

Background: Understanding of the prokaryotic N-end rule is incomplete with respect to generation of primary and secondary N-degrons.
Results: Proteomics analysis of ClpS-interacting proteins identified >100 new putative N-end rule substrates in Escherichia coli.
Conclusion: Both primary and secondary N-degrons are generated by limited endoproteolytic cleavage of native proteins.
Significance: A possible mechanism for the generation of N-end rule substrates is proposed.
Keywords: Adaptor Proteins, ATP-dependent Protease, Protein Degradation, Protein Processing, Proteomics, Aat, ClpA, ClpS, END Site, N-end Rule
Abstract
The N-end rule is a conserved mechanism found in Gram-negative bacteria and eukaryotes for marking proteins to be degraded by ATP-dependent proteases. Specific N-terminal amino acids (N-degrons) are sufficient to target a protein to the degradation machinery. In Escherichia coli, the adaptor ClpS binds an N-degron and delivers the protein to ClpAP for degradation. As ClpS recognizes N-terminal Phe, Trp, Tyr, and Leu, which are not found at the N terminus of proteins translated and processed by the canonical pathway, proteins must be post-translationally modified to expose an N-degron. One modification is catalyzed by Aat, an enzyme that adds leucine or phenylalanine to proteins with N-terminal lysine or arginine; however, such proteins are also not generated by the canonical protein synthesis pathway. Thus, the mechanisms producing N-degrons in proteins and the frequency of their occurrence largely remain a mystery. To address these issues, we used a ClpS affinity column to isolate interacting proteins from E. coli cell lysates under non-denaturing conditions. We identified more than 100 proteins that differentially bound to a column charged with wild-type ClpS and eluted with a peptide bearing an N-degron. Thirty-two of 37 determined N-terminal peptides had N-degrons. Most of the proteins were N-terminally truncated by endoproteases or exopeptidases, and many were further modified by Aat. The identities of the proteins point to possible physiological roles for the N-end rule in cell division, translation, transcription, and DNA replication and reveal widespread proteolytic processing of cellular proteins to generate N-end rule substrates.
Introduction
ATP-dependent proteolysis is an essential function carried out by all organisms to modulate the intracellular levels of functional proteins and to help maintain protein quality control. In Escherichia coli, the ATP-dependent proteases include ClpAP and ClpXP along with Lon protease, HslUV, and FtsH, each of which recognizes different chemical or structural features of proteins and targets specific proteins for degradation (1, 2). Clp proteases exist in an autoinhibited state with the active sites sequestered in an interior between the two stacked heptameric rings of ClpP (3, 4). To become fully functional, ClpP must form a complex with either of two ATP-dependent protein unfoldases, ClpA or ClpX, which bind protein substrates, unfold them, and translocate the unfolded substrate through the ATPase ring and into ClpP (5–11). Because ClpA and ClpX recognize substrates and deliver them to ClpP for degradation, they are considered “regulatory” particles, and befitting the role, ClpA and ClpX recognize different classes of proteins that vary according to short sequence motifs usually located near the N or C terminus of the substrate protein (2, 12). Substrate specificity is further amplified or modified by adapter proteins that selectively interact with either ClpA or ClpX. For example, whereas both ClpAP and ClpXP can recognize and degrade proteins co-translationally tagged with SsrA, the adapter SspB binds SsrA-tagged proteins with 8–10-fold higher affinity and preferentially delivers them to ClpXP (13). Another adapter, RssB, specifically targets the σ factor RpoS for degradation by ClpXP (14–16). ClpA on the other hand interacts with the adapter ClpS, which binds proteins bearing one of a small subset of N-terminal residues and delivers them to ClpAP for degradation (17–21).
The process by which the stability of a protein is linked to the identity of its N-terminal residue is referred to as the N-end rule degradation pathway (22). Residues are considered destabilizing if their presence at the N terminus shortens the half-life of the protein in vivo. In eukaryotic cells, the N-end rule targets proteins to the ubiquitination system, and the ubiquitinated proteins are degraded by the proteasome. In Gram-negative bacteria, N-end rule substrates are recognized by ClpS (17), which targets them directly to ClpAP. N-terminal residues that directly interact with ClpS are called primary destabilizing residues, and in E. coli, there are only four such residues: leucine, phenylalanine, tyrosine, and tryptophan (23). In addition, there are two secondary destabilizing residues, lysine and arginine, which are not directly recognized by ClpS but are modified by an amino acyltransferase (Aat)3 (24) that installs the primary destabilizing residue, leucine or phenylalanine, on the N terminus, enabling them to bind to ClpS. The binding site in ClpS has a deep hydrophobic pocket that accommodates the aromatic or hydrophobic side chain of the destabilizing N-terminal residue; two aspartate residues at the mouth of the pocket interact with the α-amino group and the amide nitrogen of the peptide bond between the first two residues (21, 25). ClpS, an 11-kDa monomer, forms a one-to-one complex with the N-domain of the ClpA subunit (18, 19). When a ClpS-substrate complex encounters the ClpA hexamer, the binding of the complex is enhanced by interaction of the flexible N-terminal peptide of ClpS with a site near or within the axial channel of the hexamer (26, 27). Because steric constraints allow only one ClpS N-terminal peptide to access the axial site, this interaction not only increases binding affinity of the complex but also ensures that only one substrate molecule is positioned for delivery through the axial channel of ClpA to ClpP.
Despite detailed knowledge of the mechanism of N-end rule substrate degradation by ClpSAP, the study of the N-end rule degradation pathway in E. coli has been limited by a general lack of data about in vivo substrates or the part played by the pathway in cellular physiology. Two confirmed substrates of the E. coli system are DNA protection during starvation (Dps) and putrescine aminotransferase (PATase) (20, 28). The N-degron for Dps, Leu at position 6 (28), is part of the primary amino acid sequence, and thus Dps is an Aat-independent substrate; how the truncated form of Dps with Leu6 at the N terminus is generated is not known. PATase is a novel Aat-dependent substrate; its modification has two unique features. Aat, which was previously thought to require an N-terminal lysine or arginine in the target protein, adds leucine to the initiating methionine of PATase (28). In addition, Aat adds multiple leucine residues rather than a single leucine residue. Methionine is not known to be a secondary destabilizing residue for any other proteins, and no other instances of polyleucylation by Aat have yet been reported.
Systematic identification of a broad set of substrates could provide valuable insight into potential regulatory roles of the N-end rule pathway. In this study, we isolated over 100 putative substrates of ClpS and the N-end rule pathway by immobilization on a ClpS affinity column and selective desorption of the proteins with a peptide bearing an N-degron. We report an extensive catalogue of ClpS-interacting proteins, which we propose are N-end rule substrates, and provide evidence that entrance to the N-end rule pathway is a multistep process for many of the proteins. N-terminal sequencing demonstrated that, with the exception of PATase, all substrates isolated required a prior proteolytic event to be generated. A large fraction was also dependent on the activity of the Leu/Phe-tRNA-protein transferase, Aat. Our data suggest that the N-end rule pathway in E. coli has regulatory roles in addition to contributing to protein quality control.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, and Growth Conditions
The E. coli K12 strains used in this study were derived from MG1655 (F− λ− ilvG− rfb-50 rph-1) and are summarized in supplemental Table S1. All bacteria were grown with shaking (200 rpm) at 37 °C in Luria-Bertani broth (LB) (KD Scientific).
Chemicals and Other Materials
Laboratory chemicals were purchased from Sigma-Aldrich unless otherwise noted. DNA oligonucleotides and PCR reagents were obtained from Invitrogen. Restriction enzymes and ligase used in cloning reactions were obtained from New England Biolabs. PCR products were checked by electrophoresis on 0.8% agarose gels stained with ethidium bromide.
Protein Purification and Quantification
GFP-SsrA (8) and LR-GFPVENUS (26) were expressed and purified as described. Wild-type ClpS protein was purified from MG1655 Δara ompT cells carrying a pBAD33-clpS plasmid (18). Expression of ClpS was induced for 3–5 h in 0.2–0.4% arabinose. Cells were harvested by centrifugation at 4000 × g for 30 min and lysed in a French pressure cell at 20,000 p.s.i. in 50 mm HEPES, 10% (v/v) glycerol, pH 7.5. Cell lysate was clarified by centrifugation at 20,000 × g for 30 min. DNA and proteins were precipitated in 0.05% PEI on ice for 30 min and collected by centrifugation. The resulting supernatant was loaded on a Q Sepharose column. ClpS was eluted from the column between 250 and 350 mm KCl. Fractions were pooled, and ClpS was precipitated in 50% saturated ammonium sulfate. Precipitates were dissolved in 50 mm HEPES, 10% glycerol, pH 7.5 and loaded on a Superdex 75 column. Fractions containing purified ClpS were tested for electrophoretic purity and pooled for use in these studies. Protein concentrations were measured by absorbance using estimated or experimentally determined extinction coefficients of purified proteins. A Bradford dye binding assay (Bio-Rad) was used for complex protein mixtures after calibration with known concentrations of a standard protein. Protein A fusions were induced in cells carrying the pSS101 plasmid (29). Cells were grown in the presence of 1 mm isopropyl β-d-1-thiogalactopyranoside in LB medium to an A600 of 0.5, collected by centrifugation, suspended in Tris-saline-Tween (50 mm Tris, pH 7.5, 150 mm NaCl, 0.05% Tween 20), and boiled for 20 min. Clarified lysates were added directly to an IgG-Sepharose column (GE Healthcare), and the protein was eluted at pH 3.2 according to previous methods (30).
Protein Pulldowns and Peptide Synthesis
FKTA-NH2 peptide was synthesized in house from Fmoc (N-(9-fluorenyl)methoxycarbonyl)-amino acids on an ABI431 peptide synthesizer following standard procedures and purified by reverse phase liquid chromatography (HPLC). Peptide purity was confirmed by HPLC and matrix-assisted laser desorption ionization (MALDI) mass spectrometry. Peptides were dissolved in 20 mm sodium phosphate containing 150 mm NaCl (PBS). ClpS was added to AminoLink Plus resin at a final concentration of 5–10 mg/ml and immobilized through the addition of 50 mm NaCNBH3 according to the manufacturer's instructions. Cross-linking was terminated, and unreacted aldehyde was blocked by the addition of 100 mm Tris, pH 7.2. The ClpS-charged resin was washed with PBS and stored at 4 °C. Immobilized ClpS columns (1-ml bed volume) were washed with 10 volumes of PBS at room temperature prior to use. E. coli cells were suspended at a ratio of 1 g of cells/10 ml of PBS and lysed by multiple passages through a French pressure cell at 20,000 p.s.i. in the presence of DNase I. Lysates were clarified by centrifugation at 15,000 rpm in an SS-34 rotor for 30 min. The lysate was passed through the immobilized ClpS column at a ratio of 250–750 mg of total cell protein/5–10 mg of immobilized ClpS. Unbound proteins were washed off the column with 20 bed volumes of PBS. ClpS bound proteins were eluded with a 0.5–1 mm concentration of the peptide FKTA-NH2. The resulting fractions were separated on 12% polyacrylamide gels and stained with Coomassie Brilliant Blue or PageBlue protein staining solution for mass spectrometry purposes.
Antibiotic Chase Experiments and Western Blotting
E. coli cells were grown overnight in LB and subcultured at 1:50 into LB containing antibiotics as required. For the chase experiments, cells were grown to an A600 of 0.6–0.8 in the presence of 1 mm isopropyl β-d-1-thiogalactopyranoside, and chloramphenicol was added to the culture at a final concentration of 50 μg/ml. Samples of 500 μl each were taken at 0, 10, 30, 60, 120, 180, and 240 min and either pelleted by centrifugation and suspended in 50 μl of SDS-PAGE sample buffer or precipitated in 10% TCA. The cell pellets were suspended in sample buffer and heated at 95 °C for 20 min; TCA precipitates were washed twice with cold 100% acetone and dissolved in 50 μl of SDS-PAGE sample buffer. After removal of insoluble material by centrifugation, aliquots of 5 μl were loaded on SDS-polyacrylamide gels. Proteins were transferred onto charged polyvinyl difluoride membranes in MOPS transfer buffer. Proteins were detected using a 1:5000 dilution of α-rabbit IgG conjugated to horseradish peroxidase (Amersham Biosciences). ECL reagent was used for detection according to the manufacturer's instructions (GE Healthcare). Bands were visualized on Kodak BioMax XAR film.
Two-dimensional Electrophoresis
Protein samples from pulldown experiments were precipitated in 10% TCA (final concentration) and washed two to five times with 100% acetone. Dried protein pellets were dissolved in 250 μl of two-dimensional gel electrophoresis rehydration buffer (8 m urea, 2% CHAPS, 50 mm dithiothreitol, 0.2% Bio-Lyte ampholytes). Readystrip immobilized pH gradient strips (pI range, 3–10 nonlinear) (Bio-Rad) were actively rehydrated for 12–24 h, and isoelectric focusing was performed at a maximum of 8000 V for a total of 25,000 V-h. The second dimension was performed using precast Criterion XT 12% Bis-Tris SDS-polyacrylamide gels (Bio-Rad) after which proteins were transferred to PVDF membranes. Protein bands were stained with dilute Coomassie Brilliant Blue prior to N-terminal sequencing.
Mass Spectrometry and N-terminal Sequencing
N-terminal sequences of stained bands on PVDF membranes were obtained on an Applied Biosciences Procise protein sequencer following standard procedures. For mass spectrometry, protein gels were stained with PageBlue protein staining solution (Fermentas). Bands were excised and destained with 100 mm NH4HCO3 in 50% (v/v) acetonitrile. In-gel digests were performed using l-1-tosylamido-2-phenylethyl chloromethyl ketone-treated trypsin (Sigma-Aldrich). Extracted peptides were dried in a speed vacuum and then dissolved in water containing 2% acetonitrile and 0.5% acetic acid. Aliquots were injected onto a 0.2 × 50-mm Magic C18AQ reverse phase column (Michrom Bioresources, Inc.) using the Paradigm MS4 HPLC instrument (Michrom Bioresources, Inc.). Peptides were separated at a flow rate of 2 μl/min followed by on-line analysis by tandem mass spectrometry using an LTQ ion trap mass spectrometer (Thermo Scientific) equipped with an ADVANCE CaptiveSpray ion source (Michrom Bioresources, Inc.). Peptides were eluted into the mass spectrometer using a linear gradient from 95% mobile phase A (2% acetonitrile, 0.5% acetic acid, 97.5% water) to 65% mobile phase B (10% water, 0.5% formic acid, 89.5% acetonitrile) over 20 min followed by 95% mobile phase B over 5 min. Peptides were detected in positive ion mode using a data-dependent method in which the nine most abundant ions detected in an initial survey scan were selected for MS/MS analysis. The raw data were converted into Mascot generic format using the Trans-Proteomic Pipeline. The transformed data were searched against the NCBI non-redundant protein database of predicted E. coli proteins. Probability-based Mascot scores were determined by a comparison of search results against estimated random match population and are reported as ∼10 × log10(p) where p is the absolute probability. Individual Mascot ion scores greater than 40 were considered to indicate identity or extensive homology (p < 0.05), and proteins with scores above this significance value were considered for inclusion by the criteria described in supplemental Table 2. MALDI mass spectrometry analysis was done on a Waters MALDI micro MX instrument with 1 μl of sample co-crystallized with 1 μl of a 20% solution of α-cyano-4-hydroxycinnamic acid in 50% acetonitrile, 1% trifluoroacetic acid on a stainless steel MALDI plate.
Spectral Counting and Relative Quantitation
Spectral counting was used to assess the relative enrichment of identified proteins. Statistical analysis was performed using the G test as described previously (31, 32). Briefly, the G statistic for each protein was calculated according to Equation 1,
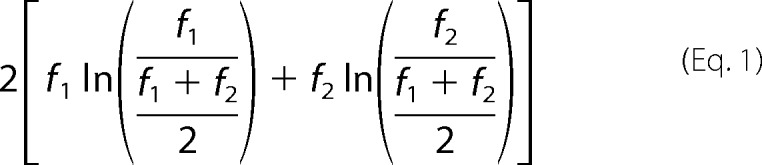 |
where f1 and f2 are the number of detected spectral counts for a specific protein in the wild-type and mutant samples, respectively. For proteins that were only detected in one sample or another, a spectral count of 1 was applied. The p value for each protein was then calculated as the probability of observing a random variable larger than G from the χ2 distribution with one degree of freedom.
RESULTS
Use of a ClpS Affinity Column for the Isolation of N-degron-containing Proteins
To optimize conditions for isolation and elution of N-degron-containing proteins on an immobilized ClpS column, we used modified green fluorescence proteins (GFPs) as model substrates. An N-degron-containing protein, LR-GFPVENUS, was applied to AminoLink resin with immobilized wild-type ClpS at a final concentration of 5–10 mg/ml. LR-GFPVENUS was quantitatively retained (not present in flow-through fractions) and remained bound after more than 10 column bed volumes of washing with buffer. When FKTA-NH2, a tetrapeptide bearing an N-degron, was added to the buffer at 1 mm final concentration, >85% of the LR-GFP was recovered after 1 bed volume had passed through the column (Fig. 1A). In contrast, no LR-GFPVENUS was retained by the column containing inactivated resin that had been cross-linked with Tris buffer and had no ClpS attached (Fig. 1B). To test the stringency for peptides with N-degrons in eluting specifically bound proteins, we compared the ability of two peptides, LRKGE and SLRKGE, to elute LR-GFPVENUS from the ClpS column. With 1 mm SLRKGE in the buffer, no bound LR-GFPVENUS was eluted from the column after several column volumes; however, when washing was continued with buffer containing 1 mm LRKGE, ∼86% of the immobilized LR-GFPVENUS was recovered (Fig. 1C). We also showed that retention of GFP by the column charged with wild-type ClpS column required an N-degron in the protein. His-GFP-SsrA, which has an N-terminal methionine, passed through the ClpS column and was recovered in >90% yield in the flow-through fraction; no additional protein was eluted when the column was washed with buffer containing FKTA-NH2 (data not shown). Finally, to confirm that the binding pocket on ClpS is required for the retention of the LR-GFPVENUS in the column, we made an affinity column with the binding pocket mutant of ClpS, ClpS-D35A,D36A (ClpSDD/AA), which has very low affinity for N-degrons and does not support degradation of N-end rule proteins in vivo (17), and tested binding of LR-GFPVENUS to the column. In contrast to the total retention seen with the wild-type ClpS column (Fig. 1A), >90% of the LR-GFPVENUS was present in the flow-through fraction from the ClpSDD/AA column, and no additional LR-GFPVENUS was recovered by the addition of peptide to the wash buffer (Fig. 1D).
FIGURE 1.

N-end rule substrate immobilization on a ClpS affinity column and elution with peptides possessing N-degrons. Small columns with 1-ml bed volumes of various AminoLink resins were equilibrated with PBS. Samples were loaded, and the columns were washed at ∼1 ml/min with PBS with and without addition of peptides. GFP-proteins were detected by fluorescence measurements. A, LR-GFPVENUS (1 mg) was applied to a column cross-linked with ClpS. The column was washed with buffer, and 1 mm FKTA was added to elute the bound LR-GFPVENUS. B, LR-GFPVENUS (1 mg) was applied to a control resin prepared by inactivation with Tris buffer in the absence of ClpS. Most of the protein (98%) was recovered in the flow-through fractions. No additional protein emerged after FKTA addition. C, LR-GFPVENUS (1 mg) was added to a ClpS column. The column was washed with buffer containing 1 mm SLRKGE followed by buffer containing 1 mm LRKGE. D, LR-GFPVENUS (1 mg) was applied to a column cross-linked to the variant ClpS-D35A,D36A. Most of the protein (>95%) was recovered in the flow-through fractions, and no additional protein was detected after adding FKTA.
Pulldown of E. coli Proteins on Columns Charged with Wild-type ClpS
Having established that the ClpS affinity column selectively binds a protein with an N-degron and that the protein can be specifically eluted with a competitive ligand, we applied this procedure to isolate N-degron-containing proteins from E. coli cell extracts. Stationary phase cultures of E. coli MG1655 ompT− grown in LB were lysed by passage through a French pressure cell, and the clarified lysates were loaded under non-denaturing conditions onto an AminoLink column with immobilized wild-type ClpS and a parallel column with immobilized ClpSDD/AA. Proteins were eluted from the columns with 1 mm FKTA-NH2 peptide. Eluted proteins were separated by SDS-PAGE and detected by staining with Coomassie Blue. Fig. 2A shows a side-by-side comparison of the protein profiles for the four major fractions from each column. Only trace amounts of proteins could be seen in the fractions from the column with ClpSDD/AA, whereas a large number of discrete protein bands were seen in the fractions from the column with wild-type ClpS. The banding profiles across the different fractions are similar, suggesting that all the proteins are bound and eluted with nearly the same efficiency and furthermore that they have relatively similar affinities and are bound by similar mechanisms. As an additional control, we used AminoLink gel that had been charged with Tris buffer but no ClpS. The Tris-inactivated resin alone only bound vanishingly small amounts of protein when subjected to the same wash and elution as the ClpS-immobilized columns (Fig. 2B). Any protein shown to interact with the resin in this manner was excluded from our list of putative ClpS-interacting proteins. Proteins specifically retained by the wild-type ClpS column that met our statistical criteria are summarized in supplemental Table S2. Previously identified highly abundant N-end rule substrates in E. coli Dps and PATase were identified in our wild-type ClpS pulldown fractions as well as more than 100 additional proteins not previously known to have any association with ClpS or the N-end rule degradation pathway.
FIGURE 2.

Differential capture of E. coli cell proteins on a wild-type ClpS affinity column. A, many E. coli proteins bind to wild-type ClpS and not to ClpSDD/AA. Affinity resins were prepared with either wild-type ClpS or mutated ClpS in which Asp35 and Asp36 were changed to alanine. Extracts of cells harvested during stationary phase were clarified, and equal portions were loaded onto columns (1-ml bed volume) with either wild-type or mutated ClpS. The columns were washed with several column volumes of buffer, and proteins were eluted with buffer containing 1 mm FKTA-NH2. Fractions of 0.5 ml were collected, and equal aliquots of four fractions that contained protein eluted from the wild-type column were mixed with SDS sample buffer and loaded onto an SDS-polyacrylamide gel (lanes labeled WT). Parallel fractions from the ClpSDD/AA were loaded in adjacent lanes as indicated (lanes labeled AA). Proteins were detected by staining with Coomassie Blue. B, pulldown of proteins from E. coli cells carrying mutations in the N-end rule pathway. Clarified cell lysates from wild-type, ΔclpSA, or Δaat strains were loaded onto a ClpS affinity column, and bound proteins were eluted with FKTA. No proteins were detected in the FKTA eluate when wild-type lysates were applied to a control column with inactivated (Inact.) resin that had no cross-linked ClpS. Stds, standards.
N-terminal Sequencing Confirms the Presence of an N-degron in Most Proteins
To establish that the proteins pulled down by ClpS contained N-degrons, Edman degradation was performed to determine the distribution of N-terminal residues in the entire population. The amino acids detected in the first round of sequencing were predominantly N-degrons. Leu, Phe, Tyr, and Trp constituted 72% of the observed amino acids with Leu and Phe as the most abundant overall (Fig. 3). Serine (14%) and alanine (4%) were also present in relatively high abundance. The amino acids obtained in the second round of sequencing were dominated by valine, threonine, arginine, lysine, and alanine. The enrichment of arginine and lysine in the second round would be expected if a significant portion of the proteins had been modified by Aat in vivo, which was confirmed by experiments reported later in the paper. The appearance of Ser and Ala at the N terminus indicated that non-N-end rule substrates were also isolated from the cell extracts. One possibility for their appearance is that they were present in homo- or heterooligomeric complexes with N-degron-containing partners as in the example of Dps. Dps exists in vivo as a stable dodecamer, and native Dps has an N-terminal serine followed by threonine. Native Dps was pulled down in relatively high abundance along with the N-degron-containing form, which has Leu6 as its N-terminal residue (see Ref. 28 and data below). We repeated the pulldown using a dps mutant strain to see whether the occurrence of serine at the N terminus in the pulldown fraction was reduced. When the pool of proteins isolated from MG1655 dps::kan was sequenced, the amount of serine released in the first round was drastically reduced to <5% (Fig. 3), whereas leucine and phenylalanine remained as the dominant N-terminal amino acids. In addition, threonine was no longer dominant in the second position, confirming that Dps contributed a disproportionate amount to the non-N-degrons observed in the pulldowns from wild-type cells.
FIGURE 3.
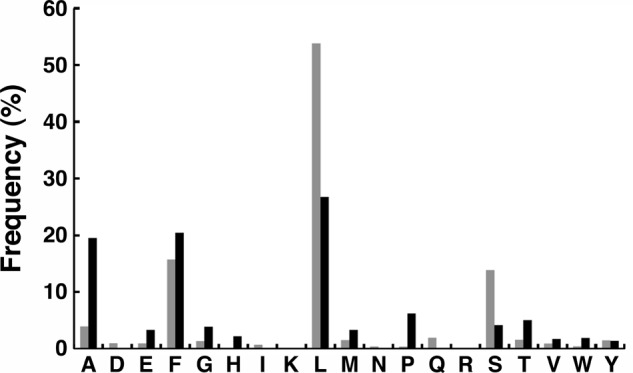
N-terminal sequencing of the total pool of proteins pulled down by ClpS. Proteins from cell lysates of MG1655 (gray bars) or MG1655 dps− (black bars) were bound and eluted from a ClpS column. They were then precipitated with TCA and dissolved in 4% SDS. The solubilized proteins were spotted on PVDF membranes and after washing the spots with methanol solutions to remove SDS subjected to a single round of Edman sequencing. Individual amino acids are expressed as a percentage of the total amino acids in the first position. The N-degrons, leucine and phenylalanine, are dominant in both samples.
To obtain a more detailed analysis of the N-terminal residues in the isolated proteins, we separated proteins eluted from the ClpS column on one-dimensional or two-dimensional gels, transferred them to PVDF membranes, and sequenced individual proteins detected by staining with Coomassie Blue. The resulting directed sequencing results for 31 proteins are summarized in Table 1. Another 10 N-terminal peptides were identified as semitryptic peptides during the mass spectroscopy experiments described later (Table 2). Nearly all (∼90%) of the N-terminal amino acids were leucine or phenylalanine. For most of the proteins, the N-terminal leucine or phenylalanine and the following residues were present in the primary sequence of a protein, although in every case except PATase, the sequence was internal and not at the start of the open reading frame. For ∼25% of the proteins, the N-terminal leucine or phenylalanine was not part of the primary sequence but was followed by a basic residue and subsequent residues that constituted an internal segment of the primary sequence. Thus, many N-terminally truncated proteins had been modified by Aat, which added a leucine or phenylalanine to the α-amino group of a lysine or arginine residue at the N terminus. Based on the sequencing results for the global pool and for individual proteins, we conclude that most of the proteins isolated are ClpS substrates and were bound by virtue of exposed N-end degrons. In the few examples of proteins that do not contain N-degrons (Dps, AccA, and gyrase subunit B (GyrB)), the partners with which they associate were also isolated in ClpS column, and the partners were found to have N-degrons (discussed in more detail below).
TABLE 1.
N-terminal sequences determined by Edman sequencing
| Protein | N-terminal sequence |
|---|---|
| AccA | S2LNFLDa |
| AccD | (L/F)K16ASIPb |
| AphA | L24ASSPS |
| DegP | F84FGDDSP |
| Dps | L6VKSK |
| FtsY | F8FSWLGFG |
| GyrB | (L/F)R394KGALDL |
| IciA | L69LRQVELL |
| InfB | (L/F)K87KRTFVc |
| (L/F)K88RTFVKRc | |
| InfB-AZ | (L/F)K64TRSTLN |
| LacI | (L/F)K33TREKV |
| L56AGKQS | |
| MreB | F103MRPSP3 |
| MreB-AZ | F94IKQVHS |
| (L/F)K96QVHSNS | |
| NusG | F102IGGTS |
| Odo1/SucA | (L/F)R68LAKDAS |
| OsmE | F31VQPVVKD |
| Pat | (F/L/L/L)M1NRLPS |
| RpsA | F130LPGSLVD |
| RsgA | (L/F)R21LKTS |
| Tig | (L/F)K46GKVP |
| TufA/B (EF-Tu) | (L/F)K5FERTK |
| UspG | M1YKTIIMP |
| YbaB | (L/F)K4GGLGNL |
| YgiC | L182TGTLAG |
| YpfJ | L57MTGQPVS |
| YtfR | F402LSGGNNQQ |
a The superscript represents the amino acid position in the open reading frame encoded by the gene for that protein.
b Amino acids in parentheses are leucine or phenylalanine residues added by Aat.
c These N-terminal sequences were obtained for truncated forms of the native protein pulled down from wild-type cells and for the cleavage products of the corresponding AZ fusion proteins.
TABLE 2.
N termini determined by MS/MS analysis of semitryptic fragments
| Protein | N-terminal peptide | Mascot | Expect |
|---|---|---|---|
| AccA | S2LNFLDFEQPIAELEAK | 117 | 3.9 × 10−10 |
| AccC | F84LSENANFAEQVER | 90 | 1 × 10−6 |
| AphA | L24ASSPSPLNPGTNVAR | 102 | 1.5 × 10−8 |
| S26SPSPLNPGTNVAR | 85 | 2.5 × 10−6 | |
| AtpA | M1QLNSTEISELIK | 95 | 4.1 × 10−7 |
| DegP | A27ETSSATTAQQMPSLAPMLEK | 91 | 6.8 × 10−8 |
| F75FGDDSPFCQEGSPFQSSPFCQGGQGGNGGGQQQK | 49 | 6.6 × 10−5 | |
| FtsY | F9FSWLGFGQK | 43 | 8.6 × 10−2 |
| LoiP | L27LSSGAEAFQAYSLSDAQVK | 106 | 2.6 × 10−8 |
| Y38SLSDAQVK | 47 | 2.4 × 10−3 | |
| RplI | L12GSLGDQVNVK | 65 | 3.5 × 10−5 |
| RplN | L86LNNNSEQPIGTR | 77 | 9.2 × 10−6 |
Proteins Pulled Down by ClpS Are More Abundant in Cells Lacking ClpAS
Model N-end rule substrates are degraded in vivo by ClpAP, and the degradation is dependent on ClpS (17, 33). Because the proteins pulled down on the ClpS affinity column have N-degrons, we expected that that most of them should be substrates for ClpSAP and would accumulate in cells lacking components of the degradation machinery, most notably ClpS and/or ClpA. To test this hypothesis, we prepared extracts of wild-type and ΔclpAS cells grown in parallel and isolated N-degron-containing proteins on the ClpS affinity column. Because of the unexpectedly high abundance of proteins with N-degrons, even wild-type cell extracts had saturated the ClpS columns under our initial conditions, resulting in similar recoveries of proteins from wild-type and ΔclpAS cells (Fig. 2B). To allow quantitative comparisons, we adjusted the loading of extracts to ensure that the columns were not saturated and that all of the proteins with N-degrons were bound. Fig. 4, A and B, show the two-dimensional SDS-PAGE profiles of proteins pulled down from wild-type and ΔclpSA cells under identical loading, eluting, and processing conditions. There are obvious differences in the relative abundance of many proteins with a number of proteins accumulating to 5–10-fold higher levels in cells lacking ClpSAP (Fig. 4B,proteins with black circles). These data indicate that many of the proteins isolated are substrates for ClpSAP. Because the steady-state level of an N-end rule protein depends on the rate at which its N-degron is exposed or generated and the rate at which the protein is degraded, the accumulation data do not allow us to estimate the relative rates of degradation in vivo of the proteins isolated on the ClpS column. Only slight differences or in some cases no difference was seen for many other proteins, suggesting that some proteins with N-degrons are not rapidly targeted to ClpSAP for degradation either because of intrinsic stability of the protein or its functional complexes or because there is an additional level of regulation controlling the accessibility of the N-degron to ClpS.
FIGURE 4.

Two-dimensional gel electrophoresis of E. coli proteins eluted from the ClpS affinity column. Cell lysates were prepared from MG1655 (A), MG1655 ΔclpSA (B), and MG1655 aat::kan (C) cells grown to stationary phase in LB medium. The ClpS columns were loaded with 200 mg of protein from clarified cell extracts. Proteins were eluted from the column with FKTA-NH2, and equal aliquots were treated with TCA to precipitate the protein. Proteins were dissolved in buffered urea and separated by two-dimensional electrophoresis. The total number of protein spots and the relative yields of many individual proteins are both increased in the ΔclpSA strain compared with the parental MG1655 (black circles in panel B). Many proteins are absent or present in lower amounts in pulldowns from cells lacking Aat as well (circled in panel A).
Retention of Many of the Proteins on the ClpS Column Is Dependent on Aat
We next asked whether the yields or the distribution of proteins bound to the ClpS column was affected by Aat, which adds primary N-degrons to proteins, enabling them to be recognized by ClpS. When wild-type and aat mutant cells extracts were passed over the ClpS column and separated by one-dimensional SDS-PAGE, the protein banding patterns were quite similar for both extracts, but a number of bands were notably absent in the fraction recovered from the mutant cells (Fig. 2B). To better estimate the number of proteins that only appeared when Aat was present in the cells, we separated the proteins by two-dimensional SDS-PAGE. Comparison of the two-dimensional gel profiles of proteins isolated from wild-type (Fig. 4A) and aat mutant strains (Fig. 4C) confirmed that 20–30% of the proteins were absent from the mutant cells and are therefore most likely Aat substrates (Fig. 4A, proteins with white circles). When proteins isolated on the ClpS affinity column were later identified by mass spectroscopy (see below), we also found that a significant fraction of the proteins was dependent on Aat (highly abundant Aat-dependent substrates are listed in Table 3). Finally, as mentioned above, the Aat dependence for several of the high abundance proteins was corroborated by direct sequencing, which revealed that the N termini of many of the proteins were modified by addition of a leucine or phenylalanine (Table 1).
TABLE 3.
Proteins significantly enriched in lysates from Aat+ cells compared with Aat− cells
Cells were grown in LB to stationary phase (≤16 h). Proteins were judged to be Aat substrates if they were present in ClpS pulldowns from wild-type cells but were absent in the pulldowns from aat mutant cells. Of the 17 proteins selected by this criterion, 13 had been independently identified as Aat substrates based on N-terminal sequencing.
| Protein ID | Spectral count |
G value | p value | N-terminal sequencea | |
|---|---|---|---|---|---|
| Aat+ | Aat− | ||||
| AccA | 8 | 0 | 6.2 | 1.3 × 10−2 | S2LNFLD |
| AccD | 5 | 0 | 3 | 8.8 × 10−2 | (L)K16ASIP |
| AldB | 11 | 0 | 9.8 | 1.8 × 10−3 | |
| AtpA | 11 | 0 | 9.8 | 1.8 × 10−3 | M1QLNSTEISELIKb |
| AtpD | 11 | 0 | 9.8 | 1.8 × 10−3 | |
| FhuF | 6 | 0 | 3.9 | 4.6 × 10−2 | |
| GrpE | 7 | 0 | 5 | 2.5 × 10−2 | |
| GyrB | 12 | 0 | 11 | 9.2 × 10−4 | (L)R394KGALDL |
| InfB | 32 | 0 | 37 | 1.3 × 10−9 | (L)K87KRTFV |
| (L)K88RTFVKR | |||||
| Odo1/SucA | 10 | 0 | 8.5 | 3.5 × 10−3 | (L)R68LAKDAS |
| PatA (PATase) | 20 | 0 | 21 | 4.4 × 10−6 | (LLL)M1NRLPS |
| RecT | 7 | 0 | 5 | 2.5 × 10−2 | |
| RsgA | 9 | 0 | 7.3 | 6.7 × 10−3 | (L)R21LKTS |
| Tig | 25 | 0 | 28 | 1.5 × 10−7 | (L)K46GKVP |
| TufA/TufB (EF-Tu) | 18 | 0 | 19 | 1.7 × 10−5 | (L)K5FERTK |
| YbaB | 9 | 0 | 7.3 | 6.7 × 10−3 | (L)K4GGLGNL |
| YggL | 12 | 0 | 11 | 9.2 × 10−4 | |
a N-terminal sequences were determined by Edman degradation with the exception of AtpA. The superscript indicates the position of the amino acid in the deduced open reading frame encoded by the corresponding gene. The (L) indicates a leucine residue added to the N terminus by Aat.
b N-terminal peptide for AtpA was determined by MS/MS.
As a control, we repeated the pulldowns with extracts of a strain lacking the outer membrane protease OmpT, which cleaves between basic residues and could potentially generate Aat substrates in vitro. Deletion of ompT did not alter the composition of the proteins isolated on the ClpS column (data not shown); nonetheless, all experiments discussed in this study were done in an ompT deletion strain as well as in the presence of the serine protease inhibitor phenylmethylsulfonyl fluoride (PMSF) during preparation of cell lysates. In addition, to confirm that Aat was not actively adding N-degrons to proteins in the extracts, we identified trigger factor (Tig) as one of the most abundant non-essential Aat-dependent substrates and performed the following experiment. Two cultures were grown: one MG1655 tig− and the other MG1655 aat−. An equal number of cells from each culture were mixed before lysis. In addition, aliquots of cells from the separate cultures were also lysed. Separate lysates or mixed cell lysates were passed over wild-type ClpS columns. No trigger factor was present in the pulldowns from the separate lysates or from the lysate of the mixed cells (data not shown), indicating that Aat did not add an N-degron to trigger factor in vitro. We also note that a number of relatively abundant E. coli proteins are known to have naturally occurring basic N-terminal residues as a result of processing during localization to the periplasm. None of the proteins, which included DppA (KTLVYC), RbsB (KDTIAL), and Sbp (KDIQLL) (34), were detected in ClpS pulldown experiments. Thus, Aat does not modify proteins in lysates under the conditions of our experiments. In summary, the results of the above experiments indicate that both Aat-dependent and Aat-independent primary N-degrons are distributed among many proteins in vivo and that cellular mechanisms exist to generate N-end rule substrates with either primary or secondary N-degrons.
Identification of Proteins Pulled Down by ClpS
The proteins isolated from the ClpS column were identified by mass spectrometry. Eluted proteins were separated by one-dimensional SDS-PAGE, and several gel slices were digested with trypsin. The tryptic peptides were analyzed by ion trap mass spectrometry (LTQ, Thermo Fisher). To detect less abundant proteins eluted from the ClpS column, samples were also precipitated with TCA and dissolved in a reduced volume of 40 mm ammonium bicarbonate and 2 m urea prior to digestion with trypsin and mass spectrometry. The masses of the resulting peptides were matched against the non-redundant Swiss-Prot database of E. coli proteins using the Mascot search engine. The criterion for enrichment in the ClpS column (therefore a ClpS-associated protein) was a Mascot score >40 with at least two unique peptide hits for each protein. Also, the protein could not be present in the ClpSDD/AA column elution, and the spectral count data had to generate a G-score greater than 7 to be considered significant (p value <0.01). Each protein on this list was isolated in a minimum of two independent experiments on top of meeting the previously stated criteria. Over 100 different proteins met these stringent criteria (supplemental Table S2). Included in this list of putative N-end rule substrates were the two previously published substrates, PATase and Dps.
Different growth conditions were examined to gain a better understanding of the nature of substrates and when they may appear within cells. Primarily, exponential and late stationary phases were compared. Two cultures of E. coli MG1655 were grown in parallel: one harvested at A600 of 0.7 and the other harvested after 24 h (late stationary phase). The protein profiles of the pulldowns were compared and revealed several significant differences (supplemental Table S3). Some proteins like elongation factor Tu (EF-Tu) (Aat-dependent substrate) showed no difference (in spectral count) in the stationary phase versus exponential phase pulldown experiments (G-score of 0.05, p value of 0.8), whereas others showed significant enrichment in one fraction over the other. Examples of exponentially enriched substrates are GyrB, translation initiation factor 2 (IF-2), IF-3, Odo1, PutA, PyrG, and others. Stationary phase-enriched substrates include Dps, PATase, LacI, OsmY, and AldB among others. Although proteins from a broad variety of functional and structural classes were represented, proteins involved in translation, DNA transactions, and cell envelope processes were somewhat overrepresented.
N-degrons Are Generated in Cells by Partial Proteolysis
All the proteins with N-degrons, except PATase, were shorter than the known or predicted gene products and appeared to be missing variably sized portions of their N-terminal polypeptides (Tables 1 and 2). The most probable mechanism for such truncations, which varied anywhere from three to four to several hundred amino acids, was partial proteolysis. Although aberrant internal translation initiation would also give rise to truncated versions of proteins, it appears far less likely and has been ruled out for a few of the proteins (see below). Subsequent discussion will take the proteolytic origin of the truncated N-end rule proteins as its premise, although the exact mechanism remains to be confirmed in most cases. Some cleavage events occurred close to the N terminus of the native protein as in the case of EF-Tu. In these cases, the action of either an endoprotease or exopeptidase could give rise to the truncated protein. Other proteins were cleaved at positions far removed from the N terminus and were almost certainly cleaved by an endoproteolytic event. For several of the proteins that appear to be cleaved internally, experimentally determined structures or structural predictions located the cut sites in accessible loops separating domains or in apparently mobile regions of the proteins that were not visible in the crystal structures (schematically shown in Fig. 6B). In general, it appears that many if not all of the N-end rule proteins are produced by cleavage by endoproteases/peptidases within accessible regions near the N terminus or within flexible surface-exposed regions of native proteins. We will refer to sites where cleavage or other modification can expose an N-degron as a “pro-N-degron” (22).
FIGURE 6.
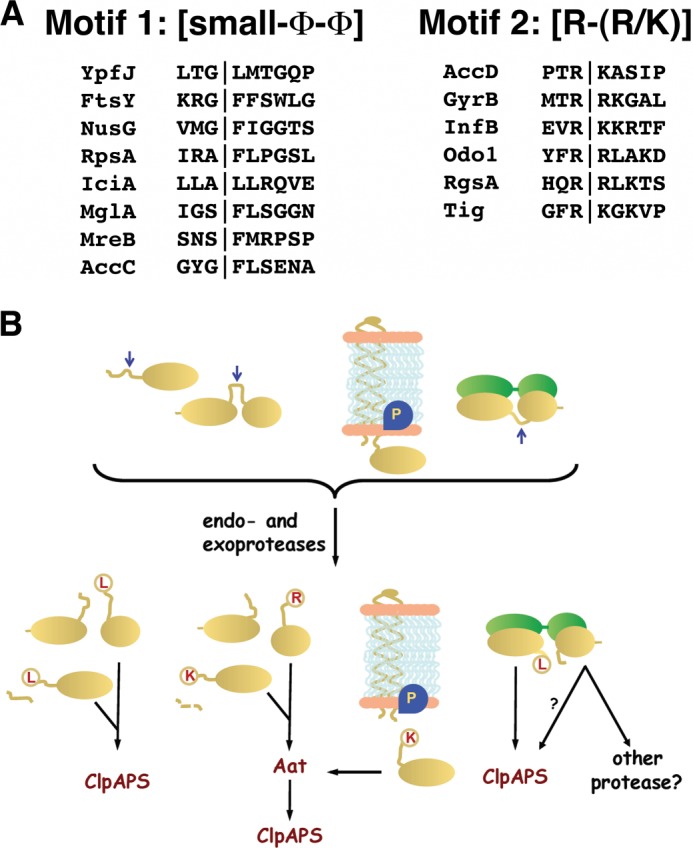
Model for the generation of N-end rule substrates by partial proteolysis of native proteins. A, pro-N-degron motifs for Aat-independent and -dependent substrates. Examination of the sequences surrounding the pro-N-degron in the N-end rule substrates revealed a possible pattern for sets of Aat-independent and Aat-dependent substrates. Motif 1, for Aat-independent pro-N-degrons, is small-Φ-Φ with the cleavage event occurring between the small and first hydrophobic amino acids (Φ). Motif 2, for Aat-dependent substrates, is Arg-(Lys/Arg) with the cleavage event occurring C-terminal of the first arginine. B, potential locations of pro-N-degrons in native proteins. Natively unstructured regions of proteins or regions that can become exposed are susceptible to cleavage by one or more proteases and peptidases, resulting in the appearance of primary or secondary N-degrons. Modification of the latter by Aat produces a form recognized by ClpS and degraded by ClpAP.
Pro-N-degrons Can Be Transferred to Fusion Proteins and Cleaved with Fidelity
To confirm the hypothesis that the N-degrons of the isolated proteins were generated as a result of proteolytic cleavage of specific sites in native proteins, several putative substrates were selected, and N-terminal fragments containing the pro-N-degron identified in pulldowns from wild-type cells were expressed as N-terminal fusions to three tandem Z domains of Staphylococcus aureus protein A (hereafter referred to as AZ) (30). Analysis of fusions of two different proteins, MreB and IF-2, revealed that several primary and secondary N-degrons could be generated within susceptible regions of the proteins. When an extract of cells in which MreB-AZ had been expressed was passed over the ClpS column, a slightly truncated form of the fusion protein was bound and was eluted with the peptide FKTA-NH2 (Fig. 5A). Sequencing confirmed that the protein had an N-end degron, demonstrating that the portion of the protein fused to AZ contained enough information to allow cleavage and generation of the primary (MreB) or secondary (IF-2) N-end degron (Table 1). A closer examination of the N-terminal sequence of the products isolated in these pulldowns revealed multiple N termini. For the IF-2 fusion, three different Aat-dependent N-terminal sequences were identified. All three N-terminal residues fell within a region of ∼30 residues that is predicted to be unstructured or structurally variable in the protein. Two of the sites, Lys87 and Lys88 (before addition of leucine by Aat), corresponded to the sites present in the IF-2 protein isolated on the ClpS column from wild-type cells. When Lys87 was mutated to alanine in the IF-2-AZ fusion and expressed in E. coli, the fusion protein was again recovered on the ClpS affinity column, but this time its N-terminal residue was Aat-modified Arg89, two residues away from the processing site in the wild-type protein. These data suggest that this segment of IF-2 is highly susceptible to cleavage by a protease that leaves an N-terminal basic residue that is subsequently modified by Aat to introduce a primary N-degron.
FIGURE 5.

Formation and degradation of the N-end rule fragment of MreB. Wild-type and ΔclpSA cells expressing the MreB-AZ fusion were grown to an A600 of 0.8, and chloramphenicol was added to prevent further synthesis. At the times indicated, aliquots of the culture were treated with TCA to stop metabolic and enzymatic activities and to precipitate the protein. After separation by SDS-PAGE, MreB-AZ was detected by Western blotting with HRP-conjugated anti-rabbit IgG, which binds to the AZ domain. A, turnover of MreB-AZ. The full-length fusion protein is indicated as well as the shorter N-end rule substrate that is formed by cleavage at the pro-N-degrons in MreB. The identity of the latter was confirmed in a separate experiment by N-terminal sequencing of the protein isolated on a ClpS affinity column, which was loaded as a reference in the lane marked PD. The cleavage site in the fragment indicated by an asterisk is not known. B, half-life of MreB-AZ in wild-type and clpSA mutant cells. The protein bands detected by Western blotting were scanned, and the protein remaining at each time point was determined from the density. Integration and calculation were performed using the program NIH ImageJ. The full-length protein was cleaved with a half-life of about 1 h in both wild-type (diamonds) and clpSA cells (squares). C, quantitation of the cleaved MreB-AZ protein. The amount of cleaved MreB-AZ at each time was measured by densitometry as above. The N-degron-containing fusion accumulated in the clpSA mutant (25–30% of the original fusion) but not in the wild-type cells. The slight stabilization of the N-end rule form at later times is due to the loss of ClpA, which is unstable and lost from the cells during the chloramphenicol chase.
The MreB-AZ fusion pulled down from cell extracts with IgG also displayed multiple species with different N termini, and multiple bands of the fusion were detected with IgG in Western blots of TCA precipitates from whole cells. Interestingly, N-terminal sequencing of the MreB recovered in a pulldown revealed both Aat-dependent and Aat-independent N-degrons. The three observed N-degrons, Phe103, Phe94, and Lys96, were all close in the primary amino acid sequence of MreB, and examination of the crystal structure of MreB shows that Phe103, which was the N-degron identified in the original pulldown of MreB, is on a solvent-exposed loop. Lys96 was subsequently modified by Aat, which added the N-terminal leucine. Multiple N-degrons were also observed with LacI, which is encoded on the pSS101 plasmid used for expression of the protein A fusions and was thus recovered in high yield from the cells. The two N-degrons were Lys33, which Aat modified by the addition of a leucine, and Leu56, which is an Aat-independent N-degron.
Cleaved Fusion Proteins with N-degrons Are Degraded by ClpAP
The AZ domain in the fusions allowed a facile means of monitoring the fusion protein in whole cell extracts by Western blotting. To determine whether the truncated fusions with exposed N-degrons were degraded in vivo by ClpAP, we induced expression of the MreB-AZ fusion in cells and monitored its decay at various times after addition of chloramphenicol to block further synthesis. Two forms of the fusion protein were observed: one corresponding to the full-length fusion and another corresponding to a truncated form. In a separate experiment, both forms were isolated on an IgG affinity column and subjected to N-terminal sequencing, which confirmed that the full-length protein had the encoded N terminus and that the truncated form had the same N-degron as that identified in the MreB fragment isolated on the ClpS column (Table 1). During the chase, the full-length fusion band disappeared with a half-life of ∼57 min (Fig. 5, A and B), whereas the truncated form accumulated briefly and was subsequently degraded (Fig. 5, A and C). In a ΔclpSA strain, the full-length protein was processed to the truncated form with similar kinetics, but the N-end rule fragment accumulated and persisted in the cell for a considerable period (Fig. 5, B and C). Exact calculation of the degradation rate of the truncated form in the wild-type cells was not possible because ClpA itself was unstable and disappeared from the cell in the same time frame as the fragment (35).
DISCUSSION
This study identifies more than 100 proteins that interact with immobilized ClpS under native conditions and are eluted by the addition of a competing peptide, FKTA-NH2. These proteins are potential N-end rule substrates. N-terminal sequencing and mass spectroscopy of several dozen proteins confirmed the presence of an N-degron in most of them, bolstering our conclusion that the vast majority of the proteins that differentially interacted with wild-type ClpS are N-end rule substrates. Further support for this conclusion comes from our observation that many of the proteins pulled down by ClpS were present at 5–10 times higher levels in cells lacking ClpSA, which would be needed for their degradation by the N-end rule pathway. Our identification of over 100 new N-end rule substrates in addition to more detailed Aat- and growth phase-dependent data, allows a more complete appreciation of the scope of the N-end rule in bacterial cells. Two earlier studies identified ClpS-interacting proteins, although only two of those proteins, PATase and Dps, were confirmed to have N-degrons (20, 28). We isolated nine of the 12 proteins identified by Schmidt et al. (20) and 13 of the 22 proteins identified by Ninnis et al. (28). Moreover, we determined the N-terminal sequence of nine of those proteins and confirmed the presence of an N-degron in seven of them, including PATase and Dps. Of the two proteins that did not have N-degrons, DnaK came down with both ClpS and ClpSDD/AA and is known to have promiscuous protein binding activity, and AphA, a periplasmic protein, had an N-terminal leucine that it acquires when its signal sequence is processed. The differences in the proteins isolated in our study and earlier studies might reflect different growth conditions (37 versus 30 °C) or the time of growth (in this study, samples were taken during logarithmic growth or ∼16 h of stationary phase compared with 26-h cultures used by others).
The list of putative substrates identified in this study (supplemental Table S2) reveals that the N-end rule pathway may play a role in central cellular functions, including cell division, DNA replication, transcription, and translation. Newly identified Aat-independent substrates of ClpS include GyrA, several ribosomal structural proteins (S1, L1, L4, L2, S2, L7/L12, L21, and L15), and subunits of RNA polymerase (β and β′). Novel Aat-dependent substrates include InfB/IF-2, Tig, GyrB, two subunits from ATP synthase (α and β), and TufAB/EF-Tu (Table 3). In earlier experiments, proteolytically inactive His-ClpPS97A (ClpPTRAP) was expressed in E. coli cells and used to pull down substrates dependent on the presence of ClpX or ClpA (12). A number of the trapped proteins overlapped with our substrates. Flynn et al. (12) identified 61 proteins as ClpX substrates trapped by ClpPTRAP. In our own laboratory, pulldown of GyrA, RNA polymerase β and β′ subunits, several ribosomal proteins, and different subunits of ATP synthase with ClpPTRAP was dependent on ClpA (data not shown). Flynn et al. (12) grouped the ClpX substrates into several classes based on putative degrons, including SsrA-like and MuA-like C-terminal peptides and three novel N-terminal binding motifs. Of proteins dependent on ClpX for trapping, 13 overlap with proteins pulled down by ClpS (DnaK is excluded for reasons stated above). Two of the latter, RplJ and RplU, have SsrA-like motifs, suggesting that the N-end rule pathway might play only a minor role in their degradation. Two other proteins, Dps and AtpD, have similar N-terminal ClpX degradation motifs, but we have shown here that this motif appears to be the target of an endopeptidase that generates an N-degron in Dps. Flynn et al. (12) also identified four proteins as being dependent on ClpA for trapping: OmpA, AceA, GapA, and TnaA. Only one was present in our ClpS pulldowns: AceA. In the absence of N-terminal sequence data, we cannot definitively conclude that AceA is a substrate for ClpSAP. The reproducible isolation of proteins in trapping and pulldown experiments with components of the degradation machinery lends confidence that these proteins are targeted for regulatory degradation by Clp proteases.
We considered the possibility that there are multiple mechanisms by which proteins are initiated into the N-end rule pathway in E. coli. The dominant mechanism appears to be cleavage of full-length proteins by as yet unknown proteases or peptidases to reveal an N-degron. We have not ruled out other mechanisms, but we note that no systematic increase in accumulation of proteins with N-degrons was observed when we used strains mutated in translation initiation factors that lead to lower fidelity of initiation (data not shown). Sequences around the apparent cleavage sites allowed the proteins to be grouped into two classes based on the potential targeting motifs (Fig. 6A), which we refer to as pro-N-degrons, because it appears that specific endoproteases recognize the sites and cleave the protein in such a way as to expose an N-end degron. Although pro-N-degrons motifs differ from one another, one common feature is the absence of negatively charged residues in the positions following the N-end degron. This restriction on cleavage specificity would correlate with the observation that negatively charged residues in the second position of peptide substrates weaken the affinity of binding to EcClpS (17). For those proteins in which cleavage resulted in a primary N-degron (leucine or phenylalanine), the following amino acid was another hydrophobic amino acid. When a secondary N-degron was generated by proteolysis, cleavage occurred before a charged amino acid, which was followed by glycine, phenylalanine, or another positively charged amino acid. Differences among the several classes of pro-N-degrons imply the existence of at least two endoproteases with different cleavage specificities that are involved in generating N-end rule substrates. In ongoing experiments, we have found several N-end substrates that accumulate in wild-type cells but are absent or present in far lower levels in E. coli mutants lacking specific proteolytic functions (data not shown).
For several of the putative ClpS substrates identified here, we have been able to locate the observed cleavage sites within published structures. In every case examined, the cut sites appear in regions that were exposed in loops on the surface of the protein and often were not visible or were highly variable in the crystal structure, implying that they were in exposed mobile regions of the protein. Our working hypothesis is that several proteases or peptidases are responsible for cleaving proteins to generate primary and secondary N-degrons by recognizing specific sites in protein regions that lack intrinsic structure or that can be destabilized and exposed in response to changes in interacting ligands or macromolecular partners or by changes in environmental conditions (Fig. 6B). The identities of the proteases or peptidases responsible for generation of N-degrons are being investigated, and further analysis of cleavage sites creating N-degrons is underway to obtain a more complete profile of their enzymatic specificities.
One issue that emerges from our findings is the relationship between the cleavage of proteins to expose an N-degron and the metabolic stability of the protein and their associated partners in many cases. Although many proteins accumulated to higher levels in clpSA-deleted cells, others did not. This finding suggests that the rate of degradation by ClpSAP is relatively slow or at least no higher than the rate at which the native protein is cleaved by the endoprotease that generates the N-degron. Degradation of the fusion proteins with pro-N-degrons from MreB and InfB bears this out. The MreB fusion disappeared with an apparent half-life of ∼40 min (implying a half-life much shorter than that), whereas the InfB fusion appeared unchanged during the chloramphenicol chase, suggesting that it was regenerated as fast as it was degraded by ClpSAP. Another possibility is that the proteolytically nicked forms continue to function, perhaps in a modified manner, and that their ultimate targeting to ClpSAP is subject to regulation by associated protein partners or other ligands.
Many of the proteins with pro-N-degrons identified here occur in large protein complexes within the cell. Examples include Dps (homomeric dodecamer), AccA/AccD (heteromeric tetramer of acetyl-CoA carboxyltransferase), (AccA)2(AccD)2, LacI (homomeric tetramer), and AtpA/AtpD (both components of the F1 ATP synthase). In each of those four cases, ClpS pulled down the complex containing subunits with an N-degron and subunits that had the canonical N terminus without an N-degron. Targeting key components of protein complexes for degradation is an established mechanism for remodeling such complexes and has been shown to play essential roles in processes such as replication of phage Mu (36) and removal of the error-prone DNA polymerase, UmuDD′, after acute DNA damage (37). The N-end rule pathway might serve a similar function either for regulatory purposes by targeting specific subunits in response to ligand-induced conformational changes or for quality control purposes by attacking N-degrons generated by peptidases or proteases that conduct surveillance of protein complexes and cleave structurally damaged regions exposing N-degrons. Maintenance of the native oligomeric structure in the proteins pulled down by ClpS implies that limited cleavage of one or more subunits did not disrupt all interactions within these complexes and points to the need for targeting to ClpAP to extract the marked subunit and to degrade that subunit and possibly the other subunits in the complex as well. Among the four examples mentioned, two were homomeric complexes in which a minority of the subunits had pro-N-degrons, whereas most remained unmodified (LacI and Dps). Intact Dps subunits are known to be degraded by ClpXP in vivo, and one possibility is that limited cleavage by an endoprotease is followed by extraction and degradation of the damaged subunit by ClpSAP, leading to complete dissolution of the dodecameric complex and turnover by ClpXP. In the heterooligomeric complex (AccA)2(AccD)2, only AccD contained an N-degron. We do not know whether AccA is degraded by ClpSAP along with AccD or whether the ClpS-ClpA complex would extract and degrade only the AccD. Further studies are needed to elucidate the role played by ClpSAP in the degradation of specific subunits of complexes.
In summary, the isolation of proteins using immobilized ClpS, elution with the N-end rule peptide, identification by mass spectrometry, and subsequent validation by N-terminal sequencing has provided an extensive set of substrates for the N-end rule pathway in E. coli. We have shown that these substrates are generated in vivo by the partial proteolysis of native proteins in variable loops or unstructured regions by unknown proteases or peptidases (Fig. 6B). The finding that cleavage occurs within unstructured regions of the proteins may point to a possible role for the N-end rule in E. coli in a quality control pathway to clear proteins damaged by unregulated proteolysis or peptidase activity as nicked proteins may need to be cleared to maintain optimal function for essential processes like translation initiation or cell division. Limited proteolysis followed by interaction with ClpSAP could also play a role in subunit remodeling of larger protein complexes. These hypotheses are yet to be tested, and much remains to be learned regarding the initiating events that lead to the partial proteolysis or cause of the partial proteolysis of presubstrates. However, the identities of the substrates and the phenotype of the clpS mutant point to the N-end rule pathway having a larger and more general role in central processes of cellular physiology than previously believed.
Acknowledgment
We thank Dr. Jan Rozycki for synthesizing the peptide FKTA-NH2.
This work was supported, in whole or in part, by the National Institutes of Health Intramural Research Program of the Center for Cancer Research, NCI.

This article contains supplemental Tables S1–S3.
- Aat
- Leu/Phe-tRNA protein transferase
- Dps
- DNA protection during starvation
- PATase
- putrescine aminotransferase
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- DD/AA
- D35A,D36A
- EF
- elongation factor
- GyrB
- gyrase subunit B
- Tig
- trigger factor
- IF
- translation initiation factor
- GyrA
- gyrase subunit A.
REFERENCES
- 1. Gottesman S., Wickner S., Maurizi M. R. (1997) Protein quality control: triage by chaperones and proteases. Genes Dev. 11, 815–823 [DOI] [PubMed] [Google Scholar]
- 2. Dougan D. A., Mogk A., Zeth K., Turgay K., Bukau B. (2002) AAA+ proteins and substrate recognition, it all depends on their partner in crime. FEBS Lett. 529, 6–10 [DOI] [PubMed] [Google Scholar]
- 3. Kessel M., Maurizi M. R., Kim B., Kocsis E., Trus B. L., Singh S. K., Steven A. C. (1995) Homology in structural organization between E. coli ClpAP protease and the eukaryotic 26 S proteasome. J. Mol. Biol. 250, 587–594 [DOI] [PubMed] [Google Scholar]
- 4. Wang J., Hartling J. A., Flanagan J. M. (1997) The structure of ClpP at 2.3 Å resolution suggests a model for ATP-dependent proteolysis. Cell 91, 447–456 [DOI] [PubMed] [Google Scholar]
- 5. Wickner S., Gottesman S., Skowyra D., Hoskins J., McKenney K., Maurizi M. R. (1994) A molecular chaperone, ClpA, functions like DnaK and DnaJ. Proc. Natl. Acad. Sci. U.S.A. 91, 12218–12222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weber-Ban E. U., Reid B. G., Miranker A. D., Horwich A. L. (1999) Global unfolding of a substrate protein by the Hsp100 chaperone ClpA. Nature 401, 90–93 [DOI] [PubMed] [Google Scholar]
- 7. Ishikawa T., Beuron F., Kessel M., Wickner S., Maurizi M. R., Steven A. C. (2001) Translocation pathway of protein substrates in ClpAP protease. Proc. Natl. Acad. Sci. U.S.A. 98, 4328–4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Singh S. K., Grimaud R., Hoskins J. R., Wickner S., Maurizi M. R. (2000) Unfolding and internalization of proteins by the ATP-dependent proteases ClpXP and ClpAP. Proc. Natl. Acad. Sci. U.S.A. 97, 8898–8903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martin A., Baker T. A., Sauer R. T. (2008) Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding. Nat. Struct. Mol. Biol. 15, 1147–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aubin-Tam M. E., Olivares A. O., Sauer R. T., Baker T. A., Lang M. J. (2011) Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell 145, 257–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maillard R. A., Chistol G., Sen M., Righini M., Tan J., Kaiser C. M., Hodges C., Martin A., Bustamante C. (2011) ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell 145, 459–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Flynn J. M., Neher S. B., Kim Y. I., Sauer R. T., Baker T. A. (2003) Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell 11, 671–683 [DOI] [PubMed] [Google Scholar]
- 13. Levchenko I., Seidel M., Sauer R. T., Baker T. A. (2000) A specificity-enhancing factor for the ClpXP degradation machine. Science 289, 2354–2356 [DOI] [PubMed] [Google Scholar]
- 14. Muffler A., Fischer D., Altuvia S., Storz G., Hengge-Aronis R. (1996) The response regulator RssB controls stability of the σ(S) subunit of RNA polymerase in Escherichia coli. EMBO J. 15, 1333–1339 [PMC free article] [PubMed] [Google Scholar]
- 15. Pratt L. A., Silhavy T. J. (1996) The response regulator SprE controls the stability of RpoS. Proc. Natl. Acad. Sci. U.S.A. 93, 2488–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou Y., Gottesman S., Hoskins J. R., Maurizi M. R., Wickner S. (2001) The RssB response regulator directly targets σ(S) for degradation by ClpXP. Genes Dev. 15, 627–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Erbse A., Schmidt R., Bornemann T., Schneider-Mergener J., Mogk A., Zahn R., Dougan D. A., Bukau B. (2006) ClpS is an essential component of the N-end rule pathway in Escherichia coli. Nature 439, 753–756 [DOI] [PubMed] [Google Scholar]
- 18. Guo F., Esser L., Singh S. K., Maurizi M. R., Xia D. (2002) Crystal structure of the heterodimeric complex of the adaptor, ClpS, with the N-domain of the AAA+ chaperone, ClpA. J. Biol. Chem. 277, 46753–46762 [DOI] [PubMed] [Google Scholar]
- 19. Zeth K., Ravelli R. B., Paal K., Cusack S., Bukau B., Dougan D. A. (2002) Structural analysis of the adaptor protein ClpS in complex with the N-terminal domain of ClpA. Nat. Struct. Biol. 9, 906–911 [DOI] [PubMed] [Google Scholar]
- 20. Schmidt R., Zahn R., Bukau B., Mogk A. (2009) ClpS is the recognition component for Escherichia coli substrates of the N-end rule degradation pathway. Mol. Microbiol. 72, 506–517 [DOI] [PubMed] [Google Scholar]
- 21. Román-Hernández G., Grant R. A., Sauer R. T., Baker T. A. (2009) Molecular basis of substrate selection by the N-end rule adaptor protein ClpS. Proc. Natl. Acad. Sci. U.S.A. 106, 8888–8893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Varshavsky A. (2011) The N-end rule pathway and regulation by proteolysis. Protein Sci. 20, 1298–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tobias J. W., Shrader T. E., Rocap G., Varshavsky A. (1991) The N-end rule in bacteria. Science 254, 1374–1377 [DOI] [PubMed] [Google Scholar]
- 24. Shrader T. E., Tobias J. W., Varshavsky A. (1993) The N-end rule in Escherichia coli: cloning and analysis of the leucyl, phenylalanyl-tRNA-protein transferase gene aat. J. Bacteriol. 175, 4364–4374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schuenemann V. J., Kralik S. M., Albrecht R., Spall S. K., Truscott K. N., Dougan D. A., Zeth K. (2009) Structural basis of N-end rule substrate recognition in Escherichia coli by the ClpAP adaptor protein ClpS. EMBO Rep. 10, 508–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Donatis G. M., Singh S. K., Viswanathan S., Maurizi M. R. (2010) A single ClpS monomer is sufficient to direct the activity of the ClpA hexamer. J. Biol. Chem. 285, 8771–8781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Román-Hernández G., Hou J. Y., Grant R. A., Sauer R. T., Baker T. A. (2011) The ClpS adaptor mediates staged delivery of N-end rule substrates to the AAA+ ClpAP protease. Mol. Cell 43, 217–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ninnis R. L., Spall S. K., Talbo G. H., Truscott K. N., Dougan D. A. (2009) Modification of PATase by L/F-transferase generates a ClpS-dependent N-end rule substrate in Escherichia coli. EMBO J. 28, 1732–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Surkov S., Nilsson H., Rasmussen L. C., Sperling-Petersen H. U., Isaksson L. A. (2010) Translation initiation region dependency of translation initiation in Escherichia coli by IF1 and kasugamycin. FEBS J. 277, 2428–2439 [DOI] [PubMed] [Google Scholar]
- 30. Björnsson A., Mottagui-Tabar S., Isaksson L. A. (1998) The analysis of translational activity using a reporter gene constructed from repeats of an antibody-binding domain from protein A. Methods Mol. Biol. 77, 75–91 [DOI] [PubMed] [Google Scholar]
- 31. Seyfried N. T., Gozal Y. M., Donovan L. E., Herskowitz J. H., Dammer E. B., Xia Q., Ku L., Chang J., Duong D. M., Rees H. D., Cooper D. S., Glass J. D., Gearing M., Tansey M. G., Lah J. J., Feng Y., Levey A. I., Peng J. (2012) Quantitative analysis of the detergent-insoluble brain proteome in frontotemporal lobar degeneration using SILAC internal standards. J. Proteome Res. 11, 2721–2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Old W. M., Meyer-Arendt K., Aveline-Wolf L., Pierce K. G., Mendoza A., Sevinsky J. R., Resing K. A., Ahn N. G. (2005) Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 4, 1487–1502 [DOI] [PubMed] [Google Scholar]
- 33. Wang K. H., Oakes E. S., Sauer R. T., Baker T. A. (2008) Tuning the strength of a bacterial N-end rule degradation signal. J. Biol. Chem. 283, 24600–24607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Link A. J., Robison K., Church G. M. (1997) Comparing the predicted and observed properties of proteins encoded in the genome of Escherichia coli K-12. Electrophoresis 18, 1259–1313 [DOI] [PubMed] [Google Scholar]
- 35. Dougan D. A., Reid B. G., Horwich A. L., Bukau B. (2002) ClpS, a substrate modulator of the ClpAP machine. Mol. Cell 9, 673–683 [DOI] [PubMed] [Google Scholar]
- 36. Abdelhakim A. H., Sauer R. T., Baker T. A. (2010) The AAA+ ClpX machine unfolds a keystone subunit to remodel the Mu transpososome. Proc. Natl. Acad. Sci. U.S.A. 107, 2437–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gonzalez M., Rasulova F., Maurizi M. R., Woodgate R. (2000) Subunit-specific degradation of the UmuD/D′ heterodimer by the ClpXP protease: the role of trans recognition in UmuD′ stability. EMBO J. 19, 5251–5258 [DOI] [PMC free article] [PubMed] [Google Scholar]