

Background: KDACis impair GR transactivation of the MMTV promoter, but their impact on cellular target genes is unknown.
Results: KDACi or KDAC depletion suppresses transactivation of about 50% of GR target genes.
Conclusion: KDAC1 is required for efficient GR transactivation in a gene-selective fashion.
Significance: Because KDACs facilitate GR transactivation, clinical KDACi use may have a major impact on GR signaling.
Keywords: Gene Transcription, Glucocorticoid Receptor, Histone Deacetylase, Histone Deacetylase Inhibitors, Transcription Coactivators
Abstract
Nuclear receptors use lysine acetyltransferases and lysine deacetylases (KDACs) in regulating transcription through histone acetylation. Lysine acetyltransferases interact with steroid receptors upon binding of an agonist and are recruited to target genes. KDACs have been shown to interact with steroid receptors upon binding to an antagonist. We have shown previously that KDAC inhibitors (KDACis) potently repress the mouse mammary tumor virus promoter through transcriptional mechanisms and impair the ability of the glucocorticoid receptor (GR) to activate it, suggesting that KDACs can play a positive role in GR transactivation. In the current study, we extended this analysis to the entire GR transcriptome and found that the KDACi valproic acid impairs the ability of agonist-bound GR to activate about 50% of its target genes. This inhibition is largely due to impaired transcription rather than defective GR processing and was also observed using a structurally distinct KDACi. Depletion of KDAC1 expression mimicked the effects of KDACi in over half of the genes found to be impaired in GR transactivation. Simultaneous depletion of KDACs 1 and 2 caused full or partial impairment of several more GR target genes. Altogether we found that Class I KDAC activity facilitates GR-mediated activation at a sizable fraction of GR-activated target genes and that KDAC1 alone or in coordination with KDAC2 is required for efficient GR transactivation at many of these target genes. Finally, our work demonstrates that KDACi exposure has a significant impact on GR signaling and thus has ramifications for the clinical use of these drugs.
Introduction
Lysine acetylation is a post-translational modification of proteins regulated directly through the actions of lysine acetyltransferases and deacetylases (also known as histone acetyltransferases and deacetylases). For many years, histones were the only proteins known to be acetylated, and the general functions of histone acetylation in transcription are well established. Long standing models of the role of histone acetylation in transcription cast lysine acetyltransferases (KATs)2 as transcriptional coactivators and lysine deacetylases (KDACs) as transcriptional corepressors that act on histones. However, recent proteomics studies have revealed that lysine acetylation occurs on well over 1000 proteins involved in a wide variety of essential cellular functions (1, 2). Many of these proteins are involved in regulation of transcription and signal transduction, indicating that KATs and KDACs participate in these processes beyond their role in regulating histone acetylation. Although the various roles of phosphorylation in signaling-regulated transcription have been established, the functions of acetylation are poorly defined for most target proteins.
KDACs comprise a family of proteins that have been divided into four classes (for a review, see Ref. 3). Class I KDACs (KDACs 1, 2, 3, and 8) are located in the nucleus and have been shown to be involved in a variety of nuclear processes. Class II KDACs have been subdivided into IIA and IIB. The IIA members have been shown to have little catalytic activity due to an amino acid change in their catalytic domains (4). In contrast, the IIB member, KDAC6, has robust catalytic activity and is largely located in the cytoplasm. Its substrates include tubulin and hsp90 (5, 6). The Class III deacetylases are the NAD+-dependent sirtuins, which are located in multiple cellular compartments (for a review, see Ref. 7). KDAC11 is the sole member of Class IV, and little is known about its activity or function.
Small molecule inhibitors of the Class I and IIB deacetylases comprise a diverse set of chemicals that inhibit these enzymes with varying potencies and specificities (8, 9). Some of these lysine deacetylase inhibitors (KDACis) such as valproic acid, vorinostat, and romidepsin are used clinically to treat epilepsy, bipolar disorder, migraines, and cancer (for a review, see Ref. 10). Vorinostat along with trichostatin A (TSA) are pan-KDACis, which can impair the activity of both Class I and Class IIB deacetylases. In contrast, KDACis such as VPA, romidepsin, and apicidin are Class I-selective and are unable to inhibit KDAC6 at doses attained in vivo. KDACis are currently being investigated for potential use in treating a variety of additional diseases, including HIV, inflammatory disorders, and neurological disorders, and thus, their use in humans may eventually expand (3, 11). VPA has been used clinically for over 30 years and has been found to cause metabolic and reproductive side effects in about 50% of users (12, 13). Our general lack of knowledge about the functions of Class I and II KDACs in signaling is an obstacle to understanding the physiological impact of these drugs and to improving their usage so that benefits are maximized and unwanted side effects are minimized.
The study of steroid receptor signaling has been at the leading edge of understanding mechanisms of transcriptional regulation through signaling. It is established that agonist-bound steroid receptors recruit KATs to target genes and that their acetyltransferase activity targeted to histones is important for activation of transcription (14, 15). Studies have also shown that steroid receptors bound to antagonists can associate with corepressor complexes that contain KDACs (16–18). These findings have led to the formulation of a model for steroid receptor action in which KDACs largely oppose transactivation of target genes. However, accumulating evidence suggests that KDACs in addition to KATs may be required for activation of transcription by the glucocorticoid receptor (GR). First, KDACis impair GR-mediated activation of target genes (19–22). In the case of the mouse mammary tumor virus (MMTV) promoter, this impairment is transcriptional in nature but independent of chromatin remodeling or histone acetylation (22). Second, glucocorticoid treatment causes a significant decrease in histone acetylation at the MMTV promoter over the time period in which transcription is activated (23). Accordingly, GR associates with KDAC1 both in solution and at the MMTV promoter (24). At the peak of GR-induced MMTV promoter activity, KDAC1 is deacetylated and active, and depletion of KDAC1 and/or KDAC2 impairs GR-activated MMTV transcription (25). Altogether these findings make a strong case that KDAC1 functions as a coactivator of GR at the MMTV promoter. However, it is unclear whether this is a general mechanism that extends to all or only selected cellular GR target genes or whether other KDACs are involved.
In the current study, the involvement of KDACs in activation of cellular GR target genes was investigated. We show that exposure to the clinically relevant KDACi VPA has a profound effect on the GR-activated transcriptome. The most prevalent effect of VPA was the impaired activation of GR target genes when co-administered with the synthetic glucocorticoid dexamethasone (Dex). We show that about half of such genes are dependent on KDAC1 expression for transcriptional activation by GR. Our results show that the models for the role of KDACs in GR signaling must be expanded to include an activating function and raise the possibility that KDACis may modulate endocrine signaling.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Anti-acetylated α-tubulin (sc-23950), α-GR (sc-1002), lamin A/C (sc-6215), GAPDH (sc-25778), HDAC3 (sc-11417), and HDAC8 (sc-17778) were obtained from Santa Cruz Biotechnology. The antibodies against α-tubulin (2144S) and HDAC2 (2540S) were obtained from Cell Signaling Technology. Anti-acetylated histone H3 (06-599) and anti-HDAC1 (05-100) antibodies are from Millipore. Anti-glucocorticoid receptor (Ab-2) mouse monoclonal antibody (BUGR2, GR32L) was obtained from Calbiochem. The secondary anti-mouse (115-035-146) and anti-rabbit (111-035-144) antibodies were purchased from Jackson ImmunoResearch Laboratories, and anti-goat (sc-2056) was purchased from Santa Cruz Biotechnology. VPA, TSA, and apicidin were obtained from Sigma-Aldrich.
Cell Culture
Murine hepatoma cells (Hepa-1c1c7) were maintained in minimum essential medium α (Invitrogen) containing 10% fetal bovine serum (FBS) (Gemini Bio-Products) and 0.1% gentamicin (Invitrogen). Murine mammary adenocarcinoma cells (1470.2) were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS and 0.1% gentamycin.
RNA Analysis
Cells were seeded in 6-well dishes at 2 × 105 cells/well. The next day cells were treated with VPA (5 mm), TSA (200 nm), or apicidin (0.5 μg/ml) for 5 h and Dex (100 nm) for 4 h followed by lysis in TRIzol (Invitrogen). Total RNA was isolated using the Nucleospin RNA II kit (Clontech). cDNA was generated using the iScript cDNA synthesis kit (Bio-Rad). qPCR was performed using the Applied Biosystems StepOne instrument with SYBR Green Master Mix (Bioline) according to the manufacturer's specifications. Exon-exon and exon-intron primer pairs for the various genes tested can be found in Table 1. In each experiment, the test gene Ct values were normalized against corresponding glyceraldehyde-3-phosphate dehydrogenase (GAPDH) Ct values to obtain ΔCt values for each sample. To determine changes in expression between treated and untreated samples (ΔΔCt), the ΔCt values for treated samples were normalized against untreated, control ΔCt values for each test gene. In each experiment, primer efficiency was calculated using standard curves and used to convert the ΔΔCt values into -fold change, which was then used to graph results and calculate S.E. The ΔΔCt values of two different treatments (Dex alone versus Dex + Drug or Dex + control siRNA versus Dex + KDAC siRNA) were compared using a paired t test (two-tailed) to determine whether changes were statistically significant (p ≤ 0.05).
TABLE 1.
PCR primers used in the study
dGRE, distal GRE; pGRE, proximal GRE.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Exon primers | ||
| Sgk1 | CTGCTCGAAGCACCCTTACC | TCCTGAGGATGGGACATTTTCA |
| Ampd3 | AAGATGATCCGGTCGCAGTC | CCAGGCTTAGAAGTAGCTCCG |
| Glu1 | GTGAGCCCAAGTGTGTGGAA | GAAGGGGTCTCGAAACATGGC |
| Tgm2 | GACAATGTGGAGGAGGGATCT | CTCTAGGCTGAGACGGTACAG |
| H6pd | GGGCCACAGTTTCAGCTTC | GAGGGTCTGATAGTCCTCCAC |
| Sdpr | CACACACTCCTGGATAAATTGGT | GCGAGACTTCTCTAGCAGCTTG |
| Ror1 | ACCGCACTGTGTATATGGAGT | TGGCGAACTGAGAGCACTTAT |
| St5 | CCAATCCCTGTATCCCTCTTCT | CAAGGAGTTCTCAGACAGTTGC |
| D8ertd82e | AGGGACCACGTACAAGACCAA | CCTCAGAGTTAAGGGATGGCTT |
| Tns1 | AGAGACCGTACCCAAGAATGT | GTAGGCTGTGATTGTGGTTGT |
| Tsc22d3 | GGTGGCCCTAGACAACAAGA | TCTTCTCAAGCAGCTCACGA |
| Fam107a | CCAGACCAGAGTACAGAGAGTG | CCATACCCAAGCCCCTTTTGT |
| Slc35d1 | ATGCTCCGGTTAAAGGAGAAGC | CAGGAACACCGTTAGCGTTTC |
| Lcn2 | TGGCCCTGAGTGTCATGTG | CTCTTGTAGCTCATAGATGGTGC |
| Nfkbia | TTGGCAATCATCCACGAAGAG | GTATTTCCTCGAAAGTCTCGGAG |
| Zfp36 | TCGAAGAGACCCTAACCAGGC | GCGTAGTCATCAGGATCGGA |
| Pfkfb3 | CCCAGAGCCGGGTACAGAA | GAGCCCCACCATCACAATCAC |
| Exon-intron primers | ||
| Ampd3 | AAGGAGCTTGCAGAGCAGAAGTC | CAGCTCCCTCAGGTCTCACAACTAT |
| Tgm2 | TGTCACCAGGGATGAGAGACGG | TCCAAATCACACCTCTCCAGGAG |
| St5 | AGAGCTGAGGATGCACAGATAGCA | TATGCCTCTTGGGTAATCGTGGCA |
| Tns1 | TTCTCTCACACGCTTCCGGACTTT | TACAGCACACACAGGCAAGGAACT |
| H6pd | GAACGCTGAAGGCAAAGCAAGACA | GCCTTGCCAGACATCAGGATGAAA |
| Ror1 | AGCGTCGTACCATTACCTTCAGCA | ATTCTTCCATGAAACGCACAGCGG |
| ChIP primers | ||
| Tns1 | TGAGAAAGTGCAGCTGTTGG | GCCAACAGGATGTGTCTGTG |
| Tsc22d3 | CTGCCTTTCTCCACCATAGC | AAATCTTGTCCCGCAGTCAC |
| Sdpr | GCAACACCACTTGCTTTGAC | GCCCCAGAGCTGACTGATAG |
| Sgk1 dGRE | CTTCCCTTATCCAGCATGTCTTGTG | TGCATCGTGCAATCTGTGGC |
| Sgk1 pGRE | ACGTGTTCTTGGCATGGCTAGGA | GGGGCGGAAATAAGTCTCTGCTCTA |
| Zfp36 | CTCTATCAAGTCCGCCCAAG | GTCCCTCCGGCTCTTGAC |
| Lcn2 | TTTGGACGCCTCACCCTGTG | AAGGGTGAGCAAGCTGAGAGTGAA |
Expression Profiling
Hepa-1c1c7 cells were seeded in 6-well plates at 5 × 105 cells/well. The next day cells were treated with VPA (5 mm) for 5 h and Dex (100 nm) for 4 h, and total RNA was isolated using the Nucleospin RNA II kit. RNA quality control, labeling, purification, and hybridization were performed by the Genomics Shared Service at the University of Arizona Cancer Center. GeneChip Mouse Gene 1.0 ST arrays (Affymetrix) were used for hybridization. Bioconductor software was used for statistical analysis of the microarray data. In the data analysis, the robust multichip algorithm was used for data preprocessing, including background correction, normalization, and perfect match correction, and then log2 -fold change analysis was performed to detect candidate genes that were either up- or down-regulated. Results of the expression profiling data are in the process of submission to the Gene Expression Omnibus (GEO).
Co-immunoprecipitation
Hepa-1c1c7 and 1470.2 cells were seeded at a density of 2 × 106 cells/150-mm plate. After 48 h, they were treated with VPA or TSA for 0, 1, and 5 h. Following treatment, cells were trypsinized, washed with Dulbecco's PBS, and pelleted by centrifugation. The cell pellet was resuspended in ice-cold HEDW buffer (10 mm HEPES, pH 7.4, 1 mm EDTA, pH 8.0, 10 mm sodium tungstate, 2 mm DTT, and protease inhibitor mixture (Roche Applied Science)). Cells were then lysed using a Dounce homogenizer, and glycerol was added to a final concentration of 10%. The lysate was centrifuged at 100,000 × g for 45min to obtain the cytosolic extract. Protein A-agarose and protein G-agarose slurry were used to preclear 500 μg of cytosolic protein diluted in HEDW buffer containing 10% glycerol. To this supernatant, 5 μg of either anti-GR (BuGR2) antibody or anti-GFP antibody and a mixture of protein A-agarose and protein G-agarose beads were added and rotated for 2 h at 4 °C. The beads were pelleted and washed three times with buffer containing 10 mm HEPES, pH 7.4, 1 mm EDTA, pH 8.0, 10 mm sodium tungstate, 50 mm NaCl, and 0.5% Tween 20. The bound proteins were eluted by addition of 2× SDS-PAGE buffer followed by incubation at 95 °C for 4–5 min prior to separation by SDS-PAGE.
Western Blotting
Cell lysates for analysis of acetylated α-tubulin and histone H3 and KDAC expression were prepared by adding 2× SDS-PAGE buffer to treated cells. Proteins were separated by SDS-PAGE and transferred onto a nitrocellulose membrane (Bio-Rad) at 400 mA for 2.5 h. The membrane was blocked with 2% nonfat dry milk for 1 h followed by exposure to primary antibodies at 4 °C overnight. After subsequent exposure to secondary antibodies, the membrane was washed three times with 1× TBS and 0.1% Tween 20 solution. The proteins were visualized using a 1:1 ratio of hydrogen peroxide and luminol (Pierce) with the ChemiDoc XRS+ molecular imager (Bio-Rad).
siRNA-mediated KDAC Knockdown
Hepa-1c1c7 cells were plated in 24-well dishes at a density of 2 × 104 cells/well in antibiotic-free minimum essential medium α. DharmaFECT Reagent 1 (Dharmacon) was used according to the manufacturer's specifications to transiently transfect the cells with siRNA. KDACs 1, 2, 3, and 8 were depleted using the corresponding ON-TARGETplus SMARTpool ORF siRNA (Dharmacon). Successful knockdown was confirmed by Western blotting. Lamin siRNA and non-targeting siRNA were used as controls.
Chromatin Immunoprecipitation
Hepa-1c1c7 cells were seeded at a density of 2.2 × 106 cells/15-cm plate. After 48 h, they were treated with either VPA for 2 h, Dex for 1 h, or a combination of both. The cells were then fixed with 1% formaldehyde at room temperature for 10 min and neutralized using 0.125 m glycine. Fixed cells were scraped using ice-cold PBS with 2% FBS, washed once with ice-cold PBS, and resuspended in lysis buffer (0.625% SDS, 10 mm EDTA, and 50 mm Tris, pH 8.0). Chromatin was then fragmented to 200–500 bp at 4 °C with 35 cycles (30 s on, 30 s off) in a Bioruptor (Diagenode). Five percent of the supernatant was removed for use as input.
The remaining sonicated chromatin was precleared with protein A- and protein G-agarose (pretreated with 1 mg/ml salmon sperm DNA and 1 mg/ml BSA) for 45 min. The chromatin equivalent of 1 × 107 cells was then rotated overnight at 4 °C with either antibody against acetylated histone H3 (Millipore 06-599) or no antibody. Protein A- and protein G-agarose beads (Pierce) were added, and incubation continued for 45 min. Following incubation, the beads were pelleted and washed sequentially once with low salt buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris, pH 8.0, and 150 mm NaCl), high salt buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris, pH 8.0, and 500 mm NaCl), and lithium chloride buffer (0.25 m LiCl, 1% Nonidet P-40, 1% sodium deoxycholate, 1 mm EDTA, and 10 mm Tris, pH 8.0) and twice with TE buffer (10 mm Tris, pH 8.0 and 1 mm EDTA). All buffers contained protease inhibitor mixture and 100 nm TSA. Bound chromatin was eluted with sequential washes of high SDS buffer (1.5% SDS and 0.1 m NaHCO3 in TE buffer) and low SDS buffer (0.5% SDS and 0.1 m NaHCO3 in TE buffer). Cross-links were reversed by incubation at 65 °C in 200 mm NaCl and 10 mg RNase A for 5 h followed by proteinase K digestion at 45 °C in 10 mm EDTA and 40 mm Tris, pH 6.8 for 2 h. DNA was extracted twice with phenol-chloroform-isoamyl alcohol (25:24:1) and ethanol-precipitated in the presence of glycogen.
Input and bound DNA was amplified in 2× SYBR buffer (Bioline) using real time qPCR (ABI StepOnePlus). Primer sequences are listed in Table 1. Ct values for the bound DNAs were normalized against the input Ct values (ΔCt), and the various treatments were normalized against control (ΔΔCt). Primer efficiency was calculated using standard curves and used to convert the ΔΔCt value into -fold change. The ΔCt values from the various treatments were compared with those of untreated control within each experiment and were used in a paired t test (one-tailed) to determine whether changes were statistically significant. Values of p ≤ 0.05 were deemed significant.
RESULTS
Effects of VPA Treatment on Expression of GR-regulated Genes
Our previous studies showed that the KDACi TSA inhibits both basal and GR-activated transcription of the MMTV promoter (22, 26). To determine whether these findings could be extended to cellular genes, we took a candidate approach, selecting known GR target genes that are activated in the presence of glucocorticoids in a variety of cell types (27). We exposed a mouse hepatoma line, Hepa-1c1c7, to the KDACi VPA in the presence and absence of Dex and measured expression of these genes by RT-qPCR. We chose VPA because it is clinically relevant and causes reproductive and metabolic side effects that suggest an impact on nuclear receptor signaling (13, 28, 29). Our analysis identified four genes at which VPA impaired Dex-activated gene expression as shown in Fig. 1A. Remarkably, three of these genes were not activated at all in the presence of Dex and VPA (Fig. 1A, compare VPA+Dex with Control). The same four genes were also activated by Dex in a mouse mammary adenocarcinoma-derived cell line (1470.2). As shown in Fig. 1B, cotreatment with Dex and VPA or the pan-KDACi TSA also resulted in impaired glucocorticoid activation.
FIGURE 1.
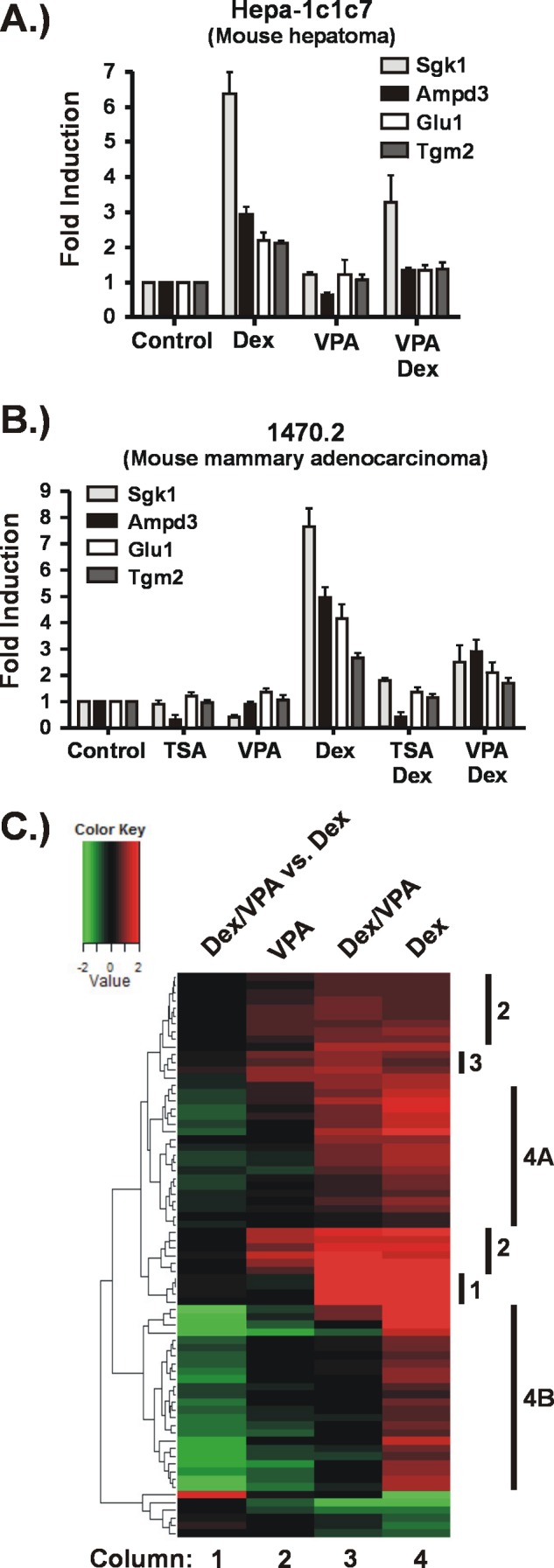
KDACis impair Dex-induced expression of GR target genes. Hepa-1c1c7 cells (A) or 1470.2 cells (B) were exposed to KDACis (VPA (5 mm) or TSA (200 nm)) for 5 h or to Dex (100 nm) for 4 h. For the combination treatments, the KDACis were added to the cells 1 h prior to addition of Dex, which continued for 4 h. A and B, analysis of selected GR target genes by RT-qPCR. The results are represented as -fold inductions relative to the control (untreated). C, effect of VPA on the GR-regulated transcriptome. Cellular RNA was processed for hybridization to microarrays (Affymetrix Mouse GeneChip ST 1.0). Genes whose expression was significantly changed relative to untreated cells were identified for each treatment condition as described under “Experimental Procedures.” The heat map shows only the genes significantly changed by Dex treatment over three biological replicates. Columns 2–4 represent -fold change relative to untreated cells. Column 1 represents the -fold change of the combination treatment compared with Dex alone, indicating the impact of VPA on Dex-regulated gene expression. Denoted to the right of the heat map are groups of genes (1–3, 4A, and 4B) that show similar expression patterns in response to the various treatments and are described under “Results.”
The fact that we quickly identified four GR-activated genes sensitive to the presence of KDACi in two cell lines of different tissue origin suggested that KDACi may have a significant impact on the GR-regulated transcriptome. Thus, we carried out expression profiling of the Hepa-1c1c7 cells treated with VPA in the presence and absence of Dex. Genes that were significantly regulated by Dex alone were extracted from the expression profiling data to identify how VPA exposure affects their expression in the presence or absence of Dex as shown by the heat map in Fig. 1C. The expression patterns of the genes activated by Dex can be organized into four groups that are denoted to the right of the heat map. The first group contains genes that are activated by Dex but unaffected by VPA treatment. The fact that this group is rather small in number indicates that VPA exposure has profound effects on expression of GR target genes.
The second group contains two clusters of genes that are activated by either Dex or VPA. At these GR target genes, VPA alone mimics the effects of Dex. In the presence of both drugs, the extent to which these genes are activated is unchanged relative to Dex treatment alone. This is evidenced by the lack of green or red signal in the first column of the heat map, which is a comparison of the -fold change in the presence of VPA plus Dex versus Dex alone. The third group of genes is very small and contains those at which the combination treatment results in more activation than either drug alone. However, the increase in activation at these genes is rather small, indicating additive rather than synergistic effects.
The fourth group of genes is the largest: ∼50% of the total number of GR-activated genes. These genes show impaired activation by Dex when VPA is present as evidenced by the green signal in the Dex/VPA versus Dex column. These genes cluster into two main groups, indicated by 4A and 4B in Fig. 1C. Dex activation of genes in group 4A is moderately impaired by VPA. In addition, VPA treatment alone has either no effect or only a weak effect on expression of these genes, suggesting that the predominant effects of VPA are manifested upon activation of the GR. In contrast, Dex activation of the genes in group 4B is strongly impaired as indicated by the bright green signal in column 1. Although VPA treatment alone shows little to no effect on basal expression of some of these genes, others are strongly repressed by VPA alone (column 2). Thus, the effects of VPA on expression of these genes may be both independent of and dependent on GR activation.
Impact of VPA on GR-Hsp90 Interaction
The impaired activation of many GR target genes in the presence of VPA indicates that GR signaling is negatively impacted in some way. In the absence of ligand, GR is complexed with the molecular chaperone hsp90. This interaction is important to maintain the GR in a ligand binding-competent conformation. Acetylation of hsp90 is known to inhibit its ability to interact with GR and other steroid receptors, resulting in impaired binding to ligand and receptor degradation (5, 30). Hsp90 is deacetylated primarily by the Class IIB KDAC6. It is possible the GR-hsp90 interactions may be disrupted by TSA or VPA so we carried out co-immunoprecipitation experiments to measure this directly. Hepa-1c1c7 cells were exposed to TSA or VPA for up to 5 h. Cytosolic extracts were prepared and subjected to immunoprecipitation with GR antibody. In the presence of the pan-KDACi TSA, GR-hsp90 complexes were disrupted quickly in Hepa-1c1c7 cells (Fig. 2A). Accordingly, GR levels declined over 5-h TSA treatment. Interestingly, we performed the same experiment in the mouse mammary adenocarcinoma-derived cell line 1470.2 in which we had shown that TSA treatment impairs GR transactivation (Fig. 1B). In contrast to the Hepa-1c1c7 results, the GR-hsp90 complex remained intact over the 5 h of treatment, and GR levels did not decline (Fig. 2C). Thus, the impaired transactivation observed in the 1470.2 cells in the presence of TSA is not likely due to negative effects on GR processing. The difference in response of the two cell lines to TSA may reflect differences in the rate at which hsp90 is acetylated.
FIGURE 2.

Effect of TSA and VPA on GR-hsp90 interactions. Hepa-1c1c7 cells (A and B) or 1470.2 cells (C) were treated with TSA (200 nm) (A and C) or VPA (5 mm) (B) for up to 5 h. Cytosolic extracts were generated. Overall levels of GR and hsp90 were measured by Western blot. GR-hsp90 interaction was measured by immunoprecipitation (IP) with GR or GFP antibody (Ab) followed by Western blotting with antibodies to GR or hsp90. The results are representative of two to three independent experiments.
VPA treatment of Hepa-1c1c7 cells (Fig. 2B) did not affect the GR-hsp90 complex nor change GR levels. This is consistent with studies showing that VPA does not inhibit KDAC6 efficiently (8). Like hsp90, α-tubulin is primarily deacetylated by KDAC6. To confirm that VPA does not inhibit KDAC6 in our system, we treated the Hepa-1c1c7 cells with either TSA or VPA and used an acetyl-specific α-tubulin antibody to measure tubulin acetylation. We observed that VPA has no effect on α-tubulin acetylation, whereas TSA potently induces it (data not shown) in accordance with the fact that it is a pan-KDACi effective against both Class I KDACs and KDAC6. Altogether these results indicate that impaired GR transactivation is not due to a disruption in GR processing caused by KDACi treatment. This is supported by our findings that there are genes at which activation by Dex is unaffected by VPA treatment (Figs. 1C and 4C). A defect in GR processing that affects ligand binding would likely impact all GR target genes.
FIGURE 4.

The Class I-selective KDACis apicidin and VPA have very similar effects on GR-activated gene expression. Hepa-1c1c7 cells were treated with VPA (5 mm) or apicidin (0.25 μg/ml) for 5 h and Dex for 4 h. In the combination treatments, the KDACis were added 1 h prior to Dex with continued treatment for 4 h. RNA was isolated and subjected to RT-qPCR. A, the distinct chemical structures of VPA and apicidin. B, effects of apicidin on GR target genes found to have impaired transactivation in the presence of VPA. C, effects of apicidin on GR target genes at which transactivation was unaffected by VPA. The graphs shown are the summary of three to five independent experiments and represent -fold changes relative to untreated cells. Asterisks indicate a significant change in -fold induction under the combination treatments relative to Dex alone as determined using the paired t test. *, p ≤ 0.05; **, p ≤ 0.01. Error bars represent S.E.
VPA Exposure Blunts Dex-induced Transcription
The expression profiling results clearly show that impaired GR transactivation in the presence of VPA is gene-selective, suggesting that VPA impacts the genes at the transcriptional level. To address this question, we measured levels of nascent, unspliced transcripts from six GR target genes at which transactivation was impaired using exon-intron primer sets in RT-qPCR (Table 1). Hepa-1c1c7 cells were treated with Dex or a combination of Dex plus VPA for up to 4 h as shown in Fig. 3. The results clearly show that at four of the six genes transcriptional activation by Dex was blunted. Transcription from the Ampd3 and Tns1 genes barely increased at all with the combination treatment over a time frame in which Dex alone caused significant increases in transcription (Fig. 3, A and D). At the Tgm2 and St5 genes, transcription did increase in response to the combination treatment, but the increase was much smaller than that induced by Dex alone (Fig. 3, B and C). Interestingly, we did not observe significant differences in transcription from the Ror1 and H6pd genes between the combination treatment and Dex alone (Fig. 3, E and F). GR transactivation of these genes was significantly repressed by KDACi as determined by measurement of mRNA (see Fig. 4B). The lack of change in transcription indicates that expression of these genes is impacted by VPA at the post-transcriptional level.
FIGURE 3.

Effects of VPA on Dex-induced transcription. Hepa-1c1c7 cells were treated with either Dex (100 nm) or a combination of VPA (5 mm) plus Dex. In the latter, cells were exposed to VPA for 1 h prior to the addition of Dex. Cells were harvested at 30, 60, 120, and 240 min after Dex addition. RNA was isolated and subjected to RT-qPCR using intron-exon primers sets to measure nascent transcripts. The graphs represent -fold changes in nascent transcripts for each treatment time relative to levels in untreated cells for the Ampd3 (A), Tgm2 (B), St5 (C), Tns1 (D), Ror1 (E), and H6pd (F) genes. The results shown were derived from three to five independent experiments. Error bars represent S.E.
A Structurally Distinct KDACi Impacts GR Transactivation Similarly to VPA
It is clear from our results that VPA can impair GR-induced transcription, but the role of its KDAC-inhibiting properties is an open question. VPA is known to have direct effects on two classes of proteins: KDACs and enzymes that breakdown the neurotransmitter γ-aminobutyric acid (GABA). However, it also modulates the activity of several signaling pathways through unknown mechanisms that could involve KDACs or unrecognized targets (for a review, see Ref. 31). One approach to investigate the role of KDAC inhibition in the negative effect of VPA on GR transactivation is to determine whether a structurally distinct KDACi has similar effects. KDACis are a structurally diverse set of small molecules. VPA is a simple aliphatic acid, whereas apicidin is a cyclic tetrapeptide (Fig. 4A). They are both Class I-selective KDACis, and their vast structural differences are likely to cause distinct off-target effects. Thus, if VPA and apicidin have distinct effects on GR target genes, then the impaired GR transactivation caused by VPA exposure may not be mediated through KDAC inhibition.
We treated Hepa-1c1c7 cells with either VPA or apicidin in the presence or absence of Dex and assayed their effects on two groups of genes. The first group consisted of 13 genes at which Dex induced mRNA levels at least 2-fold and VPA impaired Dex-induced gene expression. Apicidin treatment had remarkably similar effects on these genes when compared with VPA. Results from five such genes are shown in Fig. 4B. In the absence of Dex, both apicidin and VPA had modest repressive effects on four of the five genes. In the presence of Dex, both apicidin and VPA significantly impaired activation of all genes by Dex. The second group of genes examined consisted of four genes at which VPA had no significant effect in the expression profiling experiment. Fig. 4C shows that both apicidin and VPA had little effect on the expression of these genes. Importantly, neither significantly impaired GR transactivation as determined by statistical analysis. Altogether apicidin and VPA had similar effects on GR transactivation across 17 GR target genes, strongly suggesting that the impairment of GR transactivation we observed is mediated through the KDAC-inhibiting activity of these small molecules.
VPA Induces Histone H3 Acetylation at GRE Regions of Target Genes
Histone acetylation has been found to be very dynamic in active regions of the genome, indicating that both KATs and KDACs are present and active (for a review, see Ref. 32). This is consistent with more recent ChIP-sequencing studies that showed that KDACs are enriched at active or potentially active genes along with KATs (33–35). Chromatin immunoprecipitation was performed to determine whether short term VPA treatment increases histone H3 acetylation in the GR binding regions of six GR-activated genes. (Chromosomal sequence positions and locations of these regions relative to the target gene are shown in Table 2.)
TABLE 2.
Positions of GR binding regions and location relative to GR target gene
dGRE, distal GRE; pGRE, proximal GRE; chr, chromosome.
| Gene | Positiona | Location |
|---|---|---|
| Tns1 | chr1:73,921,071–73,921,546 | Intronic GRE |
| Tsc22d3 | chrX:135,884,061–135,884,464 | 1.6 kbp downstream of gene |
| Sdpr | chr1:51,233,235–51,233,746 | 350 bp upstream of Exon1 |
| Sgk1 dGRE | chr10:21,694,174–21,694,484 | 5.2 kbp downstream of gene |
| Sgk1 pGRE | chr10:21,682,962–21,683,307 | 1 kbp upstream of gene |
| Zfp36 | chr7:28,084,970–28,085,703 | 200 bp downstream of gene |
| Lcn2 | chr2:32,210,118–32,210,462 | 500 bp upstream of Exon1 |
a Sequence positions were derived from the UCSC Genome Browser, mouse genome build February 2006.
Fig. 5 shows that VPA induces histone H3 acetylation at five of seven GR binding regions examined. Significant Dex-induced GR binding was confirmed by ChIP assay (data not shown). The Tns1, Tsc22d3, Sdpr, and Sgk1 genes are in the group of VPA-impaired GR target genes (Figs. 1A, 6D, and 8E). As shown in Fig. 5, A and B, VPA increased the average level of H3 acetylation at GREs in all of these genes. Interestingly, two GREs in the Sgk1 gene showed different responses to VPA (Fig. 5B). Histone H3 acetylation was unaffected by VPA at the proximal GRE in the Sgk1 promoter, whereas it was increased at the distal GRE found downstream of the gene. This finding in particular shows that the VPA-induced changes in H3 acetylation are not due to nonspecific inhibition of KDACs in the nucleoplasm but reflect changes in activity of KDACs present in specific gene regions. Finally, we tested two genes at which GR transactivation was unaffected by VPA, Zfp36 and Lcn2 (Fig. 4C). Fig. 5C shows that VPA increased H3 acetylation at one but not the other. These results show that histone acetylation in the GR binding regions of most of these genes is highly dynamic, strongly indicating the presence of active KDACs.
FIGURE 5.
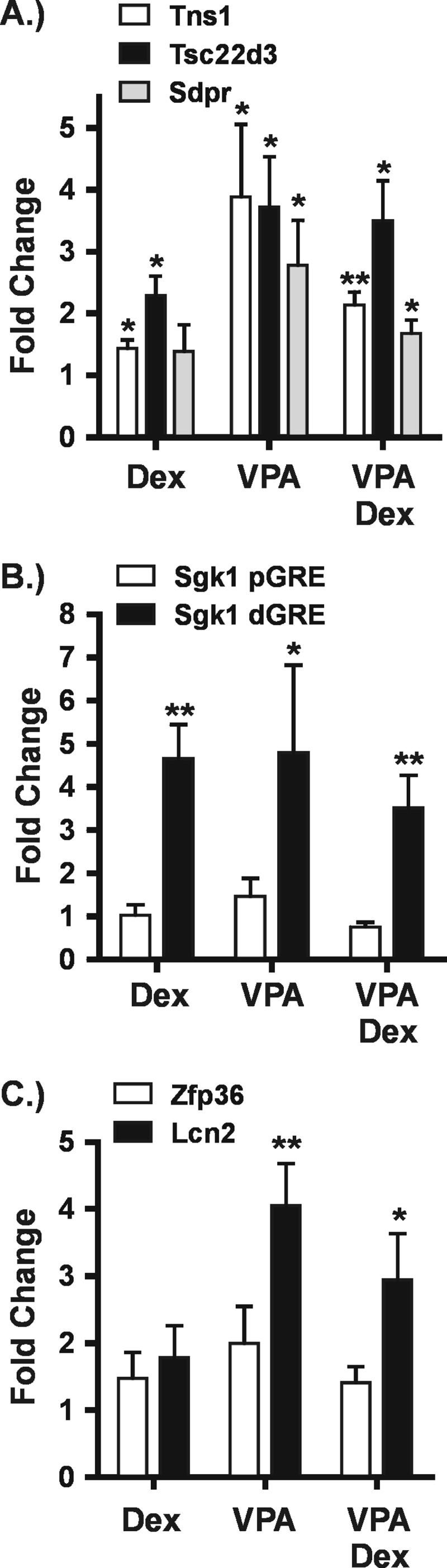
Histone H3 acetylation is sensitive to VPA treatment at GR binding regions. Hepa-1c1c7 cells were treated with Dex (100 nm) for 1 h, VPA (5 mm) for 2 h, or a combination of VPA (5 mm) plus Dex. In the latter, cells were exposed to VPA for 1 h prior to the addition of Dex for an additional hour. Chromatin immunoprecipitation was performed with an antibody against acetylated histone H3. The graphs show -fold changes in the level of histone H3 acetylation for each of the treatments relative to untreated and are a summary of three to four independent experiments. A, results for VPA-impaired GR target genes Tns1, Tsc22d3, and Sdpr. B, results for two GREs in the VPA-impaired Sgk1 gene. C, results for the VPA-unaffected genes Zfp36 and Lcn2. *, p ≤ 0.05; **, p ≤ 0.01. Error bars represent S.E.
FIGURE 6.

KDAC1 depletion fully mimics the effect of VPA at seven GR target genes. A and C, Hepa-1c1c7 cells were transfected with control (Ctrl) or KDAC1 siRNAs as described under “Experimental Procedures.” Forty eight hours after transfection, cells were treated with or without Dex (100 nm) for 4 h. RNA was isolated from cells and subjected to RT-qPCR. The graphs are a summary of four to five independent experiments and show -fold inductions in the presence of Dex relative to the corresponding untreated control for cells transfected with either control or KDAC1 siRNAs. Asterisks denote significant changes between Dex-treated cells transfected with KDAC1 siRNA relative to control siRNA. B and D, Hepa-1c1c7 cells were treated as described in the legend to Fig. 1. The graphs summarize the results of three to five independent experiments and show -fold inductions for each treatment condition relative to control, untreated cells. Asterisks represent significant changes between cells treated with Dex alone and cells treated with Dex plus VPA. *, p ≤ 0.05; **, p ≤ 0.01. Error bars represent S.E.
FIGURE 8.

Multiple KDACs cooperate in transactivation of some GR target genes. A–D, Hepa-1c1c7 cells were transfected with siRNAs and treated as described in the legend to Fig. 6. The graphs shown are a summary of four to five independent experiments and show -fold inductions in the presence of Dex relative to the corresponding untreated control for cells transfected with control or KDAC1 siRNAs (A and B) or with control or KDAC1 plus KDAC2 siRNAs (C and D). Asterisks denote significant changes between Dex-treated cells transfected with siRNAs targeted against KDACs relative to control siRNA. E, Hepa-1c1c7 cells were treated as described in the legend to Fig. 4. The graph shows a summary of three to five independent experiments and represents -fold changes relative to untreated cells. Asterisks indicate a significant change under the combination treatments relative to Dex alone as determined using the paired t test. *, p ≤ 0.05; **, p ≤ 0.01. Error bars represent S.E.
Interestingly, Dex treatment significantly increased H3 acetylation at only three of seven GREs, and with the exception of the distal Sgk1 GRE, the changes were smaller than those induced by VPA. Cotreatment with Dex and VPA did not result in higher levels of H3 acetylation than were observed with VPA alone. Steroid receptors are well established to recruit KATs to target genes. Our findings suggest that histones may not be their primary substrates in these cases.
Depletion of KDAC1 Impairs GR Transactivation at the Majority of KDACi-impaired Genes
A role for Class I KDACs in GR transactivation is implicated by the above results because VPA and apicidin are Class I-selective KDACis. To test this directly, we performed siRNA-mediated depletions of all the Class I KDACs individually. We then assayed their effects on GR transactivation at 13 target genes impaired by VPA treatment as well as four target genes that were unaffected. KDAC1 depletion (Fig. 6A, inset) had the most significant effect, and the responses of the 17 target genes could be classified into three groups.
In the first group, KDAC1 depletion fully mimicked the degree of impairment in GR transactivation observed with VPA treatment as shown in Fig. 6. This group encompasses over half of the 13 KDACi-impaired GR target genes examined. Relative to the amount of Dex activation observed in the presence of a control siRNA, -fold inductions by Dex were significantly impaired in the presence of KDAC1 siRNA (Fig. 6, A and C). For each of these genes, the magnitude of the impairment (control versus KDAC1 siRNA) was similar to that seen when comparing -fold induction by Dex in the presence and absence of VPA (compare VPA/Dex with Dex conditions in Fig. 6, B and D). In contrast, depletion of KDACs 2, 3, and 8 had no significant effect on Dex activation of any of these genes (data not shown). Thus, KDAC1 is likely to be sufficient in facilitating transactivation at these GR target genes.
At the second group of genes, KDAC1 depletion partially impaired GR transactivation relative to VPA. Depletion of KDAC1 significantly reduced the magnitude of Dex induction of the Fam107a and Ampd3 genes (Fig. 7A) but to a smaller extent than observed with VPA (Fig. 7D). In addition, like the genes described above, individual depletion of the other Class I KDACs had no effect on GR transactivation at these genes (data not shown). KDACs 1 and 2 are known to form homo- or heterodimers and are reported to be present in the same complexes (36). Thus, we considered the possibility that both KDACs 1 and 2 are required for efficient transactivation of these promoters. Fig. 7C shows that simultaneous depletion of KDACs 1 and 2 (Fig. 7B) severely impaired transactivation of the Fam107A gene, indicating that these KDACs cooperate to facilitate GR action in this context. In contrast, the co-depletion did not recapitulate the strong effects of either VPA or apicidin (Figs. 4B and 7D) on transactivation of the Ampd3 gene. This result suggests that other KDACs cooperate with KDAC1 to facilitate transactivation in this gene context.
FIGURE 7.

KDAC1 and KDAC2 cooperate in transactivation of the Fam107a gene. A–C, Hepa1-c1c7 cells were transfected with siRNAs and treated as described in the legend to Fig. 6. B, Western blot of KDAC1 and KDAC2 in cells that had been transfected with control (Ctrl) siRNA or with siRNAs against both KDAC1 and KDAC2. GAPDH levels are shown as a loading control. The graphs in A and C are a summary of four to five independent experiments and show -fold inductions in the presence of Dex relative to the corresponding untreated control for cells transfected with control or KDAC1 siRNAs (A) or with control or KDAC1 plus KDAC2 siRNAs (C). Asterisks denote significant changes between Dex-treated cells transfected with siRNAs targeted against KDACs relative to control siRNA. D, Hepa-1c1c7 cells were treated as described in the legend to Fig. 1. The graphs summarize the results of three to five independent experiments and show -fold inductions for each treatment condition relative to control, untreated cells. Asterisks represent significant changes between cells treated with Dex alone and cells treated with Dex plus VPA. *, p ≤ 0.05; **, p ≤ 0.01. Error bars represent S.E.
The third group of genes consists of those resistant to KDAC1 depletion. Of 13 genes at which VPA impaired GR transactivation, four fell into this group as shown in Fig. 8A. Individual depletion of the other Class I KDACs also had no effect on these genes (data not shown). In addition, three of the four GR target genes at which VPA had no effect on transactivation (Fig. 4C) were also resistant to KDAC1 depletion as shown in Fig. 8B. The fourth, Pfkfb3, showed a small but significant inhibition of GR transactivation upon KDAC1 depletion. This is likely to be an indirect effect of KDAC1 depletion for two reasons. First, co-depletion of KDACs 1 and 2 had no significant effect on this gene (Fig. 8D), and second, exposure to neither VPA nor apicidin had a significant effect on transactivation of Pfkfb3 (Fig. 4C).
Because GR transactivation of the Fam107a gene was dependent on both KDACs 1 and 2, we carried out simultaneous depletion of these proteins to examine their effects on the group of genes resistant to KDAC1 depletion. We observed partially impaired transactivation of three of these genes. The Nfkbia gene showed a very small but significant inhibition of GR transactivation upon co-depletion of KDACs 1 and 2 (Fig. 8D). This is again likely to be an indirect effect because KDACis did not affect transactivation of this gene (Fig. 4C). Dex activation of Sdpr and Slc35d1 (Fig. 8C) was also significantly inhibited upon co-depletion of KDACs 1 and 2. Because the magnitude of the impairment was small, it is possible that this represents an indirect effect of the co-depletion. However, unlike Nfkbia, GR transactivation of the Sdpr and Slc35d1 genes was potently suppressed by both VPA and apicidin (Fig. 8E), indicating that other KDACs in addition to KDACs 1 and 2 cooperate to directly facilitate transactivation by GR. Two genes (Ror1 and H6pd) at which GR transactivation was strongly impaired by the two KDACis were resistant to individual and simultaneous depletion of KDACs 1 and 2 (Fig. 8, A and C, and data not shown). Interestingly, these genes are also those at which Dex-induced transcription was unaffected by the presence of VPA (Fig. 3, E and F).
DISCUSSION
The action of KDACs is generally thought to be inhibitory to transcription, including that activated by steroid receptors. Our study shows that exposure to KDACis has a major impact on the GR-regulated transcriptome. Specifically, we demonstrate that Class I KDAC activity enables efficient GR-induced transcription at a sizable fraction of GR-activated cellular target genes and establish that KDAC1 is a major player in facilitating GR transactivation.
Our expression profiling showed that VPA treatment inhibited GR-induced transactivation at about 50% of Dex-induced genes, raising the interesting possibility that KDACs may generally facilitate rather than impair GR transactivation. KDAC6 has been shown to facilitate GR signaling through deacetylation of hsp90. However, VPA treatment did not disrupt the GR-hsp90 interaction nor induce GR degradation, indicating that it does not cause global defects in GR processing. Analysis of nascent transcripts showed that VPA either prevents or blunts Dex-induced transcription at the majority of genes tested. Combined with a lack of effect on GR processing, this result strongly indicates that VPA impacts GR action primarily at the transcriptional level.
Interestingly, VPA had no significant effect on Dex-induced transcription at the Ror1 and H6pd genes, but it impaired the accumulation of mRNA, suggesting an effect on post-transcriptional processes. This is noteworthy because it is often assumed that short term changes in mRNA levels in response to drugs or hormones reflect changes in transcription. A large proteomics study reported that many proteins involved in RNA processing were acetylated (1), thereby raising the possibility that KDACs regulate gene expression at the post-transcriptional level. The Ror1 and H6pd genes are further set apart from the other GR target genes examined in that they were resistant to all the KDAC depletions performed, suggesting that a distinct combination of KDACs may influence post-transcriptional regulation of gene expression.
Our study clearly shows that VPA impairs Dex-induced transcription through its ability to inhibit KDACs. First, the responses of 17 GR-activated genes to the structurally distinct KDACi apicidin were remarkably similar to the responses elicited by VPA, suggesting that the effects of VPA are not likely to be mediated through non-KDAC targets. Second, siRNA-mediated depletion of KDACs impaired GR transactivation at many genes found to be sensitive to KDACi. Furthermore, the action of KDACs in facilitating GR transactivation is likely to be mediated directly at the target genes affected. Short term VPA treatment increased levels of histone H3 acetylation in the GRE regions of five of six GR target genes tested, indicating that Class I KDACs are present and active in these gene regions prior to GR binding. These KDACs could thus potentially interact with GR or deacetylate components of GR-assembled transcription complexes to facilitate transcription. This is consistent with the findings of Qiu and co-workers (24, 25, 37) who showed that KDACs 1 and 2 are both present at the MMTV promoter prior to GR binding. KDAC1 and GR interact at the MMTV promoter but do not cycle on and off with the same kinetics (25), suggesting that their association with the promoter is not interdependent. In addition, Chng et al. (38) showed that KDACs 1–3 are associated with androgen receptor binding regions of target genes in the presence and absence of androgen. Global analyses of Class I KDAC localization have consistently shown enrichment in active or transcriptionally competent regions of the genome (33–35).
The siRNA experiments established that KDAC1 plays a major role in facilitating efficient GR transactivation. For seven of 13 genes at which VPA impaired GR transactivation, KDAC1 depletion fully mimicked the effects of VPA, indicating that it is the predominant KDAC involved. At an additional gene (Fam107a), co-depletion of KDACs1 and 2 was required to fully mimic the effects of VPA or apicidin. For three of 13 genes at which VPA or apicidin significantly impaired GR transactivation (Ampd3, Sdpr, and Slc35d1), depletion of either KDAC1 or both KDACs 1 and 2 caused a partial impairment of GR transactivation. Thus, in these gene contexts, it is possible that KDACs 1 and 2 directly facilitate GR action but do so in cooperation with at least one other KDAC. We did not attempt co-depletion of KDACs 1–3 because of potential deleterious effects on cell survival. Altogether our study shows that KDAC1, acting alone or sometimes in concert with KDAC2, makes a positive and significant contribution to GR-induced transcription in a variety of gene contexts.
KDACs 1 and 2 have been found in the same complexes, specifically the Sin3, co-repressor of RE1 silencing transcription factor (CoREST), and nucleosome remodeling and histone deacetylation (NURD) complexes (36), so it is somewhat surprising that depletion of either KDAC1 or KDAC2 resulted in different outcomes. Over 50% of the 13 selected genes at which KDACis impair GR transactivation were dependent on KDAC1 alone. It is possible that KDAC1 is expressed at a significantly higher level than KDAC2 in our cell line and thus would predominate in such complexes. However, in K562 cells, KDACs 1 and 2 show differential association with the above-mentioned complexes (9) and colocalize with distinct sets of transcriptional regulatory proteins at genomic loci containing active genes (35). These studies suggest that the functions of KDACs 1 and 2 are not entirely redundant. In addition to the large group of genes at which KDAC1 alone facilitates GR transactivation, we identified a smaller group of genes at which both KDACs 1 and 2 contribute, suggesting that more than one KDAC-containing complex cooperates with GR to allow efficient activation of transcription.
Class I KDACs have been shown to facilitate gene expression activated by other steroid receptors. Welsbie et al. (39) determined that KDACs 1 and 3 were largely responsible for promoting androgen receptor transactivation in LNCaP cells, whereas KDAC3 has been shown to potentiate mineralocorticoid receptor transactivation in HEK293 cells (40). Several studies suggest a positive relationship between KDAC activity and promoter association of RNA polymerase (pol) II. KDACi did not inhibit the association of agonist-bound androgen receptor with the prostate-specific antigen enhancer, but its ability to recruit RNA pol II to the prostate-specific antigen gene was dramatically impaired (39). This is strikingly reminiscent of reports on the MMTV promoter. In the presence of mutant KDAC1 or KDACi, GR is able to associate with the promoter and induce chromatin remodeling (22, 25). The block to transactivation was in the activation step during which RNA pol II is recruited. Accordingly, TSA treatment resulted in loss of RNA pol II from the promoter (26). This relationship may apply to other signaling-activated transcription factors. In the presence of TSA, IL3-activated Stat5 associated efficiently with target promoters but was unable to recruit RNA pol II (41).
KDACs may facilitate glucocorticoid-induced transcription by removing inhibitory acetylation from transcriptional regulatory proteins. GR acetylation has been shown to impair (42) or have no effect on DNA binding (43), suggesting that its impact may be gene-specific. Acetylation of steroid receptor coactivator 3 inhibits its ability to interact with the estrogen receptor (44). In fact, androgen receptor fails to recruit steroid receptor coactivator 1 and p300 to the prostate-specific antigen enhancer in the presence of TSA (39). Finally, a major proteomics study found that components of KAT, lysine methyltransferase, and chromatin remodeling complexes known to be recruited to steroid-activated genes are acetylated at multiple sites (1). In these cases, little is known about the functional impact of acetylation. It is clearly possible that the targets of KDAC action may be varied and dependent on context, consistent with the gene-selective effects we have documented.
Long held models of steroid receptor transactivation predict that KDAC inhibition should increase the magnitude of target gene responses because KATs, which directly facilitate transactivation through histone acetylation, would be unopposed. The expression profiling identified only a few GR target genes with moderately increased activation in the presence of Dex and VPA. The fact that a large fraction of GR target genes showed the opposite effect as a result of KDACi exposure or KDAC depletion clearly demonstrates that the current models for GR transactivation are incomplete probably because they focus on the stimulatory role of histone acetylation and do not account for acetylation of non-histone transcriptional regulatory proteins.
Our findings also have ramifications for the clinical use of KDACis, which are now used to treat mood and seizure disorders as well as cancer but are also being evaluated to treat a wide variety of disorders, including neurological and inflammatory diseases (45). VPA has a striking impact on the GR-activated transcriptional program that is not limited to a particular cell type. Few GR-activated genes were completely unaffected by VPA; the majority were impacted in two ways. First, treatment with VPA alone activated a subset of GR target genes. Second, the most prevalent effect of VPA was the moderate to severe impairment of Dex-induced activation. Overall, these effects of VPA exposure have high potential to either mimic some of the effects of glucocorticoids in their absence or change the overall cellular response in their presence. Because human exposure to KDACis is likely to increase, it is important to understand how these drugs affect endocrine pathways.
Acknowledgments
For assistance with expression profiling, we thank the staff of the Genomics Shared Service, which is jointly supported by the Southwest Environmental Health Science Center (National Institutes of Health Grant ES006694) and the Arizona Cancer Center. We are also grateful to Dr. Dean Billheimer (University of Arizona) for providing guidance with the statistical analyses of the RT-qPCR results. Finally, we thank Drs. Gordon Hager and Sam John (National Cancer Institute) for providing us with the positions of GR binding sites in Hepa-1c1c7 cells as determined by ChIP-sequencing.
This work was supported by start-up funds (to C. L. S.) and by National Science Foundation Grant MCB-1122088 (to C. L. S.).
- KAT
- lysine acetyltransferase
- KDAC
- lysine deacetylase
- KDACi
- lysine deacetylase inhibitor
- GR
- glucocorticoid receptor
- VPA
- valproic acid
- hsp90
- heat shock protein 90
- MMTV
- mouse mammary tumor virus
- Dex
- dexamethasone
- HDAC
- histone deacetylase
- TSA
- trichostatin A
- RT-qPCR
- quantitative real time polymerase chain reaction
- Ct
- threshold cycle
- pol
- polymerase
- GRE
- glucocorticoid response element.
REFERENCES
- 1. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 2. Kim S. C., Sprung R., Chen Y., Xu Y., Ball H., Pei J., Cheng T., Kho Y., Xiao H., Xiao L., Grishin N. V., White M., Yang X. J., Zhao Y. (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618 [DOI] [PubMed] [Google Scholar]
- 3. Haberland M., Montgomery R. L., Olson E. N. (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lahm A., Paolini C., Pallaoro M., Nardi M. C., Jones P., Neddermann P., Sambucini S., Bottomley M. J., Lo Surdo P., Carfí A., Koch U., De Francesco R., Steinkühler C., Gallinari P. (2007) Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 104, 17335–17340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kovacs J. J., Murphy P. J., Gaillard S., Zhao X., Wu J. T., Nicchitta C. V., Yoshida M., Toft D. O., Pratt W. B., Yao T. P. (2005) HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 18, 601–607 [DOI] [PubMed] [Google Scholar]
- 6. Zhang Y., Kwon S., Yamaguchi T., Cubizolles F., Rousseaux S., Kneissel M., Cao C., Li N., Cheng H. L., Chua K., Lombard D., Mizeracki A., Matthias G., Alt F. W., Khochbin S., Matthias P. (2008) Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell. Biol. 28, 1688–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Houtkooper R. H., Pirinen E., Auwerx J. (2012) Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 13, 225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Khan N., Jeffers M., Kumar S., Hackett C., Boldog F., Khramtsov N., Qian X., Mills E., Berghs S. C., Carey N., Finn P. W., Collins L. S., Tumber A., Ritchie J. W., Jensen P. B., Lichenstein H. S., Sehested M. (2008) Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 409, 581–589 [DOI] [PubMed] [Google Scholar]
- 9. Bantscheff M., Hopf C., Savitski M. M., Dittmann A., Grandi P., Michon A. M., Schlegl J., Abraham Y., Becher I., Bergamini G., Boesche M., Delling M., Dümpelfeld B., Eberhard D., Huthmacher C., Mathieson T., Poeckel D., Reader V., Strunk K., Sweetman G., Kruse U., Neubauer G., Ramsden N. G., Drewes G. (2011) Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 29, 255–265 [DOI] [PubMed] [Google Scholar]
- 10. Spiegel S., Milstien S., Grant S. (2012) Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene 31, 537–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chuang D. M., Leng Y., Marinova Z., Kim H. J., Chiu C. T. (2009) Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 32, 591–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Isojärvi J. I., Taubøll E., Herzog A. G. (2005) Effect of antiepileptic drugs on reproductive endocrine function in individuals with epilepsy. CNS Drugs 19, 207–223 [DOI] [PubMed] [Google Scholar]
- 13. Luef G., Rauchenzauner M., Waldmann M., Sturm W., Sandhofer A., Seppi K., Trinka E., Unterberger I., Ebenbichler C. F., Joannidis M., Walser G., Bauer G., Hoppichler F., Lechleitner M. (2009) Non-alcoholic fatty liver disease (NAFLD), insulin resistance and lipid profile in antiepileptic drug treatment. Epilepsy Res. 86, 42–47 [DOI] [PubMed] [Google Scholar]
- 14. Robyr D., Wolffe A. P., Wahli W. (2000) Nuclear hormone receptor coregulators in action: diversity for shared tasks. Mol. Endocrinol. 14, 329–347 [DOI] [PubMed] [Google Scholar]
- 15. Chen J. D., Li H. (1998) Coactivation and corepression in transcriptional regulation by steroid/nuclear hormone receptors. Crit. Rev. Eukaryot. Gene Expr. 8, 169–190 [DOI] [PubMed] [Google Scholar]
- 16. Jackson T. A., Richer J. K., Bain D. L., Takimoto G. S., Tung L., Horwitz K. B. (1997) The partial agonist activity of antagonist-occupied steroid receptors is controlled by a novel hinge domain-binding coactivator L7/SPA and the corepressors N-CoR or SMRT. Mol. Endocrinol. 11, 693–705 [DOI] [PubMed] [Google Scholar]
- 17. Wagner B. L., Norris J. D., Knotts T. A., Weigel N. L., McDonnell D. P. (1998) The nuclear corepressors NCoR and SMRT are key regulators of both ligand- and 8-bromo-cyclic AMP-dependent transcriptional activity of the human progesterone receptor. Mol. Cell. Biol. 18, 1369–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lavinsky R. M., Jepsen K., Heinzel T., Torchia J., Mullen T. M., Schiff R., Del-Rio A. L., Ricote M., Ngo S., Gemsch J., Hilsenbeck S. G., Osborne C. K., Glass C. K., Rosenfeld M. G., Rose D. W. (1998) Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proc. Natl. Acad. Sci. U.S.A. 95, 2920–2925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Plesko M. M., Hargrove J. L., Granner D. K., Chalkley R. (1983) Inhibition by sodium butyrate of enzyme induction by glucocorticoids and dibutyryl cyclic AMP. A role for the rapid form of histone acetylation. J. Biol. Chem. 258, 13738–13744 [PubMed] [Google Scholar]
- 20. Bresnick E. H., John S., Berard D. S., LeFebvre P., Hager G. L. (1990) Glucocorticoid receptor-dependent disruption of a specific nucleosome on the mouse mammary tumor virus promoter is prevented by sodium butyrate. Proc. Natl. Acad. Sci. U.S.A. 87, 3977–3981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tichonicky L., Santana-Calderon M. A., Defer N., Giesen E. M., Beck G., Kruh J. (1981) Selective inhibition by sodium butyrate of glucocorticoid-induced tyrosine aminotransferase synthesis in hepatoma tissue-cultured cells. Eur. J. Biochem. 120, 427–433 [DOI] [PubMed] [Google Scholar]
- 22. Mulholland N. M., Soeth E., Smith C. L. (2003) Inhibition of MMTV transcription by HDAC inhibitors occurs independent of changes in chromatin remodeling and increased histone acetylation. Oncogene 22, 4807–4818 [DOI] [PubMed] [Google Scholar]
- 23. Sheldon L. A., Becker M., Smith C. L. (2001) Steroid hormone receptor-mediated histone deacetylation and transcription at the mouse mammary tumor virus promoter. J. Biol. Chem. 276, 32423–32426 [DOI] [PubMed] [Google Scholar]
- 24. Qiu Y., Zhao Y., Becker M., John S., Parekh B. S., Huang S., Hendarwanto A., Martinez E. D., Chen Y., Lu H., Adkins N. L., Stavreva D. A., Wiench M., Georgel P. T., Schiltz R. L., Hager G. L. (2006) HDAC1 acetylation is linked to progressive modulation of steroid receptor-induced gene transcription. Mol. Cell 22, 669–679 [DOI] [PubMed] [Google Scholar]
- 25. Qiu Y., Stavreva D. A., Luo Y., Indrawan A., Chang M., Hager G. L. (2011) Dynamic interaction of HDAC1 with a glucocorticoid receptor-regulated gene is modulated by the activity state of the promoter. J. Biol. Chem. 286, 7641–7647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee S. C., Magklara A., Smith C. L. (2011) HDAC activity is required for efficient core promoter function at the mouse mammary tumor virus promoter. J. Biomed. Biotechnol. 2011, 416905, dx..org/ 10.1155/2011/416905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. John S., Johnson T. A., Sung M. H., Biddie S. C., Trump S., Koch-Paiz C. A., Davis S. R., Walker R., Meltzer P. S., Hager G. L. (2009) Kinetic complexity of the global response to glucocorticoid receptor action. Endocrinology 150, 1766–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Isojärvi J. (2008) Disorders of reproduction in patients with epilepsy: antiepileptic drug related mechanisms. Seizure 17, 111–119 [DOI] [PubMed] [Google Scholar]
- 29. Pylvänen V., Pakarinen A., Knip M., Isojärvi J. (2006) Insulin-related metabolic changes during treatment with valproate in patients with epilepsy. Epilepsy Behav. 8, 643–648 [DOI] [PubMed] [Google Scholar]
- 30. Murphy P. J., Morishima Y., Kovacs J. J., Yao T. P., Pratt W. B. (2005) Regulation of the dynamics of hsp90 action on the glucocorticoid receptor by acetylation/deacetylation of the chaperone. J. Biol. Chem. 280, 33792–33799 [DOI] [PubMed] [Google Scholar]
- 31. Terbach N., Williams R. S. (2009) Structure-function studies for the panacea, valproic acid. Biochem. Soc. Trans. 37, 1126–1132 [DOI] [PubMed] [Google Scholar]
- 32. Clayton A. L., Hazzalin C. A., Mahadevan L. C. (2006) Enhanced histone acetylation and transcription: a dynamic perspective. Mol. Cell 23, 289–296 [DOI] [PubMed] [Google Scholar]
- 33. Wang Z., Zang C., Cui K., Schones D. E., Barski A., Peng W., Zhao K. (2009) Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138, 1019–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kidder B. L., Palmer S. (2012) HDAC1 regulates pluripotency and lineage specific transcriptional networks in embryonic and trophoblast stem cells. Nucleic Acids Res. 40, 2925–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ram O., Goren A., Amit I., Shoresh N., Yosef N., Ernst J., Kellis M., Gymrek M., Issner R., Coyne M., Durham T., Zhang X., Donaghey J., Epstein C. B., Regev A., Bernstein B. E. (2011) Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell 147, 1628–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang X. J., Seto E. (2008) The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9, 206–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Luo Y., Jian W., Stavreva D., Fu X., Hager G., Bungert J., Huang S., Qiu Y. (2009) Trans-regulation of histone deacetylase activities through acetylation. J. Biol. Chem. 284, 34901–34910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chng K. R., Chang C. W., Tan S. K., Yang C., Hong S. Z., Sng N. Y., Cheung E. (2012) A transcriptional repressor co-regulatory network governing androgen response in prostate cancers. EMBO J. 31, 2810–2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Welsbie D. S., Xu J., Chen Y., Borsu L., Scher H. I., Rosen N., Sawyers C. L. (2009) Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res. 69, 958–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee H. A., Lee D. Y., Cho H. M., Kim S. Y., Iwasaki Y., Kim I. (2013) Histone deacetylase inhibition attenuates transcriptional activity of mineralocorticoid receptor through its acetylation and prevents development of hypertension. Circ. Res. 112, 1004–1012 [DOI] [PubMed] [Google Scholar]
- 41. Rascle A., Johnston J. A., Amati B. (2003) Deacetylase activity is required for recruitment of the basal transcription machinery and transactivation by STAT5. Mol. Cell. Biol. 23, 4162–4173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nader N., Chrousos G. P., Kino T. (2009) Circadian rhythm transcription factor CLOCK regulates the transcriptional activity of the glucocorticoid receptor by acetylating its hinge region lysine cluster: potential physiological implications. FASEB J. 23, 1572–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ito K., Yamamura S., Essilfie-Quaye S., Cosio B., Ito M., Barnes P. J., Adcock I. M. (2006) Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-κB suppression. J. Exp. Med. 203, 7–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen H., Lin R. J., Xie W., Wilpitz D., Evans R. M. (1999) Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell 98, 675–686 [DOI] [PubMed] [Google Scholar]
- 45. Dinarello C. A., Fossati G., Mascagni P. (2011) Histone deacetylase inhibitors for treating a spectrum of diseases not related to cancer. Mol. Med. 17, 333–352 [DOI] [PMC free article] [PubMed] [Google Scholar]