

Background: Mutations in the TREX1 C-terminal region (CTR) cause human autoimmune disease.
Results: The CTR directs TREX1 ubiquitination and interaction with ubiquilin 1.
Conclusion: TREX1 ubiquitination and co-localization with ubiquilin 1 are differentially affected in autoimmune disease mutants.
Significance: Multiple mechanisms of TREX1 dysfunction include altered covalent modification and diminished catalytic function, resulting in a spectrum of autoimmune diseases.
Keywords: Autoimmune Diseases, DNA, DNA Enzymes, Nucleic Acid, Ubiquitination
Abstract
TREX1 is an autonomous 3′-exonuclease that degrades DNA to prevent inappropriate immune activation. The TREX1 protein is composed of 314 amino acids; the N-terminal 242 amino acids contain the catalytic domain, and the C-terminal region (CTR) localizes TREX1 to the cytosolic compartment. In this study, we show that TREX1 modification by ubiquitination is controlled by a highly conserved sequence in the CTR to affect cellular localization. Transfection of TREX1 deletion constructs into human cells demonstrated that this sequence is required for ubiquitination at multiple lysine residues through a “non-canonical” ubiquitin linkage. A proteomic approach identified ubiquilin 1 as a TREX1 CTR-interacting protein, and this interaction was verified in vitro and in vivo. Cotransfection studies indicated that ubiquilin 1 localizes TREX1 to cytosolic punctate structures dependent upon the TREX1 CTR and lysines within the TREX1 catalytic core. Several TREX1 mutants linked to the autoimmune diseases Aicardi-Goutières syndrome and systemic lupus erythematosus that exhibit full catalytic function were tested for altered ubiquitin modification and cellular localization. Our data show that these catalytically competent disease-causing TREX1 mutants exhibit differential levels of ubiquitination relative to WT TREX1, suggesting a novel mechanism of dysfunction. Furthermore, these differentially ubiquitinated disease-causing mutants also exhibit altered ubiquilin 1 co-localization. Thus, TREX1 post-translational modification indicates an additional mechanism by which mutations disrupt TREX1 biology, leading to human autoimmune disease.
Introduction
The proper management of DNA polynucleotides in humans is critical to prevent the inappropriate activation of the immune response. There are accumulating data that the pathophysiology of nucleic acid-mediated autoimmunity functions in parallel to antiviral immunity (1–3), and there is an emerging concept that lupus-like diseases result largely from genetic variants that allow self-nucleic acids to persistently trigger antiviral immune responses erroneously directed against nuclear autoantigens (4). The discovery that TREX1 mutations cause Aicardi-Goutières syndrome (AGS)3 provides an important link between nucleic acid metabolism, autoimmune disease, and innate antiviral response (5). The additional findings of TREX1 mutations in patients diagnosed with familial chilblain lupus, Cree encephalitis, retinal vasculopathy with cerebral leukodystrophy (RVCL), and systemic lupus erythematosus (SLE) genetically link a spectrum of human autoimmune disorders with overlapping related clinical symptoms (5–21).
Approximately 40 disease-causing TREX1 missense and frameshift mutations that locate to positions throughout the gene have been identified. The disease-causing TREX1 mutant enzymes exhibit a broad range of catalytic activities, indicating that multiple mechanisms of TREX1 dysfunction contribute to the spectrum of related autoimmune disorders (10, 16, 19, 22–26). The TREX1 protein is a 314-amino acid polypeptide containing a robust 3′-exonuclease that degrades single- and double-stranded DNA polymers and is expressed in many mammalian tissues (27–29). The TREX1 N-terminal 242 amino acids contain all of the necessary structural elements for full catalytic activity (22), and the C-terminal region (CTR) 72 amino acids are required for cytosolic localization to the perinuclear space in cells (16, 19). The varied effects exhibited by TREX1 mutant alleles on catalytic function and cellular localization, coupled with the varied levels of disease pathogenesis in humans, highlight the complex biology of TREX1 in DNA degradation and immune activation.
The TREX1 exonuclease degrades DNA polynucleotides that might originate endogenously or exogenously, preventing the accumulation of these macromolecules to levels sufficient for induction of an interferon-mediated immune response. This concept is supported by the TREX1 degradation of genomic DNA in a cell death pathway (30) and by the autoimmune phenotype of the Trex1 knock-out mouse (31–34). The mouse Trex1 autoimmune pathology has been attributed to dysregulated activation of the interferon stimulatory DNA response pathway (31, 33) or to spontaneous DNA damage with chronic checkpoint activation (32, 34). TREX1 participates in a cytosolic nucleic acid-sensing pathway mediated through the adaptor protein STING (31, 33, 35–37) and has been identified as a regulator of lysosomal biogenesis in interferon-independent activation of antiviral genes (38).
The indication that TREX1 participates in a nucleic acid detection pathway (31, 33, 35–37) and evidence that ubiquitination regulates the cytosolic nucleic acid sensor pathways (39, 40) prompted our studies here revealing the CTR-directed ubiquitination of TREX1. A series of TREX1 deletion constructs was generated to show that the CTR is necessary for covalent modification by ubiquitination in human cells. A mutagenesis strategy was used to determine a “non-canonical” ubiquitin linkage at multiple TREX1 lysine residues, suggesting that this modification might regulate cellular function. A proteomic approach identified ubiquilin 1 interaction with the TREX1 CTR, facilitating the translocation of ubiquitinated TREX1 from the perinuclear region to cytosolic puncta in human cells. We show that a subset of AGS-causing TREX1 mutants located in the catalytic core and in the CTR exhibit full catalytic activity, are differentially ubiquitinated compared with WT TREX1, and exhibit altered translocation and co-localization with ubiquilin 1. These data indicate that the TREX1 CTR controls cellular trafficking of this exonuclease by ubiquitination and interaction with ubiquilin 1, providing a novel mechanism through which TREX1 mutations disrupt nucleic acid processing and aberrant immune activation.
EXPERIMENTAL PROCEDURES
Materials
The HRP-conjugated anti-FLAG (A8592) and HRP-conjugated anti-HA (H6533) antibodies were from Sigma. The anti-Myc antibody (2276) was from Cell Signaling. The HRP-conjugated anti-mouse (NA931V) and anti-rabbit (NA934V) antibodies were from Amersham Biosciences. The 5′-fluorescein 30-mer 5′-ATACGACGGTGACAGTGTTGTCAGACAGGT-3′ was from Eurofins. Plasmid 1 (10.8 kb) was nicked with the Nt.BbvCI restriction endonuclease as described (23).
Cell Culture, Transfections, and Plasmids
HEK 293T cells were grown in DMEM containing 10% FBS supplemented with 2 mm GlutaMAX at 37 °C in an atmosphere of 95% air and 5% CO2. The HEK 293T cells were transfected by the calcium phosphate precipitation method (41). Plasmids containing TREX1 deletions (pEBB-TREX1(1–314), pEBB-TREX1(1–286), pEBB-TREX1(1–242), and pEBB-TREX1(243–314)) were produced by PCR using pLM303-TREX1(1–314) as a template (19, 23). Variants were cloned into the parental pEBB-FLAG and pEBB-HA vectors (42). Plasmids containing single TREX1 mutations (K30R, K66R, K75R, K160R, K175R, K242R, K271R, K277R, E198K, V122A, P290L, T303P, and Y305C) and lysine-less TREX1 (referred to as NoK) were generated by site-directed mutagenesis as described (22). The ubiquilin 1 constructs FLAG-Ubqln (8663) and FLAG-Ubqln-112X (8664) were provided by Peter Howley through Addgene. Ubiquilin 1 and the ubiquilin 1 N-terminal fragment (NTF) were subcloned into the pEBB-NFLAG parental vector, and ubiquilin 1 was subcloned into the pEBB-NYFP vector (42). The TREX1(243–314) construct for cell-free expression reactions was subcloned into pT7CFE1-NHis-NGFP (Ambion), and the ubiquilin 1 (variant 1; OriGene SC110414) was subcloned into pT7CFE1-CHis. All constructs were confirmed by sequencing (GENEWIZ). The pCW7-His-Myc-ubiquitin plasmids (WT, K6R, K11R, K27R, K29R, K48R, and K63R) have been described (43).
Cell Lysis and Immunoprecipitation
Cell lysates were prepared in radioimmune precipitation assay buffer, urea lysis buffer, or CHAPS buffer (43, 44). All lysis buffers were supplemented with 1 mm PMSF and one Complete Mini protease inhibitor mixture tablet (Roche Applied Science) prior to use. Lysates used as input controls were normalized for protein content and separated by SDS-PAGE using 4–12% gradient SDS-polyacrylamide gels (Invitrogen). Lysates for HA immunoprecipitation (CHAPS buffer) were incubated for 1 h at 4 °C with monoclonal antibody HA.11 (Covance 14945101) bound to protein G-agarose. Beads were recovered by centrifugation and washed with CHAPS buffer, and precipitated proteins were eluted by the addition of NuPAGE lithium dodecyl sulfate sample buffer (Novex) and heating for 5 min at 95 °C. Urea lysates for nickel-nitrilotriacetic acid (Ni-NTA) precipitation were prepared as described (43). Immunoprecipitations were performed multiple times from independent transfections with similar results.
Immunoblot Analysis
After SDS-PAGE, proteins were transferred to nitrocellulose membranes (Invitrogen) and blocked with 5% milk in TBS containing 0.1% Tween 20 (TBS/Tween). Incubation with the indicated primary antibodies was for 1 h or with the indicated HRP-conjugated primary antibodies for 45 min at room temperature. After washing, membranes with non-HRP-conjugated primary antibodies were incubated with HRP-conjugated anti-mouse IgG or anti-rabbit IgG secondary antibodies for 45 min at room temperature, washed, and visualized by enhanced chemiluminescence (PerkinElmer Life Sciences). TREX1 ubiquitination was quantified using ImageQuant TL (GE Healthcare). The levels of TREX1 ubiquitination were determined for mutant relative to WT by calculating the weighted densitometric volume of ubiquitinated TREX1 bands detected on immunoblots. Weighted volumes were divided by the amount of plasmid transfected (μg), averaged, and expressed as -fold changes in mutant relative to WT TREX1 ubiquitination.
Enzyme Preparation
The human WT and mutant TREX1 enzymes were prepared from Escherichia coli lysates as described (22–24, 26). All protein concentrations were determined by A280 using the molar extinction coefficient for TREX1 protomer of ϵ = 23,950 M−1 cm−1.
Exonuclease Assays
TREX1 exonuclease assays were performed and quantified as described (22–24, 26, 45). HEK 293T cell lysates containing plasmid-expressed TREX1 were prepared in CHAPS lysis buffer and diluted in 1 mg/ml BSA. The exonuclease assays contained 20 mm Tris (pH 7.5), 5 mm MgCl2, 2 mm dithiothreitol, 100 μg/ml BSA, 50 nm fluorescein-labeled 30-mer oligonucleotide (ssDNA assay) or 10 μg/ml nicked plasmid DNA (dsDNA assay), and TREX1 protein or cell extract as indicated in the legend to Fig. 5. Reactions were incubated at 25 °C for the indicated times, quenched with the addition of 3 volumes of cold ethanol, and dried in vacuo.
FIGURE 5.

Disease-causing TREX1 V122A and E198K mutants are active exonucleases. A, exonuclease time course reactions (210 μl) were prepared containing a fluorescein-labeled 30-mer oligonucleotide and indicated TREX1 enzyme (15 nm). Samples (30 μl) were removed after incubation at 25 °C for the indicated times and subjected to electrophoresis on 23% urea-polyacrylamide gels. B, exonuclease time course reactions (160 μl) were prepared containing nicked dsDNA plasmid (10 μg/ml) and indicated TREX1 enzyme (15 nm). Samples (20 μl) were removed after incubation at 25 °C for the indicated times and subjected to electrophoresis on agarose gels. C, cells were transfected with plasmids encoding the indicated TREX1(1–314) enzymes as N-terminal HA epitope fusions. Lysates were prepared and diluted in 1 mg/ml BSA to the indicated -fold dilutions. Exonuclease reactions were prepared with a fluorescein-labeled 30-mer oligonucleotide. Cell lysates (3 μl) containing the expressed TREX1 enzymes were added to reactions (30 μl) and incubated for 20 min at 25 °C. The reaction products were subjected to electrophoresis on 23% urea-polyacrylamide gels.
Cell-free Transcription, Translation, and Immunoprecipitation
RNAs for the cell-free translation reactions were generated from linearized pT7CFE plasmids with the MEGAscript T7 high yield transcription kit (Ambion). The pT7CFE1-NHis-NGFP control and pT7CFE1-NHis-NGFP-TREX1(243–314) plasmids were linearized with SpeI. RNAs were translated using the human in vitro protein expression kit (containing [35S]methionine) according to the manufacturer's instructions (Pierce 88857). Translation reaction products were separated on 12% SDS-polyacrylamide gels and silver-stained. Excised bands were dissolved in water and analyzed by nano-LC-MS/MS (MS Bioworks) to identify proteins. The ubiquilin 1 cDNA was cloned into the pT7CFE1-CHis-ubiquilin 1 plasmid and linearized with XhoI to eliminate the C-terminal His tag. His-tagged proteins were immunoprecipitated using anti-polyhistidine antibody conjugated to magnetic beads (GenScript L00275). Immunoprecipitated 35S-labeled proteins were separated on 12% SDS-polyacrylamide gels and visualized using a Storm PhosphorImager (GE Healthcare).
Fluorescence Microscopy
HEK 293T cells (∼25,000/well) were seeded on 8-well chamber slides (Lab-Tek) and transfected for 24 h with constructs expressing YFP- or HA-tagged proteins. Cells were fixed in 4% paraformaldehyde, washed with PBS containing 0.1% Tween (PBS/Tween) and 100 mm glycine, and permeabilized with 1% Triton X-100 in PBS/Tween. Permeabilized cells were blocked for 1 h in 2% BSA in PBS/Tween and incubated with anti-HA primary antibody for 1 h at room temperature. After washing, slides were incubated with TBS/Tween containing Alexa Fluor 594-conjugated goat anti-mouse IgG (Invitrogen A-11005) for 45 min at room temperature. After washing, coverslips were mounted with ProLong Gold antifade reagent with DAPI (Invitrogen) and allowed to cure overnight at room temperature. Slides were imaged on an Olympus IX71 inverted fluorescence microscope. The most representative images from multiple transfections are included in Figs. 4 and 7.
FIGURE 4.

Ubiquilin 1 alters TREX1 localization in cells. HEK 293T cells were grown in chambered slides and transfected with plasmids encoding TREX1 and the indicated TREX1 mutants expressed as N-terminal HA fusions (A), transfected with a plasmid encoding ubiquilin 1 (Ubqln 1) expressed as an N-terminal fusion with YFP (B), and cotransfected with the TREX1 and ubiquilin 1 plasmids (C). TREX1 (A and C) was visualized in fixed cells stained with anti-HA antibody, followed by secondary detection with Alexa Fluor 596-conjugated antibody and ubiquilin 1 (B and C) by YFP fluorescence. Coverslips were mounted with DAPI-containing medium, and slides were imaged on a Nikon fluorescence microscope.
FIGURE 7.
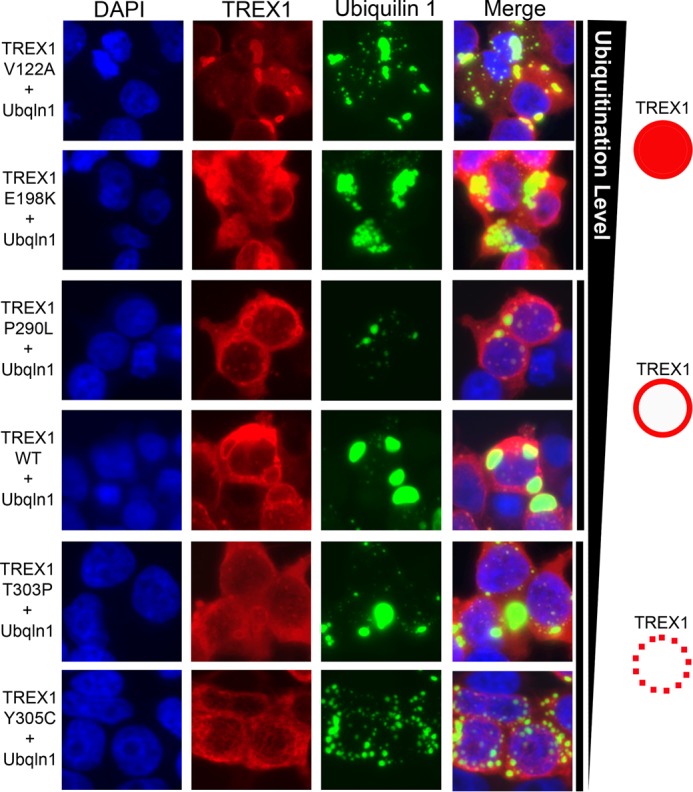
Disease-causing TREX1 mutants exhibit modified ubiquilin 1 co-localization. HEK 293T cells were grown in chambered slides and cotransfected with plasmids encoding WT TREX1 or the indicated TREX1 mutants expressed as N-terminal HA fusions and a plasmid encoding ubiquilin 1 (Ubqln 1) expressed as an N-terminal fusion with YFP. TREX1 was visualized in fixed cells stained with anti-HA antibody, followed by secondary detection with Alexa Fluor 596-conjugated antibody and ubiquilin 1 by YFP fluorescence. Coverslips were mounted with DAPI-containing medium, and slides were imaged on a Nikon fluorescence microscope. The varied levels of ubiquitination detected for the TREX1 mutants and the observed TREX1 fluorescence pattern are indicated on the right.
RESULTS
Covalent Modification of TREX1
The TREX1 CTR is required for ubiquitination (Fig. 1). A series of TREX1 deletion constructs was prepared and transfected into human cells to investigate the cellular importance of the CTR (Fig. 1A). Immunoblotting of the expressed TREX1 variants revealed multiple bands of migration consistent with ubiquitination that was detected only upon expression of the full-length TREX1 enzyme (Fig. 1B, lanes 1–4). The ∼8-kDa size difference between the multiple detected TREX1 bands using the full-length enzyme construct (amino acids 1–314) indicated covalent modification with one to four ubiquitin molecules/TREX1 monomer. Expression of TREX1 constructs containing amino acids 1–286 (Fig. 1B, lane 2), 1–242 (lane 3), and 243–314 (lane 4) indicated no detectable modification. Ubiquitination most commonly occurs at lysines, and TREX1 contains eight lysines at Lys-30, Lys-66, Lys-75, Lys-160, Lys-175, Lys-242, Lys-271, and Lys-277. TREX1(1–286) was not detectably ubiquitinated despite possessing all eight of the candidate lysine residues, like the full-length TREX1 enzyme (Fig. 1B, compare lanes 1 and 2). Ubiquitination of TREX1(1–242) and TREX1(243–314) was also not detected despite containing six and two lysines, respectively. These data highlight the dramatic reduction in ubiquitin modification of TREX1 in the absence of the highly conserved C-terminal 30 amino acids to facilitate this process.
FIGURE 1.

The TREX1 CTR is required for ubiquitination. A, TREX1 contains the catalytic core (black boxes) and a CTR of ∼72 amino acids with a less well conserved region (gray boxes) and a highly conserved 30 residues at the end (white boxes). TREX1(1–314) and three deletion constructs encoding amino acids 1–286, 1–242, and 243–314 were prepared as N-terminal FLAG epitope fusions. B, the plasmids were transfected into HEK 293T cells, and lysates were fractionated by SDS-PAGE and immunoblotted with anti-FLAG antibody (TREX1). The positions of migration of the TREX1 polypeptides are indicated (arrows). The slower migrating bands visible in lane 1 indicate ubiquitination of TREX1(1–314), and slower migrating bands were not apparent using TREX1(1–286), TREX1(1–242), and TREX1(243–314) (lanes 2–4). A sufficient chemiluminescence exposure was used to verify the presence of four higher molecular mass species of TREX1(1–314) from whole cell lysates (lane 1), resulting in overexposure of unmodified TREX1 constructs (lanes 1–3) and detection of some minor nonspecific bands (lanes 1–4). C, plasmids encoding TREX1 and the deletion variants expressed as N-terminal HA epitope fusions were cotransfected into cells with a His6-Myc-ubiquitin plasmid (lanes 1, 3, 5, and 7) and alone (lanes 2, 4, 6, and 8). His-Myc-ubiquitin was recovered from cell lysates with Ni-NTA agarose, and eluted samples were fractionated by SDS-PAGE and immunoblotted with anti-HA antibody (TREX1; upper panel). Ubiquitinated TREX1 polypeptides are bands present in lanes 1, 3, 5, and 7 that are not in lanes 2, 4, 6, and 8. Nonspecific binding of TREX1(1–314), TREX1(1–286), and TREX1(1–242) to the Ni-NTA resin resulted in capture of some unmodified TREX1 in the pulldown assays (upper panel, lanes 1–6). The farthest migrating bands in the Ni-NTA pulldown panel in lanes 1–6 are not ubiquitinated. TREX1 polypeptides (middle panel) and ubiquitin (lower panel) in cell lysates are shown.
TREX1 modification by ubiquitination was confirmed by cotransfecting the TREX1 constructs with a His-tagged ubiquitin-expressing plasmid. Ubiquitinated proteins were recovered from lysates with Ni-NTA resin, and TREX1 was detected by immunoblotting (Fig. 1C). The multiple bands detected using the full-length TREX1 plasmid construct (amino acids 1–314) indicated modification by covalent linkage of multiple ubiquitin molecules (Fig. 1C, lane 1). The increased sensitivity of this cotransfection analysis also revealed the presence of a single ubiquitin modification using the TREX1(1–286) and TREX1(1–242) constructs (Fig. 1C, lanes 3 and 5) and multiple ubiquitin molecules using the TREX1(243–314) CTR construct (lane 7). Thus, the presence of multiple ubiquitination events was detected only in TREX1 constructs containing the C-terminal 30 amino acids.
Nature of TREX1 Ubiquitination
The TREX1 protein is ubiquitinated by a non-canonical linkage at multiple lysine residues that does not appear to target the enzyme for proteasomal degradation. The most common signal for proteasomal recognition is covalent modification of lysine in a target protein with a polyubiquitin chain containing four or more ubiquitin molecules branched through Lys-48 in ubiquitin (46). However, polyubiquitin chains linked to all seven ubiquitin lysine residues have been described in proteasomal targeting (47–49). To identify the target site for TREX1 ubiquitin linkage, a series of mutant constructs was prepared in which the eight TREX1 lysines were mutated to arginine individually, in combination, or in total (lysine-less TREX1). The TREX1 Lys-to-Arg mutant constructs were cotransfected with the His-tagged ubiquitin plasmid, ubiquitinated proteins were recovered from lysates, and TREX1 was detected by immunoblotting (Fig. 2A). The TREX1 K66R mutant was ubiquitinated identically to WT TREX1 (Fig. 2A, compare lanes 1 and 3), indicating that this residue may not be targeted for modification. All of the other Lys-to-Arg mutants exhibited reduced levels of ubiquitination to varying degrees (Fig. 2A, compare lane 1 with lanes 2 and 4–9). These results suggest that the remaining TREX1 lysine residues are potential targets for ubiquitination, although the relative extent is not possible to determine. The TREX1 K271R/K277R double mutant exhibited a dramatically reduced level of ubiquitination (Fig. 2A, compare lanes 1 and 10), and lysine-less TREX1 was not covalently modified (lane 11). These data demonstrate that TREX1 is ubiquitinated at lysine residues and that multiple TREX1 lysines are candidates for ubiquitination.
FIGURE 2.
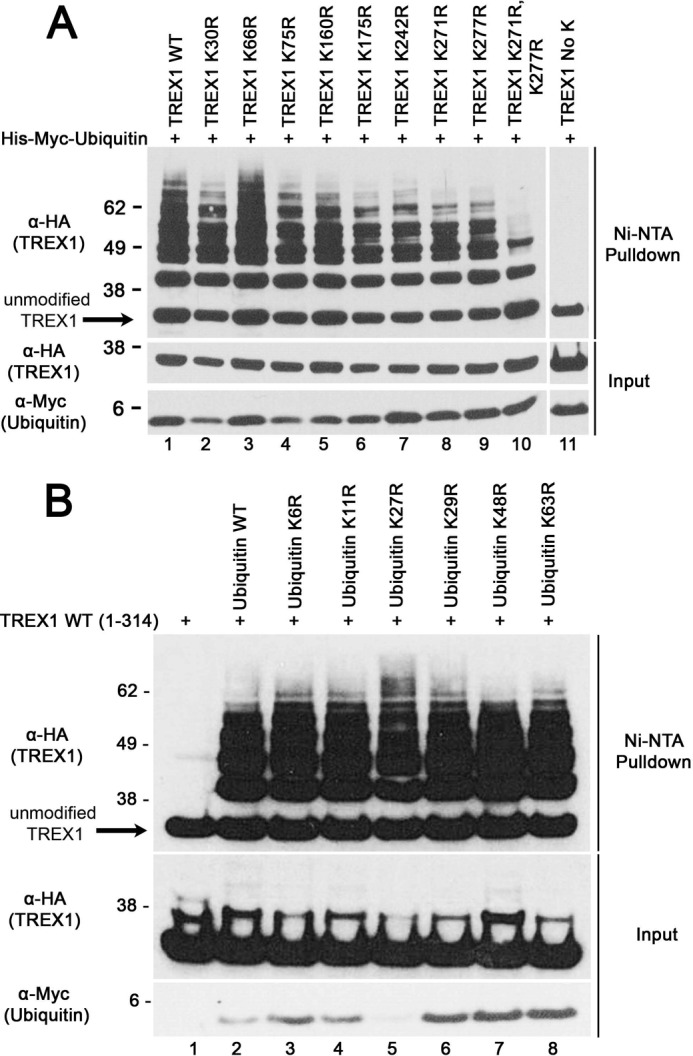
Ubiquitination of TREX1 at multiple lysine residues. A, the TREX1(1–314) construct encoding WT TREX1 (lane 1) and the indicated TREX1 mutants (lanes 2–11) expressed as N-terminal HA epitope fusions were cotransfected into cells with a His6-Myc-ubiquitin plasmid. B, the WT TREX1(1–314) plasmid was transfected alone (lane 1) and with plasmids encoding His6-Myc-WT ubiquitin (lane 2) and the indicated mutants (lanes 3–8). His-Myc-ubiquitin was recovered from cell lysates with Ni-NTA agarose, and eluted samples were fractionated by SDS-AGE, and immunoblotted with anti-HA antibody (TREX1) (upper panels). TREX1 (middle panels) and ubiquitin (lower panels) in cell lysates are shown.
To determine the nature of the ubiquitin linkage, a collection of His-ubiquitin mutant constructs containing one of the seven Lys-to-Arg mutations (K6R, K11R, K27R, K29R, K48R, and K63R) was tested for their ability to support TREX1 modification. The TREX1(1–314) and His-tagged ubiquitin Lys-to-Arg mutant constructs were cotransfected, ubiquitinated proteins were recovered from lysates, and TREX1 was detected by immunoblotting (Fig. 2B). TREX1 ubiquitination using all of the ubiquitin mutants was similar to that using the WT ubiquitin construct (Fig. 2B, compare lane 2 with lanes 3–8). These data support a linkage pattern distinctly different from the canonical Lys-48 branching frequently associated with targeting proteins for proteasomal degradation.
Identification of the TREX1 CTR Interaction with Ubiquilin 1
The TREX1 C-terminal 30 amino acids are highly conserved in mammalian species. The dependence of this region in TREX1 ubiquitination prompted us to develop an unbiased approach to identify proteins that physically associate with this region of TREX1. An expression construct was prepared to generate a His-tagged GFP fused with the TREX1 CTR 72 amino acids to identify TREX1 CTR-interacting proteins in cell-free lysates. The His-GFP-TREX1 CTR and His-GFP control were translated in vitro and recovered by immunoprecipitation with anti-His antibody (Fig. 3A). Recovered proteins were separated on polyacrylamide gels and visualized by silver staining to reveal proteins uniquely present in the TREX1 CTR-containing samples. A protein band corresponding to ∼70 kDa in size was identified, excised, and determined by mass spectrometry analysis to be ubiquilin 1 (Fig. 3A). Ubiquilin 1 (also known as PLIC-1, DA41, DSK2, and XDRP1) is a multidomain protein that interacts with ubiquitinated proteins (44, 50–59).
FIGURE 3.

The TREX1 CTR interacts with ubiquilin 1. A, left panel, the in vitro translation reactions were performed in human reticulocyte lysates with RNA transcripts encoding the His-GFP-TREX1 CTR (lane 1) and His-GFP (lane 2). His-tagged proteins were recovered from reactions using anti-His antibody, separated by SDS-PAGE, and visualized by silver staining. Right panel, ×3.5 magnification of the 55–95-kDa region in the left panel. A band of ∼70 kDa (right and left panels, lane 1) was excised from the gel and identified as ubiquilin 1 by nano-LC-MS/MS. IP, immunoprecipitation. B, the in vitro translation reactions were performed in human reticulocyte lysates containing [35S]methionine with RNA transcripts encoding the His-GFP-TREX1 CTR (lane 1), His-GFP (lane 2), ubiquilin 1 (lane 3), the His-GFP-TREX1 CTR + ubiquilin 1 (lane 4), and His-GFP + ubiquilin 1 (lane 5) to generate radiolabeled proteins. The His-GFP proteins were recovered from reactions using anti-His antibody, separated by SDS-PAGE, and detected by phosphorimaging (upper panel). The radiolabeled proteins generated in the translation reactions are shown (lower panel). C, cells were transfected with plasmids encoding TREX1(1–314) expressed as a C-terminal HA epitope fusion (lane 1), ubiquilin 1 (lane 2) and the ubiquilin 1 NTF (ubiquitin-like domain; lane 3) expressed as N-terminal FLAG epitope fusions, TREX1(1–314) + ubiquilin 1 (lane 4), and TREX1(1–314) + the ubiquilin 1 NTF (lane 5). TREX1 was recovered from cell lysates using anti-HA antibody, separated by SDS-PAGE, and immunoblotted for HA (TREX1) and FLAG (ubiquilin 1) (upper panel). TREX1 and ubiquilin 1 in cell lysates are shown (lower panel).
The TREX1 CTR interacts with ubiquilin 1 in vitro and in vivo. The TREX1 CTR-ubiquilin 1 interaction was verified in a series of in vitro translation reactions performed in the presence of [35S]methionine (Fig. 3B). The His-GFP-TREX1 CTR, His-GFP, and ubiquilin 1 were translated individually (Fig. 3B, lower panel, lanes 1–3), and ubiquilin 1 was cotranslated with the His-GFP-TREX1 CTR (lane 4) and His-GFP (lane 5). The translation products were recovered using anti-His-GFP antibody. The physical association of the TREX1 CTR with ubiquilin 1 was apparent by the quantitative recovery of the radiolabeled ubiquilin 1 as indicated by the presence of an ∼70-kDa band only in the presence of the His-GFP-TREX1 CTR (Fig. 3B, upper panel, lane 4) and not His-GFP (lane 5).
The interaction of the TREX1 CTR with ubiquilin 1 was also demonstrated in cells. Plasmids containing full-length TREX1, ubiquilin 1, and the ubiquilin 1 NTF (ubiquitin-like domain, residues 1–112) were transfected into cells individually (Fig. 3C, lower panel, lanes 1–3) and cotransfected with TREX1 (lanes 4 and 5). TREX1 was recovered from cell lysates by immunoprecipitation using anti-HA antibody, samples were separated by SDS-PAGE, and the presence of ubiquilin 1 and TREX1 was determined by immunoblotting using anti-FLAG and anti-HA antibodies. The TREX1 CTR-ubiquilin 1 interaction is apparent by the presence of full-length ubiquilin 1 detected upon immunoprecipitation of TREX1 (Fig. 3C, upper panel, lane 4) and, to a lesser extent, the detection of the ubiquilin 1 NTF (lane 5). Neither ubiquilin 1 nor the ubiquilin 1 NTF was recovered in the absence of TREX1 (Fig. 3C, upper panel, lanes 2 and 3). These in vitro and in vivo analyses support the physical association of the TREX1 CTR with ubiquilin 1.
Ubiquilin 1 Alters the Localization of TREX1 in Cells
The interaction between the TREX1 CTR and ubiquilin 1 re-localizes TREX1 from the perinuclear space to cytosolic puncta in human cells. The TREX1 CTR is necessary to localize the enzyme to the perinuclear region of the cytosolic compartment (16, 19). The effect of ubiquilin 1 expression on TREX1 cell localization was determined by microscopy in a series of cotransfection experiments detecting HA-TREX1 by red fluorescence and YFP-ubiquilin 1 by yellow fluorescence (Fig. 4). As demonstrated previously, full-length TREX1 (amino acids 1–314) localized to the cytosolic compartment in a distinct perinuclear region (Fig. 4A, row 1), and the absence of the CTR resulted in a more diffusely dispersed TREX1(1–286) throughout the cytosolic compartment (row 2). TREX1(243–314) also localized to the perinuclear space despite the absence of the TREX1 exonuclease-containing core (Fig. 4A, row 3). TREX1(1–314) in which all eight lysines were mutated to arginine (TREX1 NoK(1–314)) also localized to the perinuclear space (Fig. 4A, row 4). Together, these data highlight the requirement of the TREX1 CTR in perinuclear targeting and further suggest that ubiquitination is not required for this localization.
Ubiquilin 1 has been shown to localize in cells in varied size puncta that include the autophagosome marker LC3, suggesting its involvement in autophagy (51, 60). Thus, as expected, when the ubiquilin 1-containing plasmid was transfected into cells, the expressed protein was detected in cytosolic puncta (Fig. 4B). When the ubiquilin 1 and TREX1(1–314) plasmids were cotransfected, the expressed TREX1 protein was clearly detected localized at the membrane that surrounds the ubiquilin 1-containing puncta (Fig. 4C, row 1). Cotransfection of the TREX1(1–286) construct with ubiquilin 1 eliminated the TREX1-ubiquilin 1 co-localization (Fig. 4C, row 2), whereas cotransfection of the TREX1(243–314) construct retained co-localization (row 3). These results demonstrate the requirement of the TREX1 CTR in co-localization with ubiquilin 1. Also, co-localization of TREX1 and ubiquilin 1 was not detected using the lysine-less TREX1 NoK(1–314) plasmid (Fig. 4C, row 4), suggesting that TREX1 ubiquitination is required for this interaction.
Exonuclease Activities of AGS and SLE TREX1 Mutants
Approximately 40 TREX1 mutations have been identified in a spectrum of related autoimmune diseases, and we previously demonstrated the extremely broad range of catalytic activities exhibited among TREX1 mutants that yield similar human pathologies (10, 19, 22, 25). The TREX1 V122A and E198K mutations cause AGS (11, 61), and these mutations map within the TREX1 catalytic core. The TREX1 V122A/V122A and E198K/E198K homodimer enzymes were prepared, and the ssDNA and dsDNA exonuclease activities were measured in time course experiments (Fig. 5, A and B). The TREX1 V122A/V122A and E198K/E198K enzymes exhibited ssDNA and dsDNA exonuclease activities that were indistinguishable from those of the WT TREX1 enzyme (Fig. 5, A and B). Purified WT TREX1(1–286) also exhibited wild-type levels of ssDNA and dsDNA exonuclease activities (data not shown). However, full-length WT TREX1 (amino acids 1–314) could not be generated recombinantly in E. coli. Therefore, the effects of disease-causing mutations located in the TREX1 CTR on exonuclease activity were determined by expressing these mutants in human cells. Plasmids containing WT TREX1(1–314) and the P290L, T303P, Y305C, and D200N mutations were transfected into cells, and ssDNA exonuclease activities were measure in dilutions of cell lysates (Fig. 5C). The TREX1 P290L-, T303P-, and Y305C-containing lysates exhibited exonuclease activities similar to the WT enzyme-containing lysate as indicated by the pattern of ssDNA degradation present in the 100- and 1000-fold diluted samples. These results contrast with the lysate containing the catalytically deficient TREX1 D200N enzyme, which exhibited only background exonuclease activity, as expected. Thus, a subset of disease-causing TREX1 mutants that exhibited full enzymatic activity was identified, suggesting alternative mechanisms of TREX1 dysfunction in disease. Our attempts to purify ubiquitinated TREX1 from unmodified TREX1 in a buffer solution compatible with measuring catalytic function have not been successful. Thus, the effect of ubiquitination on TREX1 exonuclease activity is not known.
Ubiquitination of Disease-causing TREX1 Mutants
Mutations in the TREX1 CTR linked to AGS, SLE, and RVCL can result in prematurely truncated protein and exhibit altered cellular localization (16, 19, 62). However, the TREX1 mutants E198K, V122A, P290L, T303P, and Y305C are full-length proteins that exhibit wild-type levels of exonuclease activity. Therefore, we tested these TREX1 mutants to determine whether ubiquitination levels might be altered (Fig. 6). The TREX1-containing plasmids were cotransfected at two different concentrations with a His-ubiquitin plasmid, and ubiquitinated proteins were recovered with Ni-NTA resin. Samples were separated by SDS-PAGE and immunoblotted for TREX1. The amount of ubiquitinated TREX1 was quantified and expressed relative to the total TREX1 recovered to determine differential levels of ubiquitination. These data indicate that TREX1 E198K exhibited an ∼2.0-fold increase and TREX1 V122A an ∼3.5-fold increase in ubiquitination relative to WT TREX1. As expected, the lysine-less TREX1 NoK construct was not detectably modified by ubiquitination (Fig. 6A). In contrast, the TREX1 D18N, D200N, and D200H dominant mutants, which were catalytically deficient, were ubiquitinated at levels similar to WT TREX1 (data not shown). TREX1 T303P exhibited a 6.0-fold lower level and TREX1 Y305C exhibited a 2.9-fold lower level of ubiquitination relative to WT TREX1, whereas TREX1 P290L exhibited a 2.9-fold higher level of ubiquitination (Fig. 6B). These results show that a subset of disease-causing TREX1 mutations altered ubiquitination levels and suggest a novel mechanism for altered DNA degradation dysfunction and disease.
FIGURE 6.

Ubiquitination of disease-causing TREX1 mutants. A and B, the indicated amounts of TREX1(1–314) plasmids encoding WT TREX1 or the indicated TREX1 mutants expressed as N-terminal HA epitope fusions were cotransfected into cells with a His6-Myc-ubiquitin plasmid. His-Myc-ubiquitin was recovered from cell lysates with Ni-NTA agarose, and eluted samples were fractionated by SDS-PAGE and immunoblotted with anti-HA antibody (TREX1). The amount of WT and mutant TREX1 ubiquitination was quantified as described under “Experimental Procedures” (upper panels). TREX1 (middle panels) and ubiquitin (lower panels) in cell lysates are shown.
Disease-causing TREX1 Mutants Exhibit Modified Ubiquilin 1 Co-localization
The TREX1 CTR mutants that exhibited altered levels of ubiquitination also exhibited distinctly different properties of ubiquilin 1 co-localization compared with WT TREX1. Like full-length WT TREX1 (amino acids 1–314), the TREX1(1–314) T303P, P290L, Y305C, E198K, and V122A mutants localized to the perinuclear region in cells (data not shown). When the disease-causing TREX1 mutants were cotransfected with ubiquilin 1, the cellular localization indicated varied co-localization dependent upon the TREX1 mutation (Fig. 7). Similar to WT TREX1, TREX1 P290L localized at the membrane surrounding the ubiquilin 1-containing puncta but with less expanded ubiquilin 1 compartments (Fig. 7). The TREX1 T303P and Y305C mutants exhibited considerably lower levels of co-localization with ubiquilin 1 as indicated by the less distinct TREX1-associated fluorescence surrounding the ubiquilin 1-containing puncta (Fig. 7). The TREX1 E198K and V122A mutants exhibited greater levels of co-localization with ubiquilin 1 as indicated by the more intense TREX1-associated fluorescence in the ubiquilin 1-containing puncta (Fig. 7). Thus, the varied levels of TREX1 ubiquitination detected upon transfection of the mutants into human cells (Fig. 6) correlated directly with the levels of TREX1 co-localization with ubiquilin 1 (Fig. 7).
DISCUSSION
The TREX1 exonuclease is a 314-amino acid polypeptide. The exonuclease domain is positioned in the N-terminal 242 amino acids and forms stable dimers such that residues in one protomer contribute to DNA degradation in the active site of the opposing protomer (22, 24, 26). The TREX1 CTR of 72 amino acids has no homology to other proteins, and the last 30 amino acids of this region are highly conserved in mammalian species, indicating a specific function in TREX1 biology. The TREX1 CTR is necessary for localization of the exonuclease to the cytosolic perinuclear space, and mutations in the CTR that alter cellular localization have been identified in patients with AGS, SLE, and RVCL (16, 18, 19, 30, 33). The studies presented here show that the CTR controls ubiquitination of TREX1 and its interaction with ubiquilin 1. The TREX1 interaction with ubiquilin 1 localizes to cytosolic punctate structures and is altered in a subset of catalytically competent disease-causing TREX1 mutants. These data help to provide an alternative mechanistic explanation for TREX1 dysfunction in nucleic acid-mediated disease that cannot be readily attributed to loss of enzymatic activity but results in DNA accumulation and immune activation.
The TREX1 CTR directs ubiquitination of the exonuclease for attachment of single ubiquitin molecules to multiple lysines (multiple monoubiquitination), a ubiquitination pattern often associated with regulation of endocytosis and signaling processes (63, 64). Although the ubiquitination machinery targeting TREX1 is not known, it is clear that the last 30 amino acids of the TREX1 CTR are critical for this modification process. Our results show that ubiquitin is linked to TREX1 lysine residues as evidenced by the lack of modification of the lysine-less TREX1 enzyme. It is also apparent that a single lysine residue is not exclusively targeted, as seven of the eight TREX1 Lys-to-Arg mutants exhibit lower levels of ubiquitination compared with WT TREX1. TREX1 Lys-66 does not appear to be targeted for ubiquitination. TREX1 Lys-66 is positioned at the dimer interface hydrogen-bonded across the interface to Glu-198 of the opposing protomer, likely rendering the Lys-66 side chain unavailable for ubiquitination. Thus, the higher level of ubiquitination detected for the TREX1 E198K mutant might be explained by the additional lysine residue at position 198 or by the available Lys-66 side chain in the opposing protomer. The relatively modest effect on ubiquitination levels detected with the ubiquitin Lys-to-Arg mutants supports a multiple monoubiquitination pattern. The last 30 amino acids of the TREX1 CTR are not targeted for ubiquitination, as there are no lysines in this region. Also, our results show that the TREX1 catalytic core (amino acids 1–242) and the TREX1(1–286) enzyme are modified by the addition of a single ubiquitin molecule. Ubiquitination is dramatically increased in the presence of the last 30 amino acids of the TREX1(1–314) enzyme, suggesting that this region is critical for a strong interaction with the ubiquitin ligase machinery.
It is apparent that the TREX1 CTR plays a central role in localization to the cytosolic space. The non-catalytic function of the TREX1 CTR and the linkage of this region to disease-causing mutations make understanding the cellular function of this unique amino acid sequence an important challenge. Computational analysis predicts a transmembrane helix in the last 30 amino acids, leading to our original proposal that TREX1 monomers are anchored in the endoplasmic reticulum via the C-terminal transmembrane domain (19). However, TREX1 lacks an identifiable signal sequence for membrane insertion, and fractionation studies suggest instead an interaction with the endoplasmic reticulum or with resident proteins (33). Using an unbiased approach, we determined that the TREX1 CTR facilitates association with ubiquilin 1 and confirmed this association by immunoprecipitation with TREX1 in cotranslation studies in vitro and in coexpression analyses in human cells.
Ubiquilin 1 is a multidomain protein that contains an N-terminal ubiquitin-like domain, multiple heat shock chaperonin domains, and a C-terminal ubiquitin-associated domain (44, 65, 66). Ubiquilin 1 is a member of a subfamily of ubiquitin-binding proteins that recognize ubiquitinated proteins through the ubiquitin-associated domain, promoting monoubiquitination and shuttling proteins during relocation in a variety of cellular processes (44, 64, 67, 68). Ubiquilin 1 also interacts with the regulator kinase mTOR to control cellular homeostasis and autophagy in the lysosomal degradation of cellular components (51, 55, 57, 58). Recently, TREX1 has been identified as a regulator of lysosomal biogenesis operating through the transcription factor TFEB and the regulator mTORC1 (38).
The association of TREX1 with ubiquilin 1 in cells indicates a previously unknown linkage to autophagy. Ubiquilin 1 forms cytosolic puncta indicative of autophagosomes, and the interaction of ubiquilin 1 with TREX1 causes redistribution of TREX1 from the perinuclear space to these cytosolic puncta dependent upon ubiquitination levels. TREX1 appears to surround ubiquilin 1, indicating its association with the autophagosomal membrane. The last 30 amino acids of the TREX1 CTR are required for the TREX1-ubiquilin 1 interaction, which is likely mediated by ubiquitination. The interaction of TREX1 with ubiquilin 1 could be a mechanism to recruit TREX1 and possibly cytosolic DNA to vesicles such as those generated in the autophagy process. TREX1 activity might be required for degradation of DNA that could otherwise promote the progression of autophagy and lysosomal expansion. The role for TREX1 in regulating lysosomal biogenesis might be in the balance between autophagy and lysosomal expansion. If TREX1 is successfully recruited to autophagosomes for DNA degradation, the autophagic signal could be turned off. In the case that TREX1 is not successfully recruited or dysfunctional TREX1 is recruited, the autophagosomes might continue to mature and fuse with lysosomes, leading to self-consumption and cell death. Our data show that some catalytically competent disease-causing TREX1 variants exhibit altered levels of ubiquitination and cellular localization, implicating these processes in aberrant nucleic acid processing and immune activation.
Acknowledgment
We thank Dr. David A. Ornelles (Wake Forest School of Medicine) for excellent guidance in fluorescence microscopy.
This work was supported, in whole or in part, by National Institutes of Health Grant GM069962 (to F. W. P.). This work was also supported by Alliance for Lupus Research Grant 179222 (to F. W. P.), American Heart Association Grant 10GRNT3650033 (to T. H.), and American Cancer Society Research Scholar Award RSG-09-166-01-CCG (to J. C. W.).
- AGS
- Aicardi-Goutières syndrome
- RVCL
- retinal vasculopathy with cerebral leukodystrophy
- SLE
- systemic lupus erythematosus
- CTR
- C-terminal region
- NTF
- N-terminal fragment
- Ni-NTA
- nickel-nitrilotriacetic acid.
REFERENCES
- 1. Münz C., Lünemann J. D., Getts M. T., Miller S. D. (2009) Antiviral immune responses: triggers of or triggered by autoimmunity? Nat. Rev. Immunol. 9, 246–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poole B. D., Templeton A. K., Guthridge J. M., Brown E. J., Harley J. B., James J. A. (2009) Aberrant Epstein-Barr viral infection in systemic lupus erythematosus. Autoimmun. Rev. 8, 337–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rekvig O. P., Bendiksen S., Moens U. (2006) Immunity and autoimmunity induced by polyomaviruses: clinical, experimental and theoretical aspects. Adv. Exp. Med. Biol. 577, 117–147 [DOI] [PubMed] [Google Scholar]
- 4. Migliorini A., Anders H. J. (2012) A novel pathogenetic concept-antiviral immunity in lupus nephritis. Nat. Rev. Nephrol. 8, 183–189 [DOI] [PubMed] [Google Scholar]
- 5. Crow Y. J., Hayward B. E., Parmar R., Robins P., Leitch A., Ali M., Black D. N., van Bokhoven H., Brunner H. G., Hamel B. C., Corry P. C., Cowan F. M., Frints S. G., Klepper J., Livingston J. H., Lynch S. A., Massey R. F., Meritet J. F., Michaud J. L., Ponsot G., Voit T., Lebon P., Bonthron D. T., Jackson A. P., Barnes D. E., Lindahl T. (2006) Mutations in the gene encoding the 3′ → 5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet. 38, 917–920 [DOI] [PubMed] [Google Scholar]
- 6. Black D. N., Watters G. V., Andermann E., Dumont C., Kabay M. E., Kaplan P., Meagher-Villemure K., Michaud J., O'Gorman G., Reece E. (1988) Encephalitis among Cree children in northern Quebec. Ann. Neurol. 24, 483–489 [DOI] [PubMed] [Google Scholar]
- 7. Crow Y. J., Black D. N., Ali M., Bond J., Jackson A. P., Lefson M., Michaud J., Roberts E., Stephenson J. B., Woods C. G., Lebon P. (2003) Cree encephalitis is allelic with Aicardi-Goutières syndrome: implications for the pathogenesis of disorders of interferon α metabolism. J. Med. Genet. 40, 183–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stephenson J. B. (2008) Aicardi-Goutières syndrome (AGS). Eur. J. Paediatr. Neurol. 12, 355–358 [DOI] [PubMed] [Google Scholar]
- 9. Lee-Kirsch M. A., Gong M., Schulz H., Rüschendorf F., Stein A., Pfeiffer C., Ballarini A., Gahr M., Hubner N., Linné M. (2006) Familial chilblain lupus, a monogenic form of cutaneous lupus erythematosus, maps to chromosome 3p. Am. J. Hum. Genet. 79, 731–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee-Kirsch M. A., Chowdhury D., Harvey S., Gong M., Senenko L., Engel K., Pfeiffer C., Hollis T., Gahr M., Perrino F. W., Lieberman J., Hubner N. (2007) A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J. Mol. Med. 85, 531–537 [DOI] [PubMed] [Google Scholar]
- 11. Rice G., Patrick T., Parmar R., Taylor C. F., Aeby A., Aicardi J., Artuch R. (2007) Clinical and molecular phenotype of Aicardi-Goutières syndrome. Am. J. Hum. Genet. 81, 713–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hedrich C. M., Fiebig B., Hauck F. H., Sallmann S., Hahn G., Pfeiffer C., Heubner G., Lee-Kirsch M. A., Gahr M. (2008) Chilblain lupus erythematosus–a review of literature. Clin. Rheumatol. 27, 1341. [DOI] [PubMed] [Google Scholar]
- 13. Crow Y. J., Rehwinkel J. (2009) Aicardi-Goutières syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum. Mol. Genet. 18, R130–R136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Günther C., Meurer M., Stein A., Viehweg A., Lee-Kirsch M. A. (2009) Familial chilblain lupus–a monogenic form of cutaneous lupus erythematosus due to a heterozygous mutation in TREX1. Dermatology 219, 162–166 [DOI] [PubMed] [Google Scholar]
- 15. Prendiville J. S., Crow Y. J. (2009) Blue (or purple) toes: chilblains or chilblain lupus-like lesions are a manifestation of Aicardi-Goutières syndrome and familial chilblain lupus. J. Am. Acad. Dermatol. 61, 727–728 [DOI] [PubMed] [Google Scholar]
- 16. Richards A., van den Maagdenberg A. M., Jen J. C., Kavanagh D., Bertram P., Spitzer D., Liszewski M. K., Barilla-Labarca M. L., Terwindt G. M., Kasai Y., McLellan M., Grand M. G., Vanmolkot K. R., de Vries B., Wan J., Kane M. J., Mamsa H., Schäfer R., Stam A. H., Haan J., de Jong P. T., Storimans C. W., van Schooneveld M. J., Oosterhuis J. A., Gschwendter A., Dichgans M., Kotschet K. E., Hodgkinson S., Hardy T. A., Delatycki M. B., Hajj-Ali R. A., Kothari P. H., Nelson S. F., Frants R. R., Baloh R. W., Ferrari M. D., Atkinson J. P. (2007) C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat. Genet. 39, 1068–1070 [DOI] [PubMed] [Google Scholar]
- 17. Stam A. H., Haan J., van den Maagdenberg A. M., Ferrari M. D., Terwindt G. M. (2009) Migraine and genetic and acquired vasculopathies. Cephalalgia 29, 1006–1017 [DOI] [PubMed] [Google Scholar]
- 18. Gruver A. M., Schoenfield L., Coleman J. F., Hajj-Ali R., Rodriguez E. R., Tan C. D. (2011) Novel ophthalmic pathology in an autopsy case of autosomal dominant retinal vasculopathy with cerebral leukodystrophy. J. Neuroophthalmol. 31, 20–24 [DOI] [PubMed] [Google Scholar]
- 19. Lee-Kirsch M. A., Gong M., Chowdhury D., Senenko L., Engel K., Lee Y. A., de Silva U., Bailey S. L., Witte T., Vyse T. J., Kere J., Pfeiffer C., Harvey S., Wong A., Koskenmies S., Hummel O., Rohde K., Schmidt R. E., Dominiczak A. F., Gahr M., Hollis T., Perrino F. W., Lieberman J., Hübner N. (2007) Mutations in the gene encoding the 3′–5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 39, 1065–1067 [DOI] [PubMed] [Google Scholar]
- 20. de Vries B., Steup-Beekman G. M., Haan J., Bollen E. L., Luyendijk J., Frants R. R., Terwindt G. M., van Buchem M. A., Huizinga T. W., van den Maagdenberg A. M., Ferrari M. D. (2010) TREX1 gene variant in neuropsychiatric systemic lupus erythematosus. Ann. Rheum. Dis. 69, 1886–1887 [DOI] [PubMed] [Google Scholar]
- 21. Namjou B., Kothari P. H., Kelly J. A., Glenn S. B., Ojwang J. O., Adler A., Alarcón-Riquelme M. E., Gallant C. J., Boackle S. A., Criswell L. A., Kimberly R. P., Brown E., Edberg J., Stevens A. M., Jacob C. O., Tsao B. P., Gilkeson G. S., Kamen D. L., Merrill J. T., Petri M., Goldman R. R., Vila L. M., Anaya J. M., Niewold T. B., Martin J., Pons-Estel B. A., Sabio J. M., Callejas J. L., Vyse T. J., Bae S. C., Perrino F. W., Freedman B. I., Scofield R. H., Moser K. L., Gaffney P. M., James J. A., Langefeld C. D., Kaufman K. M., Harley J. B., Atkinson J. P. (2011) Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. 12, 270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Silva U., Choudhury S., Bailey S. L., Harvey S., Perrino F. W., Hollis T. (2007) The crystal structure of TREX1 explains the 3′ nucleotide specificity and reveals a polyproline II helix for protein partnering. J. Biol. Chem. 282, 10537–10543 [DOI] [PubMed] [Google Scholar]
- 23. Lehtinen D. A., Harvey S., Mulcahy M. J., Hollis T., Perrino F. W. (2008) The TREX1 double-stranded DNA degradation activity is defective in dominant mutations associated with autoimmune disease. J. Biol. Chem. 283, 31649–31656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Orebaugh C. D., Fye J. M., Harvey S., Hollis T., Perrino F. W. (2011) The TREX1 exonuclease R114H mutation in Aicardi-Goutières syndrome and lupus reveals dimeric structure requirements for DNA degradation activity. J. Biol. Chem. 286, 40246–40254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rice G., Newman W. G., Dean J., Patrick T., Parmar R., Flintoff K., Robins P., Harvey S., Hollis T., O'Hara A., Herrick A. L., Bowden A. P., Perrino F. W., Lindahl T., Barnes D. E., Crow Y. J. (2007) Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutières syndrome. Am. J. Hum. Genet. 80, 811–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fye J. M., Orebaugh C. D., Coffin S. R., Hollis T., Perrino F. W. (2011) Dominant mutation of the TREX1 exonuclease gene in lupus and Aicardi-Goutières syndrome. J. Biol. Chem. 286, 32373–32382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Höss M., Robins P., Naven T. J., Pappin D. J., Sgouros J., Lindahl T. (1999) A human DNA editing enzyme homologous to the Escherichia coli DnaQ/MutD protein. EMBO J. 18, 3868–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mazur D. J., Perrino F. W. (1999) Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3′ → 5′ exonucleases. J. Biol. Chem. 274, 19655–19660 [DOI] [PubMed] [Google Scholar]
- 29. Mazur D. J., Perrino F. W. (2001) Structure and expression of the TREX1 and TREX2 3′ → 5′ exonuclease genes. J. Biol. Chem. 276, 14718–14727 [DOI] [PubMed] [Google Scholar]
- 30. Chowdhury D., Beresford P. J., Zhu P., Zhang D., Sung J. S., Demple B., Perrino F. W., Lieberman J. (2006) The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol. Cell 23, 133–142 [DOI] [PubMed] [Google Scholar]
- 31. Gall A., Treuting P., Elkon K. B., Loo Y. M., Gale M., Jr., Barber G. N., Stetson D. B. (2012) Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity 36, 120–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morita M., Stamp G., Robins P., Dulic A., Rosewell I., Hrivnak G., Daly G., Lindahl T., Barnes D. E. (2004) Gene-targeted mice lacking the Trex1 (DNase III) 3′ → 5′ DNA exonuclease develop inflammatory myocarditis. Mol. Cell. Biol. 24, 6719–6727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stetson D. B., Ko J. S., Heidmann T., Medzhitov R. (2008) Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang Y. G., Lindahl T., Barnes D. E. (2007) Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131, 873–886 [DOI] [PubMed] [Google Scholar]
- 35. Yan N., Lieberman J. (2011) Gaining a foothold: how HIV avoids innate immune recognition. Curr. Opin. Immunol. 23, 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yan N., Regalado-Magdos A. D., Stiggelbout B., Lee-Kirsch M. A., Lieberman J. (2010) The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 11, 1005–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barber G. N. (2011) Cytoplasmic DNA innate immune pathways. Immunol. Rev. 243, 99–108 [DOI] [PubMed] [Google Scholar]
- 38. Hasan M., Koch J., Rakheja D., Pattnaik A. K., Brugarolas J., Dozmorov I., Levine B., Wakeland E. K., Lee-Kirsch M. A., Yan N. (2013) Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat. Immunol. 14, 61–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gack M. U., Shin Y. C., Joo C. H., Urano T., Liang C., Sun L., Takeuchi O., Akira S., Chen Z., Inoue S., Jung J. U. (2007) TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920 [DOI] [PubMed] [Google Scholar]
- 40. Tsuchida T., Zou J., Saitoh T., Kumar H., Abe T., Matsuura Y., Kawai T., Akira S. (2010) The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33, 765–776 [DOI] [PubMed] [Google Scholar]
- 41. Duckett C. S., Nava V. E., Gedrich R. W., Clem R. J., Van Dongen J. L., Gilfillan M. C., Shiels H., Hardwick J. M., Thompson C. B. (1996) A conserved family of cellular genes related to the baculovirus iap gene and encoding apoptosis inhibitors. EMBO J. 15, 2685–2694 [PMC free article] [PubMed] [Google Scholar]
- 42. Wilkinson J. C., Wilkinson A. S., Galbán S., Csomos R. A., Duckett C. S. (2008) Apoptosis-inducing factor is a target for ubiquitination through interaction with XIAP. Mol. Cell. Biol. 28, 237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lewis E. M., Wilkinson A. S., Davis N. Y., Horita D. A., Wilkinson J. C. (2011) Nondegradative ubiquitination of apoptosis inducing factor (AIF) by X-linked inhibitor of apoptosis at a residue critical for AIF-mediated chromatin degradation. Biochemistry 50, 11084–11096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beverly L. J., Lockwood W. W., Shah P. P., Erdjument-Bromage H., Varmus H. (2012) Ubiquitination, localization, and stability of an anti-apoptotic BCL2-like protein, BCL2L10/BCLb, are regulated by Ubiquilin1. Proc. Natl. Acad. Sci. U.S.A. 109, E119–E126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Perrino F. W., Harvey S., McMillin S., Hollis T. (2005) The human TREX2 3′ → 5′-exonuclease structure suggests a mechanism for efficient nonprocessive DNA catalysis. J. Biol. Chem. 280, 15212–15218 [DOI] [PubMed] [Google Scholar]
- 46. Pickart C. M., Eddins M. J. (2004) Ubiquitin: structures, functions, mechanisms. Biochim. Biophys. Acta 1695, 55–72 [DOI] [PubMed] [Google Scholar]
- 47. Kravtsova-Ivantsiv Y., Ciechanover A. (2012) Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 125, 539–548 [DOI] [PubMed] [Google Scholar]
- 48. Saeki Y., Kudo T., Sone T., Kikuchi Y., Yokosawa H., Toh-e A., Tanaka K. (2009) Lysine 63-linked polyubiquitin chain may serve as a targeting signal for the 26S proteasome. EMBO J. 28, 359–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thrower J. S., Hoffman L., Rechsteiner M., Pickart C. M. (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J. 19, 94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bedford F. K., Kittler J. T., Muller E., Thomas P., Uren J. M., Merlo D., Wisden W., Triller A., Smart T. G., Moss S. J. (2001) GABAA receptor cell surface number and subunit stability are regulated by the ubiquitin-like protein Plic-1. Nat. Neurosci. 4, 908–916 [DOI] [PubMed] [Google Scholar]
- 51. Rothenberg C., Srinivasan D., Mah L., Kaushik S., Peterhoff C. M., Ugolino J., Fang S., Cuervo A. M., Nixon R. A., Monteiro M. J. (2010) Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum. Mol. Genet. 19, 3219–3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Funakoshi M., Geley S., Hunt T., Nishimoto T., Kobayashi H. (1999) Identification of XDRP1; a Xenopus protein related to yeast Dsk2p binds to the N-terminus of cyclin A and inhibits its degradation. EMBO J. 18, 5009–5018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gao L., Tu H., Shi S. T., Lee K. J., Asanaka M., Hwang S. B., Lai M. M. (2003) Interaction with a ubiquitin-like protein enhances the ubiquitination and degradation of hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 77, 4149–4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim T. Y., Kim E., Yoon S. K., Yoon J. B. (2008) Herp enhances ER-associated protein degradation by recruiting ubiquilins. Biochem. Biophys. Res. Commun. 369, 741–746 [DOI] [PubMed] [Google Scholar]
- 55. Levine B., Kroemer G. (2008) Autophagy in the pathogenesis of disease. Cell 132, 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lim P. J., Danner R., Liang J., Doong H., Harman C., Srinivasan D., Rothenberg C., Wang H., Ye Y., Fang S., Monteiro M. J. (2009) Ubiquilin and p97/VCP bind erasin, forming a complex involved in ERAD. J. Cell Biol. 187, 201–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ravikumar B., Futter M., Jahreiss L., Korolchuk V. I., Lichtenberg M., Luo S., Massey D. C., Menzies F. M., Narayanan U., Renna M., Jimenez-Sanchez M., Sarkar S., Underwood B., Winslow A., Rubinsztein D. C. (2009) Mammalian macroautophagy at a glance. J. Cell Sci. 122, 1707–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Xie Z., Klionsky D. J. (2007) Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 9, 1102–1109 [DOI] [PubMed] [Google Scholar]
- 59. Nakayasu E. S., Ansong C., Brown J. N., Yang F., Lopez-Ferrer D., Qian W. J., Smith R. D., Adkins J. N. (2013) Evaluation of selected binding domains for the analysis of ubiquitinated proteomes. J. Am. Soc. Mass Spectrom. 24, 1214–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Biswas N., Liu S., Ronni T., Aussenberg S. E., Liu W., Fujita T., Wang T. (2011) The ubiquitin-like protein PLIC-1 or ubiquilin 1 inhibits TLR3-Trif signaling. PLoS ONE 6, e21153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ramantani G., Kohlhase J., Hertzberg C., Innes A. M., Engel K., Hunger S., Borozdin W., Mah J. K., Ungerath K., Walkenhorst H., Richardt H. H., Buckard J., Bevot A., Siegel C., von Stülpnagel C., Ikonomidou C., Thomas K., Proud V., Niemann F., Wieczorek D., Häusler M., Niggemann P., Baltaci V., Conrad K., Lebon P., Lee-Kirsch M. A. (2010) Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutières syndrome. Arthritis Rheum. 62, 1469–1477 [DOI] [PubMed] [Google Scholar]
- 62. Kavanagh D., Spitzer D., Kothari P. H., Shaikh A., Liszewski M. K., Richards A., Atkinson J. P. (2008) New roles for the major human 3′–5′ exonuclease TREX1 in human disease. Cell Cycle 7, 1718–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mukhopadhyay D., Riezman H. (2007) Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 315, 201–205 [DOI] [PubMed] [Google Scholar]
- 64. Husnjak K., Dikic I. (2012) Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem. 81, 291–322 [DOI] [PubMed] [Google Scholar]
- 65. Kleijnen M. F., Shih A. H., Zhou P., Kumar S., Soccio R. E., Kedersha N. L., Gill G., Howley P. M. (2000) The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol. Cell 6, 409–419 [DOI] [PubMed] [Google Scholar]
- 66. Wu A. L., Wang J., Zheleznyak A., Brown E. J. (1999) Ubiquitin-related proteins regulate interaction of vimentin intermediate filaments with the plasma membrane. Mol. Cell 4, 619–625 [DOI] [PubMed] [Google Scholar]
- 67. Zhang D., Raasi S., Fushman D. (2008) Affinity makes the difference: nonselective interaction of the UBA domain of Ubiquilin-1 with monomeric ubiquitin and polyubiquitin chains. J. Mol. Biol. 377, 162–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Walters K. J., Kleijnen M. F., Goh A. M., Wagner G., Howley P. M. (2002) Structural studies of the interaction between ubiquitin family proteins and proteasome subunit S5a. Biochemistry 41, 1767–1777 [DOI] [PubMed] [Google Scholar]