

Background: Although temperature-sensitive (ts) mutations of TrmD exist, deficient in converting G37- to m1G37-tRNA, the basis of their phenotype is unknown.
Results: The ts-S88L mutation, while conferring thermal lability, caused a stronger defect on catalysis.
Conclusion: The catalytic defect of the ts-S88L mutation reduced the quantity and quality of tRNA methylation.
Significance: ts mutations leading to catalytic defects are useful for studying enzyme mechanism.
Keywords: Bacterial Metabolism, Protein Folding, RNA Methyltransferase, RNA Modification, Transfer RNA (tRNA), TrmD, m1G37-tRNA Modification, Methyl Transfer, Trefoil Knot Fold
Abstract
Conditional temperature-sensitive (ts) mutations are important reagents to study essential genes. Although it is commonly assumed that the ts phenotype of a specific mutation arises from thermal denaturation of the mutant enzyme, the possibility also exists that the mutation decreases the enzyme activity to a certain level at the permissive temperature and aggravates the negative effect further upon temperature upshifts. Resolving these possibilities is important for exploiting the ts mutation for studying the essential gene. The trmD gene is essential for growth in bacteria, encoding the enzyme for converting G37 to m1G37 on the 3′ side of the tRNA anticodon. This conversion involves methyl transfer from S-adenosyl methionine and is critical to minimize tRNA frameshift errors on the ribosome. Using the ts-S88L mutation of Escherichia coli trmD as an example, we show that although the mutation confers thermal lability to the enzyme, the effect is relatively minor. In contrast, the mutation decreases the catalytic efficiency of the enzyme to 1% at the permissive temperature, and at the nonpermissive temperature, it renders further deterioration of activity to 0.1%. These changes are accompanied by losses of both the quantity and quality of tRNA methylation, leading to the potential of cellular pleiotropic effects. This work illustrates the principle that the ts phenotype of an essential gene mutation can be closely linked to the catalytic defect of the gene product and that such a mutation can provide a useful tool to study the mechanism of catalytic inactivation.
Introduction
Essential genes encode critical cellular functions that are not supported by redundant pathways. Because of their indispensability, essential genes have been studied and functionally controlled by using temperature-sensitive (ts)4 alleles. Such alleles are typically mis-sense mutations, which preserve the gene function at permissive and low temperatures but inactivate the gene function upon temperature upshifts. The study of ts phenotypes is fundamental and important for insight into gene essentiality. However, the molecular basis of ts phenotypes of essential genes remains poorly understood; although it is generally assumed that such phenotypes result from thermal inactivation of gene products, the possibility that they arise from inherent functional defects that become lethal at higher temperatures is often overlooked. In the latter case, ts mutations giving rise to severe functional defects at the permissive temperature can be powerful tools to study essential genes and to correlate phenotypes with mechanisms of gene inactivation.
In Escherichia coli, systematic exploration and profiling of single-gene knockouts has suggested that ∼7% of the ∼4,000 protein-coding genes are essential (1, 2). One example of an essential gene is trmD, broadly conserved in all bacterial species, encoding the tRNA methyltransferase for conversion of G37 to m1G37 necessary to prevent frameshift errors on the 3′ side of the anticodon (3–6). In addition to E. coli, the growth essentiality of trmD has also been shown for Streptococcus pneumoniae and Bacillus subtilis (7, 8). In one genetic study of Salmonella typhimurium, several ts mutants of trmD were isolated for the deficiency in m1G37-tRNA synthesis, resulting in changes in the thiamine metabolism flux (9). It was suggested that the trmD deficiency at the restrictive temperature of these mutants reduced the m1G37-tRNA level, such that ribosome translation at codons normally read by the modified tRNA was slow, thus inactivating gene expression in the normal thiamine biosynthesis pathway while activating expression in an alternative pathway. This study showed that the deficiency of trmD had altered the profile of global gene expression, consistent with increasing evidence indicating that tRNA modifications often have regulatory and stress-response roles at genome-wide levels (10).
Although the isolated ts-trmD mutants had been confirmed for the deficiency in m1G37-tRNA synthesis (9), none had been characterized at the structural level for locations, at the molecular level for thermal instability, or at the enzyme level for catalytic defect. To gain insight into these mutations, we have mapped their locations onto the known structure of E. coli TrmD (EcTrmD) in complex with S-adenosyl homocysteine (AdoHcy), the product after methyl transfer (11). From this mapping, we selected for further analysis of the S88L mutation, which is placed closely adjacent to the S-adenosyl methionine (AdoMet)-binding site. Remarkably, TrmD binds AdoMet in a topologically knotted trefoil fold, involving the protein backbone making three passes in and out of a tightly folded loop (11, 12). The location of the S88L mutation within the trefoil knot is attractive for interrogating the molecular basis of its temperature sensitivity.
We report here that although the S88L mutation indeed confers susceptibility to thermal denaturation, the effect is rather minor. Instead, the mutation markedly decreases the enzyme activity relative to the native sequence at the permissive temperature, and it causes further deterioration of activity upon temperature upshifts. We further show that the decrease in the enzyme activity is correlated with decrease in the level of tRNA methylation and the quality of tRNA recognition, suggesting the possibility of accumulating frameshift errors during protein synthesis and altering global gene expression. We suggest that it is the loss of the catalytic efficiency of TrmD by the S88L mutation, and consequently the increase of protein synthesis errors, that is the driving force for the lethal phenotype at the restrictive temperature. This work demonstrates that ts phenotypes can be closely correlated with catalytic defects of an essential gene product and that such correlation can provide unique insight into the function of the essential gene in vivo and the mechanism of action in vitro.
MATERIALS AND METHODS
Construction of the E. coli S88L-trmD-Kan Strain
The S88L-trmD mutation was isolated with a ts phenotype in Salmonella (25). We introduced this mutation to the chromosomal trmD gene of E. coli Xac strain, using the λ Red recombinase-mediated gene disruption method (16). We inserted the E. coli wt-trmD gene to the pQE-30 vector and introduced the ts-S88L mutation, by QuikChange site-directed mutagenesis (Stratagene). The modified coding sequence was amplified by PCR using the following primer set: 5′-ATGTGGATTGGCATAATTAGCCTGTTTCC-3′ (forward) and 5′-GGAACTTCGAAGCAGCTCCAGCCTACACTTACGCCATCCCATCATGTTTATG-3′ (reverse). The PCR product was purified and amplified again according to the megaprimer method (34) in a mixture containing the first PCR product, the kanamycin resistance cassette plasmid pKD4, and the third primer, 5′-ATCCTGGGTAAACTGATATCTCGGGGGCATGGGAATTAGCCATGGTCCATATG-3′. The final PCR product, together with pKD46 (the λ Red recombinase plasmid), was introduced to E. coli Xac strain and colonies were selected on LB-Kan plates. The mutation was verified by sequence analysis, and the temperature sensitivity and curing of pKD46 was confirmed. In parallel, a wt-trmD-Kan construct was made as a control.
Measurement of Kd (AdoMet) by Fluorescence
The quenching of the intrinsic tryptophan fluorescence of EcTrmD by AdoMet binding was determined as described (17). The WT enzyme was titrated at 0.2 μm with AdoMet ranging from 0.2 to 4.5 μm, whereas the S88L mutant enzyme was titrated at 2.0 μm with AdoMet ranging from 1.5 to 22 μm. Enzyme fluorescence was excited at 280 nm in the standard buffer (100 mm Tris-HCl, pH 8.0, 4 mm DTT, 0.1 mm EDTA, 6 mm MgCl2, and 100 mm KCl), and emission was monitored at 320–340 nm at room temperature. Inner filter corrections were calculated by the equation: Fc/Fo = anti-log [(Aex + Aem)/2], where Fc is the dilution-corrected fluorescence, Fo is the observed fluorescence, and Aex and Aem are the absorbencies at the excitation and emission wavelengths, respectively. Nonspecific quenching was measured by titrating AdoMet against a solution of l-Trp. Data corrected after inner filter effects and nonspecific quenching were fit to a hyperbolic equation: y = A × S/(S + Kd), where y is the change in fluorescence from the reference point, A is the maximum change in amplitude, and S is the AdoMet concentration.
Measurement of Cell Growth and Viability
E. coli cells harboring wt or S88L-trmD were grown in LB medium overnight at 30 °C. A fresh culture was inoculated with the overnight culture at 1:100-fold dilution and continued to grow for 3–4 h at 30 °C until A600 = ∼0.3. The culture was split into two, with one shifted to 43 °C by mixing with an equal volume of LB at 55 °C while the other was maintained at 30 °C by mixing with an equal volume of LB at 30 °C. Aliquots of each culture were sampled up to 24 h. Cell viability was determined by directly plating the cell culture at each time point with appropriate dilution. The plates were incubated at 30 or 43 °C, respectively, for additional 12–14 h. The number of colonies formed was normalized by A600 and shown as relative to the time of split.
Measurement of Intracellular TrmD Activity
E. coli cells were grown as above. Each aliquot of cell culture was spun down, and the cells were disrupted by sonication to separate the lysate from debris. The lysate was directly used to measure enzyme activity in the TrmD reaction buffer (100 mm Tris-HCl, pH 8.0, 24 mm NH4Cl, 4 mm DTT, 100 μm EDTA, 6 mm MgCl2, 0.024 mg/ml BSA, 40 units/sample RNasin (Promega)) at 30 or 43 °C, using EctRNAPro as the substrate at 5-fold or lower concentration of Km. The A37 mutant of EctRNAPro, which was not a TrmD substrate, was used to measure methyl transfer to non-G37 positions. [3H-methyl]AdoMet (25 μm) was used, and the synthesis of [3H]m1G37-tRNA was monitored by acid precipitation (35). The initial rate of m1G37-tRNA synthesis was normalized by A600 and shown as relative to the time of culture split.
Western Blot Analysis
Because the endogenous level of TrmD is very low in E. coli (28), we used E. coli strain SG13009 overexpressing the WT or S88L-TrmD from the plasmid pQE30. Cells were grown in LB medium at 30 °C for an hour and then split into three cultures, each at 30, 37, or 43 °C. After 8 h of growth, when the mutant TrmD was inactivated at the restrictive temperatures (see Fig. 5), cells were harvested and sonicated, the soluble cell lysates were collected, and protein concentrations were determined by the Bradford method. Total protein (10 μg) of each cell lysate was loaded to a 12% SDS-PAGE, transferred to Immobilon PVDF membrane (Millipore), and reacted with the primary antibody raised against StTrmD (given by Dr. Glenn Björk). The membrane was incubated with the anti-rabbit IgG secondary antibody conjugated with peroxidase (Sigma-Aldrich), and the signal was detected using SuperSignal West Pico (Thermo Scientific) and quantified by ImageQuant (GE Healthcare). Two independent experiments were performed to report the average signal of TrmD at 37 and 43 relative to 30 °C. Errors are standard deviations.
FIGURE 5.
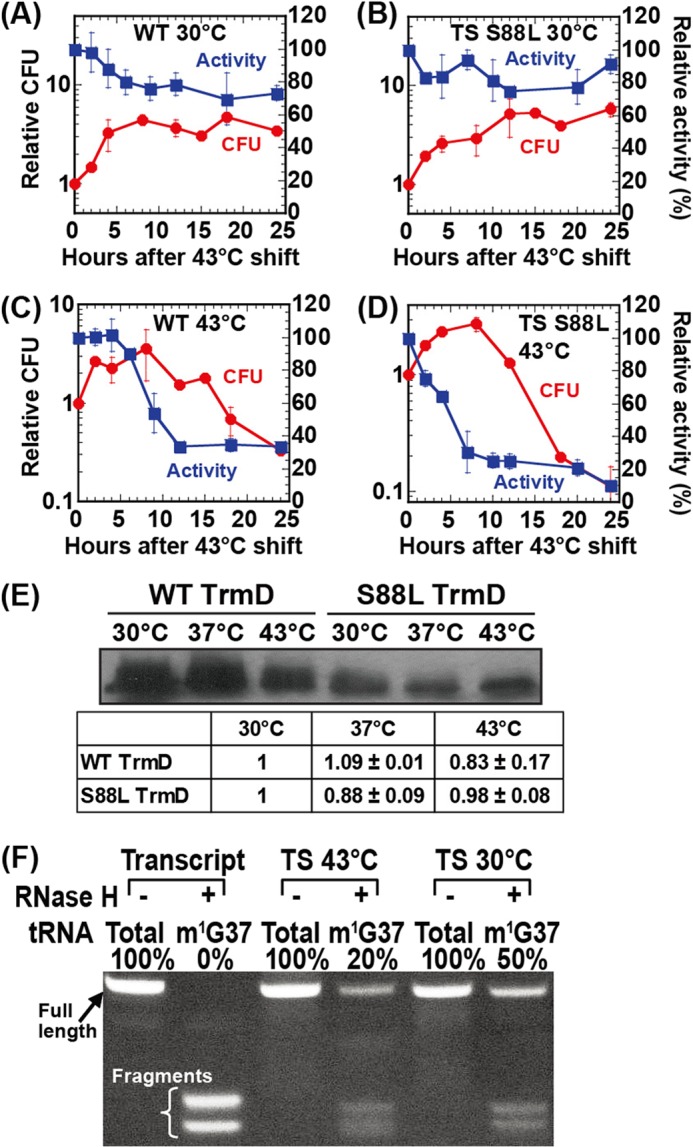
Parameters of E. coli cells harboring wt-trmD and S88L-trmD alleles. A–D, colony forming unit (CFU) analysis of growth (in red) and intracellular activity analysis of TrmD (in blue) of the wt-trmD allele at 30 °C (A), the S88L-trmD allele at 30 °C (B), the wt-trmD allele at 43 °C (C), and the S88L-trmD allele at 43 °C (D). The data are shown as average of two independent experiments. The error bars represent standard deviations. E, Western analysis of cell lysates prepared from E. coli cells overexpressing WT or S88L TrmD. Cells were first grown at 30 °C for 1 h (A600 = ∼0.2) and then shifted to 30, 37, and 43 °C, respectively, for 8 h, and cell lysates with 10 μg of total protein were loaded to a 12% SDS-PAGE and probed with an anti-TrmD antibody, followed by detection with a secondary antibody. The signal of TrmD co-migrated with a purified enzyme in a separate 12% SDS-PAGE stained by Coomassie Blue. The intensity of each signal was quantified using imaging analysis and was calculated relative to the level of each enzyme at 30 °C. The reported ratios were the average of two independent measurements. F, intracellular level of m1G37-tRNA as determined by the resistance to RNaseH digestion. EctRNALeu/CAG was purified from total E. coli tRNA using a biotin-tagged oligonucleotide specific to the D-loop region. Total refers to the combined pool of both G37- and m1G37-tRNA, whereas m1G37 refers to the fraction of only m1G37-tRNA in the combined pool. Transcript refers to the unmodified tRNALeu/CAG prepared from transcription of the tRNA gene. TS 43 °C refers to the tRNA isolated from E. coli S88L-trmD cells grown at 43 °C for 10 h, whereas TS 30 °C refers to the tRNA isolated from E. coli S88L-trmD cells grown at 30 °C for 10 h. The full-length and nuclease-cleaved fragments of the tRNA were separated on a denaturing 12% PAGE, 7 m urea, and the quantities were estimated by ethidium bromide stain.
Measurement of Intracellular Level of m1G37-tRNA
E. coli wt- and S88L-trmD cells were grown at 30 °C to A600 of 0.3 and then shifted to 43 °C or maintained at 30 °C for 10 h. Total RNA was isolated from each cell culture, and the native E. coli tRNALeu/CAG was purified by a biotin-tagged oligonucleotide specific to the D loop region (36). The purified native tRNALeu/CAG was tested by the RNaseH assay (24), using a DNA oligonucleotide complementary to the region of nucleotides from positions 31 to 46. RNaseH-treated tRNAs were separated on a 12% PAGE, 7 m urea. The fractions of intact tRNA and cleaved fragments were estimated based on ethidium bromide staining.
Measurement of CD Spectra
Enzyme was purified by affinity resin and by FPLC, and concentration was determined by UV absorption for CD analysis. Spectra were measured on a JASCO spectrometer J-810 equipped with JASCO Peltier thermo controller. For the wavelength scan, a cuvette with 1-mm path length was used. Each sample (1 μm dimer) in the CD buffer (50 mm Tris-HCl, pH 8.0, 3 mm MgCl2, and 24 mm NH4Cl) was preincubated at 30 or 43 °C for 10 min, and then the spectrum was recorded from 260 to 200 nm at each temperature. Scan speed was 10 nm/min, response was 32 s, and bandwidth was 2 nm. Each spectrum was the average of four runs, and the average of two spectra for each sample was presented. For the temperature scan, a cuvette with 10-mm path length was used. The spectrum of each sample (0.1 μm) in the CD buffer was recorded from 15 to 75 °C at 222 nm. Temperature raise was 24 °C/hour, response was 32 s, and bandwidth was 1 nm. Calculation of α-helical content was based on the equation: % helix = (Δϵ220 − 0.25)/0.105, where Δϵ220 is the measured circular dichroism at 220 nm derived from [θ] = 3330 Δϵ as previously reported (37).
Kinetic Analysis of EcTrmD
WT and the S88L mutant of EcTrmD enzymes, each with an N-His tag, were expressed from pQE30 in E. coli SG13009. Enzymes from one liter of culture were purified using HisLink (Promega). Steady-state kinetic assays were performed as described (35). Each tRNA sample (ranging in final concentration from 1.0 to 128.0 μm) was heat-denatured and annealed before use. Each reaction contained tRNA and saturating [3H-methyl]AdoMet (25 μm) in the standard buffer (0.1 m Tris-HCl, pH 8.0, 4 mm DTT, 0.1 mm EDTA, 6 mm MgCl2, 24 mm NH4Cl, and 0.024 mg/ml BSA) and was initiated by addition of the WT or mutant enzyme at final concentrations 0.025 to 1.5 μm. Aliquots were taken at every 2 min up to 10 min and precipitated by 5% (w/v) TCA on filter pads. Methyl transfer was determined by quantification of [3H] incorporation into acid precipitable counts. After correction of the filter quenching factor, the data of initial rate as a function of tRNA concentration were fit to the Michaelis-Menten equation using the KaleidaGraph software (Synergy software) to determine the Km, kcat, and kcat/Km values. Each value was the average of at least three independent measurements. Errors are standard deviations.
Discrimination analysis between G36- and C36-EctRNALeu was performed in the same buffer. Data for the G36-tRNA was obtained from Table 1. For the C36-tRNA, the WT enzyme was assayed at 1.0 μm with 16.0 and 20.0 μm of tRNA at 30 and 43 °C, respectively, whereas the S88L mutant enzyme was assayed at 1.0 μm with 16 μm tRNA at 30 °C and at 2.6 μm with 50 μm tRNA at 43 °C. Each enzyme-tRNA condition was in the kcat/Km range. Aliquots were taken at every 5 min up to 25 min at 30 °C and at every 10 min up to 50 min at 43 °C. Samples were processed as above, and the initial rate of each reaction (pmol/s) was obtained. The value of kcat/Km (μm−1 s−1) was calculated by dividing the initial rate by the enzyme amount and the tRNA concentration. For all kinetic assays, each value was the average of at least two to three independent measurements, and standard deviations are reported.
TABLE 1.
Steady-state kinetic parameters of TrmD on EctRNALeu
Ratio of kcat/Km was calculated relative to the value of WT-TrmD at 30 °C. Each value is the average of two independent measurements and is reported with standard deviations.
| WT-TrmD |
S88L-TrmD |
|||||
|---|---|---|---|---|---|---|
| 30 °C | 37 °C | 43 °C | 30 °C | 37 °C | 43 °C | |
| Km (μm) | 2.4 ± 0.4 | 3.1 ± 0.3 | 21.7 ± 4.3 | 13.0 ± 1.1 | 52.2 ± 4.6 | 62.5 ± 12.8 |
| kcat (s−1) | (4.9 ± 0.2) × 10−2 | (9.3 ± 0.9) × 10−2 | (1.3 ± 0.1) × 10−1 | (3.2 ± 0.3) × 10−3 | (6.7 ± 0.4) × 10−3 | (1.5 ± 0.2) × 10−3 |
| kcat/Km (μm−1 s−1) | (2.0 ± 0.4) × 10−2 | (3.0 ± 0.4) × 10−2 | (5.9 ± 1.2) × 10−3 | (2.4 ± 0.3) × 10−4 | (1.3 ± 0.1) × 10−4 | (2.5 ± 0.6) × 10−5 |
| kcat/Km ratio | 1.00 | 1.46 | 0.29 | 0.012 | 0.0064 | 0.0012 |
RESULTS
Structural Mapping of ts Mutations of trmD
A genetic study led by Björk and Nilsson (9) isolated a group of ts mutations in S. typhimurium TrmD (StTrmD) that conferred altered thiamine metabolism at a restrictive temperature (P58L/L94F, S88L, G117S, G117N, G117Q, S165L, P184L, G199R, G214D, W217D, and E243K). A separate study by Li and Björk (13) isolated additional ts mutants (E243K, L94F, P184L, G140S, and A145T) that displayed elevated levels of frameshift errors caused by the deficiency in m1G37-tRNA. The two groups of mutants showed overlapping amino acid substitutions at the protein level and collectively occupied 12 positions in the StTrmD enzyme structure. Because these mutations were isolated before the structure of TrmD was available, nothing was known about their structural context.
We now mapped these mutations onto the crystal structure of EcTrmD in complex with AdoHcy (11), which was a logical model based on the over 92% homology in the primary sequence between the S. typhimurium and E. coli enzymes. Although TrmD exists as an obligated homodimer, with each subunit featuring an N-terminal domain (residues 1–159), a flexible linker (residues 160–169), and a C-terminal domain (residues 170–250), the active site is built between the N-terminal domain of one subunit and the flexible linker and the C-terminal domain of the other. In this intriguing cross-subunit active site, AdoMet is bound to the trefoil knot fold in the N-terminal domain, whereas the target G37 is predicted to bind to the flexible linker (11). However, the active site cannot be precisely depicted, because the flexible linker is disordered and invisible in structures that lack tRNA (12), including the EcTrmD structure (11). This prevented the mapping of the S165L mutation. To the rest of the mutations, we found that those localized to the N-terminal domain were near the AdoMet binding site, whereas those localized to the C-terminal domain were in helical regions (Fig. 1).
FIGURE 1.
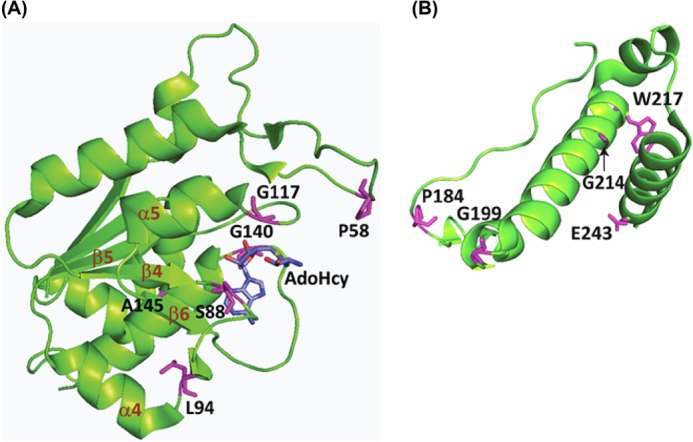
Mapping of ts mutations onto the structure of EcTrmD in complex with AdoHcy (Protein Data Bank code 1P9P) drawn in PyMOL. The ts mutations were isolated from two previous genetic studies (9, 13), and each is marked in purple; AdoHcy is marked in blue by elements. A, ts mutations localized to the N-terminal domain (residues 1–159). B, ts mutations localized to the C-terminal domain (residues 170–250). Note that A and B were made separately in different orientations to clearly display residues of interest. The flexible linker between the two domains is not visible in the structure. The trefoil knot fold in the N-terminal domain is marked with the initiating β4, followed by α4, β5, and α5, and ending with β6 making a circular insertion into the loop created by β4, α4, β5, and α5.
We focused on mutations near the AdoMet binding site to provide a structural framework for interpreting mutational effects. In this framework, AdoMet is bound at the deep end of the trefoil knot fold, which is initiated with a β strand (β4) in the central β sheet structure of the N-terminal domain. The β4 curves around through α4 and β5 and turns into α5 and then into β6, which makes a circular insertion into the loop (Fig. 1A). Within this knot, AdoMet adopts a distinctively bent conformation that places the adenosine and methionine moieties facing each other, which is rare among AdoMet-dependent methyltransferases (12, 14). During methyl transfer, the AdoMet-binding site is presumably “capped” by the flexible linker provided by the C-terminal domain of the other monomer. Proteins with a knotted fold, such as the trefoil knot in TrmD, are rare but are known to have an inherent rigidity not present in unknotted proteins (15).
Of the mutations localized to near the AdoMet site, some were discovered together in one mutant (e.g., P58L and L94F), whereas others were discovered in multiple mutants each with a distinct substitution (e.g., G117S, G117N, and G117Q). We chose to study the S88L mutation, which was discovered in one mutant at a single position. The natural S88 residue is at a position highly conserved among Gram-negative TrmD enzymes but not conserved in Gram-positive (Fig. 2A), indicating the possibility of differential roles in the two classes of bacteria. In the EcTrmD structure, S88 is placed in the beginning of the loop emanating from the central β4 that stabilizes the knot (Fig. 2B). The S88L mutation is therefore attractive for interrogating the molecular basis of its ts phenotypes, because on the one hand it is expected to interfere with AdoMet binding, because of the replacement of the smaller side chain of serine with the bulkier side chain of leucine, whereas on the other hand it is located at a key position of the knot such that the mutation might disrupt the rigidity of the knot and confer sensitivity to thermal denaturation.
FIGURE 2.

The S88L mutation in EcTrmD. A, sequence alignment of the region in TrmD harboring S88L. The annotation is as follows: E. coli (YP_490830), Enterobacter cloacae (YP_004953267), Haemophilus influenzae (NP_438371), Helicobacter pylori (NP_207939), Pseudomonas aeruginosa (NP_252432), Salmonella enterica (NP_461604), B. subtilis (ZP_03591325), Clostridium botulinum (YP_001254944), Enterococcus faecalis (ZP_07107430), Mycobacterium tuberculosis (YP_005309161), Staphylococcus aureus (YP_040627), and Streptococcus pneumoniae (YP_816173). The alignment was generated using the ClustalW program (38). B, the position of S88 in the binary crystal structure of EcTrmD in complex with AdoHcy, showing secondary structural elements β4, α4, β5, α5, and β6 that form the trefoil knot fold (Protein Data Bank code 1P9P). The fold is localized in the N-terminal domain of TrmD, which is connected to the C-terminal domain via an invisible flexible linker.
Introduction of the ts-S88L-trmD Mutation to E. coli
The S88L-TrmD mutation was originally isolated from chemical mutagenesis of S. typhimurium (9). The mutation was identified and confirmed in the trmD gene by complementation analysis using plasmids. Because the mutation occurred at a highly conserved position in Gram-negative TrmD enzymes (Fig. 2A), we tested its broader significance by introducing it to E. coli. This was achieved by replacing the chromosomal wt-trmD locus in E. coli with a plasmid carrying a cassette of the S88L-trmD gene linked to the Kan resistance marker (S88L-trmD-Kan), using the λ Red approach (16), such that the reading frame of trmD was preserved. This consideration was necessary because trmD is present in an operon, consisting of four genes in the order of rpsP (encoding ribosomal protein S16), rimM (encoding a 21-kDa protein that processes 16 S rRNA), trmD, and rplS (encoding ribosomal protein L19) (Fig. 3A). As a control, the Kan resistance cassette of the native trmD sequence (wt-trmD-Kan) was introduced to E. coli to create an isogenic wt strain. This wt-trmD and ts-S88L-trmD pair was compared for the rest of the study. Growth analysis by serial dilution revealed thermal sensitivity of the ts-S88L-trmD allele at 43 °C (Fig. 3B), demonstrating that the S88L mutation also gave rise to a ts phenotype in E. coli. Thus, consistent with the broad sequence conservation of trmD between S. typhimurium and E. coli, the S88L mutation conferred a thermal sensitivity of growth in both bacteria.
FIGURE 3.
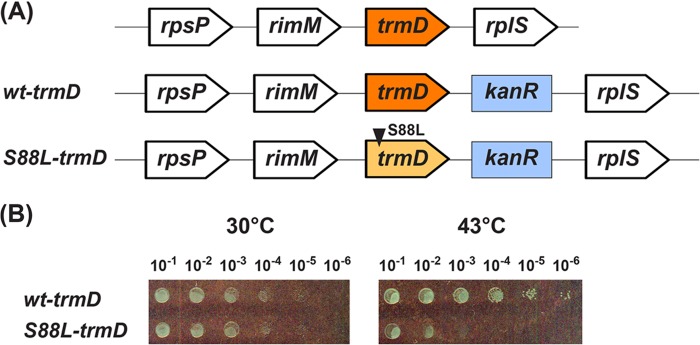
The E. coli wt and S88L-trmD alleles. A, the operon structure harboring wt-trmD-Kan or S88L-trmD-Kan. The operon consists of four genes in the order of rpsP (the ribosomal protein S16), rimM (a 21-kDa protein that processes 16 S rRNA), trmD, and rplS (the ribosomal protein L19). A Kan-resistant cassette containing wt-trmD-Kan or S88L-trmD-Kan was introduced to replace the chromosomal trmD in the operon. B, growth analysis by a dilution series. E. coli cells harboring wt-trmD-Kan or S88L-trmD-Kan were spotted on an LB-Kan plate (Kan at 50 μg/ml) through serial dilutions and incubated at 30 or 43 °C overnight.
The structural and functional significance of the S88L mutation, which was mapped to near the AdoMet-binding site, was confirmed by a fluorescence analysis of enzyme binding to the methyl donor. Both the WT and S88L enzymes were purified from E. coli by a metal affinity resin, followed by anion exchange on an FPLC column to remove residual nucleic acids. Using a fluorescence titration assay developed previously, showing that AdoMet binding quenches the intrinsic fluorescence of EcTrmD in a concentration-dependent manner (17), we monitored fluorescence changes, corrected for inner filter effects and nonspecific quenching, and determined the equilibrium Kd (AdoMet) for these enzymes. Although the Kd (AdoMet) for the WT enzyme was 0.8 ± 0.3 μm (Fig. 4A), closely similar to the value obtained previously (1.1 ± 0.2 μm) (17), the Kd (AdoMet) for the S88L mutant was 7 ± 1 μm (Fig. 4B), almost 10-fold higher. Thus, the S88L mutant was indeed deficient for AdoMet binding, possibly because of the bulkier leucine blocking the methyl donor from accessing the active site. Although this AdoMet binding deficiency of the mutant was detected at room temperature by fluorescence analysis, the effect of temperature shifts on the deficiency is unknown.
FIGURE 4.
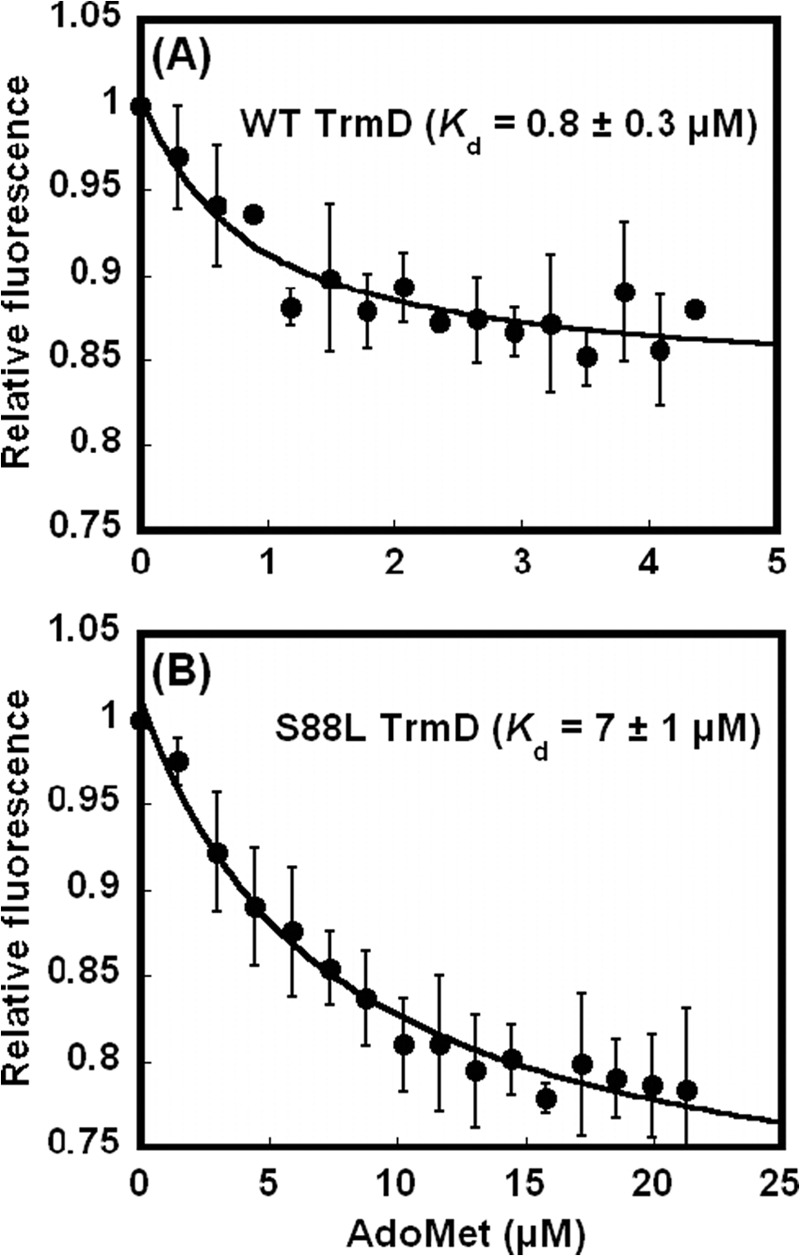
Determination of Kd (AdoMet) by fluorescence. A, intrinsic fluorescence of WT TrmD enzyme upon titration of AdoMet (0.2–4.5 μm), showing the Kd (AdoMet) of 0.8 ± 0.3 μm. B, intrinsic fluorescence of S88L TrmD enzyme upon titration of AdoMet (1.5–22 μm), showing the Kd (AdoMet) of 7 ± 1 μm. Each trace was the average of at least two independent measurements at room temperature. The error bars are standard deviations.
Cellular Consequences of the S88L-trmD Mutation
The arrest of cell growth by the S88L-trmD mutation at the restrictive temperature prompted us to determine the timing of cell death to correlate with intracellular activity of TrmD and levels of m1G37-tRNA at the time of cell death. This correlation was necessary to gain insight into the mechanism of cell death. The timing of cell death was determined by monitoring cell viability over time rather than cell density, because the latter lacked the sensitivity once cells entered the stationary phase. E. coli cells harboring the wt-trmD or ts-trmD allele were first grown at 30 °C to a mid-log phase and were then split into two cultures, with one continuing growth at 30 °C while the other shifted to growth at 43 °C. After this split, analysis of colony-forming units (colony-forming units/ml) over time relative to the time of split showed that although both wt-trmD and ts-trmD cells were viable at 30 °C (Fig. 5, A and B), they gradually lost viability at 43 °C (Fig. 5, C and D). However, although the viability of both began to decay after 7 h at 43 °C, the mutant decayed significantly faster than the wt (Fig. 5, C and D). We marked the onset of the decay, at 7 h after the shift to 43 °C, as the beginning of cell death.
To determine the intracellular TrmD activity, we measured the intracellular enzyme activity in cell lysates as we described (18), using [3H-methyl]AdoMet as the methyl donor to monitor the synthesis of [3H]m1G37-tRNA in acid-precipitable counts. The transcript of EctRNAPro was used as the substrate, because the synthesis of m1G37 on this tRNA had been shown to be essential for bacterial cell growth (19), and key features of the tRNA for the synthesis had been identified (20). The tRNA substrate was maintained at low concentrations relative to the respective Km for the WT and mutant enzymes (Table 1), so that the methyl transfer activity was measured near the kcat/Km condition. The measured acid-precipitable counts were then corrected by removing counts from the methyl transfer activity to non-G37 positions on the tRNA, such as methyl transfer to synthesize m7G46 by the enzyme TrmB, which was also present in cell lysates (21). For the correction, we used the single-substitution mutant A37-EctRNAPro as the substrate, which would not be recognized by TrmD (22) but would allow AdoMet-dependent methylation to all but G37 positions on the tRNA. Analysis of the G37-specific activity in each cell lysate relative to the activity at the time of split showed that, although the enzyme remained active in both wt-trmD and ts-trmD cells at 30 °C (Fig. 5, A and B), it began to deteriorate at 43 °C (Fig. 5, C and D). This deterioration occurred instantly for the mutant but with a 4-h delay in the wt cells (Fig. 5, C and D), showing the temperature sensitivity of the mutant enzyme in cell lysates. After 7 h at 43 °C, when the cell viability began to decline in both cultures, the mutant enzyme retained 30% activity, whereas the wt enzyme retained ∼80% (Fig. 5, C and D). After 24 h at 43 °C, when the cell viability stalled at 30% for the wt cells and at 10% for the mutant, the wt enzyme retained 30% activity, whereas the mutant retained 10%. These results demonstrate a clear correlation between the decline of cell viability and the decrease of intracellular TrmD activity.
We confirmed that the rapid decline of intracellular TrmD activity in the mutant was not due to the instability of the enzyme as the temperature shifted upward. Western analysis using an anti-TrmD antibody showed that both enzymes maintained similar protein levels throughout the temperature range (Fig. 5E). Specifically, whole cell lysates from cells overexpressing WT or mutant enzymes at 30, 37, and 43 °C were prepared and probed at a constant amount of total protein. Although the signal for the WT enzyme was generally stronger than that of the mutant at a specific temperature, the relative level of each enzyme remained within 83% for the WT and 88% for the mutant. Thus, despite the possibility of activating proteases by higher temperatures (23), both the WT and mutant enzyme were considerably resistant to proteolysis.
Upon inactivation of TrmD, cells would contain a mixture of pre-existing m1G37-tRNA and newly synthesized unmodified G37-tRNA. Although the m1G37 state would support normal protein synthesis, the G37 state would introduce frameshift errors; the ratio of the two states would be a determinant of cell death. We measured this ratio using an RNaseH assay that we developed (24), in which the m1G37-tRNA was resistant to hybridization to a complementary DNA oligonucleotide, caused by disruption of W-C base pairing by the modification, whereas the unmodified G37-tRNA was accessible to hybridization and as a result was sensitive to RNaseH cleavage in the hybrid. The fractions of each tRNA resistant or sensitive to cleavage were then resolved by denaturing PAGE. Using EctRNALeu/CAG as an example, we showed that the transcript form, lacking any modifications, was completely cleaved by RNaseH to two fragments, whereas the native form isolated from ts-trmD cells contained a mixture of both intact and cleaved fractions (Fig. 5F). This native form was isolated from total E. coli tRNA using a biotin-tagged oligonucleotide specific to the D-loop, which was nondiscriminative of the G37- and m1G37 states; it was termed “total” because of the presence of both states and was then subjected to RNaseH cleavage using an oligonucleotide specific to the anticodon loop. The result showed that the m1G37 state in the tRNA isolated from the mutant at 43 °C was only 20%, whereas it was 50% from the mutant at 30 °C (Fig. 5F). The lower level of the m1G37 state correlated with the reduced TrmD activity at the restrictive temperature. In contrast, the m1G37 state isolated from wt cells at 30 °C or at 43 °C was similar (∼50%, data not shown). Overall, the level of the m1G37 state decreased from 50% in the permissive condition to 20% in the nonpermissive condition, representing a decrease to 40%. This result was striking, indicating that the reduction of the m1G37 state by merely 2.5-fold was sufficient to initiate the onset of cell death. An earlier study of the S88L-trmD mutation in S. typhimurium had shown that a temperature shift reduced the m1G37 level by 20-fold (25). The discrepancy is due to the fact that our analysis focused on changes of the m1G37 level in a specific tRNA, whereas the earlier analysis examined changes in bulk tRNA, where the m1G37 deficiency in one tRNA might induce alterations in other modifications or in more than one tRNA or accelerate tRNA degradation (26).
In summary, we have established cellular parameters of E. coli ts-S88L-trmD cells relative to the isogenic wt cells, showing that upon temperature shift to 43 °C, although both types of cells decline viability and intracellular TrmD activity, the rate of decline is faster for mutant than for wt cells, indicating ts phenotypes of the S88L-trmD mutation. Importantly, for both types of cells, the rate of viability decline is closely correlated with the rate of TrmD activity decline, indicating that TrmD activity is the major determinant of cell viability in the restricted condition. Our data further show that, after 7 h at 43 °C, marking the onset of cell death; intracellular TrmD activity in mutant cells was reduced to 30% of the normal level, and intracellular m1G37-tRNA level was reduced to 40% of the normal level.
Thermal Denaturation of TrmD in Vitro
The decrease of the intracellular TrmD activity in both wt and S88L mutant cells raised the possibility of structural changes in both enzymes. This possibility was addressed by CD analysis of these enzymes at 30 and 43 °C. CD spectra of a buffer-only sample scanned from 260 to 200 nm revealed stable signals and a sufficiently low dynode voltage (<500 V) to permit detecting signal with high sensitivity. These qualities were maintained in a temperature scan of 15–75 °C at 222 nm and throughout experiments.
CD spectra of the WT enzyme displayed peaks at 208 and 222 nm at both temperatures, indicative of the presence of α-helices (Fig. 6A). The degree of α-helices, determined from the degree of ellipticity, was 24% at 30 °C, similar to values obtained for Thermus thermophilus TrmH, a close structural homolog of TrmD for AdoMet-dependent synthesis of Gm18 (27). This degree of α-helices dropped to 21% at 43 °C, indicating the loss of 3% helical structure. These features were reproduced in the mutant enzyme; CD spectra revealed stable α-helical structure at 26% at 30 °C, which was dropped to 18% at 43 °C (Fig. 6B), indicating the loss of 8% helical structure upon the shift. Thus, whereas both the WT and mutant enzymes were subject to a small degree of thermal denaturation, the mutant was ∼3-fold more sensitive. This was confirmed in a temperature scan analysis of helical elements, showing that the mutant enzyme was generally more thermal labile than the WT and that as the temperature shifted from 30 to 43 °C, the mutant was more prone to lose helical structure by 3-fold (Fig. 6C), consistent with results of spectral analysis.
FIGURE 6.
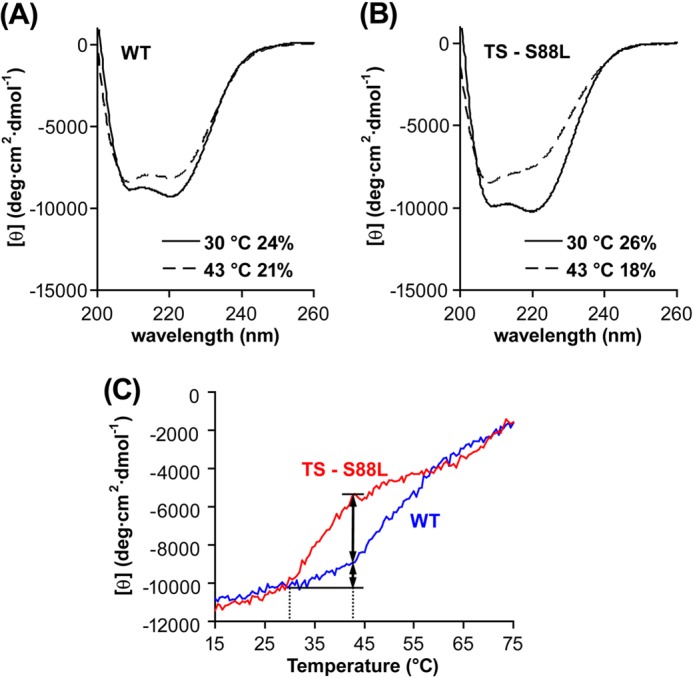
CD spectra of TrmD enzymes. A, CD spectra of the WT enzyme at 30 °C (solid line) and 43 °C (dotted line). B, CD spectra of the S88L mutant enzyme at 30 °C (solid line) and 43 °C (dotted line). C, CD spectra of the WT and mutant enzymes over a temperature scan. The spectrum of each enzymes (at 0.1 μm) in the CD buffer was recorded from 15 to 75 °C at 222 nm, with temperature rise at 24 °C/h, response time at 32 s, and bandwidth at 1 nm. In A and B, the content of α-helix at each temperature was calculated and shown as a percentage.
Enzyme Activity of TrmD in Vitro
To understand the basis of the thermal inactivation, we performed kinetic analysis of the mutant relative to the WT enzyme at three temperatures: 30, 37, and 43 °C. Native gel analysis confirmed that both enzymes migrated to a molecular weight corresponding to a dimer (data not shown), indicating that the S88L mutation, although localized near the dimer interface of TrmD, did not disrupt the dimer structure. The transcript of EctRNALeu/CAG, after heat denaturation and reannealing, was used as the substrate and shown with a capacity of methylation to 80–90% levels in extended time courses. Steady-state parameters were determined and summarized in Table 1. AdoMet was maintained at 25 μm, which was saturating for both the WT and mutant enzymes (Kd = 0.8 ± 0.3 and 7 ± 1 μm; Fig. 4). For the WT enzyme, the parameters at 37 °C (Km (tRNA), kcat, and kcat/Km (tRNA)) were closely similar to published data (11, 17). As the temperature shifted downward or upward, the kcat value maintained relatively constant, whereas the Km value varied, showing a small decrease when measured at 30 °C relative to 37 °C and a significant increase at 43 °C. These variations resulted in changes in the kcat/Km value for catalytic efficiency. Using the kcat/Km value at 30 °C as the reference, the catalytic efficiency of the WT enzyme increased by 1.5-fold at 37 °C but decreased by 3.4-fold at 43 °C. However, the S88L mutant enzyme was severely defective even at 30 °C, showing a 100-fold decrease in kcat/Km relative to WT because of a defect in both Km (tRNA) and kcat. As the temperature increased to 37 and 43 °C, the Km defect worsened progressively, whereas the kcat defect remained relatively unchanged, a trend consistent with the WT enzyme (Table 1). Overall, the S88L mutant showed a decrease in kcat/Km by 1.8-fold at 37 °C and by 10-fold at 43 °C. Thus, except for the 100-fold difference in their catalytic efficiency at 30 °C, the WT and mutant enzymes reacted similarly to thermal inactivation, both displaying a temperature-dependent increase in Km (tRNA) and an overall decrease in kcat/Km by 3–10-fold as the temperature shifted from 30 to 43 °C. As a result of the inherent catalytic defect and the temperature-dependent decline of activity, the mutant at 43 °C was only acting at 0.1% capacity of the WT capacity at 30 °C.
Discrimination of tRNA by TrmD in Vitro
The localization of the S88L mutation to near the AdoMet-binding site of TrmD raised the question of whether it would interfere with tRNA discrimination for methyl transfer and, if so, whether the interference of tRNA discrimination would be amplified upon temperature upshifts. The key feature of tRNA discrimination by TrmD is recognition of G36, the nucleotide preceding the target G37 (22). Substitutions of the G36 determinant with base analogs decrease methyl transfer by 10–100-fold (18). To more specifically probe the significance of G36, we evaluated the ability of the WT and mutant enzyme to discriminate between G36 and the C36 variant of EctRNALeu/CAG transcript. Because of the substantially large increases of Km (tRNA) for both enzymes upon temperature shift to 43 °C (Table 1), indicating the difficulty to reach substrate saturation, we did not determine individual values of kcat or Km (tRNA) but instead evaluated the enzyme activity by estimating the kcat/Km parameter, with each tRNA measured at a sub-Km concentration (Table 2).
TABLE 2.
Steady-state discrimination of EctRNALeu by TrmD
Each value is the average of two independent measurements and is reported with standard deviations.
| WT-TrmD |
S88L-TrmD |
|||
|---|---|---|---|---|
| 30 °C | 43 °C | 30 °C | 43 °C | |
| kcat/Km (μm−1s−1) | ||||
| G36-tRNA | (2.0 ± 0.4) × 10−2 | (6 ± 1) × 10−3 | (2.4 ± 0.3) × 10−4 | (2.5 ± 0.6) × 10−5 |
| C36-tRNA | (3.3 ± 0.2) × 10−5 | (2.1 ± 0.1) × 10−5 | (1.6 ± 0.1) × 10−6 | (2.8 ± 0.6) × 10−7 |
| Fold discrimination G36/C36 | 615 | 287 | 158 | 87 |
| Relative to 30 °C | 1.0 | 0.47 | 1.0 | 0.55 |
The WT enzyme discriminated G36- from C36-tRNA effectively by a factor of 615 at 30 °C, supporting the notion that the G36 nucleotide plays a major role in tRNA discrimination. This discrimination dropped by ∼2-fold to a factor of 287 at 43 °C, indicating that thermal denaturation of the enzyme had relaxed the stringency of tRNA discrimination. Interestingly, the mutant enzyme indeed displayed less discrimination relative to the WT, by a factor of only 158 at 30 °C, supporting the notion that the mutation near the AdoMet-binding site had an adverse effect on the quality of tRNA discrimination. The 4-fold decrease of discrimination from 615- to 158-fold at 30 °C indicated an energetic penalty of ∼1 kcal/mol. At 43 °C, the mutant further relaxed the stringency of tRNA discrimination, but this relaxation was similar to the 2-fold relaxation of the WT enzyme. However, despite the lack of amplification of the adverse effect by temperature shift, the mutant was acting with a 7-fold less stringency at 43 °C relative to WT at 30 °C, representing a reduction to ∼15% in the quality of tRNA discrimination.
DISCUSSION
Insight into the mechanism of temperature sensitivity of macromolecules caused by a mutation has been limited by the lack of biochemical assays to directly correlate the loss of activity with the susceptibility to thermal denaturation. In many studies, ts phenotypes are inferred only from thermal instability. Here we use the S88L mutation of the essential enzyme TrmD as an example to illustrate the correlation of thermal instability with loss of activity for both the WT and mutant enzymes. We show by CD analysis that although the mutation does confer a higher degree of thermal instability relative to the native sequence, the effect is rather mild on the order of ∼3-fold from 30 to 43 °C. This mild effect is not detected at protein levels by Western analysis, and it is consistent with activity analysis of TrmD upon the temperature shift (Table 1), showing that although both the WT and mutant enzymes decrease in activity, the extent of decrease of the mutant (10-fold) is 3-fold more severe relative to the WT (3-fold). However, whereas the mutant is indeed more prone to thermal inactivation both in structure and in activity, it has catalytic deficiency acting at 1% of the WT capacity at 30 °C, which deteriorates to 0.1% capacity at 43 °C (Table 1). This decreased catalytic efficiency is accompanied by the reduced quality of tRNA discrimination to 15% of the WT (Table 2). For TrmD, the residual 0.1% capacity of catalytic efficiency and 15% quality of tRNA recognition is likely the major cause of cell death at 43 °C. This model is supported by the observation that the decline of intracellular TrmD activity over time is closely correlated with the decline of cell viability. At the onset of cell death, we show that cells with S88L-TrmD maintain m1G37-tRNA only at 40% level of the normal.
Is the 0.1% catalytic capacity and 15% quality of TrmD lethal to cells? We have shown that the intracellular concentration of EcTrmD in wt cells is already low (∼0.1 μm) (18) relative to its cellular tRNA substrates (∼60 μm). The low enzyme level is consistent with a previous study showing that translation of trmD is the lowest among the four genes in the operon (28). This presents a challenging situation, implying that the enzyme must possess high levels of efficiency and quality to rapidly screen for, bind to, and catalyze methyl transfer to all of its tRNA substrates to provide m1G37-tRNA for cell survival. Given that the concentration difference between TrmD and its tRNA substrates inside an E. coli cell is already large (∼600-fold), a decrease of the enzyme catalytic efficiency to 0.1% and quality to 15% will further aggravate the difference by leaving many substrate tRNA molecules unmodified and in the highly error-prone G37 state.
Is the residual 40% level of m1G37-tRNA lethal to cells? Some tRNA substrates for TrmD have a strong propensity to induce frameshift errors on the ribosome, such as EctRNAPro/GGG (GGG: the anticodon sequence), which reads CC(C/U) codons (29). Deficiency of m1G37 in this tRNA increases +1 frameshift errors, particularly at slippery sequences such as CC(C/U)(C/U), which we estimate occurs at a notable frequency of ∼0.4% among E. coli protein-coding sequences. A larger impact, however, is that accumulation of frameshift errors can generate translational stress at global levels, such as the earlier observation that TrmD deficiency forced bacteria to switch to an alternative thiamine biosynthesis pathway (9). An emerging theme has been that tRNA modifications are intimately involved in cellular stress response. For example, the deficiency of Trm9 affects cellular response to DNA damage, because translation of damage response genes requires Trm9-dependent tRNA modification for the last step of synthesis of mcm5U34 in tRNAArg/UCU and mcm5s2U34 in tRNAGlu/UUC (30, 31). Similarly, the deficiency of Trm4 affects cellular response to oxidative stress, because translation of stress-response genes requires Trm4-dependent methylation in m5C34 of tRNALeu/CAA (32). These examples suggest a role of tRNA modifications in translational control of gene expression. For the deficiency of m1G37-tRNA, whereas the full extent of global impact on protein synthesis is unknown, the deficiency may affect the modification pattern of other tRNA molecules, based on a genome wide analysis showing that the synthesis of m1G37, m7G46, and Gm18 appears interdependent (33). This interdependence can further expand the impact of m1G37 deficiency to levels of gene expression involving m7G46 and Gm18. The potential of multiplicity and collateral damage may explain the observation of cell death at 40% level of m1G37-tRNA.
The isolation of S88L-trmD by ts phenotypes has identified a mutation that induces a mild thermal denaturation relative to the wt sequence while more importantly generating a significant catalytic defect that further aggravates upon temperature upshifts. The catalytic defect of this mutation at the permissive temperature now provides a basis to probe its perturbation of the structure and mechanism of TrmD and to link the perturbation to cellular deficiencies. This work demonstrates the benefits of isolating ts mutations of an essential gene and using the mutations to correlate gene functions in vivo and in vitro.
Acknowledgments
We thank Dr. Anshul Bhardwaj and Dr. Michael Root for guidance and discussion of CD analysis, Dr. Glenn Björk for anti-TrmD antibody, and Tom Christian for creating the C36 mutant tRNA.
This work was supported, in whole or in part, by National Institutes of Health Grant GM081601 (to Y. M. H.).
- ts
- temperature-sensitive
- AdoMet
- S-adenosyl methionine
- AdoHcy
- S-adenosyl homocysteine
- Kan
- kanamycin.
REFERENCES
- 1. Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K. A., Tomita M., Wanner B. L., Mori H. (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nichols R. J., Sen S., Choo Y. J., Beltrao P., Zietek M., Chaba R., Lee S., Kazmierczak K. M., Lee K. J., Wong A., Shales M., Lovett S., Winkler M. E., Krogan N. J., Typas A., Gross C. A. (2011) Phenotypic landscape of a bacterial cell. Cell 144, 143–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Byström A. S., Björk G. R. (1982) The structural gene (trmD) for the tRNA(m1G)methyltransferase is part of a four polypeptide operon in Escherichia coli K-12. Mol. Gen. Genet. 188, 447–454 [DOI] [PubMed] [Google Scholar]
- 4. Byström A. S., Björk G. R. (1982) Chromosomal location and cloning of the gene (trmD) responsible for the synthesis of tRNA (m1G) methyltransferase in Escherichia coli K-12. Mol. Gen. Genet. 188, 440–446 [DOI] [PubMed] [Google Scholar]
- 5. Björk G. R., Wikström P. M., Byström A. S. (1989) Prevention of translational frameshifting by the modified nucleoside 1-methylguanosine. Science 244, 986–989 [DOI] [PubMed] [Google Scholar]
- 6. Hagervall T. G., Tuohy T. M., Atkins J. F., Björk G. R. (1993) Deficiency of 1-methylguanosine in tRNA from Salmonella typhimurium induces frameshifting by quadruplet translocation. J. Mol. Biol. 232, 756–765 [DOI] [PubMed] [Google Scholar]
- 7. O'Dwyer K., Watts J. M., Biswas S., Ambrad J., Barber M., Brulé H., Petit C., Holmes D. J., Zalacain M., Holmes W. M. (2004) Characterization of Streptococcus pneumoniae TrmD, a tRNA methyltransferase essential for growth. J. Bacteriol. 186, 2346–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kobayashi K., Ehrlich S. D., Albertini A., Amati G., Andersen K. K., Arnaud M., Asai K., Ashikaga S., Aymerich S., Bessieres P., Boland F., Brignell S. C., Bron S., Bunai K., Chapuis J., Christiansen L. C., Danchin A., Débarbouille M., Dervyn E., Deuerling E., Devine K., Devine S. K., Dreesen O., Errington J., Fillinger S., Foster S. J., Fujita Y., Galizzi A., Gardan R., Eschevins C., Fukushima T., Haga K., Harwood C. R., Hecker M., Hosoya D., Hullo M. F., Kakeshita H., Karamata D., Kasahara Y., Kawamura F., Koga K., Koski P., Kuwana R., Imamura D., Ishimaru M., Ishikawa S., Ishio I., Le Coq D., Masson A., Mauël C., Meima R., Mellado R. P., Moir A., Moriya S., Nagakawa E., Nanamiya H., Nakai S., Nygaard P., Ogura M., Ohanan T., O'Reilly M., O'Rourke M., Pragai Z., Pooley H. M., Rapoport G., Rawlins J. P., Rivas L. A., Rivolta C., Sadaie A., Sadaie Y., Sarvas M., Sato T., Saxild H. H., Scanlan E., Schumann W., Seegers J. F., Sekiguchi J., Sekowska A., Seror S. J., Simon M., Stragier P., Studer R., Takamatsu H., Tanaka T., Takeuchi M., Thomaides H. B., Vagner V., van Dijl J. M., Watabe K., Wipat A., Yamamoto H., Yamamoto M., Yamamoto Y., Yamane K., Yata K., Yoshida K., Yoshikawa H., Zuber U., Ogasawara N. (2003) Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. U.S.A. 100, 4678–4683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Björk G. R., Nilsson K. (2003) 1-Methylguanosine-deficient tRNA of Salmonella enterica serovar Typhimurium affects thiamine metabolism. J. Bacteriol. 185, 750–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yi C., Pan T. (2011) Cellular dynamics of RNA modification. Acc. Chem. Res. 44, 1380–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Elkins P. A., Watts J. M., Zalacain M., van Thiel A., Vitazka P. R., Redlak M., Andraos-Selim C., Rastinejad F., Holmes W. M. (2003) Insights into catalysis by a knotted TrmD tRNA methyltransferase. J. Mol. Biol. 333, 931–949 [DOI] [PubMed] [Google Scholar]
- 12. Ahn H. J., Kim H. W., Yoon H. J., Lee B. I., Suh S. W., Yang J. K. (2003) Crystal structure of tRNA(m1G37)methyltransferase. Insights into tRNA recognition. EMBO J. 22, 2593–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li J. N., Björk G. R. (1999) Structural alterations of the tRNA(m1G37)methyltransferase from Salmonella typhimurium affect tRNA substrate specificity. RNA 5, 395–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schubert H. L., Blumenthal R. M., Cheng X. (2003) Many paths to methyltransfer. A chronicle of convergence. Trends Biochem. Sci. 28, 329–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. King N. P., Jacobitz A. W., Sawaya M. R., Goldschmidt L., Yeates T. O. (2010) Structure and folding of a designed knotted protein. Proc. Natl. Acad. Sci. U.S.A. 107, 20732–20737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Datsenko K. A., Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Christian T., Lahoud G., Liu C., Hou Y. M. (2010) Control of catalytic cycle by a pair of analogous tRNA modification enzymes. J. Mol. Biol. 400, 204–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sakaguchi R., Giessing A., Dai Q., Lahoud G., Liutkeviciute Z., Klimasauskas S., Piccirilli J., Kirpekar F., Hou Y. M. (2012) Recognition of guanosine by dissimilar tRNA methyltransferases. RNA 18, 1687–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Näsvall S. J., Nilsson K., Björk G. R. (2009) The ribosomal grip of the peptidyl-tRNA is critical for reading frame maintenance. J. Mol. Biol. 385, 350–367 [DOI] [PubMed] [Google Scholar]
- 20. Christian T., Hou Y. M. (2007) Distinct determinants of tRNA recognition by the TrmD and Trm5 methyl transferases. J. Mol. Biol. 373, 623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. De Bie L. G., Roovers M., Oudjama Y., Wattiez R., Tricot C., Stalon V., Droogmans L., Bujnicki J. M. (2003) The yggH gene of Escherichia coli encodes a tRNA (m7G46) methyltransferase. J. Bacteriol. 185, 3238–3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Redlak M., Andraos-Selim C., Giege R., Florentz C., Holmes W. M. (1997) Interaction of tRNA with tRNA (guanosine-1)methyltransferase. Binding specificity determinants involve the dinucleotide G36pG37 and tertiary structure. Biochemistry 36, 8699–8709 [DOI] [PubMed] [Google Scholar]
- 23. Guisbert E., Herman C., Lu C. Z., Gross C. A. (2004) A chaperone network controls the heat shock response in E. coli. Genes Dev. 18, 2812–2821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hou Y. M., Li Z., Gamper H. (2006) Isolation of a site-specifically modified RNA from an unmodified transcript. Nucleic Acids Res. 34, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Björk G. R., Jacobsson K., Nilsson K., Johansson M. J., Byström A. S., Persson O. P. (2001) A primordial tRNA modification required for the evolution of life? EMBO J. 20, 231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alexandrov A., Chernyakov I., Gu W., Hiley S. L., Hughes T. R., Grayhack E. J., Phizicky E. M. (2006) Rapid tRNA decay can result from lack of nonessential modifications. Mol. Cell 21, 87–96 [DOI] [PubMed] [Google Scholar]
- 27. Watanabe K., Nureki O., Fukai S., Ishii R., Okamoto H., Yokoyama S., Endo Y., Hori H. (2005) Roles of conserved amino acid sequence motifs in the SpoU (TrmH) RNA methyltransferase family. J. Biol. Chem. 280, 10368–10377 [DOI] [PubMed] [Google Scholar]
- 28. Wikström P. M., Björk G. R. (1988) Noncoordinate translation-level regulation of ribosomal and nonribosomal protein genes in the Escherichia coli trmD operon. J. Bacteriol. 170, 3025–3031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qian Q., Björk G. R. (1997) Structural alterations far from the anticodon of the tRNAProGGG of Salmonella typhimurium induce +1 frameshifting at the peptidyl-site. J. Mol. Biol. 273, 978–992 [DOI] [PubMed] [Google Scholar]
- 30. Begley U., Dyavaiah M., Patil A., Rooney J. P., DiRenzo D., Young C. M., Conklin D. S., Zitomer R. S., Begley T. J. (2007) Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol. Cell 28, 860–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Patil A., Dyavaiah M., Joseph F., Rooney J. P., Chan C. T., Dedon P. C., Begley T. J. (2012) Increased tRNA modification and gene-specific codon usage regulate cell cycle progression during the DNA damage response. Cell Cycle 11, 3656–3665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chan C. T., Pang Y. L., Deng W., Babu I. R., Dyavaiah M., Begley T. J., Dedon P. C. (2012) Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 3, 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chan C. T., Dyavaiah M., DeMott M. S., Taghizadeh K., Dedon P. C., Begley T. J. (2010) A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 6, e1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen J. R., Lü J. J., Wang H. F. (2008) Rapid and efficient gene splicing using megaprimer-based protocol. Mol. Biotechnol. 40, 224–230 [DOI] [PubMed] [Google Scholar]
- 35. Christian T., Evilia C., Williams S., Hou Y. M. (2004) Distinct origins of tRNA(m1G37) methyltransferase. J. Mol. Biol. 339, 707–719 [DOI] [PubMed] [Google Scholar]
- 36. Yokogawa T., Kitamura Y., Nakamura D., Ohno S., Nishikawa K. (2010) Optimization of the hybridization-based method for purification of thermostable tRNAs in the presence of tetraalkylammonium salts. Nucleic Acids Res. 38, e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Clark D. J., Hill C. S., Martin S. R., Thomas J. O. (1988) α-Helix in the carboxy-terminal domains of histones H1 and H5. EMBO J. 7, 69–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thompson J. D., Higgins D. G., Gibson T. J. (1994) CLUSTAL W. Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]