

Background: Increased calcium uptake can rescue congenital cardiomyopathies.
Results: Decreasing tropomyosin phosphorylation increases phospholamban phosphorylation and improves cardiac function and morphology in a cardiomyopathic mouse.
Conclusion: Changing tropomyosin phosphorylation can influence calcium handling to adverse cardiac remodeling.
Significance: This is the first report that altering tropomyosin phosphorylation can rescue a cardiomyopathic phenotype.
Keywords: Calcium, Cardiac Hypertrophy, Muscle, Phosphorylation, Transgenic Mice, Tropomyosin
Abstract
Studies indicate that tropomyosin (Tm) phosphorylation status varies in different mouse models of cardiac disease. Investigation of basal and acute cardiac function utilizing a mouse model expressing an α-Tm protein that cannot be phosphorylated (S283A) shows a compensated hypertrophic phenotype with significant increases in SERCA2a expression and phosphorylation of phospholamban Ser-16 (Schulz, E. M., Correll, R. N., Sheikh, H. N., Lofrano-Alves, M. S., Engel, P. L., Newman, G., Schultz Jel, J., Molkentin, J. D., Wolska, B. M., Solaro, R. J., and Wieczorek, D. F. (2012) J. Biol. Chem. 287, 44478–44489). With these results, we hypothesized that decreasing α-Tm phosphorylation may be beneficial in the context of a chronic, intrinsic stressor. To test this hypothesis, we utilized the familial hypertrophic cardiomyopathy (FHC) α-Tm E180G model (Prabhakar, R., Boivin, G. P., Grupp, I. L., Hoit, B., Arteaga, G., Solaro, R. J., and Wieczorek, D. F. (2001) J. Mol. Cell. Cardiol. 33, 1815–1828). These FHC hearts are characterized by increased heart:body weight ratios, fibrosis, increased myofilament Ca2+ sensitivity, and contractile defects. The FHC mice die by 6–8 months of age. We generated mice expressing both the E180G and S283A mutations and found that the hypertrophic phenotype was rescued in the α-Tm E180G/S283A double mutant transgenic animals; these mice exhibited no signs of cardiac hypertrophy and displayed improved cardiac function. These double mutant transgenic hearts showed increased phosphorylation of phospholamban Ser-16 and Thr-17 compared with the α-Tm E180G mice. This is the first study to demonstrate that decreasing phosphorylation of tropomyosin can rescue a hypertrophic cardiomyopathic phenotype.
Introduction
Tropomyosin (Tm)2 is an α-helical coiled coil protein involved in the Ca2+-dependent regulation of the thin filament of the sarcomere. Once Ca2+ binds to troponin C, a conformational change occurs allowing the Tm to move away from the myosin head binding site on the sarcomeric actin filament, resulting in muscle contraction. α-Tm is the predominant Tm isoform found in cardiovascular muscle, making up ∼95% of total myofibrillar Tm (1). All striated muscle isoforms, including α-Tm, are phosphorylated at a single site, serine 283, by several potential kinases (2–7). Recent studies indicate that mice expressing transgenic (TG) α-Tm with Ser-283 mutated to alanine (S283A) show no major alterations in cardiac function at basal levels with striking increases in the expression of sarcoplasmic reticulum Ca2+-ATPase 2a (SERCA2a) and an increase in phospholamban (PLN) Ser-16 phosphorylation (8). Additionally, these mice maintain a compensated hypertrophic phenotype throughout their lifetime.
Previous studies demonstrate that TG animals expressing Tm mutations that lead to cardiac disease show changes in Tm phosphorylation (9–11). It is also established that increasing SERCA2a expression and/or activity by decreasing PLN expression or increasing PLN phosphorylation at Ser-16 and/or Thr-17 can rescue hypertrophic cardiomyopathy or heart failure in mice (12–15). In addition, SERCA2a gene therapy has been shown to be efficacious in human patients with end stage heart failure (10, 16–18).
Familial hypertrophic cardiomyopathy (FHC) is considered a disease of contractile proteins of the cardiac sarcomere, z-disc proteins, and Ca2+-handling proteins (19). α-Tm has been found to encode 11 mutations that lead to FHC (20). Previous work in our laboratories established a transgenic mouse model expressing mutant FHC α-Tm E180G protein. This mouse model develops severe concentric cardiac hypertrophy with significant ventricular fibrosis and atrial enlargement. Significant physiological alterations in cardiac function, including diastolic dysfunction, occur in addition to myofilaments that demonstrate an increased activation of the thin filament through enhanced calcium sensitivity of steady-state force (21, 22). Recent studies show that Tm phosphorylation levels change from 1.5 to 15 months of age; however, by 4–5 months, the levels of Tm phosphorylation are higher in α-Tm E180G transgenic hearts than in their controls (8). Furthermore, these results of increased Tm phosphorylation in hypertrophic cardiomyopathy mice were confirmed with another FHC mouse model, namely the α-Tm D175N mice (23). Interestingly, a dilated cardiomyopathy mouse model (α-Tm E54K) shows a 40% decrease in Tm phosphorylation in the heart; myofilaments from these mice exhibit a decrease in Ca2+ sensitivity (9).
Unknown issues concerning the FHC Tm mice are whether the increased Tm phosphorylation levels are a contributing cause or consequence of the disease phenotype. To address this question, we hypothesized that decreasing Tm phosphorylation through a Tm S283A mutation may attenuate cardiac hypertrophy and improve cardiac function in mice expressing the α-Tm E180G mutation alone. To that end, we generated double mutant transgenic (DMTG) mice expressing Tm encoding both the α-Tm E180G and S283A mutations on the same molecule; several TG mouse lines were generated and analyzed. DMTG animals had no change in heart:body weight ratios and no deposition of fibrotic material characteristic of the α-Tm E180G hypertrophic cardiomyopathy phenotype. Also, echocardiographic studies indicated rescued cardiac function with improved performance in the DMTG animals compared with non-transgenic (NTG) mice. Also, the DMTG myofilaments exhibited intermediate levels of Ca2+ sensitivity between those found in NTG and α-Tm E180G hearts. Interestingly, the DMTG hearts did not show the dramatic increase in SERCA2a expression seen in the α-Tm S283A hearts but did exhibit increased phosphorylation of PLN at both Ser-16 and Thr-17 when compared with the α-Tm E180G levels. This is the first study demonstrating that decreasing phosphorylation of α-Tm can rescue a hypertrophic cardiac disease phenotype.
EXPERIMENTAL PROCEDURES
Generation of TG Mice
Mouse striated muscle α-Tm cDNA was subjected to QuikChange II site-directed mutagenesis (Agilent Technologies) utilizing the primer 5′-CAC GCT CTC AAC GAT ATG ACT GCC ATA TAA GTT TCT TTG CTT CAC-3′ mutating the penultimate serine to an alanine and the primer 5′-ACG TGC AGA GGG GCG GGC TGA-3′ mutating glutamic acid 180 to a glycine. The mutation was verified through sequencing of the construct by Genewiz. The α-Tm E180G/S283A DMTG construct was then cloned into a vector containing the cardiac-specific α-myosin heavy chain (α-MHC) promoter and a human growth hormone poly(A) tail sequence (see Fig. 1A) (24). DMTG mice were generated using the FVB/N strain, and founder mice were identified using PCR (25). Nucleotide sequencing of DMTG mouse tail DNA verified the sequence of the α-Tm E180G/S283A transgene.
FIGURE 1.

A, α-Tm E180G/S283A DMTG construct. The α-MHC promoter drives cardiac-specific expression of the striated muscle α-Tm with encoded substitutions at amino acids (aa) 180 (E180G) and 283 (S283A) on the same molecule. B, myofibrillar preparations resolved by SDS-PAGE and stained with Coomassie Blue. Note the small shift in migration between the α-Tm E180G and DMTG Tm samples. Arrows indicate DMTG endogenous and TG protein. C, quantification of endogenous Tm protein remaining in NTG, α-Tm E180G, α-Tm S283A, and DMTG samples. Error bars represent S.E.
The α-Tm E180G TG mice and the α-Tm S283A mice were generated previously and extensively characterized with respect to their cardiac phenotype and function (8, 21, 22). We confined our study to male mice because previous results showed sex-specific differences in the development of a compensated hypertrophic phenotype in the α-Tm S283A mice (8).
Genotyping
DNA samples were obtained from 5-day-old mice, and PCR was utilized to determine which animals carried the transgene. The following primers specific for the transgene were used: α-MHC Forward, 5′-GCC CAC ACC AGA AAT GAC AGA-3′; α-Tm Reverse, 5′-TCC AGT TCA TCT TCA GTG CCC-3′. GAPDH was used as an internal control, and the following primer set was used: GAPDH Forward, 5′-AGC GAG CTC AGG ACA TTC TGG-3′; GAPDH Reverse, 5′-CTC CTA ACC ACG CTC CTA GCA-3′.
Transgenic Protein Quantification and Western Blot Analyses
Myofibrillar proteins were extracted from NTG, α-Tm E180G, α-Tm S283A, and DMTG mouse ventricles as described previously (25). 25 μg of the myofibrillar protein preparations were separated by 12% SDS-PAGE and stained with Coomassie Blue. The presence of the α-Tm E180G mutation results in differential mobility in SDS-PAGE (23, 25), allowing quantification of endogenous and DMTG proteins in the sample. Measurements were performed using ImageQuant 5.1. To confirm that the double mutant Tm was properly assembled into the sarcomere, cytoplasmic protein fractions (25 μg) isolated from 3-month-old NTG and DMTG male mice were separated by 12% SDS-PAGE and visualized with Coomassie Blue stain.
Western blot analyses on myofibrillar protein preparations (4 μg) from 3-month-old male NTG, α-Tm E180G, α-Tm S283A, and DMTG hearts were conducted using the Tm-specific antibody CH1 (Sigma-Aldrich), Tm Ser-283 phosphorylation-specific antibody generated for our laboratory (YenZyme), and sarcomeric α-actin antibody 5C5 (Sigma-Aldrich) as a loading control.
Whole ventricular homogenates from 3-month-old NTG, α-Tm E180G, α-Tm S283A, and DMTG mice were utilized to visualize Ca2+-handling protein expression levels. Western blots were used to visualize SERCA2a (Abcam), troponin I (TnI) (Cell Signaling Technology), pTnI23/24 (Cell Signaling Technology), PLN (Thermo Scientific), phosphorylated PLN Ser-16 (Badrilla), and phosphorylated PLN Thr-17 (Badrilla). Sarcomeric actin (Sigma) was used as a loading control. Phosphorylated PLN levels are given as a ratio of the phosphorylated form over total PLN expression. Total PLN expression was calculated by adding the monomeric and pentameric species of PLN for each sample and normalizing to actin.
Two-dimensional Isoelectric Focusing PAGE
Two-dimensional isoelectric focusing PAGE was performed on mouse hearts as described previously (9). 3 μg of myofibrillar preparations were resolved on a 24-cm 4.0–5.0 immobilized pH gradient isoelectric focusing strip. The samples were then resolved in the second dimension by 10% SDS-PAGE, transferred to nitrocellulose, and subjected to immunoblotting. Tm muscle-specific antibody CH1 was used to visualize both the unphosphorylated and phosphorylated NTG and α-Tm E180G TG protein species.
Histopathological Analyses and Cardiomyocyte Cross-sectional Area Analyses
Mouse hearts from 3 to 13 months were isolated, weighed, and examined for histopathological changes. Heart weight to body weight ratios were calculated to determine evidence of cardiac hypertrophy. For histological analyses, the hearts were stained with hematoxylin/eosin (H&E) or Masson's trichrome and evaluated for the presence of fibrosis, myocyte disarray, and calcification. Images were taken on a Nikon SM2–2T dissecting microscope and an Olympus BX4C compound microscope.
To quantify changes in cardiomyocyte cross-sectional area, tissue sections were stained with wheat germ agglutinin conjugated with Texas Red (Sigma-Aldrich) to visualize cardiomyocyte membranes. DAPI was used to stain the nuclei of cardiomyocytes. Randomized images of the left ventricular free wall were taken using a fluorescence camera mounted on a Zeiss Axioskop, and cardiomyocyte cross-sectional area was measured using NIH ImageJ.
Quantitative Real Time PCR Analyses
RNA was isolated from 3-month-old NTG, α-Tm E180G, α-Tm S283A, and DMTG mouse ventricular tissue using TRIzol reagent (Invitrogen). cDNA was generated using the Superscript III kit (Invitrogen). Real time RT-PCR was performed using an Opticon 2 real time RT-PCR machine (MJ Research). Each sample was measured in triplicate, and each experiment was repeated twice. Target mRNA was normalized to GAPDH expression as described by Pfaffl (26). The following specific primers were used: atrial natriuretic peptide (ANP) Forward, 5′-GCTTCCAGGCCATATTGGAG-3′; ANP Reverse, 5′-GGGGGCATGACCTCATCTT-3; β-MHC Forward, 5′-TCATCCGAATCCATTTTGGG-3′; β-MHC Reverse, 5′-CATAATCGTAGGGGTTGTTG-3′; brain natriuretic peptide (BNP) Forward, 5′-GAGGTCACTCCTATCCTCTGG-3′; BNP Reverse, 5′-GCCATTTCCTCCGACTTTTCTC-3′; GAPDH Forward, 5′-TGACCACAGTCCATGCCATC-3′; GAPDH Reverse, 5′-GACGGACACATTGGGGGTAG-3′.
Echocardiographic Measurements
Echocardiographic measurements were performed utilizing a high resolution transducer (Vevo 770 High Resolution Imaging System with a center frequency of 30 MHz) after anesthetization of 3-month-old mice as described previously (27). M-mode images of the left ventricle (LV), LV outflow tract, and left atrium were taken from the left parasternal long axis view. The parasternal short axis view at the level of the papillary muscles was used to measure the LV internal dimension, anterior wall thickness, and posterior wall thickness. NTG and TG 12–16-week-old mice were examined. Pulsed Doppler was performed with the apical four-chamber view. The mitral inflow was recorded with the Doppler sample volume at the tip of the mitral valve leaflets. To measure time intervals, the Doppler sample volume was moved toward the LV outflow tract, and both the mitral inflow and LV outflow were obtained in the same recording. Three parameters of the LV diastolic function were evaluated: 1) E/A ratio, which is the maximal velocity of blood flow in the early diastole (E)/maximal velocity of blood flow in the late diastole (A); 2) E wave deceleration time, which is the time from E to the end of the early diastole; and 3) LV isovolumic relaxation time, which is the time measured from the aortic valve closure to the mitral valve opening. Additional information about the diastolic function was obtained with tissue Doppler imaging. Peak myocardial velocities in the early (Em) diastole were obtained with the sample volume at the septal side of the mitral annulus in the four-chamber view. All measurements and calculations were averaged from three consecutive cycles and performed according to the American Society of Echocardiography guidelines (28, 29). Data analysis was performed with Vevo 770 analytic software.
Measurements of Ca2+-dependent Activation of Tension
Fiber bundles from papillary muscles of 5-month-old male NTG and TG hearts were detergent-extracted in high relaxing buffer as described previously (27) and mounted between a force transducer and a micromanipulator. The sarcomeric length was adjusted to 2.3 μm using laser diffraction patterns, and isometric tension was measured. Fiber bundles were then subjected to sequential Ca2+ solutions (pCa), and isometric tension was again measured. All experiments were carried out at 22 °C.
Statistics
All statistics are presented as mean ± S.E. Where appropriate, paired and unpaired t tests, analysis of variance with Bonferroni correction, and analysis of variance with repeated measures were used to detect significance. Significance was set at p < 0.05.
RESULTS
Generation of α-Tm E180G/S283A DMTG Mice
To determine the role of α-Tm phosphorylation in the context of a progressive, genetic cardiac disease, we generated a DMTG mouse model that expressed a non-phosphorylatable alanine at amino acid 283, the location of the sole known phosphorylation site in striated muscle α-Tm (2, 4, 5). On the same construct, we introduced the α-Tm E180G FHC mutation (22). The transgene construct used to generate these DMTG mice is shown in Fig. 1A. Four DMTG lines were generated and studied.
Cardiac α-Tm E180G/S283A DMTG Protein Expression
DMTG protein expression was determined as the percentage of total Tm protein expression in all four DMTG lines. These lines exhibited a range of 50–64% DMTG protein expression when compared with total Tm protein. Line 325 and Line 335 (64 and 50% DMTG protein expression, respectively) did not differ in their morphological or physiological results; for that reason, we focused on Line 325. Line 325 mice also exhibited a similar level of Tm phosphorylation as the α-Tm S283A mice. As seen in Fig. 1B, the α-Tm E180G mutation conferred differential migration to the DMTG protein when visualized by SDS-PAGE, allowing quantification of endogenous and TG protein levels (Fig. 1C). Of particular interest is a feedback mechanism present in cardiac muscle when striated muscle Tms are expressed exogenously, whereas the endogenous Tm protein level decreases concomitantly with increasing DMTG Tm protein expression; there are no changes in total Tm protein (25, 30). Examination of the cytoplasmic preparations from these DMTG mice showed no significant accumulation of DMTG protein in the cytoplasm, indicating that the DMTG protein is being properly incorporated into the sarcomere (data not shown).
Phosphorylation Status of α-Tm E180G and α-Tm E180G/S283A DMTG Mice
Utilizing a Tm phosphorylation-specific antibody (8), we investigated the phosphorylation status of both the α-Tm E180G and DMTG cardiac myofibers. Interestingly, at 3 months of age, α-Tm E180G hearts showed a significant increase in Tm phosphorylation when compared with NTG levels, whereas Line 325 DMTG hearts exhibited a significant decrease in phosphorylation (Fig. 2). Both the NTG protein and the TG α-Tm E180G protein were phosphorylated when samples were separated using two-dimensional isoelectric focusing, indicating that there is no strong bias toward phosphorylating endogenous or TG protein incorporated into the sarcomere (Fig. 2E). Additionally, the phosphorylation status of the DMTG hearts was very similar to the phosphorylation status of the α-Tm S283A mouse hearts.
FIGURE 2.

A, immunoblot of phosphorylation status of α-Tm E180G hearts at 3 months of age. B, immunoblot of phosphorylation status of α-Tm S283A hearts at 3 months of age. C, immunoblot of phosphorylation status of DMTG hearts at 3 months of age. D, quantification of α-Tm phosphorylation in hearts from 3-month-old NTG, α-Tm E180G, α-Tm S283A, and DMTG mice. *, versus NTG, p < 0.05. E, two-dimensional SDS-PAGE of Tm from α-Tm E180G TG myofibers. Error bars represent S.E. pTm, phosphorylated Tm; α-Tm-p, phosphorylated α-Tm.
Histopathological, Gravimetric, and Cardiomyocyte Cross-sectional Area Analyses of α-Tm E180G and α-Tm E180G/S283A DMTG Mice
To ascertain whether there were histological abnormalities in the hearts of the DMTG animals, we conducted a detailed morphological analysis of the left ventricular free wall of 3-month-old mice. Results show that the DMTG hearts exhibited a phenotype that is very similar to age-matched NTG mice with no cardiomyocyte disarray, enlargement, and excessive fibrosis, which are pathological changes characteristic of the α-Tm E180G hearts (Fig. 3A). Wheat germ agglutinin staining of the cell membrane of the cardiomyocytes showed no significant differences between cross-sectional areas from NTG and DMTG left ventricular cardiomyocytes (Fig. 3B). As expected, given the degree of hypertrophy that occurs in the α-Tm E180G model, there was a significant increase in cross-sectional area compared with both NTG and DMTG. There were also no differences in heart weight:body weight ratios between NTG and DMTG animals. In contrast and as we reported previously, the α-Tm E180G animals exhibited significant increases in heart weight:body weight ratios compared with NTG littermates (21, 22, 31, 32). We hypothesize that the presence of the S283A mutation is responsible for normalizing the cardiac pathological phenotype in the DMTG animals. Additionally, DMTG animals maintained the same heart weight:body weight ratios as NTG littermates for at least 18 months of age. Importantly, although α-Tm E180G animals do not typically survive past 6–8 months of age (22, 31), the addition of the S283A mutation considerably extended the life expectancy of these mice to that of NTG animals (data not shown).
FIGURE 3.

A, histological studies of NTG, α-Tm E180G, and DMTG hearts at 3 months of age stained with H&E, Masson's trichrome, and wheat germ agglutinin (WGA). Images were taken at 40×, and scale bars indicate 50 μm. B, cardiomyocyte cross-sectional area measurements (n = 6). *, versus NTG, p < 0.05; #, versus α-Tm E180G, p < 0.05. C, heart weight to body weight (HW:BW) ratios of 3-month-old mice (n = 6). Error bars represent S.E.
Cardiac Function in α-Tm E180G and α-Tm E180G/S283A DMTG Animals
To assess whether decreasing phosphorylation of α-Tm in a genetic model of hypertrophic cardiomyopathy improves cardiac function, we performed echocardiographic analysis on 3-month-old NTG, α-Tm E180G, and DMTG mice (Table 1). DMTG mice showed only a slight increase in the size of the left atria; however, the increase in size was not as large as the increase in atrial size typically found in α-Tm E180G animals (21, 22, 32). Most striking is the significant increase in ejection fraction, fractional shortening, and velocity of circumferential fiber shortening in the DMTG animals compared with both NTG and α-Tm E180G animals, indicating that the DMTG hearts have improved systolic function and are hypercontractile compared with NTG littermates. Additionally, diastolic function (E/Em and E/A ratios) was significantly improved in DMTG animals and showed rescue of the extreme diastolic dysfunction seen in the α-Tm E180G mice (Table 1). In short, the DMTG animals demonstrate the prevention of the dysfunctional cardiac phenotype seen in the α-Tm E180G FHC model and actually show improved cardiac performance. Recent studies have shown that there are no physiological differences in cardiac function between α-Tm S283A mice and controls (8).
TABLE 1.
Cardiac function of NTG, α-Tm E180G, and α-Tm E180G/S283A DMTG at 3 months of age as assessed by echocardiography
LVIDd, left ventricular internal dimension in diastole; EF, ejection fraction; FS, fractional shortening; Vcf, velocity of circumferential shortening; IVRT, isovolumetric relaxation time; DT, deceleration time; E/A ratio, ratio of early-to-late ventricular filling velocities; E/Em ratio, ratio of early pulse Doppler filling velocity to early tissue Doppler velocity; bpm, beats/min.
| Parameters | NTG (n = 9) | α-Tm E180G (n = 5) | α-Tm E180G/S283A (n = 13) |
|---|---|---|---|
| Left atrium (mm) | 1.93 ± 0.09 | 3.88 ± 0.23a | 2.16 ± 0.13a |
| LVIDd (mm) | 4.08 ± 0.13 | 3.97 ± 0.10 | 3.90 ± 0.06 |
| LV mass (mg) | 92.39 ± 5.78 | 95.29 ± 10.84 | 85.21 ± 2.56 |
| EF (%) | 68.96 ± 1.37 | 66.08 ± 4.38 | 80.52 ± 1.81a,b |
| FS (%) | 38.41 ± 1.09 | 36.33 ± 3.14 | 49.33 ± 1.92a,b |
| Vcf (circumference/s) | 6.99 ± 0.30 | 6.33 ± 0.85 | 8.13 ± 0.26a,b |
| IVRT (ms) | 12.97 ± 0.38 | 13.13 ± 1.38 | 13.73 ± 0.57 |
| DT (ms) | 22.10 ± 1.59 | 26.04 ± 0.84 | 24.2 ± 0.77 |
| E/A ratio | 1.67 ± 0.14 | 6.72 ± 1.03a | 2.07 ± 0.14b |
| E/Em ratio | 38.54 ± 3.67 | 64.6 ± 3.17a | 38.42 ± 1.95b |
| Heart rate (bpm) | 456.2 ± 10.82 | 422.9 ± 14.92 | 431.0 ± 17.88 |
a Versus NTG, p < 0.05.
b Versus α-Tm E180G, p < 0.05.
To determine whether the myofilament Ca2+ sensitivity can contribute to the observed improved phenotype in DMTG mice, we measured force-Ca2+ relations in skinned, detergent-extracted fiber bundles from the papillary muscle of 3-month-old NTG, α-Tm E180G, and DMTG hearts. Although myofilaments from DMTG mice showed an increased Ca2+ sensitivity compared with NTG controls, the Ca2+ sensitivity of the DMTG was significantly lesser than that of α-Tm E180G myofilaments (Fig. 4). Myofilaments exhibited a pCa50 of 5.94 ± 0.01 for DMTG, 5.81 ± 0.02 for NTG, and 6.01 ± 0.03 for α-Tm E180G (Table 2). The maximum tension and Hill coefficient (nH) were not significantly different among the groups.
FIGURE 4.
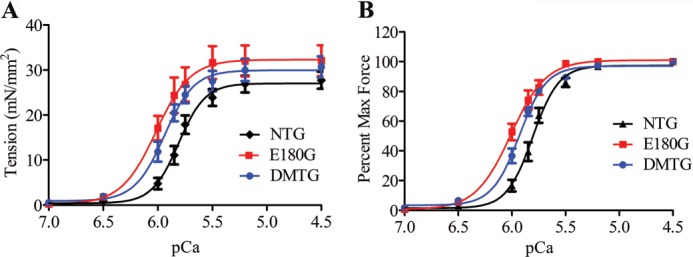
A, Ca2+-tension relations in skinned fiber bundles from NTG, α-Tm E180G, and DMTG hearts. B, normalized force relations in skinned fiber bundles from NTG, α-Tm E180G, and DMTG hearts. Error bars represent S.E. mN, millinewtons.
TABLE 2.
Myofilament properties in NTG, α-Tm E180G, and α-Tm E180G/S283A mice
| Group | pCa50 | nH | n |
|---|---|---|---|
| NTG | 5.807 ± 0.02 | 3.58 ± 0.47 | 12 |
| α-Tm E180G | 6.012 ± 0.03a | 2.65 ± 0.42 | 5 |
| α-Tm E180G/S283A | 5.940 ± 0.01a,b | 3.44 ± 0.34 | 10 |
a Versus NTG, p < 0.05.
b Versus α-Tm E180G, p < 0.05.
Gene Expression Changes in α-Tm E180G and α-Tm E180G/S283A DMTG Hearts
With the improvement in cardiac function in the DMTG hearts, we determined whether the rescued hearts also show improvements at the molecular level by assessing gene expression of cardiomyopathy markers. Real time RT-PCR analysis of RNA isolated from ventricular tissue showed that α-Tm E180G mice exhibited significant increases in β-MHC, BNP, and ANP as expected given the level of hypertrophy present in the hearts of those animals (Fig. 5, A–C). Interestingly, DMTG hearts showed a significant decrease in β-MHC compared with both NTG and α-Tm E180G hearts. Moreover, DMTG hearts exhibited similar levels of ANP and BNP in comparison with NTG hearts and significant decreases when compared with α-Tm E180G hearts, whereas levels in the α-Tm S283A hearts did not significantly differ from normal values.
FIGURE 5.

A–C, quantitative RT-PCR analysis of expression of the cardiomyopathy marker genes in 3-month-old NTG, α-Tm E180G, α-Tm S283A, and DMTG hearts. For NTG, n = 12; for E180G, n = 8; for S283A, n = 8; and for DMTG, n = 10. *, versus NTG, p < 0.05; #, versus α-Tm E180G, p < 0.05; @, versus α-Tm S283A, p < 0.05. Error bars represent S.E.
Changes in Proteins Involved in Ca2+ Handling in α-Tm E180G and α-Tm E180G/S283A DMTG Hearts
To determine whether there are changes in Ca2+-handling proteins in the DMTG hearts, we conducted Western blot analyses. Results show that there were significant decreases in phosphorylated PLN in the α-Tm E180G and DMTG animals compared with NTG controls (Fig. 6). However, the DMTG samples showed significant increases in phosphorylation at both Ser-16 and Thr-17 compared with α-Tm E180G samples (Fig. 6B). The α-Tm S283A animals exhibited a significant increase in PLN phosphorylation at Ser-16 as reported previously (8). Surprisingly, there was no increase in SERCA2a expression in the DMTG animals unlike that seen in the α-Tm S283A hearts. There was an increase in total PLN expression in both the α-Tm E180G and DMTG hearts when compared with α-Tm S283A (Fig. 6C). With the increase in PLN phosphorylation, it is possible that there was a relief of the inhibition by PLN on SERCA2a, thus increasing Ca2+ resequestration into the sarcoplasmic reticulum and helping rescue the FHC phenotype in the DMTG hearts.
FIGURE 6.

A, Western blots of sarcoplasmic Ca2+ flux proteins in NTG, α-Tm E180G, α-Tm S283A, and DMTG hearts. B, quantification of phosphorylation levels of PLN Ser-16 and PLN Thr-17. C, quantification of expression of PLN and SERCA2a. D, Western blots of TnI and phosphorylated TnI (pTnI) from NTG, α-Tm E180G, α-Tm S283A, and DMTG hearts. E, quantification of data from D. *, versus NTG, p < 0.05; #, versus α-Tm E180G, p < 0.05; @, versus α-Tm S283A, p < 0.05; n = 8. Error bars represent S.E.
We also examined phosphorylation of TnI, as this protein can be considered a master regulator of sarcomeric function and Ca2+ sensitivity. Although there was an upward trend in TnI amino acid 23/24 phosphorylation in the DMTG mice, there were no significant differences in TnI expression or TnI phosphorylation at amino acids 23 and 24 (Fig. 6, D and E). The absence of alterations indicates that the shift of DMTG myofiber Ca2+ sensitivity toward NTG levels was not mediated by TnI phosphorylation but occurred through some other mechanism.
DISCUSSION
Previous work has indicated that in hearts of two FHC mouse models that exhibit severe cardiomyopathic disease α-Tm phosphorylation levels are significantly higher than in NTG age-matched controls (11). To test whether the increased Tm phosphorylation levels may be contributing to the disease, we generated a cardiac-specific construct expressing a known human FHC mutation (α-Tm E180G) and a mutation responsible for decreasing Tm phosphorylation at the sole known phosphorylation site (α-Tm S283A) (8). This DMTG approach was taken to ensure equivalent levels of both modifications in the same Tm molecule, which may not occur in sarcomeres of α-Tm E180G and α-Tm S283A TG crossed F1 generation mice. Four DMTG lines were generated expressing both the E180G and S283A mutations on the same α-Tm molecule. These lines showed alterations in α-Tm phosphorylation without significant differences in cardiac phenotype. DMTG Line 325 was chosen for further study given the similarity in Tm phosphorylation status with the α-Tm S283A animals. We reported previously that α-Tm E180G TG animals have a short life span (6–8 months) compared with NTG controls (21, 22, 31). Interestingly, DMTG animals had a longer life span, measured out to 18 months of age, indicating a significant improvement in overall cardiac health of the DMTG animals. Although the DMTG hearts did not exhibit the striking increases in SERCA2a expression and PLN Ser-16 phosphorylation evident in the α-Tm S283A mice, the DMTG animals had significant increases in PLN phosphorylation at Ser-16 and Thr-17 compared with the α-Tm E180G mice, suggesting that DMTG mice may have more active SERCA2a. The DMTG hearts expressed NTG levels of the cardiac hypertrophic markers β-MHC, BNP, and ANP. Increased re-expression of these genes in adults is associated with the cardiac fetal gene program often found with cardiac remodeling and hypertrophy. Our data show that α-Tm E180G hearts expressed very high levels of these transcripts, which were reduced to NTG levels in the DMTG mice in the case of all three markers studied. The decrease in β-MHC levels may partially account for the improvement in cardiac function in the DMTG versus the α-Tm E180G hearts. Specifically, the improvement in diastolic function and increased speed of ventricular shortening could result from a larger α- to β-MHC ratio. These finding are in agreement with previous work showing that improved ventricular function is associated with increased α- to β-MHC expression (33, 34). That decreasing Tm phosphorylation can result in decreasing expression of these hypertrophy markers illustrates the comprehensive rescue of the hypertrophic phenotype at the biochemical level.
Investigation of cardiac performance in NTG, α-Tm E180G, and DMTG mice showed that the α-Tm E180G mice exhibited severe cardiac dysfunction, specifically in relaxation parameters such as E/Em and E/A ratios, possibly due to alterations in SERCA2a activity and PLN expression/phosphorylation that can lead to a reduction in the rate of Ca2+ transit decay, which is known to contribute to a slower relaxation rate (35). Interestingly, the introduction of the S283A dephosphorylation mutation returned E/Em and E/A ratios to NTG levels, rescuing the relaxation defect found in α-Tm E180G animals. These findings are similar to those observed in TG mice expressing both chimeric α-/β-Tm and α-Tm E180G proteins (36). The most striking result shown in the echocardiographic studies is the significant increase in ejection fraction, velocity of circumferential fiber shortening, and fractional shortening (%) in the DMTG animals compared with NTG littermates, indicating that the DMTG hearts exhibited improved contractile parameters possibly due to an increase in SERCA2a activity. The results of our skinned fiber study indicate that some of the rescue is attributable to the sarcomere because of the decrease in the pCa-tension measurements when compared with increased Ca2+ sensitivity of α-Tm E180G myofilaments. Also, there were no differences in nH from NTG values.
Tm and the Tn complex function as a Ca2+-sensitive switching mechanism in the thin filament. Ca2+ binding to TnC causes Tm to move from a position blocking strong cross-bridge binding to actin to a position facilitating strong cross-bridge binding. Strongly bound cross-bridges enhance activation by pushing Tm away from the myosin binding sites, and activation is cooperatively transmitted through the thin filament via Tm overlap regions. The fact that the α-Tm E180G mutation involves a charge change suggests that it may disrupt normal Tm-actin binding, which is dependent on polar and hydrophobic interactions. Our results show that by changing the Tm phosphorylation site (S283A) in the context of α-Tm E180G the DMTG myofilaments exhibit a decrease in the pCa-tension curve. This change occurs without affecting maximum tension and is in agreement with increased relaxation (as measured by echocardiography) when compared with the α-Tm E180G values. Furthermore, because no changes in cooperativity were found among the NTG, α-Tm E180G, and DMTG myofilaments, it appears that there are no differences in Tm interactions with actins under the control of Tm and that there is no effect on end-to-end interactions linking near neighbor functional units consisting of actin-Tm-Tn.
The carboxyl end of Tm is involved in multiple protein-protein interactions, including the head-to-tail overlap of Tm molecules, binding to actin, and binding to troponin T. Previous studies show that the carboxyl end of Tm is involved in the regulation of Ca2+ sensitivity (37, 38). Recent in vitro data examining the properties of recombinant, non-phosphorylatable α-Tm (S283A), and a phosphomimetic (α-Tm S283D) indicate that although the S283D mutation slows deactivation of the thin filament the S283A mutation does not alter myofilament function (39); these data support our in vivo myofilament Ca2+ sensitivity work regarding the effect of the S283A mutation. Additional studies show that when fiber bundles from α-Tm S283D mice are analyzed there are no changes in myofilament Ca2+ sensitivity.3 Thus, myofilament Ca2+ sensitivity may involve interactions between the Tm 180 and 283 amino acid regions probably through TnT binding. We find it surprising that both in vivo and in vitro studies indicate that α-Tm S283A myofilaments did not exhibit a change in Ca2+ sensitivity, yet the α-Tm E180G/S283A did exhibit a decrease in Ca2+ sensitivity from the values obtained with Tm E180G alone; additional studies are in progress to address this issue.
Tm is phosphorylated at amino acid Ser-283 located in the overlap region of Tm; this region interacts with the T1 conserved region of the amino terminus of cTnT (40). The α-Tm E180G mutation would be expected to influence T2 binding in cTnT. Recent work demonstrates that the interplay between Tm and cTnT in the overlap region effectuates different states of Tm on the actin filament (41, 42). These interactions facilitate the binding of Tm to actin, promote Tm-Tm polymerization on the actin filament, and regulate cooperative activation of the thin filament. Based on our observations, reducing phosphorylation of Tm together with the α-Tm E180G mutation offsets the cardiac dysfunction/impairment of the FHC α-Tm E180G mutation by itself possibly by altering interactions of Tm with cTnT in both the T1 and T2 regions. This hypothesis is supported by previous work showing that FHC α-Tm E180G mice can also be rescued by modifications in the Tm carboxyl terminus (36).
Recent studies have indicated that the α-Tm E180G mutation results in a strikingly more flexible striated muscle α-Tm compared with WT α-Tm (43). Moreover, it has been suggested that this mutation leads to an increase in local flexibility, likely partially unwinding or relaxing the coiled coil around amino acid 180 (44, 45). The greater global flexibility of mutant α-Tm indicates that a lower concentration of Ca2+ is necessary to induce conformational changes needed to move Tm off the myosin head binding sites on actin, leading to increased Ca2+ sensitivity of the thin filament (46). Also, recent work by Tardiff suggests that mutations in Tm can elicit functional effects by propagation of structural effects through the Tm helix (47). As such, it is possible that the S283A mutation at the carboxyl terminus restores α-Tm to normal levels of flexibility, which may account for the near complete rescue at the level of the sarcomere, although this possibility demands further study.
Previous studies have indicated that normalization of Ca2+ flux dynamics and altered Ca2+ uptake by the sarcoplasmic reticulum may play a role in the rescue of the FHC phenotype in the DMTG mice. Although there was no significant increase in SERCA2a expression in the DMTG animals, unlike the S283A animals, there were alterations in the phosphorylation status of PLN, which may reduce the inhibition of SERCA2a by PLN and result in increased activity. It is also possible that the decrease in SERCA2a in DMTG animals compared with the α-Tm S283A animals may be related to the presence of the disease-causing E180G mutation as previous studies have shown that decreases in SERCA2a expression occur in the α-Tm E180G mice at different ages (21). Indeed, previous studies that altered proteins involved in Ca2+ fluxes in the context of the α-Tm E180G mice have shown that Ca2+ buffering using parvalbumin or knocking out PLN in the α-Tm E180G TG animals can rescue the severe cardiomyopathic phenotype (16, 48). Similar to the DMTG animals studied here, PLN knock-out/Tm E180G (PLNKO/α-Tm E180G) mice show reversals in hypertrophic marker gene expression compared with the significant increase seen in α-Tm E180G mice. The PLNKO/α-Tm E180G animals also show rescue of relaxation parameters by echocardiography. On the other hand, the PLNKO/α-Tm E180G animals did not display the improved functional performance seen in the DMTG animals (10). Additionally, rescue of the α-Tm E180G phenotype via adenoviral delivery of SERCA2a normalizes heart weight to body weight ratios and improves cardiac function, similar to our observations in the DMTG animals (16).
This report clearly demonstrates that alterations in the phosphorylation status of α-Tm can improve a genetically induced cardiomyopathic phenotype. Other studies have also demonstrated that modifications in contractile protein phosphorylation can improve cardiac and/or sarcomeric function. In vivo cardiac function was improved in a restrictive cardiomyopathy mouse model (cTnI193His) when these mice were crossed with a TG mouse expressing an amino-terminal truncated cTnI (cTnI-ND); two phosphorylation sites on cTnI (Ser-23 and Ser-24) are deleted in the cTnI-ND mice (49). In addition, deletion of the cTnI amino-terminal domain also improves cardiac contractility in aged mice (50). Furthermore, studies show that myosin light chain kinase-induced phosphorylation of skinned muscle fibers from an FHC regulatory light chain model (D166V) reverses the overly sensitive responsiveness to calcium (51). Thus, altering phosphorylation of sarcomeric protein may offer a potential therapeutic approach to improve cardiac function under specific cardiac stress conditions.
The mechanism by which decreasing α-Tm phosphorylation improves function in the context of the α-Tm E180G mutation is unknown. Gaffin et al. (10) have shown that α-Tm E180G animals exhibit significant decreases in ERK1/2 phosphorylation that are normalized when the animals are crossed with PLNKO mice. ERK1/2 is one of the MAPK pathways involved in cardiac hypertrophy, and MEK1, an upstream effector of ERK1/2, has been shown to regulate increased levels of ERK1/2 phosphorylation in a compensated hypertrophic phenotype, similar to what is seen in the α-Tm S283A animals (52). It is possible that the alterations occurring at the level of the sarcomere, specifically changes in Tm nearest neighbor interactions, may result in alterations in the activation of proteins such as protein kinase C ϵ (PKCϵ), which binds actin and is an upstream activator of the MEK1-ERK1/2 pathway via c-Raf (53–55). These questions will be addressed in future investigations.
Another signaling pathway that might possibly be activated to prevent the disease phenotype in the DMTG mice involves protein phosphatase PP2a and casein kinase-2-interacting protein (CKIP-1). Previous work shows that α-Tm E180G mice have increased levels of PP2a and casein kinase-2 (31, 56). More recent work by Ling et al. (57) demonstrates that CKIP-1 and PP2a directly interact, which facilitates the binding of PP2a to HDAC4 and promotes HDAC4 dephosphorylation. This HDAC4 dephosphorylation suppresses cardiac hypertrophy and the fetal cardiac gene program. If decreased α-Tm phosphorylation leads to increased levels of CKIP-1 expression, then this signaling pathway may be activated to rescue the α-Tm E180G/S283A mice and suppress the cardiac fetal gene program.
The pathways by which the α-Tm E180G mutation promotes a pathological phenotype have not been well elucidated, but myofibers from FHC models exhibit increased Ca2+ sensitivity and diastolic dysfunction possibly leading to stress-sensitive pathways of hypertrophic growth. It has been suggested that decreasing myofilament sensitivity would be a straightforward approach to treating FHC, although no pharmacological interventions are currently available. Targeting Tm phosphorylation specifically, given the rescue of the FHC phenotype shown in this study, may provide a starting point for research into possible therapeutics. However, significant preliminary studies would first need to address the appropriateness of decreasing Tm phosphorylation in treating other cardiomyopathies, the specific phosphatase involved in Tm dephosphorylation, and a means of targeting that phosphatase to Tm while restricting potential secondary targets. To conclude, our data show that expression of an α-Tm protein expressing both the E180G FHC mutation and the phosphorylation-reducing S283A mutation rescues a cardiomyopathic hypertrophic phenotype. Changes in Ca2+-handling proteins may additionally be responsible for the improved functional performance found in the DMTG hearts. Changes in local flexibility of the Tm molecule conferred by the replacement of Ser-283 with an Ala residue and the significant loss of phosphorylation may be responsible for the restoration of Tm to proper flexibility. Although alterations in actin-Tn-Tm interactions could play a vital role, the precise mechanism whereby decreased Tm phosphorylation rescues cardiac hypertrophy remains to be elucidated.
Acknowledgments
We thank Hannah Yaejee Hong for technical assistance and Maureen Bender for excellent animal husbandry skills.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 HL-081680 (to D. F. W.), RO1 HL-064035 (to R. J. S. and B. M. W.), PO1 HL-062426 (to R. J. S.), T32 HL-07382 (to E. M. S.), and T32 HL-07692 (to T. W.).
B. J. Biesiadecki, R. J. Solaro, and D. F. Wieczorek, unpublished data.
- Tm
- tropomyosin
- MHC
- myosin heavy chain
- Tn
- troponin
- SERCA2a
- sarcoplasmic reticulum Ca2+-ATPase 2a
- PLN
- phospholamban
- FHC
- familial hypertrophic cardiomyopathy
- TG
- transgenic
- DMTG
- double mutant transgenic
- NTG
- non-transgenic
- LV
- left ventricle
- E/A ratio
- maximal velocity of blood flow in the early diastole (E)/maximal velocity of blood flow in the late diastole (A)
- Em
- myocardial velocity in the early diastole
- BNP
- brain natriuretic peptide
- ANP
- atrial natriuretic peptide
- cTn
- cardiac troponin.
REFERENCES
- 1. Muthuchamy M., Pajak L., Howles P., Doetschman T., Wieczorek D. F. (1993) Developmental analysis of tropomyosin gene expression in embryonic stem cells and mouse embryos. Mol. Cell. Biol. 13, 3311–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mak A., Smillie L. B., Bárány M. (1978) Specific phosphorylation at serine-283 of α tropomyosin from frog skeletal and rabbit skeletal and cardiac muscle. Proc. Natl. Acad. Sci. U.S.A. 75, 3588–3592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Montgomery K., Mak A. S. (1984) In vitro phosphorylation of tropomyosin by a kinase from chicken embryo. J. Biol. Chem. 259, 5555–5560 [PubMed] [Google Scholar]
- 4. Wu S. C., Solaro R. J. (2007) Protein kinase Cζ. A novel regulator of both phosphorylation and de-phosphorylation of cardiac sarcomeric proteins. J. Biol. Chem. 282, 30691–30698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yuan C., Solaro R. J. (2008) Myofilament proteins: from cardiac disorders to proteomic changes. Proteomics Clin. Appl. 2, 788–799 [DOI] [PubMed] [Google Scholar]
- 6. Reddy Y. S., Ballard D., Giri N. Y., Schwartz A. (1973) Phosphorylation of cardiac native tropomyosin and troponin: inhibitory effect of actomyosin and possible presence of endogenous myofibrillar-located cyclic-AMP-dependent protein kinase. J. Mol. Cell. Cardiol. 5, 461–471 [DOI] [PubMed] [Google Scholar]
- 7. deBelle I., Mak A. S. (1987) Isolation and characterization of tropomyosin kinase from chicken embryo. Biochim. Biophys. Acta 925, 17–26 [DOI] [PubMed] [Google Scholar]
- 8. Schulz E. M., Correll R. N., Sheikh H. N., Lofrano-Alves M. S., Engel P. L., Newman G., Schultz Jel J., Molkentin J. D., Wolska B. M., Solaro R. J., Wieczorek D. F. (2012) Tropomyosin dephosphorylation results in compensated cardiac hypertrophy. J. Biol. Chem. 287, 44478–44489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Warren C. M., Arteaga G. M., Rajan S., Ahmed R. P., Wieczorek D. F., Solaro R. J. (2008) Use of 2-D DIGE analysis reveals altered phosphorylation in a tropomyosin mutant (Glu54Lys) linked to dilated cardiomyopathy. Proteomics 8, 100–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gaffin R. D., Peña J. R., Alves M. S., Dias F. A., Chowdhury S. A., Heinrich L. S., Goldspink P. H., Kranias E. G., Wieczorek D. F., Wolska B. M. (2011) Long-term rescue of a familial hypertrophic cardiomyopathy caused by a mutation in the thin filament protein, tropomyosin, via modulation of a calcium cycling protein. J. Mol. Cell. Cardiol. 51, 812–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sheikh H. N. (2009) Tropomyosin Phosphorylation in Cardiac Health and Disease. M.Sc. thesis, University of Cincinnati [Google Scholar]
- 12. Chu G., Lester J. W., Young K. B., Luo W., Zhai J., Kranias E. G. (2000) A single site (Ser16) phosphorylation in phospholamban is sufficient in mediating its maximal cardiac responses to β-agonists. J. Biol. Chem. 275, 38938–38943 [DOI] [PubMed] [Google Scholar]
- 13. Hoshijima M., Ikeda Y., Iwanaga Y., Minamisawa S., Date M. O., Gu Y., Iwatate M., Li M., Wang L., Wilson J. M., Wang Y., Ross J., Jr., Chien K. R. (2002) Chronic suppression of heart-failure progression by a pseudophosphorylated mutant of phospholamban via in vivo cardiac rAAV gene delivery. Nat. Med. 8, 864–871 [DOI] [PubMed] [Google Scholar]
- 14. MacLennan D. H., Kranias E. G. (2003) Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 4, 566–577 [DOI] [PubMed] [Google Scholar]
- 15. Brittsan A. G., Carr A. N., Schmidt A. G., Kranias E. G. (2000) Maximal inhibition of SERCA2 Ca2+ affinity by phospholamban in transgenic hearts overexpressing a non-phosphorylatable form of phospholamban. J. Biol. Chem. 275, 12129–12135 [DOI] [PubMed] [Google Scholar]
- 16. Peña J. R., Szkudlarek A. C., Warren C. M., Heinrich L. S., Gaffin R. D., Jagatheesan G., del Monte F., Hajjar R. J., Goldspink P. H., Solaro R. J., Wieczorek D. F., Wolska B. M. (2010) Neonatal gene transfer of Serca2a delays onset of hypertrophic remodeling and improves function in familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 49, 993–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schmidt U., Hajjar R. J., Kim C. S., Lebeche D., Doye A. A., Gwathmey J. K. (1999) Human heart failure: cAMP stimulation of SR Ca2+-ATPase activity and phosphorylation level of phospholamban. Am. J. Physiol. Heart Circ. Physiol. 277, H474–H480 [DOI] [PubMed] [Google Scholar]
- 18. del Monte F., Hajjar R. J., Harding S. E. (2001) Overwhelming evidence of the beneficial effects of SERCA gene transfer in heart failure. Circ. Res. 88, E66–E67 [DOI] [PubMed] [Google Scholar]
- 19. Force T., Bonow R. O., Houser S. R., Solaro R. J., Hershberger R. E., Adhikari B., Anderson M. E., Boineau R., Byrne B. J., Cappola T. P., Kalluri R., LeWinter M. M., Maron M. S., Molkentin J. D., Ommen S. R., Regnier M., Tang W. H., Tian R., Konstam M. A., Maron B. J., Seidman C. E. (2010) Research priorities in hypertrophic cardiomyopathy: report of a Working Group of the National Heart, Lung, and Blood Institute. Circulation 122, 1130–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wieczorek D. F., Jagatheesan G., Rajan S. (2008) The role of tropomyosin in heart disease. Adv. Exp. Med. Biol. 644, 132–142 [DOI] [PubMed] [Google Scholar]
- 21. Prabhakar R., Petrashevskaya N., Schwartz A., Aronow B., Boivin G. P., Molkentin J. D., Wieczorek D. F. (2003) A mouse model of familial hypertrophic cardiomyopathy caused by a α-tropomyosin mutation. Mol. Cell. Biochem. 251, 33–42 [PubMed] [Google Scholar]
- 22. Prabhakar R., Boivin G. P., Grupp I. L., Hoit B., Arteaga G., Solaro R. J., Wieczorek D. F. (2001) A familial hypertrophic cardiomyopathy alpha-tropomyosin mutation causes severe cardiac hypertrophy and death in mice. J. Mol. Cell. Cardiol. 33, 1815–1828 [DOI] [PubMed] [Google Scholar]
- 23. Muthuchamy M., Pieples K., Rethinasamy P., Hoit B., Grupp I. L., Boivin G. P., Wolska B., Evans C., Solaro R. J., Wieczorek D. F. (1999) Mouse model of a familial hypertrophic cardiomyopathy mutation in α-tropomyosin manifests cardiac dysfunction. Circ. Res. 85, 47–56 [DOI] [PubMed] [Google Scholar]
- 24. Subramaniam A., Jones W. K., Gulick J., Wert S., Neumann J., Robbins J. (1991) Tissue-specific regulation of the α-myosin heavy chain gene promoter in transgenic mice. J. Biol. Chem. 266, 24613–24620 [PubMed] [Google Scholar]
- 25. Muthuchamy M., Grupp I. L., Grupp G., O'Toole B. A., Kier A. B., Boivin G. P., Neumann J., Wieczorek D. F. (1995) Molecular and physiological effects of overexpressing striated muscle β-tropomyosin in the adult murine heart. J. Biol. Chem. 270, 30593–30603 [DOI] [PubMed] [Google Scholar]
- 26. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rajan S., Jagatheesan G., Karam C. N., Alves M. L., Bodi I., Schwartz A., Bulcao C. F., D'Souza K. M., Akhter S. A., Boivin G. P., Dube D. K., Petrashevskaya N., Herr A. B., Hullin R., Liggett S. B., Wolska B. M., Solaro R. J., Wieczorek D. F. (2010) Molecular and functional characterization of a novel cardiac-specific human tropomyosin isoform. Circulation 121, 410–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lang R. M., Bierig M., Devereux R. B., Flachskampf F. A., Foster E., Pellikka P. A., Picard M. H., Roman M. J., Seward J., Shanewise J. S., Solomon S. D., Spencer K. T., Sutton M. S., Stewart W. J. (2005) Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J. Am. Soc. Echocardiogr. 18, 1440–1463 [DOI] [PubMed] [Google Scholar]
- 29. Nagueh S. F., Appleton C. P., Gillebert T. C., Marino P. N., Oh J. K., Smiseth O. A., Waggoner A. D., Flachskampf F. A., Pellikka P. A., Evangelisa A. (2009) Recommendations for the evaluation of left ventricular diastolic function by echocardiography. Eur. J. Echocardiogr. 10, 165–193 [DOI] [PubMed] [Google Scholar]
- 30. Schevzov G., Fath T., Vrhovski B., Vlahovich N., Rajan S., Hook J., Joya J. E., Lemckert F., Puttur F., Lin J. J., Hardeman E. C., Wieczorek D. F., O'Neill G. M., Gunning P. W. (2008) Divergent regulation of the sarcomere and the cytoskeleton. J. Biol. Chem. 283, 275–283 [DOI] [PubMed] [Google Scholar]
- 31. Al Moamen N. J., Prasad V., Bodi I., Miller M. L., Neiman M. L., Lasko V. M., Alper S. L., Wieczorek D. F., Lorenz J. N., Shull G. E. (2011) Loss of the AE3 anion exchanger in a hypertrophic cardiomyopathy model causes rapid decompensation and heart failure. J. Mol. Cell. Cardiol. 50, 137–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Evans C. C., Pena J. R., Phillips R. M., Muthuchamy M., Wieczorek D. F., Solaro R. J., Wolska B. M. (2000) Altered hemodynamics in transgenic mice harboring mutant tropomyosin linked to hypertrophic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 279, H2414–H2423 [DOI] [PubMed] [Google Scholar]
- 33. Krenz M., Robbins J. (2004) Impact of β-myosin heavy chain expression on cardiac function during stress. J. Am. Coll. Cardiol. 44, 2390–2397 [DOI] [PubMed] [Google Scholar]
- 34. Abraham W. T., Gilbert E. M., Lowes B. D., Minobe W. A., Larrabee P., Roden R. L., Dutcher D., Sederberg J., Lindenfeld J. A., Wolfel E. E., Shakar S. F., Ferguson D., Volkman K., Linseman J. V., Quaife R. A., Robertson A. D., Bristow M. R. (2002) Coordinate changes in myosin heavy chain isoform gene expression are selectively associated with alterations in dilated cardiomyopathy phenotype. Mol. Med. 8, 750–760 [PMC free article] [PubMed] [Google Scholar]
- 35. Hasenfuss G., Pieske B. (2002) Calcium cycling in congestive heart failure. J. Mol. Cell. Cardiol. 34, 951–969 [DOI] [PubMed] [Google Scholar]
- 36. Jagatheesan G., Rajan S., Petrashevskaya N., Schwartz A., Boivin G., Arteaga G. M., Solaro R. J., Liggett S. B., Wieczorek D. F. (2007) Rescue of tropomyosin-induced familial hypertrophic cardiomyopathy mice by transgenesis. Am. J. Physiol. Heart Circ. Physiol. 293, H949–H958 [DOI] [PubMed] [Google Scholar]
- 37. Jagatheesan G., Rajan S., Petrashevskaya N., Schwartz A., Boivin G., Vahebi S., DeTombe P., Solaro R. J., Labitzke E., Hilliard G., Wieczorek D. F. (2003) Functional importance of the carboxyl-terminal region of striated muscle tropomyosin. J. Biol. Chem. 278, 23204–23211 [DOI] [PubMed] [Google Scholar]
- 38. Jagatheesan G., Rajan S., Ahmed R. P., Petrashevskaya N., Boivin G., Arteaga G. M., Tae H. J., Liggett S. B., Solaro R. J., Wieczorek D. F. (2010) Striated muscle tropomyosin isoforms differentially regulate cardiac performance and myofilament calcium sensitivity. J. Muscle Res. Cell Motil. 31, 227–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nixon B. R., Liu B., Scellini B., Tesi C., Piroddi N., Ogut O., Solaro R. J., Ziolo M. T., Janssen P. M., Davis J. P., Poggesi C., Biesiadecki B. J. (2013) Tropomyosin Ser-283 pseudo-phosphorylation slows myofibril relaxation. Arch. Biochem. Biophys. 535, 30–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jin J. P., Chong S. M. (2010) Localization of the two tropomyosin-binding sites of troponin T. Arch. Biochem. Biophys. 500, 144–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mamidi R., Michael J. J., Muthuchamy M., Chandra M. (2013) Interplay between the overlapping ends of tropomyosin and the N terminus of cardiac troponin T affects tropomyosin states on actin. FASEB J. 10.1096/fj.13-232363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gollapudi S. K., Gallon C. E., Chandra M. (2013) The tropomyosin binding region of cardiac troponin T modulates crossbridge recruitment dynamics in rat cardiac muscle fibers. J. Mol. Biol. 425, 1565–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Loong C. K., Zhou H. X., Chase P. B. (2012) Familial hypertrophic cardiomyopathy related E180G mutation increases flexibility of human cardiac α-tropomyosin. FEBS Lett. 586, 3503–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ly S., Lehrer S. S. (2012) Long-range effects of familial hypertrophic cardiomyopathy mutations E180G and D175N on the properties of tropomyosin. Biochemistry 51, 6413–6420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li X. E., Suphamungmee W., Janco M., Geeves M. A., Marston S. B., Fischer S., Lehman W. (2012) The flexibility of two tropomyosin mutants, D175N and E180G, that cause hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 424, 493–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Loong C. K., Zhou H. X., Chase P. B. (2012) Persistence length of human cardiac α-tropomyosin measured by single molecule direct probe microscopy. PLoS One 7, e39676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tardiff J. C. (2011) Thin filament mutations: developing an integrative approach to a complex disorder. Circ. Res. 108, 765–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Coutu P., Bennett C. N., Favre E. G., Day S. M., Metzger J. M. (2004) Parvalbumin corrects slowed relaxation in adult cardiac myocytes expressing hypertrophic cardiomyopathy-linked α-tropomyosin mutations. Circ. Res. 94, 1235–1241 [DOI] [PubMed] [Google Scholar]
- 49. Li Y., Charles P. Y., Nan C., Pinto J. R., Wang Y., Liang J., Wu G., Tian J., Feng H. Z., Potter J. D., Jin J. P., Huang X. (2010) Correcting diastolic dysfunction by Ca2+ desensitizing troponin in a transgenic mouse model of restrictive cardiomyopathy. J. Mol. Cell. Cardiol. 49, 402–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Biesiadecki B. J., Tachampa K., Yuan C., Jin J. P., de Tombe P. P., Solaro R. J. (2010) Removal of the cardiac troponin I N-terminal extension improves cardiac function in aged mice. J. Biol. Chem. 285, 19688–19698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Muthu P., Kazmierczak K., Jones M., Szczesna-Cordary D. (2012) The effect of myosin RLC phosphorylation in normal and cardiomyopathic mouse hearts. J. Cell. Mol. Med. 16, 911–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bueno O. F., De Windt L. J., Tymitz K. M., Witt S. A., Kimball T. R., Klevitsky R., Hewett T. E., Jones S. P., Lefer D. J., Peng C. F., Kitsis R. N., Molkentin J. D. (2000) The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 19, 6341–6350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Prekeris R., Hernandez R. M., Mayhew M. W., White M. K., Terrian D. M. (1998) Molecular analysis of the interactions between protein kinase C-ϵ and filamentous actin. J. Biol. Chem. 273, 26790–26798 [DOI] [PubMed] [Google Scholar]
- 54. Hamilton M., Liao J., Cathcart M. K., Wolfman A. (2001) Constitutive association of c-N-Ras with c-Raf-1 and protein kinase Cϵ in latent signaling modules. J. Biol. Chem. 276, 29079–29090 [DOI] [PubMed] [Google Scholar]
- 55. Robia S. L., Ghanta J., Robu V. G., Walker J. W. (2001) Localization and kinetics of protein kinase C-ϵ anchoring in cardiac myocytes. Biophys. J. 80, 2140–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rajan S., Sheikh H. N., Schulz E. M., Jagatheesan G., Wieczorek D. (2010) AHA Basic Cardiovascular Sciences 2010 Scientific Sessions: Technological and Conceptual Advances in Cardiovascular Disease, Rancho Mirage, July 19–22, 2010, Abstr. 17a, American Heart Association, Dallas, TX [Google Scholar]
- 57. Ling S., Sun Q., Li Y., Zhang L., Zhang P., Wang X., Tian C., Li Q., Song J., Liu H., Kan G., Cao H., Huang Z., Nie J., Bai Y., Chen S., Li Y., He F., Zhang L., Li Y. (2012) CKIP-1 inhibits cardiac hypertrophy by regulating class II histone deacetylase phosphorylation through recruiting PP2A. Circulation 126, 3028–3040 [DOI] [PubMed] [Google Scholar]