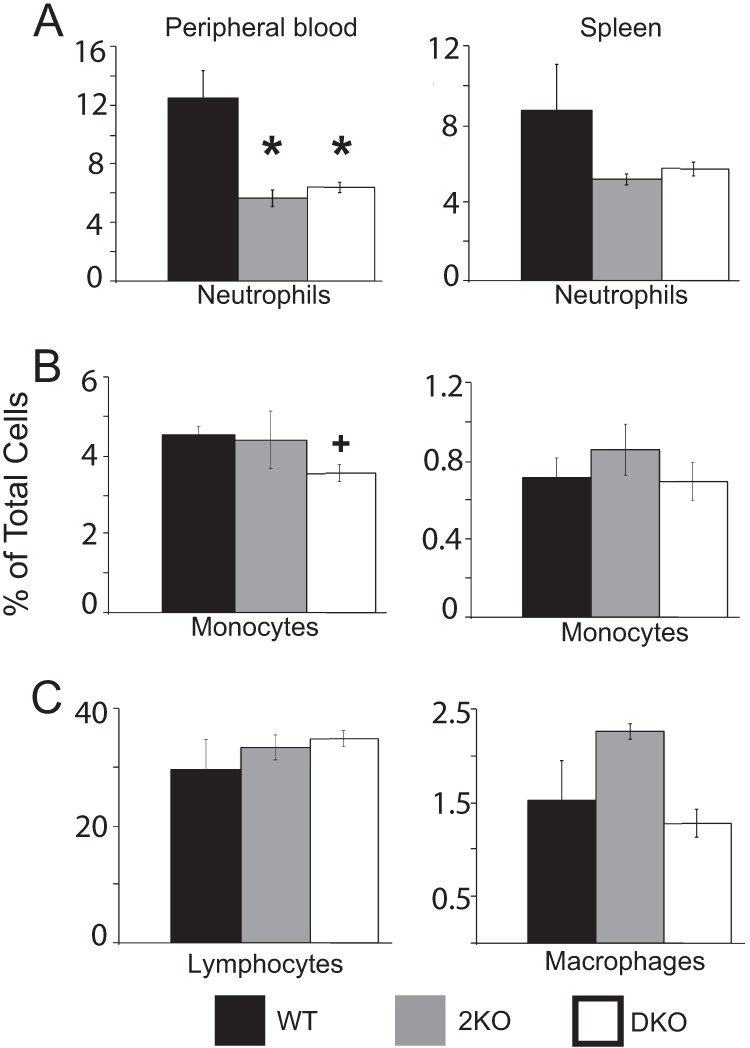
2KO mice are neutropenic. Flow cytometry was performed on peripheral blood and spleen cells collected from WT, 2KO, and DKO mice. A, GR1-positive neutrophil numbers were assessed, demonstrating that 2KO and DKO mice are significantly neutropenic. B, DKO animals have significantly fewer CD115-positive monocytes as a fraction of peripheral blood. C, histogram showing CD3-positive lymphocytes as a fraction of whole blood. D, histogram showing F4/80-positive macrophages as a fraction of total spleen cells. *, p ≤ 0.01; +, p ≤ 0.02. All data are independent of mouse age.
We have shown that mice deficient in MAGP1 are monocytopenic with fewer circulating monocytes and fewer tissue macrophages.3 2KO mice, in contrast, have CD115+ monocyte numbers comparable with WT controls (4.5% versus 4.4% blood and 0.7% versus 0.8% spleen, respectively) (Fig. 6). DKO animals have reduced monocytes as a fraction of peripheral blood (3.6% blood and 0.7% spleen), as would be expected for 1KO animals. F4/80 antibody reactivity was used as a marker for macrophages in spleen tissue. 2KO and DKO mice had macrophage numbers comparable with WT controls. These data show that loss of MAGP2 does not impact monocyte number alone or in conjunction with loss of MAGP1. This demonstrates that both 1KO and 2KO mice have unique alterations in the myeloid cell lineage.
CD3 reactivity was used to assess lymphocytes in WT, 2KO, and DKO mice. All three genotypes have equivalent lymphocyte numbers as a percentage of peripheral blood (Fig. 6). Similar results were shown for 1KO animals.3 Taken together, these data show that loss of MAGP2 causes neutropenia in vivo and demonstrate that this hematopoietic defect is MAGP1-independent. Further, loss of MAGP2 on a 1KO background does not impact the monocytopenia phenotype observed in 1KO animals. Thus, MAGP1 and MAGP2 have discrete functions in hematopoiesis.
Loss of MAGP2 Alone Does Not Significantly Alter Bone Mass or Architecture
Mice lacking MAGP1 are osteopenic by 6 months of age, with a high incidence of spontaneous fractures (13, 20). Spontaneous fractures are not observed in 2KO or DKO mice, and the skeleton appears grossly normal. μCT of left tibias in 6-month-old 2KO mice shows trabecular and cortical bone to be comparable in all aspects with age-matched WT controls (Fig. 7). Further, loss of MAGP2 on a 1KO background (DKO) does not exacerbate changes in bone mass and architecture associated with MAGP1 loss of function (Fig. 7). It is interesting to note that DKO mice share the abnormal cortical bone morphology of 1KO mice characterized by a smaller marrow cavity, or medullary area, than WT or 2KO mice and, therefore, have an increased bone volume fraction (Fig. 7B). These data demonstrate that loss of MAGP2 by itself has a minimal effect on bone character and suggest that, unlike MAGP1, MAGP2 has little or no role in normal bone homeostasis.
FIGURE 7.
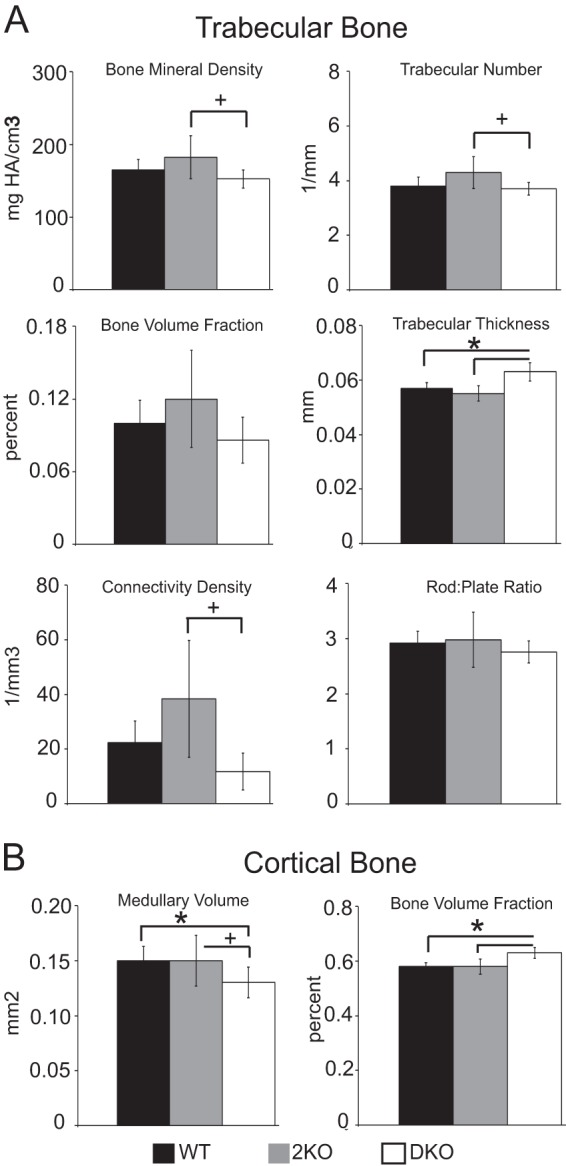
Bone density and architecture are not significantly affected by loss of MAGP2. μCT assessment of left tibias from 6-month-old WT, 2KO, and DKO mice. A, measurements of trabecular bone indicate no significant changes in 2KO trabecular bone compared with the WT. B, measurements of cortical bone indicate no changes in 2KO bone compared with the WT. A smaller marrow cavity (medullary area), associated with loss of MAGP1, persists in DKO mice, and this increases bone volume fraction in these animals. n = 5 (WT), 7 (2KO), and 5 (DKO) mice. HA, hydroxyapatite. *, p ≤ 0.01; +, p < 0.05.
MAGP1 and MAGP2 Double-null (DKO) Mice Have a Larger Aorta at Physiological Pressures
In vitro studies suggest MAGP2 expression may be necessary for normal elastic fiber assembly (30). This idea is supported by the expression pattern of MAGP2 in elastin-rich structures such as the aorta, aortic root, atrialis of the mitral valve, bronchioles of the lung, large vessels of the heart, lung, and kidney, as well as skin (Figs. 8 and 5B) (7). Previous work determined that 1KO mice are normotensive with normal aortic and left carotid pressure outer diameter curves at 14 weeks of age (13). The cardiopulmonary system in 2KO, 1KO, and DKO mice appears grossly normal. These mice are normotensive with systolic, diastolic, and mean blood pressures comparable with age-matched WT animals (Fig. 9A). However, compliance studies show that the diameter of the ascending aorta in 6-month-old DKO mice is significantly increased compared with WT controls, 1KO, and 2KO mice at physiologic pressures of 75–125 millimeters of mercury (Fig. 9B). Pressure measurements of the outer diameter taken at 4 months of age indicate that the ascending aorta outer diameter is not significantly different from the WT at this time point (data not shown). These data show that, in DKO animals, the loss of MAGP1 and MAGP2 have a combinatorial effect in increasing the aorta outer diameter at physiologic pressures and that this change in tissue architecture becomes apparent with increasing age.
FIGURE 8.
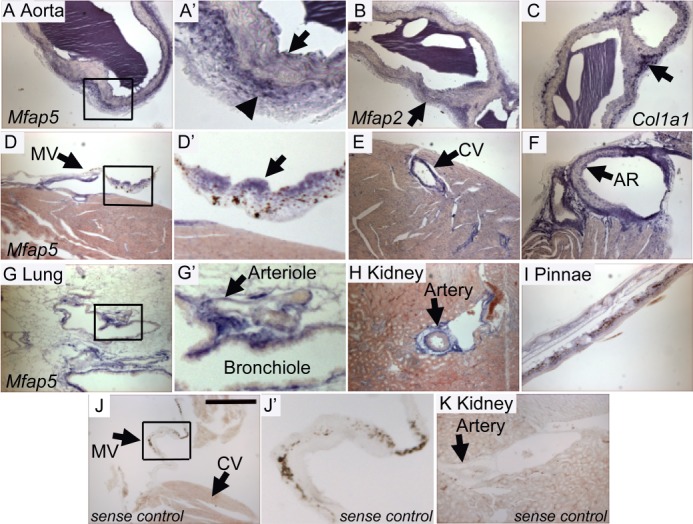
Mfap5 mRNA is expressed by cells of the aorta, mitral valve, coronary vessels, kidney, lung, and pinnae. In situ hybridization to detect Mfap5 (A and D–I), Mfap2 (B), and Col1a1 (C), expression was performed on 14-μm sections from 4-month-old wild-type mice. A and A′, arrows demonstrate digoxigenin-labeled mRNA (blue) localized to the intima (arrow) and adventitia (arrowhead) of the aorta. A′ is a magnification of the boxed region in A. B, Mfap2 mRNA is expressed throughout the aorta (arrow). C, Col1a1 mRNA is heavily expressed in the aortic adventitia (arrow). D and D′, Mfap5 is expressed in the atrialis of the mitral valve (MV, arrow). E, Mfap5 mRNA is found in the coronary vessel (CV, arrow). F, significant Mfap5 expression is localized to the aortic root (AR, arrow). G and G′, large airways and arterioles (G′, arrow) are positive for Mfap5 expression. H, large vessels of the kidney express Mfap5 (arrow). J, J′, and K, no nonspecific reactivity is detected in Mfap5 sense control sections. Scale bar = 300 μm. Micrographs were taken using identical camera settings and image processing.
FIGURE 9.
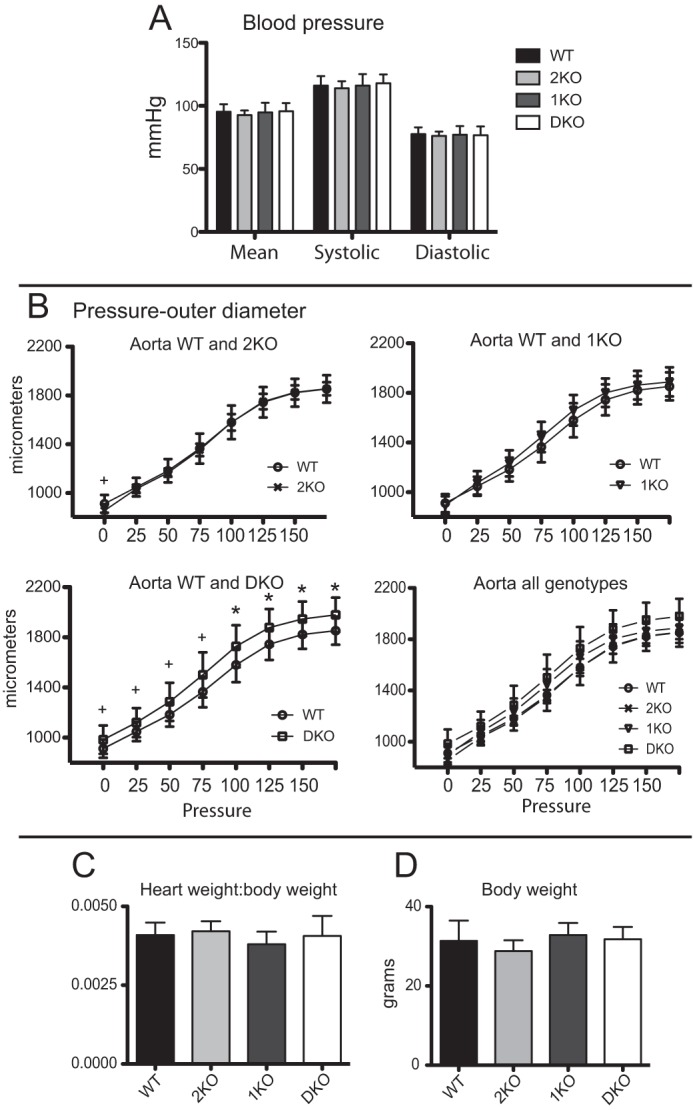
The outer diameter of aortic vessels is increased at physiological pressures in DKO mice. A, histogram of mean blood pressure measurements. B, pressure myography performed on aortae from 6-month-old male mice. A significant increase in aortic outer diameter at physiologic pressures is seen in DKO animals. C, graph indicating mean body weight of mice at the time of sacrifice. D, graph depicting mean heart weight to body weight ratios. *, p ≤ 0.01; +, p = 0.02.
To determine whether changes in the outer diameter of aortic vessels could be explained by changes in heart size, heart weight to body weight ratios were calculated and found to be comparable among all genotypes (Fig. 9, C and D). Taken together, these data demonstrate that loss of both MAGP1 and MAGP2 expression increases aortic vessel diameter in vivo. These changes are age-dependent and not secondary to changes in body size, composition, or heart size. This suggests that MAGP1 and MAGP2 have shared primary functions in maintaining large vessel integrity.
MAGP2 Protein Binds Active TGFβ and BMP Growth Factors
MAGP1 has an acidic TGFβ-binding domain located in the N terminus of the protein, and loss of MAGP1 increases TGFβ signaling in multiple tissues (13, 19, 31). Although the amino-terminal sequences are different in MAGP1 and MAGP2, both regions are highly acidic (Fig. 3, A, C, and D), suggesting that MAGP2 may have the ability to bind TGFβ family ligands (8). Surface plasmon resonance and BLI were performed with immobilized MAGP2 and active TGFβ1, TGFβ2, or BMP2 in solution. TGFβ1, TGFβ2, and BMP2 bound reversibly to MAGP2 (Fig. 10, A–C). Steady-state analysis of BMP2 binding to bacterially expressed and refolded or native mammalian cell-secreted MAGP2 showed similar kinetics. The equilibrium dissociation constant (KD) for BMP2 binding of refolded, bacterially expressed MAGP2 is 210 nm, whereas the KD for binding to mammalian MAGP2 is 190 nm. These data demonstrate that MAGP2 binds active TGFβ superfamily members and suggests that these interactions may have biological relevance.
FIGURE 10.

MAGP2 binds active growth factor cytokines. A, surface plasmon resonance was used to monitor real-time binding of immobilized, bacterially expressed, and refolded murine MAGP2 to active human TGFβ1 at concentrations of 100, 50, 50, 25, 25, and 12.5 nm. B, BLI was used to monitor real-time binding of immobilized murine MAGP2 to murine TGFβ2 at concentrations of 1000, 500, 250, 125, 62.5, 31.3, 15.6, and 7.8 nm. C, BLI was used to monitor real-time binding of immobilized refolded murine MAGP2 to murine BMP2 at concentrations of 1000, 500, 250, 125, 62.5, 31.3, 15.6, and 7.8 nm. D, equilibrium analysis of refolded MAGP2 binding BMP2 as measured by BLI. E, equilibrium analysis of natively secreted MAGP2 binding BMP2 as measured by BLI. Response units at equilibrium (Req) values from BLI data are plotted against cytokine concentration with a non-linear fit superimposed.
DISCUSSION
MAGP2 is a protein component of fibrillin-containing microfibrils where it partners with MAGP1 and other microfibril-associated proteins to define microfibril function (18). Mutations in fibrillin lead to Marfan syndrome, characterized by long bone overgrowth and abnormalities in the cardiovascular and pulmonary systems (3). It is interesting that none of those changes are evident in MAGP2-deficient mice. MAGP2 deficiency also shows little overlap with phenotypes found in mice lacking MAGP1, which supports unique functions for the individual proteins and agrees with the more limited distribution of MAGP2 compared with MAGP1.
Microfibrils serve to regulate growth factor availability, particularly the activity of TGFβ, BMP, and Notch family members (32–34). Because these growth factors are produced by and signal to many cell types, signaling must be tightly regulated. The fibrillins sequester the TGFβ large latent complex in the ECM, where it can be activated when needed. Mutations in fibrillin lead to elevated active TGFβ levels, which accounts for the phenotypes associated with Marfan syndrome. The role of MAGPs in growth factor signaling may be more complex in that they may function to sequester ligands from, or present them to, signaling receptors in a tissue-specific manner (33, 34).
MAGP1 binds TGFβs, BMPs, and Notch receptors and ligands (13, 33). Much like the classical fibrillinopathies, MAGP1 loss of function leads to increased TGFβ signaling and subsequent disease in some tissues (4, 13, 19, 20). Like MAGP1, MAGP2 also binds TGFβ superfamily members as well as Notch receptors and ligands. This suggests that MAGP2 may regulate these growth factors in vivo. Interestingly, proper levels of Notch signaling are required for definitive hematopoiesis, whereas TGFβ and BMP are master regulators of hematopoietic stem cell quiescence and osteoblastic niche size, respectively (35–37). TGFβ signaling can differentially affect most hematopoietic stem cell types according to signal strength, signal localization, and differentiation state of the target cell (36). Thus, misregulation of Notch, TGFβ, or BMP signaling because of loss of MAGPs is consistent with alterations in cells of the myeloid lineage. Both MAGP1 and MAGP2 are expressed in trabecular bone, consistent with a potential role for these proteins in hematopoiesis (16). Differences in MAGP1 and MAGP2 expression patterns and growth factor binding affinities may explain the neutropenia in 2KO mice and monocytopenia in 1KO animals. Interestingly, loss of MAGP2 function does not modify the monocytopenia seen in MAGP1-deficient mice because DKO animals exhibit both neutropenia and reduced monocyte cell numbers.
In addition to influencing cells within the bone marrow, TGFβ/BMP signaling coordinates bone anabolism (via osteoblast activation) with bone catabolism (via differentiation of osteoclasts from hematopoietic stem cell precursors) (38–41). MAGP1-deficient mice develop osteopenia with age because of increased osteoclast number (20). A study of these animals revealed increased circulating levels of the osteoclast differentiation factor receptor activator of NFκB ligand (RANKL) (19). Treatment of cultured MAGP1-deficient osteoblasts with TGFβ neutralizing antibodies reduced RANKL production by these cells to base-line levels, suggesting increased TGFβ signaling because of loss of MAGP1 function as the primary defect in bone homeostasis. The studies detailed here indicate that, unlike MAGP1, loss of MAGP2 function does not negatively affect bone mineral density. Furthermore, DKO mice do not develop spontaneous fractures and lack severe osteopenia associated with MAGP1 deficiency. This suggests that loss of MAGP2 in tandem with loss of MAGP1 restores bone mineral density.
An explanation for why loss of MAGP2 is protective against bone loss may lie in the ability of MAGP2 to manipulate Notch signaling. MAGP1 and MAGP2 both have the ability to bind Notch1, Jagged1, Jagged2, and Delta1 (33). Both proteins mediate ectodomain shedding of Notch1 and activate the Notch intracellular domain. MAGP2, however, has the added ability to activate Notch signaling through release of Jagged1 from the cell surface (42). The activated Notch intracellular domain inhibits osteoclast differentiation both from hematopoietic precursors and from preosteoclasts (43). Jagged1/Notch intracellular domain signaling also impedes osteoclast formation by inhibiting RANKL expression in mature osteoblasts (43). Inhibition of Notch signaling in the context of bone formation increases osteoblast differentiation, thereby increasing bone formation (43). The studies detailed here indicate that loss of MAGP2 alone has a slight net anabolic effect on bone mass. Thus, changes in Notch signaling associated with MAGP2 deficiency may provide a mechanistic explanation for why MAGP1-induced increases in osteoclastogenesis are not as prominent in DKO animals.
Previous work demonstrates that fibrillin-1 deficiency leads to increased aortic diameter pressure curves, hypotension, fragmentation of elastin, and changes in aortic compliance that becomes apparent with age (44, 45). Our findings also show increases in aortic diameter pressure curves in 6-month-old MAGP DKO mice but not in the single knockouts. As with the fibrillinopathies, changes in large vessel architecture and function in DKO animals may occur because of several contributing factors. Firstly, changes in MAGP expression may cause developmental changes in ECM architecture that become more apparent with age. This idea is supported by studies in zebrafish, where loss of MAGP1 causes distension and loss of vessel integrity in cranial and caudal blood vessels during development (46, 47). Secondly, ECM remodeling and homeostasis may be altered by loss of MAGPs. Increased TGFβ signaling in aortic tissue occurs with loss of growth factor binding by microfibril proteins (3, 44, 48). This leads to extensive pathological remodeling, loss of vessel integrity, and, ultimately, aortic dissection in mice and humans in vivo (3, 48, 49). Loss of MAGP results in a similar, albeit blunted pattern of pathologic remodeling, leading to loss of vessel integrity but not aortic dissection. Because MAGPs are highly expressed in large vessels and bind active TGFβ ligands, it is possible that loss of MAGPs causes small changes in TGFβ signaling that, over time, produce a fibrillinopathy-like phenotype in large vessels (7, 13).
An Arg-Gly-Asp (RGD)-containing sequence in fibrillin-1, when applied to cells, was found to increase the expression of matrix metalloproteinases 1 and 3 (50). Increased proteolytic activity by matrix metalloproteinases is a characteristic feature of fibrillinopathies such as Marfan syndrome (51). MAGP2 also contains an RGD sequence that binds αvβ3 integrins, although the function of this domain in MAGP2 is not understood (52). Because RGD-containing sequences can be important regulators of proteolytic activity, cell adhesion, and growth factor presentation to cells, one must consider mutations in fibrillins or MAGPs that impair MAGP-fibrillin interaction as potentially important. As stated previously, MAGPs covalently bind fibrillins on the microfibril. Disease-causing mutations in fibrillin-1 in mice are associated with increases in MAGP2 expression in vivo, suggesting a regulatory link between these microfibrillar proteins (24, 53, 54). Increased gene expression and/or mutations in MAGPs or fibrillins may result in unbound MAGPs in the ECM with unknown effects on growth factor signaling and matrix metalloproteinases expression. When taken together, these data demonstrate that loss of MAGPs shares pathological features of fibrillinopathies and raise the question as to how much of the pathology attributed to loss of fibrillin function is directly linked to loss of MAGP-fibrillin interaction and subsequent MAGP-dependent ECM changes.
Interestingly, MAGP2 expression is negatively correlated with patient survival from advanced-stage ovarian cancer (55). Mok et al. (55) suggest that MAGP2-dependent changes in cancer cell biology may be conferred via multiple mechanisms. The authors correlate MAGP2-mediated αvβ3 integrin interaction with chemotherapy resistance and cell survival (55). Also integral to tumor survival is development of a blood supply through angiogenesis. MAGP2 negatively regulates Notch signaling in endothelial tip cells and promotes angiogenic sprouting, potentially providing another mechanism for MAGP2-mediated tumor growth and survival (34). Previous studies demonstrate a high metastatic potential in human melanoma cell clones with expression of peanut agglutinin-reactive glycosylation on MAGP1 and MAGP2 (56). Analysis of MAGP proteins with the NetOGlyc server demonstrates two highly conserved and one non-conserved predicted O-linked glycosylation site in the N terminus of MAGP1 (12). MAGP2 has five highly conserved and one non-conserved O-linked carbohydrate site, all within the amino-terminal portion of the molecule (Fig. 3B). Additional glycosylation domains on MAGP2 suggest evolutionary acquisition of unique protein-protein interactions. It is currently known that protein glycosylation patterns change in the context of cancer, with the net effect of altering cell signaling and nutrient availability (57, 58). Moreover, MAGP2 physically interacts with active TGFβs, BMPs, and Notch receptors and ligands. It is not difficult to imagine that MAGP2 expression may allow transformed cells to harness several powerful mechanisms to promote tumor growth, metastasis, and chemotherapy resistance. Research directed at uncovering the molecular mechanisms by which MAGP2 promotes tumor growth and metastasis is clinically important.
These studies demonstrate that MAGP2 plays a role in the biology of the hematopoietic and vascular systems and suggest that MAGP2 is capable of regulating growth factor bioavailability. Work by other authors also links MAGP2 expression status to cancer outcome (34). Clearly, further understanding of MAGP2 function not only increases our understanding of ECM biology but has the potential to significantly impact human health.
Acknowledgments
We thank Megan Curtis and Chris Ciliberto for technical assistance, Fernando Segade for helpful discussion, Terese Hall for administrative support, and the Washington University Musculoskeletal and Morphology Cores for assistance with specimen analysis.
This work was supported, in whole or in part, by National Institutes of Health Grants HL53325, HL74138, and HL105314 (to R. P. M.), National Institutes of Health Developmental Cardiology and Pulmonary Training Grant T32 HL007873 (to M. D. C.), and National Institutes of Health Grant P30-AR057235 (to the Musculoskeletal and Morphology Cores).

This article contains supplemental Figs. S1–S3 and Table S1.
R. P. Mecham, unpublished results.
- ECM
- extracellular matrix
- MAGP
- microfibril-associated glycoprotein
- BMP
- bone morphogenic protein
- DKO
- MAGP1/2 double knockout
- 2KO
- MAGP2 knockout
- 1KO
- MAGP1 knockout
- μCT
- microcomputed tomography
- BLI
- biolayer interferometry
- Neo
- neomycin resistance cassette
- gDNA
- genomic DNA.
REFERENCES
- 1. Mecham R. P. (2011) The Extracellular Matrix. An Overview. Springer-Verlag, Berlin [Google Scholar]
- 2. Jensen S. A., Robertson I. B., Handford P. A. (2012) Dissecting the fibrillin microfibril. Structural insights into organization and function. Structure 20, 215–225 [DOI] [PubMed] [Google Scholar]
- 3. Kielty C. M. (2006) Elastic fibres in health and disease. Expert Rev. Mol. Med. 8, 1–23 [DOI] [PubMed] [Google Scholar]
- 4. Ramirez F., Sakai L. Y. (2010) Biogenesis and function of fibrillin assemblies. Cell Tissue Res. 339, 71–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yanagisawa H., Davis E. C. (2010) Unraveling the mechanism of elastic fiber assembly. The roles of short fibulins. Int. J. Biochem. Cell Biol. 42, 1084–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gibson M. A., Hughes J. L., Fanning J. C., Cleary E. G. (1986) The major antigen of elastin-associated microfibrils is a 31-kDa glycoprotein. J. Biol. Chem. 261, 11429–11436 [PubMed] [Google Scholar]
- 7. Gibson M. A., Finnis M. L., Kumaratilake J. S., Cleary E. G. (1998) Microfibril-associated glycoprotein-2 (MAGP-2) is specifically associated with fibrillin-containing microfibrils but exhibits more restricted patterns of tissue localization and developmental expression than its structural relative MAGP-1. J. Histochem. Cytochem. 46, 871–886 [DOI] [PubMed] [Google Scholar]
- 8. Segade F. (2009) Functional evolution of the microfibril-associated glycoproteins. Gene 439, 43–54 [DOI] [PubMed] [Google Scholar]
- 9. Segade F., Trask B. C., Broekelmann T. J., Pierce R. A., Mecham R. P. (2002) Identification of a matrix-binding domain in MAGP1 and MAGP2 and intracellular localization of alternative splice forms. J. Biol. Chem. 277, 11050–11057 [DOI] [PubMed] [Google Scholar]
- 10. Hatzinikolas G., Gibson M. A. (1998) The exon structure of the human MAGP-2 gene. J. Biol. Chem. 273, 29309–29314 [DOI] [PubMed] [Google Scholar]
- 11. Donovan L. J., Cha S. E., Yale A. R., Dreikorn S., Miyamoto A. (2013) Identification of a functional proprotein convertase cleavage site in microfibril-associated glycoprotein 2. Matrix Biol. 32, 117–122 [DOI] [PubMed] [Google Scholar]
- 12. Trask B. C., Broekelmann T., Ritty T. M., Trask T. M., Tisdale C., Mecham R. P. (2001) Post-translational modifications of microfibril associated glycoprotein-1 (MAGP-1). Biochemistry 40, 4372–4380 [DOI] [PubMed] [Google Scholar]
- 13. Weinbaum J. S., Broekelmann T. J., Pierce R. A., Werneck C. C., Segade F., Craft C. S., Knutsen R. H., Mecham R. P. (2008) Deficiency in microfibril-associated glycoprotein-1 leads to complex phenotypes in multiple organ systems. J. Biol. Chem. 283, 25533–25543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davis E. C. (1994) Immunolocalization of microfibril and microfibril-associated proteins in the subendothelial matrix of the developing mouse aorta. J. Cell Sci. 107, 727–736 [DOI] [PubMed] [Google Scholar]
- 15. Henderson M., Polewski R., Fanning J. C., Gibson M. A. (1996) Microfibril-associated glycoprotein-1 (MAGP-1) is specifically located on the beads of the beaded-filament structure of fibrillin-containing microfibrils as visualized by the rotary shadowing technique. J. Histochem. Cytochem. 44, 1389–1397 [DOI] [PubMed] [Google Scholar]
- 16. Kumaratilake J. S., Gibson M. A., Fanning J. C., Cleary E. G. (1989) The tissue distribution of microfibrils reacting with a monospecific antibody to MAGP, the major glycoprotein antigen of elastin-associated microfibrils. Eur. J. Cell Biol. 50, 117–127 [PubMed] [Google Scholar]
- 17. Visel A., Thaller C., Eichele G. (2004) GenePaint.org. An atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 32, D552–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gibson M. A., Hatzinikolas G., Kumaratilake J. S., Sandberg L. B., Nicholl J. K., Sutherland G. R., Cleary E. G. (1996) Further characterization of proteins associated with elastic fiber microfibrils including the molecular cloning of MAGP-2 (MP25). J. Biol. Chem. 271, 1096–1103 [DOI] [PubMed] [Google Scholar]
- 19. Craft C. S., Broekelmann T. J., Zou W., Chappel J. C., Teitelbaum S. L., Mecham R. P. (2012) Oophorectomy-induced bone loss is attenuated in MAGP1-deficient mice. J. Cell. Biochem. 113, 93–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Craft C. S., Zou W., Watkins M., Grimston S., Brodt M. D., Broekelmann T. J., Weinbaum J. S., Teitelbaum S. L., Pierce R. A., Civitelli R., Silva M. J., Mecham R. P. (2010) Microfibril-associated glycoprotein-1, an extracellular matrix regulator of bone remodeling. J. Biol. Chem. 285, 23858–23867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ehrman L. A., Yutzey K. E. (1999) Lack of regulation in the heart forming region of avian embryos. Dev. Biol. 207, 163–175 [DOI] [PubMed] [Google Scholar]
- 22. Lincoln J., Alfieri C. M., Yutzey K. E. (2006) BMP and FGF regulatory pathways control cell lineage diversification of heart valve precursor cells. Dev. Biol. 292, 292–302 [DOI] [PubMed] [Google Scholar]
- 23. Shelton E. L., Yutzey K. E. (2007) Tbx20 regulation of endocardial cushion cell proliferation and extracellular matrix gene expression. Dev. Biol. 302, 376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lemaire R., Farina G., Kissin E., Shipley J. M., Bona C., Korn J. H., Lafyatis R. (2004) Mutant fibrillin 1 from tight skin mice increases extracellular matrix incorporation of microfibril-associated glycoprotein 2 and type I collagen. Arthritis Rheum. 50, 915–926 [DOI] [PubMed] [Google Scholar]
- 25. Faury G., Pezet M., Knutsen R. H., Boyle W. A., Heximer S. P., McLean S. E., Minkes R. K., Blumer K. J., Kovacs A., Kelly D. P., Li D. Y., Starcher B., Mecham R. P. (2003) Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J. Clin. Invest. 112, 1419–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wagenseil J. E., Nerurkar N. L., Knutsen R. H., Okamoto R. J., Li D. Y., Mecham R. P. (2005) Effects of elastin haploinsufficiency on the mechanical behavior of mouse arteries. Am. J. Physiol. Heart Circ. Physiol. 289, H1209–1217 [DOI] [PubMed] [Google Scholar]
- 27. Swiecki M., Scheaffer S. M., Allaire M., Fremont D. H., Colonna M., Brett T. J. (2011) Structural and biophysical analysis of BST-2/tetherin ectodomains reveals an evolutionary conserved design to inhibit virus release. J. Biol. Chem. 286, 2987–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aricescu A. R., Lu W., Jones E. Y. (2006) A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. D. Biol. Crystallogr. 62, 1243–1250 [DOI] [PubMed] [Google Scholar]
- 29. Maquat L. E. (2004) Nonsense-mediated mRNA decay. Splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 5, 89–99 [DOI] [PubMed] [Google Scholar]
- 30. Lemaire R., Bayle J., Mecham R. P., Lafyatis R. (2007) Microfibril-associated MAGP-2 stimulates elastic fiber assembly. J. Biol. Chem. 282, 800–808 [DOI] [PubMed] [Google Scholar]
- 31. Massam-Wu T., Chiu M., Choudhury R., Chaudhry S. S., Baldwin A. K., McGovern A., Baldock C., Shuttleworth C. A., Kielty C. M. (2010) Assembly of fibrillin microfibrils governs extracellular deposition of latent TGF β. J. Cell Sci. 123, 3006–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramirez F., Sakai L. Y., Rifkin D. B., Dietz H. C. (2007) Extracellular microfibrils in development and disease. Cell Mol. Life Sci. 64, 2437–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miyamoto A., Lau R., Hein P. W., Shipley J. M., Weinmaster G. (2006) Microfibrillar proteins MAGP-1 and MAGP-2 induce Notch1 extracellular domain dissociation and receptor activation. J. Biol. Chem. 281, 10089–10097 [DOI] [PubMed] [Google Scholar]
- 34. Albig A. R., Becenti D. J., Roy T. G., Schiemann W. P. (2008) Microfibril-associate glycoprotein-2 (MAGP-2) promotes angiogenic cell sprouting by blocking notch signaling in endothelial cells. Microvasc. Res. 76, 7–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bigas A., Espinosa L. (2012) Hematopoietic stem cells. To be or Notch to be. Blood 119, 3226–3235 [DOI] [PubMed] [Google Scholar]
- 36. Blank U., Karlsson S. (2011) The role of Smad signaling in hematopoiesis and translational hematology. Leukemia 25, 1379–1388 [DOI] [PubMed] [Google Scholar]
- 37. Munger J. S., Sheppard D. (2011) Cross talk among TGF-β signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb. Perspect. Biol. 3, a005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen G., Deng C., Li Y. P. (2012) TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 8, 272–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nistala H., Lee-Arteaga S., Carta L., Cook J. R., Smaldone S., Siciliano G., Rifkin A. N., Dietz H. C., Rifkin D. B., Ramirez F. (2010) Differential effects of alendronate and losartan therapy on osteopenia and aortic aneurysm in mice with severe Marfan syndrome. Hum. Mol. Genet. 19, 4790–4798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nistala H., Lee-Arteaga S., Smaldone S., Siciliano G., Carta L., Ono R. N., Sengle G., Arteaga-Solis E., Levasseur R., Ducy P., Sakai L. Y., Karsenty G., Ramirez F. (2010) Fibrillin-1 and -2 differentially modulate endogenous TGF-β and BMP bioavailability during bone formation. J. Cell Biol. 190, 1107–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nistala H., Lee-Arteaga S., Smaldone S., Siciliano G., Ramirez F. (2010) Extracellular microfibrils control osteoblast-supported osteoclastogenesis by restricting TGFβ stimulation of RANKL production. J. Biol. Chem. 285, 34126–34133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nehring L. C., Miyamoto A., Hein P. W., Weinmaster G., Shipley J. M. (2005) The extracellular matrix protein MAGP-2 interacts with Jagged1 and induces its shedding from the cell surface. J. Biol. Chem. 280, 20349–20355 [DOI] [PubMed] [Google Scholar]
- 43. Mead T. J., Yutzey K. E. (2012) Notch signaling and the developing skeleton. Adv. Exp. Med. Biol. 727, 114–130 [DOI] [PubMed] [Google Scholar]
- 44. Mariko B., Pezet M., Escoubet B., Bouillot S., Andrieu J. P., Starcher B., Quaglino D., Jacob M. P., Huber P., Ramirez F., Faury G. (2011) Fibrillin-1 genetic deficiency leads to pathological ageing of arteries in mice. J. Pathol. 224, 33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Carta L., Wagenseil J. E., Knutsen R. H., Mariko B., Faury G., Davis E. C., Starcher B., Mecham R. P., Ramirez F. (2009) Discrete contributions of elastic fiber components to arterial development and mechanical compliance. Arterioscler. Thromb. Vasc. Biol. 29, 2083–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Alvarez Y., Cederlund M. L., Cottell D. C., Bill B. R., Ekker S. C., Torres-Vazquez J., Weinstein B. M., Hyde D. R., Vihtelic T. S., Kennedy B. N. (2007) Genetic determinants of hyaloid and retinal vasculature in zebrafish. BMC Dev. Biol. 7, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen E., Larson J. D., Ekker S. C. (2006) Functional analysis of zebrafish microfibril-associated glycoprotein-1 (Magp1) in vivo reveals roles for microfibrils in vascular development and function. Blood 107, 4364–4374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Habashi J. P., Judge D. P., Holm T. M., Cohn R. D., Loeys B. L., Cooper T. K., Myers L., Klein E. C., Liu G., Calvi C., Podowski M., Neptune E. R., Halushka M. K., Bedja D., Gabrielson K., Rifkin D. B., Carta L., Ramirez F., Huso D. L., Dietz H. C. (2006) Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 312, 117–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ramirez F., Dietz H. C. (2004) Therapy insight. Aortic aneurysm and dissection in Marfan's syndrome. Nat. Clin. Pract. Cardiovasc. Med. 1, 31–36 [DOI] [PubMed] [Google Scholar]
- 50. Booms P., Pregla R., Ney A., Barthel F., Reinhardt D. P., Pletschacher A., Mundlos S., Robinson P. N. (2005) RGD-containing fibrillin-1 fragments upregulate matrix metalloproteinase expression in cell culture. A potential factor in the pathogenesis of the Marfan syndrome. Hum. Genet. 116, 51–61 [DOI] [PubMed] [Google Scholar]
- 51. Xiong W., Meisinger T., Knispel R., Worth J. M., Baxter B. T. (2012) MMP-2 regulates Erk1/2 phosphorylation and aortic dilatation in Marfan syndrome. Circ. Res. 110, e92-e101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gibson M. A., Leavesley D. I., Ashman L. K. (1999) Microfibril-associated glycoprotein-2 specifically interacts with a range of bovine and human cell types via αVβ3 integrin. J. Biol. Chem. 274, 13060–13065 [DOI] [PubMed] [Google Scholar]
- 53. Ito S., Bartolák-Suki E., Shipley J. M., Parameswaran H., Majumdar A., Suki B. (2006) Early emphysema in the tight skin and pallid mice. Roles of microfibril-associated glycoproteins, collagen, and mechanical forces. Am. J. Respir. Cell Mol. Biol. 34, 688–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lemaire R., Korn J. H., Shipley J. M., Lafyatis R. (2005) Increased expression of type I collagen induced by microfibril-associated glycoprotein 2. Novel mechanistic insights into the molecular basis of dermal fibrosis in scleroderma. Arthritis Rheum. 52, 1812–1823 [DOI] [PubMed] [Google Scholar]
- 55. Mok S. C., Bonome T., Vathipadiekal V., Bell A., Johnson M. E., Wong K. K., Park D. C., Hao K., Yip D. K., Donninger H., Ozbun L., Samimi G., Brady J., Randonovich M., Pise-Masison C. A., Barrett J. C., Wong W. H., Welch W. R., Berkowitz R. S., Birrer M. J. (2009) A gene signature predictive for outcome in advanced ovarian cancer identifies a survival factor. Microfibril-associated glycoprotein 2. Cancer Cell 16, 521–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zebda N., Bailly M., Brown S., Doré J. F., Berthier-Vergnes O. (1994) Expression of PNA-binding sites on specific glycoproteins by human melanoma cells is associated with a high metastatic potential. J. Cell. Biochem. 54, 161–173 [DOI] [PubMed] [Google Scholar]
- 57. Hanover J. A., Krause M. W., Love D. C. (2012) Bittersweet memories. Linking metabolism to epigenetics through O-GlcNAcylation. Nat. Rev. Mol. Cell Biol. 13, 312–321 [DOI] [PubMed] [Google Scholar]
- 58. Hart G. W., Slawson C., Ramirez-Correa G., Lagerlof O. (2011) Cross talk between O-GlcNAcylation and phosphorylation. Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 80, 825–858 [DOI] [PMC free article] [PubMed] [Google Scholar]