

Background: Thebaine 6-O-demethylase (T6ODM) and codeine O-demethylase (CODM) catalyze late steps of morphine biosynthesis in opium poppy.
Results: New dealkylation reactions, including O,O-demethylenation, have been detected for T6ODM, CODM, and other 2-oxoglutarate/Fe(II)-dependent dioxygenases (ODDs). Virus-induced gene silencing supports physiological functions.
Conclusion: Certain ODDs are multifunctional dealkylating enzymes with widespread roles in benzylisoquinoline alkaloid metabolism.
Significance: Enzymes responsible for O-demethylation and O,O-demethylenation in plant alkaloid biosynthesis have been discovered.
Keywords: Dioxygenase, Gene Silencing, Mass Spectrometry (MS), Metabolic Regulation, Plant Biochemistry, Plant Physiology, Secondary Metabolism
Abstract
In opium poppy, the antepenultimate and final steps in morphine biosynthesis are catalyzed by the 2-oxoglutarate/Fe(II)-dependent dioxygenases, thebaine 6-O-demethylase (T6ODM) and codeine O-demethylase (CODM). Further investigation into the biochemical functions of CODM and T6ODM revealed extensive and unexpected roles for such enzymes in the metabolism of protopine, benzo[c]phenanthridine, and rhoeadine alkaloids. When assayed with a wide range of benzylisoquinoline alkaloids, CODM, T6ODM, and the functionally unassigned paralog DIOX2, renamed protopine O-dealkylase, showed novel and efficient dealkylation activities, including regio- and substrate-specific O-demethylation and O,O-demethylenation. Enzymes catalyzing O,O-demethylenation, which cleave a methylenedioxy bridge leaving two hydroxyl groups, have previously not been reported in plants. Similar cleavage of methylenedioxy bridges on substituted amphetamines is catalyzed by heme-dependent cytochromes P450 in mammals. Preferred substrates for O,O-demethylenation by CODM and protopine O-dealkylase were protopine alkaloids that serve as intermediates in the biosynthesis of benzo[c]phenanthridine and rhoeadine derivatives. Virus-induced gene silencing used to suppress the abundance of CODM and/or T6ODM transcripts indicated a direct physiological role for these enzymes in the metabolism of protopine alkaloids, and they revealed their indirect involvement in the formation of the antimicrobial benzo[c]phenanthridine sanguinarine and certain rhoeadine alkaloids in opium poppy.
Introduction
Benzylisoquinoline alkaloids (BIAs)3 are a large and structurally diverse group of nitrogenous specialized metabolites mainly produced in a restricted number of plants in the order Ranunculales, including the family Papaveraceae. Opium poppy (Papaver somniferum) produces several BIAs many of which have important pharmacological properties, including the narcotic analgesics morphine and codeine, the cough suppressant and promising anticancer drug noscapine, the vasodilator and antispasmodic papaverine, and the antimicrobial agent sanguinarine (1). BIA biosynthesis begins with the conversion of two molecules of l-tyrosine to dopamine and 4-hydroxyphenylacetaldehyde, which undergo Pictet-Spengler condensation to yield (S)-norcoclauine (2), the central intermediate to an estimated 2,500 alkaloids. Successive 6-O-methylation, N-methylation, 3′-hydroxylation, and 4′-O-methylation reactions result in the formation of the central branch point intermediate (S)-reticuline. Intramolecular carbon-carbon and carbon-oxygen coupling of (S)-reticuline and downstream pathway intermediates generate a plethora of structural backbones that are differentially modified through oxidation, reduction, and the addition of various functional groups. In opium poppy, major BIA subgroups include 1-benzylisoquinoline (e.g. papaverine), morphinan (e.g. morphine), phthalideisoquinoline (e.g. noscapine), and benzo[c]phenanthridine (e.g. sanguinarine) alkaloids. A restricted number of enzyme families and superfamilies are known to be involved in BIA metabolism, including oxidoreductases such as cytochromes P450 (CYPs) and FAD-dependent oxidases (1). Recently, a nonbiased comparative genomics approach revealed the involvement of an additional oxidoreductase family, namely 2-oxoglutarate/Fe(II)-dependent dioxygenases (ODDs), in morphinan alkaloid metabolism (3). Thebaine 6-O-demethylase (T6ODM) and codeine O-demethylase (CODM) are ODD enzymes responsible for the penultimate step in codeine biosynthesis and the final conversion of codeine to morphine, respectively. Prior to their discovery, the longstanding assumption was that morphinan alkaloid O-demethylation in opium poppy was achieved by CYPs based on the participation of such enzymes in human morphine metabolism (4). T6ODM and CODM represent two of only a small number of plant enzymes capable of catalyzing O-demethylation. Moreover, the recruitment of these two highly similar enzymes represents an apparently isolated evolutionary event that conferred the unique ability to synthesize codeine and morphine to opium poppy.
The discovery of T6ODM and CODM provided a rational basis for hypothesizing the occurrence of additional O-demethylation events in BIA metabolism and prompted a reevaluation of established biosynthetic pathways (5). Such consideration revealed several alkaloid end products with functional group substitution patterns that suggested the O-demethylation of upstream pathway intermediates. For example, numerous protoberberine and aporphine alkaloids lack one or both methyl groups presumably derived from the 6-O- and 4′-O-methyl moieties of the central intermediate (S)-reticuline. The existence of several alkaloids with such landmark substitution patterns suggests that O-demethylation could play a widespread role in BIA biosynthesis. The original isolation and characterization of T6ODM and CODM targeted their roles in morphine biosynthesis. However, the possible relevance of these enzymes in other aspects of BIA metabolism has not yet been considered.
In this paper, we extend the catalytic functions of T6ODM and CODM in opium poppy beyond the biosynthesis of morphine and assign an activity to the previously uncharacterized enzyme DIOX2, which was renamed PODA. All three enzymes differentially accepted, in some cases with considerable catalytic efficiency, BIA substrates belonging to various structural subgroups. Certain protopine and protoberberine alkaloids were particularly effective substrates, in addition to the previously reported acceptance of morphinan alkaloids (3). Interestingly, although several substrates undergo regiospecific O-demethylation similar to the late steps in morphine biosynthesis, CODM and PODA catalyze the O,O-demethylenation of methylenedioxy bridges on protopine alkaloids and, in the case of PODA, on protoberberine alkaloids containing this functional group. The metabolic roles of ODDs in the O-demethylation and O,O-demethylenation of protopine compounds and downstream derivatives (Fig. 1), such as sanguinarine and rhoeadine alkaloids, was demonstrated by suppressing specific gene transcript levels using virus-induced gene silencing (VIGS). The removal and perceived reformation of O-linked methyl groups and methylenedioxy bridges appear to play a substantial role in the direction of flux though highly branched BIA metabolism especially beyond the formation of the protoberberine backbone, from which the protopine, benzo[c]phenanthridine, and rhoeadine subgroups are derived. Although CYP-mediated O,O-demethylenation of amphetamine analogs (6–8) and BIAs (9, 10) has been reported in mammalian systems, this is the first reported occurrence of O,O-demethylenation in plants and the only one in which ODDs are involved. Plant ODDs perform a variety of reactions, including hydroxylation of aliphatic or aromatic moieties, dehydration, O- and N-demethylation, γ-ring formation, and oxidative removal of methylsulfinyl groups (5). We extend the function of ODDs to include O,O-demethylenation and provide evidence for an expanded role of ODDs in BIA metabolism.
FIGURE 1.

Biosynthesis of major protopine alkaloids and sanguinarine in opium poppy. Establishment of a methylene bridge in (S)-reticuline by the berberine bridge enzyme (BBE) yields the protoberberine alkaloid (S)-scoulerine, which can undergo O-methylation, incorporation of up to two methylenedioxy bridges, and N-methylation yielding quaternary alkaloids suitable for the formation of the protopine backbone via 14-hydroxylation. Subsequent 6-hydroxylation and tautomerization yields the benzo[c]phenanthridine alkaloid dihydrosanguinarine, which is further oxidized to sanguinarine. Abbreviations used are as follows: CFS, cheilanthifoline synthase; SPS, stylopine synthase; TNMT, tetrahydroberberine N-methyltransferase; MSH, N-methylstylopine 14-hydroxylase; P6H, protopine-6-hydroxylase; DBOX, dihydrobenzophenenthridine oxidase; NCS, norcoclaurine synthase; SOMT, scoulerine O-methyltransferase; CAS, canadine synthase.
EXPERIMENTAL PROCEDURES
Plant Material
The cultivation of opium poppy (P. somniferum L. cultivar Bea's Choice) and the collection of plant material used for analysis have been described previously (11).
Chemicals and Reagents
Allocryptopine and (S)-scoulerine were purchased from ChromaDex (Irvine, CA). Berberine, papaverine, and sanguinarine were from Sigma. Canadine was from Latoxan (Valence, France). (S)-Isocorydine, berbamine, (S)-boldine, (S)-corytuberine, (S)-glaucine, and (S)-isothebaine were from Sequoia Research Products (St. James Close, UK). Protopine was from Idofine (Hillsborough, NJ). Cryptopine was purchased from MP Biomedicals (Santa Ana, CA). (R,S)-Tetrahydropalmatine was from Ethnogarden Botanicals (Barrie, Ontario, Canada). Codeine and morphine were gifts from Sanofi-Aventis (Paris, France). (S)-Reticuline was a gift from Tasmanian Alkaloids (Westbury, Australia). Oripavine and thebaine were isolated as described previously (3). (R,S)-Stylopine (12) and dihydrosanguinarine (13) were prepared as described previously. Tetrahydropapaverine and pavine were isolated from commercial (±)-pavine (Sigma) as described previously (11).
Restriction endonucleases and Phusion DNA polymerase were from New England Biolabs (Ipswich, MA). Ex Taq DNA polymerase and Talon resin were from Clontech. Green Taq DNA polymerase was from Genscript (Piscataway, NJ). T4 DNA ligase and Moloney murine leukemia virus reverse transcriptase were from Invitrogen. All other chemicals were purchased from Sigma, VWR (Mississauga, Ontario, Canada), ThermoFisher (Ottawa, Ontario, Canada), or BioShop (Burlington, Ontario, Canada).
Preparation of O-Demethylcryptopine
Cryptopine (1 mm) was incubated overnight at 30 °C with 100 μg of PODA, 1 mm FeSO4, 1 mm α-ketoglutaric acid (α-KG), and 10 mm sodium l-ascorbate in a total volume of 1 ml. The reaction was then boiled for 5 min at 100 °C and centrifuged at 10,000 × g to remove debris. The supernatant was transferred to a new tube, basified with 1 m ammonium hydroxide, partitioned with ethyl acetate, and subsequently centrifuged at 10,000 × g. The ethyl acetate fraction was transferred to a new tube, and the aqueous portion was re-extracted. Pooled ethyl acetate fractions were reduced to dryness. The concentration of O-demethylcryptopine in methanol was estimated using the molar extinction coefficient of cryptopine (14).
Gene Isolation, Phylogenetic Analysis, and Expression Vector Construction
Cloning of CODM, T6ODM, and PODA was described previously (3). Translated opium poppy transcriptome databases (15) were searched using T6ODM protein sequence as a query to identify three additional full-length ODD candidates. Amino acid sequence alignments were performed with ClustalX (16), and phylogenetic relationships were analyzed using Geneious 6 (Biomatters Ltd., Auckland, New Zealand). DIOX4 and DIOX5 cDNAs amplified by RT-PCR with Phusion DNA polymerase using primers with flanking restriction sites (DIOX4, 5′-GCGGATCCATGGAGGCACCAAAACTA-3′ and 5′-GCCTGCAGCTAGTCAACCTGAACAAT-3′; DIOX5, 5′-GCGCATGCATGGAGACATCAAAACTA-3′ and 5′-GCCTGCAGATTGCTTATATGGGTTGA-3′) were A-tailed using green Taq DNA polymerase. A novel ODD candidate (DIOX6) was amplified from root cDNA using Ex Taq DNA polymerase and primers designed for a related sequence found only in assembled transcript databases (5′-GCGGATCCATGGAGACACCAACACTTATG-3′ and 5′-GCCTCGAGTCACATCCTCATGCAGTC-3′). PCR products were ligated into pQE30 (Qiagen, Hilden, Germany), and the expression vector was used to transform Escherichia coli expression strain SG13009 (Qiagen) grown on lysogeny broth LB medium containing 100 mg ml−1 of both ampicillin and kanamycin.
Recombinant Gene Expression and Protein Purification
Production of recombinant proteins was performed as described previously (3). Bacteria producing DIOX4 were grown at 28 °C, whereas DIOX5 and DIOX6 were cultured at 4 °C. For purification, bacterial pellets were resuspended in protein extraction buffer (100 mm Tris-HCl, pH 7.4, 10% (v/v) glycerol, 14 mm β-mercaptoethanol (BME)), and cells were lysed at 4 °C using a French pressure cell (500 p.s.i.). After centrifugation to remove insoluble debris, the supernatant containing soluble protein was combined with a buffer-equilibrated Talon resin and shaken at 60 rpm on ice for 45 min. The protein-charged resin was washed twice with cold (4 °C) extraction buffer, and protein was eluted stepwise with increasing concentrations (2.5, 10, 100, and 200 mm) of imidazole in extraction buffer. Total proteins (2.5 μg) from the 100 mm imidazole elution were separated by SDS-PAGE to assess protein yield and purity. The 100 mm imidazole fractions for each protein were dialyzed overnight using Spectro/Por (Spectrum, Gardena, CA) membranes (15,000 molecular weight cutoff) in 4 liters of extraction buffer. Final protein concentrations used for enzyme assays were estimated using a Bio-Rad protein assay.
Enzyme Assays
A selection of 26 available BIAs was screened in triplicate as potential substrates for opium poppy ODDs using a standardized assay. For each screen, 100 ng μl−1 of purified and desalted protein was incubated for 16 h at 30 °C with 100 μm of candidate substrate, 500 μm α-KG, 500 μm iron sulfate, and 10 mm sodium ascorbate in 100 mm Tris-HCl, pH 7.4, containing 10% (v/v) glycerol, 14 mm BME, and 5% (v/v) ethanol in a total reaction volume of 100 μl. Denatured proteins were used as negative controls. Assays were quenched with 5 volumes of 1 m acetic acid in methanol. Samples were reduced to dryness in a vacuum concentrator and resuspended in 1.5 ml of HPLC running buffer (10 mm ammonium acetate (pH 5.5)/acetonitrile, 95:5 (v/v)).
BIAs accepted as substrates were used to determine relative conversion rates using identical assay conditions except for an incubation time of 45 min, which was within the linear range of each assay, a final alkaloid substrate concentration of 50 μm, and a total reaction volume of 50 μl. Samples were diluted to 1.25 ml with HPLC running buffer. (S)-Scoulerine and cryptopine were used as substrates to determine kinetic parameters for CODM and PODA, respectively, at concentrations up to 500 μm. Samples were diluted with running buffer to a final substrate concentration of 1 μm for analysis.
Analysis of Enzyme Assays
Enzyme assays were analyzed by liquid chromatography-mass spectrometry (LC-MS) using a 1200 HPLC coupled with an Agilent (Santa Clara, CA) 6410 triple-quadrupole MS. For substrate range and relative activity analyses, 1 and 10 μl of sample, respectively, was injected onto a Zorbax SB C18 HPLC column (1.8 μm, 2.1 mm, 50 mm; Agilent), and analytes were eluted with a gradient of running buffer (10 mm ammonium acetate (pH 5.5)/acetonitrile, 95:5 (v/v); solvent A) and acetonitrile (solvent B). The flow rate was 700 μl min−1, and the gradient began with 0% solvent B and increased linearly to 99% solvent B by 7 min. The mobile phase composition remained constant until 8.1 min at which time it returned immediately to 0% solvent B followed by a 2-min re-equilibration period. Analytes were subjected to positive electrospray ionization(+) using source conditions optimized for BIAs (gas temperature, 350 °C; gas flow rate, 10 liters min−1; nebulizer gas pressure, 50 p.s.i.; capillary voltage 4,000 V) and were subsequently detected by full scan MS operating in positive polarity. Quadrupole 1 and 2 were set to RF only with quadrupole 3 scanning from 200 to 700 mass-to-charge (m/z). Relative enzyme activity was calculated as the percent turnover of each substrate using the following formula: product peak area ÷ (substrate peak area + product peak area) × 100. Subsequently, the compound with the highest turnover for each ODD was set to 100%, and the detected activity with all other substrates was expressed as a percentage of the maximum. For kinetic parameter analyses, a five-point calibration curve of cryptopine and (S)-scoulerine (500 pm to 5 μm) was established to semi-quantitatively determine the amount of product formed per unit of time. Kinetic constants were calculated based on Michaelis-Menten kinetics using Prism 5 (GraphPad Software, La Jolla, CA).
Reaction Product Identification
Comparison of collision-induced dissociation (CID) spectra with empirical spectra either from authentic standards or available in the literature (17–19) was used to identify or characterize each enzymatic reaction product (supplemental Table S1). CID was performed at 25 eV for each compound, and fragment ions were detected between 40 m/z and 5 atomic mass units above the m/z of the quasi-molecular ion. In some cases, unequivocal product identification was not possible because of the occurrence of identical mass spectra for compounds with an O-linked methyl group at one of two positions on either the isoquinoline or benzyl moiety.
LTQ-Orbitrap XL Analysis
High resolution MS and MS2 analysis was performed using an LTQ-Orbitrap XL equipped with a syringe pump (ThermoFisher). Allocryptopine and enzymatic reaction products (1 μg ml−1 in acetonitrile, 0.1% (v/v) acetic acid, 50:50) were introduced continuously with a syringe pump (5 μl min−1) into the electrospray ionization(+) source, and positive ions were generated using the following parameters: sheath gas, 10 arbitrary units; auxiliary gas, 0 arbitrary units; sweep gas, 0; spray voltage, 4,500 V. Ion interface settings were 275 °C and 40 V (capillary) and 255 V (tube lens). MS2 experiments were performed by conducting CID on target ions isolated in the linear ion trap followed by high resolution (60,000 full-width half-mass) mass analysis of the resulting product ions in the Orbitrap. Full scan data were collected in centroid mode over mass ranges bracketing the precursor ion (Δ20 atomic mass units) or product ions (from 165 to 220 m/z). External and internal instrument calibrations ensured an error of <2 ppm.
Nash Assay
Production of formaldehyde as a by-product of ODD-catalyzed O-demethylation was monitored using a fluorescence-based Nash assay (3). Nash reagent was prepared by adding 0.3 ml of acetic acid and 0.2 ml of acetylacetone to 100 ml of 2 m ammonium acetate. Enzyme assays were performed at 30 °C for 45 min using 500 μm of BIA substrate, 500 μm α-KG, 500 μm iron sulfate, 10 mm sodium l-ascorbate, and various quantities of up to 200 μg of CODM and PODA in 100 mm Tris-HCl, pH 7.4, containing 10% (v/v) glycerol, 14 mm BME, and 5% (v/v) in a total reaction volume of 500 μl. Assays were quenched with 2 volumes of Nash reagent followed by a 10-min incubation at 60 °C to convert formaldehyde to diacetyldihydrolutidine, which was detected by fluorescence using a Cary Eclipse fluorescence spectrophotometer (Varian, Palo Alto, CA) at λex = 412 nm and λem = 505 nm.
Formate Dehydrogenase Assay
Cryptopine (500 μm) was incubated at 30 °C for 16 h with 200 μg of CODM with 500 μm α-KG, 500 μm iron sulfate, and 10 mm sodium l-ascorbate in 100 mm Tris-HCl, pH 7.4, containing 10% (v/v) glycerol, 14 mm BME, and 5% (v/v) in a total reaction volume of 500 μl. The release of formic acid was monitored by adding 1 unit of formate dehydrogenase and 10 mm NAD+ to the enzyme assays after 16 h of incubation. In the presence of formate dehydrogenase and NAD+, formic acid undergoes stoichiometric conversion to CO2 and NADH, the latter of which was monitored at 30 °C using a Cary Eclipse UV spectrophotometer (Varian) at λ = 340 nm.
Virus-induced Gene Silencing
Construction of vectors to perform VIGS targeting T6ODM (pTRV-T6ODM), CODM (pTRV-CODM), and both genes (pTRV-DIOX) has been described previously (3). Vector delivery was performed by infiltration of Agrobacterium tumefaciens strains harboring pTRV1 and specific pTRV2 constructs into the apical meristem and the first two emerging leaves of each seedling. After 10–12 weeks, latex, three stem segments below the flower bud, and root tissue were harvested ∼1 day before anthesis. RNA was extracted from stem segments as described previously (20) and converted to cDNA using Moloney murine leukemia virus reverse transcriptase. PCR was performed using primers specific for tobacco rattle virus coat protein transcripts as described previously (21).
Real Time Quantitative PCR
Six plants infiltrated with A. tumefaciens harboring pTRV1 and each pTRV2 construct and that tested positive for TRV coat protein transcripts were subjected to real time quantitative PCR to determine the abundance of T6ODM and CODM mRNAs compared with control plants infiltrated with pTRV1 and empty pTRV2 vector. Real time quantitative PCR was performed as described previously (22) using SYBR Green detection, 300 nm each of each gene-specific forward and reverse primers (T6ODM, 5′-TTGAGGCACAAATGAGAAAATTGA-3′ and 5′-CACAACGCACTTTCGAGAAATTAC-3′; CODM, 5′-TTGTGCTTAAATTTCGTGGATGAC-3′ and 5′-TGATTACATCACTTGACCCAAACAG-3′), and 1 μl of cDNA. Relative transcript levels were determined using the 2−ΔΔCt method (23) and were expressed as a percentage relative to control plants.
Alkaloid Extraction
Opium poppy latex was collected in a pre-weighed 2-ml tube, centrifuged at 14,000 × g for 1 min at 4 °C, and immediately flash-frozen in liquid nitrogen. Samples were lyophilized for 2 days, and the dry weight of the latex was determined. Alkaloids were extracted in cold methanol (30 ml mg−1 dry weight) and subjected to vigorous vortex mixing and sonication for 1 min. Extracts were incubated for 16 h at −20 °C and subsequently centrifuged at 14,000 × g for 10 min at 4 °C to remove debris. The supernatant was transferred to a new tube and 1:1,000 and 1:10,000 dilutions were prepared in 2-ml glass auto-sampler vials using HPLC running buffer for LC-MS analysis. Opium poppy roots were flash-frozen in liquid nitrogen, lyophilized, and ground to a fine powder with a TissueLyser II (Qiagen) at 30 Hz for 1 min using a 30-ml grinding jar pre-cooled in liquid nitrogen and a 10-mm stainless steel grinding ball. Approximately 200 mg of powdered root was extracted using the protocol described for latex, and a 1:50 dilution was prepared for LC-MS analysis.
VIGS Analysis
Samples (10 μl) were injected onto a Zorbax SB C18 HPLC column (1.8 μm, 2.1 mm, 50 mm; Agilent), and compounds were separated using the HPLC mobile phase solutions listed above with a modified gradient. The flow rate was 500 μl min−1, and the gradient began with 0% solvent B, which was increased linearly to 7.5% by 1.5 min and was changed immediately to 25% at 1.6 min. Solvent B was increased linearly to 33% by 5 min, 92% by 7 min, and 98% by 7.6 min. Solvent B remained constant at 98% until 8.6 min and was changed immediately to initial conditions at 8.7 min for a 4-min re-equilibration period. Analytes were detected by LC-MS as described for the analysis of enzyme assays. Quantification was performed using calibration curves of peak areas versus alkaloid concentration for available authentic standards. Compounds not reliably detected in full scan mode were analyzed by multiple-reaction monitoring using the 1:1,000 diluted latex samples and parameters listed in Table 1. For compounds not available as authentic standards, reported values represent relative abundance. Compounds were identified or annotated based on retention time and CID spectrum compared with authentic standards or reference data (15, 17–19, 24).
TABLE 1.
Parameters used for multiple reaction monitoring
| Compound | Precursor ion | Production | Fragmentor voltage | Collision energy |
|---|---|---|---|---|
| m/z | m/z | V | eV | |
| Allocryptopine | 370.2 | 188.2 | 110 | 25 |
| Cryptopine | 370.2 | 205.2 | 110 | 35 |
| Protopine | 354.2 | 189.2 | 113 | 30 |
| Cheilanthifoline | 326.2 | 178.2 | 113 | 25 |
| Canadine | 340.2 | 176.2 | 113 | 25 |
| O-Demethylcryptopine | 356.2 | 190.2 | 110 | 35 |
| Tetrahydrocolumbamine | 342.2 | 178.2 | 128 | 25 |
| Stylopine | 324.2 | 176.2 | 113 | 32 |
| Laudanosine | 358.2 | 206.1 | 110 | 25 |
| N-Methylporphyroxine | 386.2 | 206.2 | 110 | 25 |
| Papaverubine B | 386.2 | 354.2 | 110 | 25 |
| Glaudine | 400.2 | 206.2 | 110 | 25 |
| Sinactine | 340.2 | 192.2 | 113 | 25 |
| N-Methylstylopine | 338.2 | 190.2 | 110 | 35 |
| Tetrahydropalmatine | 356.2 | 192.2 | 134 | 28 |
RESULTS
Phylogenetic Analysis of Opium Poppy ODDs
Three novel and full-length cDNAs encoding previously uncharacterized ODDs (DIOX4, DIOX5, and DIOX6) with substantial amino acid sequence identity to T6ODM, CODM, and PODA were isolated from opium poppy. Alignment of opium poppy ODD amino acid sequences with a variety of characterized and uncharacterized plant ODDs showed several absolutely conserved residues, including those forming a canonical HXDXnH catalytic triad presumably required for coordinating Fe(II) and an RXS motif implicated in α-KG binding. A phylogenetic tree (Fig. 2) derived from this alignment showed high bootstrap support for two ODD subclades, one containing T6ODM, PODA, CODM, and DIOX6 and another consisting of DIOX4 and DIOX5, distinct from other plant ODDs. Interestingly, the most similar plant proteins were all functionally uncharacterized.
FIGURE 2.

Rooted neighbor-joining phylogenetic tree for opium poppy ODDs and others reported from plants. Bootstrap frequencies for each clade are percentages of 1,000 iterations. Abbreviations and GenBankTM accession numbers are as follows: AtF3H, Arabidopsis thaliana flavanone 3-hydroxylase (AY116957); AtFLS, A. thaliana flavonol synthase 5 (NM_001037054); AtGSOH, A. thaliana putative ODD (AY114055); AtJRG21, A. thaliana putative leucoanthocyanidin dioxygenase (AJ298225); AtSRG1, A. thaliana senescence-related gene 1 (X79052); CjNCS1, Coptis japonica norcoclaurine synthase 1 (AB267398); CmG20O, Cucurbita maxima gibberellin 20-oxidase (U61385); CmG7O, C. maxima gibberellin 7-oxidase (U61386); CrD4H, Catharanthus roseus desacetoxyvindoline 4-hydroxylase (U71605); HnH6H, Hyoscyamus niger hyoscyamine 6 β-hydroxylase (DQ812529); MdACCO, Malus × domestica ACC oxidase (DQ137848); Os_uncharacterized1, Oryza sativa putative ODD (NM_001049521); Os_uncharacterized2, O. sativa putative ODD (CB663889); PcFS, Petroselinum crispum flavone synthase (AY230247); PhF3H, Petunia × hybrida flavanone 3-β-hydroxylase (X60512); PhFLS, Petunia × hybrida flavonol synthase (Z22543); PsCODM, Papaver somniferum codeine O-demethylase (GQ500141); PsT6ODM, P. somniferum thebaine 6-O-demethylase (GQ500139); PsPODA, P. somniferum protopine O,O-demethylenase (GQ500140); PsDIOX4, P. somniferum putative ODD (KC854329); PsDIOX5, P. somniferum putative ODD (KC854330); PsDIOX6, P. somniferum putative ODD (KC854331); Pt_XP002300453, Populus trichocarpa putative ODD (XM_002300417); Rc_EEF42734, Ricinus communis flavonol synthase/flavanone 3-hydroxylase (XM_002519715); SlACCO, Solanum lycopersicum ethylene-forming enzyme (NM_001246999); Ss_uncharacterized1, Citrus sinensis putative ODD (CV886305); St_uncharacterized1, Solanum tuberosum putative ODD (PGSC0003DMT400018663*); Tc_uncharacterized1, Theobroma cacao putative ODD (Thecc1EG029575t1*); Vv_uncharacterized1, Vitis vinifera putative ODD (XM_002269051); ZmBX6, Zea mays 2,4-dihydroxy-1,4-benzoxazin-3-one-glucoside dioxygenase (NM_001111630); ZmFLS/F3H, Z. mays flavonol synthase/flavanone 3-hydroxylase (EU972786). The search tags for sequences marked with an asterisk are available on line.
Biochemical Characterization of Opium Poppy ODDs
Six soluble His-tagged ODDs from opium poppy were produced in E. coli and purified using Co2+-affinity chromatography. All recombinant enzymes exhibited a molecular weight consistent with the theoretical masses of the predicted translation products. Each ODD was screened for enzymatic activity with one of 26 BIAs exhibiting a wide variety of structural backbones and functional group modifications. Full scan LC-MS detected two different modifications, loss of either 12 or 14 atomic mass units, in the m/z of reaction products compared with the corresponding substrates incubated overnight with certain ODDs. No other reaction products were detected. DIOX4 and DIOX5 failed to convert any tested BIA to a detectable product, and only trace levels of products reduced by 14 atomic mass units were detected when DIOX6 was incubated overnight with (S)-reticuline or (S)-scoulerine. In contrast, T6ODM, CODM, and PODA converted a substantial number of tested BIAs to products reduced by either 12 or 14 atomic mass units (Figs. 3 and 4). Protopine and morphinan alkaloids showed the highest apparent turnover rates among potential substrates belonging to a variety of BIA structural categories. Because the efficient acceptance of protopine alkaloids was unexpected and the reaction products included compounds reduced by both 12 and 14 atomic mass units, the biochemistry of these conversions was investigated in more detail. Incubation of T6ODM, CODM, or PODA with allocryptopine resulted in products reduced by 14, 12, and 12 atomic mass units, respectively (Fig. 3A). Incubation of CODM or PODA with cryptopine yielded products reduced by 12 and 14 atomic mass units, respectively, whereas T6ODM catalyzed no reaction (Fig. 3B). Finally, incubation of PODA with protopine reduced the m/z of the substrate by 12 atomic mass units and again, T6ODM catalyzed no reaction (Fig. 3C). CODM also reduced the m/z of protopine by 12 atomic mass units, but two distinct peaks at m/z 342 were detected (Fig. 3C). Because no authentic standards were available, O-demethylation and O,O-demethylenation reaction products were identified through interpretation of CID spectra (Fig. 5). O-demethylation of allocryptopine by T6ODM was characterized by a reaction product with a quasi-molecular ion ([M + H]+) reduced by 14 atomic mass units compared with the substrate (Fig. 5, A and B), whereas O,O-demethylenation by PODA and CODM resulted in a quasi-molecular ion reduced by 12 atomic mass units (Fig. 5, A, C, and D). Comparison of CID spectra for allocryptopine and these reaction products confirmed the reactions that occurred. The allocryptopine standard (Fig. 5A) and the product of assays containing allocryptopine and T6ODM (Fig. 5B) produce a typical even-numbered a-type fragment ion at m/z 206 consisting of the isoquinoline moiety and oxygen at C-1 (18). The a-type ion can lose water to form the b-type ion at m/z 188. For products of assays containing allocryptopine and CODM or PODA (Fig. 5, C and D), the a-type ion lost 12 atomic mass units (m/z 194) resulting from loss of the alkyl on the methylenedioxy bridge of the substrate. This a-type ion can lose water to yield the b-type ion, which is also reduced by 12 atomic mass units (m/z 176) compared with the equivalent ions (Figs. 5A and 3B). Parallel interpretations were applied to the reaction products in PODA or CODM assays containing cryptopine or protopine. CODM was the only enzyme capable of the O,O-demethylenation of either the A- or B-rings.
FIGURE 3.
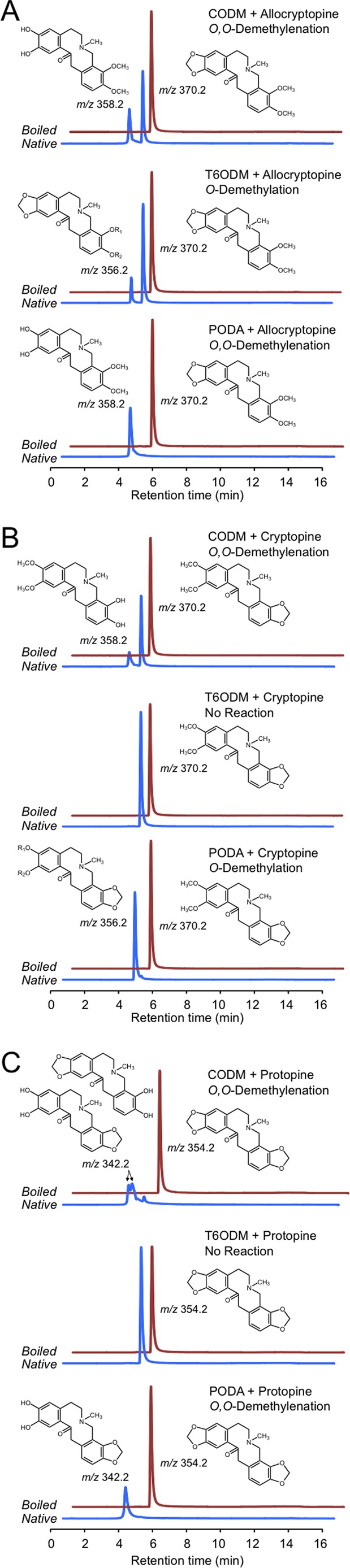
LC-MS-extracted ion chromatograms of enzyme assays using CODM, T6ODM, or PODA with the protopine alkaloids allocryptopine (A), cryptopine (B), or protopine (C) as substrates showing either O-demethylation or O,O-demethylenation. Loss of 14 atomic mass units indicates O-demethylation, whereas loss of 12 atomic mass units corresponds to O,O-demethylenation. The figure indicates when no reaction was detected.
FIGURE 4.

Relative activity, reaction catalyzed, and regiospecificity of T6ODM, CODM, and PODA with different benzylisoquinoline alkaloids as substrates. Values represent the mean percent maximum activity ± S.E. of four independent replicates for each compound compared with the substrate showing the highest turnover, which was set to 100%. Trace activity indicates less than 1% turnover. For O-demethylcryptopine, R1 = CH3 and R2 = H, or R1 = H and R2 = CH3. Abbreviation used is as follows: n.d., not detected.
FIGURE 5.

Identification of ODD reaction products using allocryptopine as the substrate by collision-induced dissociation mass spectrometry. A, fragmentation of allocryptopine yielded a characteristic isoquinoline product ion at m/z 206 (a-type ion), and loss of water from m/z 206 yielded m/z 188 (b-type ion). B, fragmentation of the reaction product at m/z 356 from assays with T6ODM and allocryptopine yielded the same characteristic isoquinoline product ions indicating that O-demethylation occurred on the B-ring. Fragmentation of the reaction product at m/z 358 from assays with PODA (C) or CODM (D) and allocryptopine yielded the same characteristic isoquinoline product ions as the allocryptopine standard, but reduced by 12 atomic mass units (m/z 194 and 176) indicating that O,O-demethylenation occurred on the A-ring.
Triple-quadrupole MS/MS strongly suggested O-demethylation and O,O-demethylenation of allocryptopine and other alkaloids. However, because authentic standards were not available and compound quantities were insufficient to perform NMR, reaction product identification was supported by high resolution MS and MS2 analysis using an LTQ-Orbitrap XL. Allocryptopine had a measured mass of 370.164461, corresponding to an elemental formula of C21H24NO5 (−0.782 ppm error). The O-demethylation product of T6ODM and allocryptopine had a measured mass of 356.14926, corresponding to an elemental formula of C20H22NO5 (0.030 ppm error). The O,O-demethylenation product of CODM or PODA and allocryptopine had a measured mass of 358.16486, corresponding to an elemental formula of C20H24NO5 (−0.11 ppm error). These values support the initial identification of substrates and reaction products obtained by triple-quadrupole MS, but for further confirmation these compounds were subjected to MS2 exact mass analysis. Allocryptopine yielded characteristic (18) isoquinoline product ions at 206.08102 and 188.07040 m/z corresponding to the elemental formulae C11H12NO3 (−0.872 ppm error) and C11H10NO2 (−1.091 ppm error) consistent with the identification of the substrate. The T6ODM reaction product of allocryptopine yielded the same characteristic isoquinoline fragments as the substrate 206.08102 and 188.07042 m/z, corresponding to elemental formulae C11H12NO3 (−0.727 ppm error) and C11H10NO2 (−0.984 ppm error), which is consistent with a B-ring O-demethylation of the substrate. The CODM reaction product of allocryptopine yielded characteristic isoquinoline fragments at 194.08098 and 176.07042 m/z corresponding to elemental formulae of C10H12NO3 (−0.978 ppm error) and C10H10NO2 (−1.051 ppm error), which is consistent with an A-ring O,O-demethylenation of the substrate.
Substrate Range
The percent maximum conversion of T6ODM was highest with the morphinans oripavine (100%) and thebaine (97%), but modest activity was also detected with the protoberberine (R,S)-canadine (32%). Conversion of the protopine allocryptopine (10%) and the 1-benzylisoquinoline papaverine (2%) was relatively low. Trace O-demethylation activities below 1% were detected with a small number of other 1-benzylisoquinoline, protoberberine, and protopine substrates (Fig. 4). CODM catalyzed the 3-O-demethylation of the morphinans codeine (90%) and thebaine (81%) but showed the highest turnover rate with protopine (100%). Other protopines, allocryptopine (50%), cryptopine (37%), and O-demethylcryptopine (41%), were also accepted by CODM with modest efficiency for A- or B-ring O,O-demethylenation, depending on the position of the methylenedioxy bridge. The 1-benzylisoquinoline (S)-reticuline (16%), the aporphine isocorydine (6%), and the protoberberines (S)-scoulerine (70%) and (R,S)-tetrahydropalmatine (17%) also exhibited low to relatively efficient conversion rates (Fig. 4).
Previously, no enzymatic function was detected for PODA (DIOX2) using a relatively limited range of substrates (3). However, several BIA substrates were either O-demethylated or O,O-demethylenated by PODA (Fig. 4). The most efficient O-demethylation reactions were observed with cryptopine (100%), (R,S)-tetrahydropalmatine (14%), and (R,S)-tetrahydropapaverine (2%), but papaverine, thebaine, and oripavine were also O-demethylated on the A-ring at trace levels. The most efficient substrate for O,O-demethylenation by PODA was protopine (79%) followed by allocryptopine (40%). Several protoberberines with methylenedioxy bridges, including (R,S)-canadine (38%), berberine (19%), and (R,S)-stylopine (16%) were also converted to corresponding hydroxyl-containing derivatives. For all reactions involving protopine, protoberberine, and 1-benzylisoquinoline, PODA displayed regiospecificity for the A-ring, except for (R,S)-tetrahydropalmatine, which was O-demethylated on the B-ring. Several tested BIAs with O-linked methyl groups and/or methylenedioxy bridges were not accepted as substrates, including (±)-pavine the benzo[c]phenenthridines dihydrosanguinarine and sanguinarine, the phthalideisoquinolines narcotoline and noscapine, the aporphines (S)-isocorydine, (S)-boldine, (S)-corytuberine, (S)-glaucine, and (S)-isothebaine, and the bisbenzylisoquinoline berbamine.
Reaction Kinetics
CODM exhibited a Km value of 198 ± 47 μm with (S)-scoulerine as the substrate, whereas PODA displayed a Km value of 27 ± 15 μm with cryptopine (Fig. 6; Table 2). Approximate Vmax values of 4.2 ± 0.7 picokatals for PODA and 11.6 ± 1.4 picokatals for CODM were calculated using (S)-scoulerine and cryptopine, respectively, as substrates. The catalytic rate and catalytic efficiency were 0.095 s−1 and 1291.4 s−1 m−1, respectively, for PODA and 0.034 s−1 and 480.1 s−1 m−1, respectively, for CODM. The insolubility in aqueous solution at saturating concentrations (>500 μm) precluded a reliable Km value determination for cryptopine as a substrate of CODM. Kinetic constants for other protopine alkaloids could not be measured because of insufficient availability of allocryptopine and the insolubility of protopine in aqueous solution at saturating concentrations.
FIGURE 6.
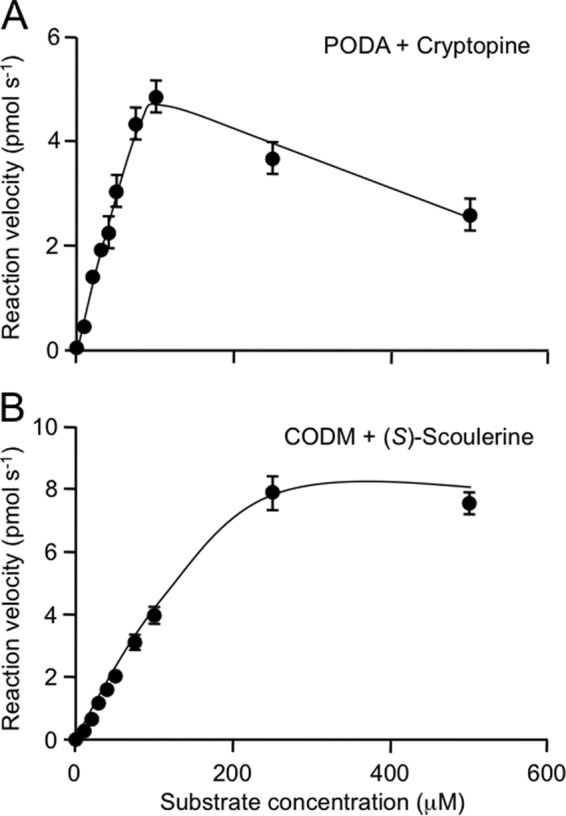
Steady-state enzyme kinetics of purified recombinant PODA (A) and CODM (B) using cryptopine and (S)-scoulerine, respectively, as substrates. Incubation time (45 min) and protein concentration (100 ng μl−1) were optimized prior to enzyme kinetic analyses. Values represent mean product formation (μm s−1) ± S.D. of three independent measurements. Maximum velocity (Vmax) and substrate affinity (Km), catalytic rate (Kcat), and catalytic efficiency (Kcat/Km) were determined based on Michaelis-Menten kinetics.
TABLE 2.
Kinetic values for PODA and CODM with cryptopine and (S)-scoulerine, respectively
| Enzyme | Substrate | Km | Vmax | kcat | kcat/Km |
|---|---|---|---|---|---|
| μm | picokatal | s−1 | s−1 m−1 | ||
| PODA | Cryptopine | 27 ± 15 | 4.2 ± 0.7 | 0.034 | 1291.4 |
| CODM | (S)-Scoulerine | 198 ± 48 | 11.6 ± 1.4 | 0.095 | 480.1 |
Reaction Mechanics
The mechanisms of O-demethylation and O,O-demethylenation were investigated using a fluorescence-based Nash assay for formaldehyde detection, and a coupled reaction with formate dehydrogenase for formic acid detection. As expected, the production of formaldehyde as the result of O-demethylation (3) showed a direct increase in response to enzyme quantity in assays containing CODM and codeine (Fig. 7A) or PODA and cryptopine (Fig. 7B). In contrast, formaldehyde was not detected in assays containing CODM and cryptopine (Fig. 7A) highlighting a potentially important difference between the O-demethylation and O,O-demethylenation reaction mechanisms. The lack of formaldehyde release in this case was comparable with incubating PODA with codeine, where no reaction product was expected (Fig. 7B). Limited substrate solubility and/or availability precluded an examination of O,O-demethylenation catalyzed by PODA. Production of formic acid was not detected in assays containing any substrate and enzyme combination.
FIGURE 7.

Formaldehyde is a by-product of the O-demethylation but not the O,O-demethylenation of benzylisoquinoline alkaloids. A, formaldehyde production increased in response to the amount of CODM used in assays containing codeine, which undergoes O-demethylation, but formaldehyde was not detected in assays containing cryptopine, which undergoes O,O-demethylenation. B, formaldehyde production increased in response to the amount of PODA used in assays containing cryptopine, which undergoes O-demethylation, but formaldehyde was not detected in assays containing codeine, which is not a substrate.
Virus-induced Gene Silencing
Mining of an Illumina-based transcriptome library of opium poppy cultivar Bea's Choice, which was used for VIGS analysis, revealed expression of T6ODM and CODM at levels comparable with other BIA biosynthetic genes. In contrast, transcripts encoding PODA were not detected in the Bea's Choice cultivar. Therefore, gene-silencing experiments were limited to T6ODM and CODM using the previously described vectors pTRV2-T6ODM and pTRV2-CODM, respectively (3). T6ODM and CODM transcript levels were suppressed simultaneously using pTRV2-DIOX. Mature opium poppy plants infiltrated as seedlings with mixed cultures of A. tumefaciens harboring pTRV1 and pTRV2, the latter as an empty vector or containing fragments of T6ODM or CODM, were screened for the occurrence of viral coat protein transcripts by RT-PCR. For each utilized pTRV2 construct, six plants that tested positive for viral coat protein transcripts were analyzed by RT quantitative PCR for mean relative abundance of T6ODM and CODM transcripts compared with empty pTRV2 vector as the control. As described previously (3), plants exposed to A. tumefaciens harboring pTRV2-DIOX resulted in a significant suppression of both T6ODM and CODM transcript levels. In contrast, plants exposed to A. tumefaciens harboring pTRV2-T6ODM resulted in the significant suppression of only T6ODM transcript levels, whereas plants exposed to A. tumefaciens harboring pTRV2-CODM resulted in the significantly reduced abundance of only CODM transcripts (3).
Plants with suppressed T6ODM and/or CODM transcript levels showed a significant alteration in the accumulation of several alkaloids. As reported previously (3), reduced transcript levels of T6ODM alone, or T6ODM and CODM together, resulted in a substantial decrease in the accumulation of codeine and morphine, and an increase in the accumulation of thebaine (Fig. 8A). In contrast, suppression of only CODM transcript levels resulted in a significant increase in the accumulation of codeine at the expense of reduced levels of morphine and oripavine. Unexpectedly, sanguinarine accumulation in the roots of plants with reduced levels of T6ODM and/or CODM transcripts was also significantly lower (Fig. 9A). Although less pronounced, noscapine and papaverine levels were also reduced in plants with reduced levels of T6ODM and/or CODM transcripts. In contrast, accumulation of the protopine alkaloids cryptopine, protopine, and allocryptopine increased significantly and in several cases substantially in response to the suppression of T6ODM and/or CODM transcript levels (Fig. 9A). The less abundant protopine, O-demethylcryptopine, also increased in abundance in CODM-silenced plants but was unaffected in plants in which T6ODM transcript levels were reduced. Similarly, accumulation of the protoberberines N-methystylopine and N-methylcanadine was also elevated in response to the suppression of T6ODM and/or CODM transcript levels (Fig. 9B). Interestingly, stylopine (Fig. 8B) and canadine (Fig. 9A) levels were unaffected or, in some cases, reduced in T6ODM- and/or CODM-silenced plants. Accumulation of other protoberberines showed variable effects in response to the suppression of T6ODM and/or CODM transcript levels. With a few exceptions, cheilanthifoline, tetrahydrocolumbamine, and tetrahydropalmatine levels were unaffected, whereas sinactine accumulation increased significantly in all cases (Fig. 8B). Levels of the rhoeadine N-methylporphyroxine (Fig. 9B) also increased significantly in T6ODM- and/or CODM-silenced plants, whereas the related compounds papaverubine B and glaudine (Fig. 8B) were more abundant only in some cases. Accumulation of the 1-benzylisoquinoline laudanosine also decreased in response to the suppression of T6ODM and/or CODM (Fig. 8B).
FIGURE 8.

Effect of virus-induced gene silencing on the accumulation of selected benzylisoquinoline alkaloids using pTRV2 constructs designed to suppress the transcript levels in opium poppy of all ODDs (pTRV2-DIOX), T6ODM (pTRV2-T6ODM), or CODM (pTRV2-CODM) compared with empty vector controls. Values represent the mean ± S.D. of six independent replicates. A, morphinan alkaloids for which authentic standards are available allowing quantitative determination of alkaloid content. B, other compounds annotated using reference spectra for which only relative abundance could be determined. Statistical significance (*) at p < 0.01 was calculated using Student's t test.
FIGURE 9.

Effect of virus-induced gene silencing on the accumulation of selected benzylisoquinoline alkaloids using pTRV2 constructs designed to suppress the transcript levels in opium poppy of all ODDs (pTRV2-DIOX), T6ODM (pTRV2-T6ODM), or CODM (pTRV2-CODM) compared with empty vector controls. All alkaloids were isolated from latex except sanguinarine, which was extracted from roots. Values represent the mean ± S.D. of six independent replicates. A, compounds for which authentic standards are available allowing quantitative determination of alkaloid content. B, compounds annotated using reference spectra for which only relative abundance could be determined. Statistically significant differences (p < 0.01) relative to values for the empty pTRV2 vector (*) were calculated using Student's t test.
DISCUSSION
The discovery of T6ODM and CODM as the first reported O-demethylases involved in plant-specialized metabolism provided rational support for the suggestion that O-demethylation is a widespread occurrence in BIA biosynthesis (3, 5). T6ODM was initially isolated using DNA microarrays and based on the differential analysis of transcripts found in high morphine and morphine-free opium poppy cultivars. DIOX2 (PODA) and CODM, which showed 85 and 72% amino acid sequence identity, respectively, with T6ODM were isolated through homology-based queries of opium poppy EST libraries. The targeted objective of our previous work was to specifically identify the enzymes conferring the unique ability of opium poppy to produce codeine and morphine. The physiological roles of T6ODM and CODM as the enzymes catalyzing the antepenultimate and ultimate conversions in morphine biosynthesis were firmly established in opium poppy plants using VIGS (3). However, with such a focus on morphinan alkaloid metabolism, only a limited number of potential alkaloid substrates were tested in vitro, and the analysis of alkaloid perturbations in T6ODM- and CODM-silenced plants was not comprehensive. Furthermore, roots were not analyzed in gene-silenced plants because they are not the major sites of morphinan alkaloid accumulation. However, opium poppy roots are a major site for the accumulation of the benzo[c]phenanthridine alkaloid sanguinarine. We previously noted that (S)-scoulerine is an effective substrate for O-demethylation by CODM, which was somewhat surprising because of the structural dissimilarity of this protoberberine alkaloid compared with morphinan alkaloids. Moreover, a widespread role for O-demethylation was initially unexpected in the context of general BIA metabolism outside of morphine biosynthesis. This result, combined with the occurrence of other BIA end products exhibiting substitution patterns indicative of O-demethylation events affecting upstream pathway intermediates, suggested that ODDs were potentially multifunctional enzymes operating at numerous points along the branched BIA biosynthetic network. We therefore pursued a more comprehensive investigation of ODDs, which included an expanded and structurally diverse selection of potential alkaloid substrates along with an enhanced detection system based on LC-MS for the analysis of both enzyme assays and VIGS experiments. Such refined approaches not only revealed that ODDs catalyze O-demethylation on a substantially broader array of substrates than previously indicated (3) but also facilitated the discovery of O,O-demethylenation as an entirely novel and unexpected activity in BIA metabolism.
The recent availability of deep transcriptome resources for opium poppy (15) permitted the isolation of three new ODD homologs, DIOX4, DIOX5, and DIOX6. Phylogenetic analyses revealed greater similarity between DIOX6, T6ODM, CODM, and PODA compared with the more distant DIOX4 and DIOX5 (Fig. 2). Catalytic functions for these new clones were not detected using a wide range of potential BIA substrates, although DIOX6 exhibited trace O-demethylase activity with (S)-reticuline and (S)-scoulerine. Based on the apparent lack of activity on available BIAs, DIOX4, DIOX5, and DIOX6 were not subjected to VIGS analysis. Assays of recombinant T6ODM and CODM using morphinan alkaloid substrates were consistent with previous results (3), whereby strict regiospecificity was detected for either the T6ODM or CODM groups of thebaine, oripavine, and/or codeine (Fig. 4). Although not reported previously (3), a trace level of 6-O-demethylase activity was detected for PODA with thebaine and oripavine. This discrepancy resulted from using a more sensitive and analytically accurate assay method compared with our prior approach. Previously, routine assays were carried out using an indirect method (25) whereby 14CO2 released via the O-demethylation-coupled decarboxylation of [1-14C]oxoglutarate was captured and measured by scintillation counting. Spontaneous, uncoupled decomposition of [1-14C]oxoglutarate in the presence of recombinant enzyme preparations caused high background measurements, which precluded the confident assignment of trace activities. In this study, the detection and identification of reaction products by LC-MS, combined with the inclusion of a substantially expanded array of BIA substrates, facilitated the discovery of several new catalytic functions (Fig. 4). The most striking result was the capacity of CODM and PODA to efficiently catalyze the O,O-demethylenation of several compounds. In particular, protopine alkaloids, a structural subclass of BIAs not previously considered, were among the best substrates together with a variety of protoberberine alkaloids. PODA displayed a preference for O,O-demethylenation of the A-ring (isoquinoline moiety) of the protopine or protoberberine backbone, whereas CODM targeted methylenedioxy bridges on either the A- or B-ring (benzyl moiety) of only protopine alkaloids. Despite the extensive sequence similarity among opium poppy ODDs, only CODM and PODA were capable of catalyzing O,O-demethylenation on protopine and/or protoberberine substrates in vitro. Remarkably, T6ODM and PODA, which display 85% amino acid sequence identity, catalyzed different reactions on the same substrate. For example, (R,S)-canadine and allocryptopine were O-demethylated by T6ODM but O,O-demethylenated by PODA. The O,O-demethylenation activity of CODM was also restricted to protopine alkaloids, whereas PODA was also able to catalyze the O,O-demethylenation of protoberberine substrates. BIAs containing methylenedioxy bridges, but belonging to the benzo[c]penanthridine (e.g. sanguinarine) and the phthalideisoquinoline (e.g. noscapine) structural subgroups, were not accepted as substrates by any of the ODDs.
The high relative activity of CODM with respect to both the O,O-demethylenation of protopine and the O-demethylation of morphinan substrates (Fig. 4) suggested an important role for both reaction types in opium poppy plants. Unfortunately, the relative insolubility of protopine and cryptopine and the limited availability of allocryptopine as the preferred protopine alkaloid substrates for O,O-demethylenation precluded the determination of kinetic constants for CODM. However, the measured Km value of CODM for the O-demethylation of (S)-scoulerine (198 ± 48 μm) (Table 2) was about 10-fold higher than the previously reported Km value for codeine (21 ± 8 μm) as a substrate for O-demethylation (3) indicating that the enzyme has a higher affinity for morphinan substrates. In contrast, PODA displayed a relatively high affinity for cryptopine (27 ± 15 μm) as a substrate for O-demethylation, and kcat/Km comparisons suggested that PODA is a more efficient enzyme than either CODM or T6ODM (3).
Enzymatic O,O-demethylenation has not been reported frequently, and no other plant enzymes capable of catalyzing this reaction have been reported. CODM and PODA are the first examples of ODDs in either plants or animals that catalyze O,O-demethylenation. In humans various CYPs, including the general xenobiotic metabolizing enzyme CYP2D6, found in the liver have been shown to O,O-demethylenate amphetamine analogs such as 3,4-methylenedioxymethamphetamine commonly known as “ecstasy” (7, 8, 26). Similarly, protopine and californine, a BIA belonging to the pavine subgroup, are O,O-demethylenated by rat liver enzymes CYP2D1 and CYP2C11 (10). O-Demethylation by ODDs, and probably by CYPs, proceeds via hydroxylation of the O-linked methyl group followed by the elimination of formaldehyde (27, 28). In agreement with known mechanistic features of ODDs and previously acquired data (3), our current Nash assay results indicated formaldehyde release associated with the O-demethylation of both codeine and cryptopine by CODM and cryptopine by PODA (Fig. 7). Interestingly, the release of formaldehyde was not detected in association with the O,O-demethylenation of cryptopine by CODM. Early investigations of the CYP-catalyzed O,O-demethylenation of 3,4-methylenedioxymethamphetamine suggested a mechanism involving formate ester hydrolysis, whereby the leaving group is formic acid (6, 29). However, the release of formic acid was also not detected in association with the action CODM on cryptopine indicating that the ODD-catalyzed O,O-demethylenation of BIAs proceeds via a distinct and as yet unknown mechanism.
The discovery of novel catalytic functions for recombinant T6ODM and CODM prompted an expanded investigation of their physiological roles using VIGS, which has proven effective for the characterization of numerous BIA biosynthetic genes in opium poppy (3, 11, 22, 30). PODA was not targeted for VIGS analysis because of the lack of corresponding transcripts in the Bea's Choice cultivar. The PODA cDNA was isolated from a fungal elicitor-treated opium poppy cell culture library (3, 31) and does not display expression in the plant under normal physiological conditions. Expression of PODA in response to fungal elicitor treatment suggests that the gene is inducible by certain environmental factors. However, the lack of transcripts in the stem or root transcriptomes indicates that PODA does not contribute to the basal BIA profile in opium poppy. The use of LC-MS, including CID, as a platform to analyze T6ODM- and/or CODM-silenced plants provided an enhanced sensitivity and a robust metabolite identification capacity compared with our previous work, whereby only HPLC coupled with UV detection was used (3). Broad scope analysis of plants with reduced T6ODM and CODM transcript levels, compared with controls, revealed remarkable, diverse, and unexpected effects on alkaloid phenotypes. Suppression of T6ODM and/or CODM transcript levels affected morphinan alkaloid accumulation as reported previously (Fig. 8A) (3). Briefly, suppression of T6ODM transcripts caused an increase in thebaine and oripavine levels and a corresponding decrease in codeine and morphine levels. In contrast, suppression of CODM caused an increase in codeine accumulation at the expense of morphine and oripavine. Extended analysis showed that similarly profound changes also occurred in protopine alkaloid levels, which significantly increased in response to the silencing of T6ODM, CODM, or both genes simultaneously (Fig. 9A). The accumulation of protopine alkaloids in CODM-silenced plants was generally in agreement with the catalytic functions and substrate range of recombinant CODM, which showed high relative activity with protopine, allocryptopine, cryptopine, and O-demethylcryptopine (Fig. 4). However, the accumulation of these compounds in response to T6ODM-silencing was unexpected because recombinant T6ODM exhibited only trace to relatively minor activity with some protopine alkaloids tested as substrates. Off-target silencing of CODM in T6ODM-silenced plants was ruled out (3). However, it is possible that an additional, as yet uncharacterized ODD with CODM activity was co-silenced. Alternatively, recombinant T6ODM might display different catalytic properties compared with the native plant enzyme. In this regard, two-dimensional gel electrophoretic analysis of opium poppy latex suggests the occurrence of multiple charge isoforms of T6ODM and/or CODM (32), which might reflect the extensive post-translational modification of ODDs represented by only singular known transcripts. Consequently, the catalytic functions of native ODDs might differ from the detected activities of the corresponding recombinant enzymes (33).
Interestingly, levels of papaverine and noscapine, which are regarded as metabolic end products in opium poppy (1), were reduced in silenced plants. It is conceivable that the substantial increases in the accumulation of protopines and other alkaloids reduced flux through other branch pathways requiring common upstream intermediates such as (S)-reticuline. However, the reduction in sanguinarine levels was remarkable because benzo[c]phenanthridine biosynthesis involves protopine as a key pathway intermediate (Fig. 10). Unexpectedly, the elevated protopine level in silenced plants did not cause an increase in sanguinarine accumulation, but instead it was associated with a substantial decrease in sanguinarine content in roots (Fig. 9A). Possible explanations include the existence of unknown regulatory mechanisms governing benzo[c]phenanthridine metabolism, such as transcriptional responses to elicitation by small molecules or substrate inhibition of enzymes. Coordinate transcriptional regulation of sanguinarine biosynthesis has been noted in opium poppy cell cultures (31), affecting enzymes such as berberine bridge enzyme, tetrahydroprotoberberine N-methyltransferase (12), and N-methylstylopine hydroxylase (Fig. 1) (34). The expression characteristics of the gene encoding protopine 6-hydroxylase has not been characterized in opium poppy, and the biochemical aspects of protoberberine, protopine, and benzo[c]phenanthridine pathway regulation have not been investigated. However, substrate inhibition of protopine 6-hydroxylase resulting from the elevated cellular pool of protopine or related alkaloids could potentially result in the decreased accumulation of sanguinarine. Substrate inhibition has been reported for salutaridine reductase involved in morphine biosynthesis in opium poppy (35) and is common in mammalian CYPs (36, 37).
FIGURE 10.

Summary of enzymatic reactions catalyzed in vitro by T6ODM, CODM, and PODA using protopine alkaloid substrates and putative metabolic relationships with the benzo[c]phenanthridine sanguinarine and the rhoeadine alkaloids N-methylporphyroxine and glaudine in opium poppy. Compounds highlighted in yellow were detected, and their levels were generally induced in plants subjected to virus-induced gene silencing resulting in the suppression of specific ODD transcripts. Levels of sanguinarine, highlighted in orange, were reduced in plants displaying lower ODD transcript abundance compared with controls. Enzymes in black were shown to catalyze the indicated conversion. Putative enzymatic reactions not tested empirically because of a lack of substrate availability are shown in gray. Dashed arrows represent multiple uncharacterized conversions. For O-demethylcryptopine, O-demethylallocryptopine, and O,O-demethylenated derivatives, R1 = CH3 and R2 = H, or R1 = H and R2 = CH3. Abbreviations: P6H, protopine 6-hydroxylase; DBOX, dihydrosanguinarine oxidase.
Silencing of T6DOM and/or CODM also resulted in elevated levels of N-methylporphyroxine (Fig. 9B), whereas T6ODM-silenced plants accumulated more glaudine compared with controls (Fig. 8B). N-Methylporphyroxine and glaudine are rhoeadine alkaloids, which are postulated to derive from protopine intermediates (38–40). The elevated accumulation of rhoeadine alkaloids could result from the increased availability of cryptopine (Fig. 9A) and O-demethylcryptopine (Fig. 9B) in silenced plants (Fig. 10). The possible recycling of protopine alkaloids must also be considered. For example, O-demethylation of allocryptopine by T6ODM could potentially provide a mono-substituted methyl ether substrate for reformation of the methylenedioxy bridge by relevant CYP enzymes (Fig. 10) (41, 42). Similarly, the potential action of relevant O-methyltransferases (43, 44) on one of the two adjacent hydroxyl groups resulting from O,O-demethylenation would provide the required mono-substituted methyl ether moiety to reform the methylenedioxy bridge. Such recycling could regulate the cellular pool size of pathway intermediates, such as protopine, possibly involved in biochemical regulation via, for example, substrate inhibition.
O,O-Demethylenated benzo[c]phenanthridine, protoberberine, and protopine alkaloids have been detected as minor alkaloids in several members of the Papaveraceae, including Fummaria vaillantii (45), Macleaya cordata (46), and Thalictrum javanicum (47). Previously, the detection of O,O-demethylenated BIAs was suggested as an artifact of extraction processes, although their natural occurrence could not be discounted (46). Discovery of the O,O-demethylenation activity of PODA and CODM provides a biochemical basis for the natural occurrence of such compounds in plants. However, O,O-demethylenated BIAs were not detected in opium poppy plants subjected to VIGS suggesting their rapid recycling or redistribution into other products, such as rhoeadine alkaloids.
In vitro profiling of recombinant enzyme activity supported by corresponding gene silencing in planta demonstrates that specific ODDs are extensively involved at multiple points in BIA biosynthesis. VIGS studies unequivocally demonstrated the widespread contribution of T6ODM and CODM to BIA metabolism beyond the formation of morphine (3) and suggested that the removal of O-linked methyl groups added early in the pathway plays a key role in the overall regulation of alkaloid biosynthesis. The discovery of new catalytic functions for ODDs, especially O,O-demethylenation, establishes a more complete appreciation for the complexities of BIA metabolism as a metabolic network with multiple interactive regulatory features, rather than the generally depicted linear routes (1). Moreover, our data provoke important, interesting questions regarding the evolution of BIA metabolism. In particular, the apparently exclusive emergence of morphine biosynthesis in opium poppy (3) might have resulted from key mutations in ODDs originally participating in the formation of protopine and, indirectly, protoberberine, benzo[c]phenanthridine, and rhoeadine alkaloids. Protopines and derivatives are taxonomically more widely distributed than morphinan alkaloids (48, 49). In this context, the substitution of only four amino acids in CODM resulted in the exclusive acceptance of codeine for 3-O-demethylation, and the elimination of thebaine as a substrate (50). As suggested (5) previously, and despite the metabolic cost associated with the extensive O-methylation of upstream BIA pathway intermediates, O-linked methyl groups and methylenedioxy bridges appear transient and replaceable in the context of the formation of end products, including morphine and, surprisingly, sanguinarine (Fig. 10). The catalytic functions of homologs in plants related to opium poppy will provide more insight into the evolutionary origin and broad biochemical roles of ODDs in BIA biosynthesis.
Acknowledgments
We thank Dr. Jillian Hagel for critical comments on the manuscript.
This work was supported in part by funding from Genome Canada, Genome Alberta, the Government of Alberta, the Natural Sciences and Engineering Research Council of Canada, and the Canada Foundation for Innovation-Leaders Opportunity Fund.

This article contains supplemental Table S1.
- BIA
- benzylisoquinoline alkaloid
- CID
- collision-induced dissociation
- CODM
- codeine O-demethylase
- CYP
- cytochrome P450
- α-KG
- α-ketoglutaric acid
- ODD
- 2-oxoglutarate/Fe(II)-dependent dioxygenase
- PODA
- protopine O-dealkyase
- T6ODM
- thebaine 6-O-demethylase
- VIGS
- virus-induced gene silencing
- BME
- β-mercaptoethanol.
REFERENCES
- 1. Hagel J. M., Facchini P. J. (2013) Benzylisoquinoline alkaloid metabolism–a century of discovery and a brave new world. Plant Cell Physiol. 54, 647–672 [DOI] [PubMed] [Google Scholar]
- 2. Samanani N., Liscombe D. K., Facchini P. J. (2004) Molecular cloning and characterization of norcoclaurine synthase, an enzyme catalyzing the first committed step in benzylisoquinoline alkaloid biosynthesis. Plant J. 40, 302–313 [DOI] [PubMed] [Google Scholar]
- 3. Hagel J. M., Facchini P. J. (2010) Dioxygenases catalyze the O-demethylation steps of morphine biosynthesis in opium poppy. Nat. Chem. Biol. 6, 273–275 [DOI] [PubMed] [Google Scholar]
- 4. Zhu W. (2008) CYP2D6: A key enzyme in morphine synthesis in animals. Med. Sci. Monit. 14, SC15–SC18 [PubMed] [Google Scholar]
- 5. Hagel J. M., Facchini P. J. (2010) Biochemistry and occurrence of O-demethylation in plant metabolism. Front. Physiol. 1, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fukuto J. M., Kumagai Y., Cho A. K. (1991) Determination of the mechanism of demethylenation of (methylenedioxy)phenyl compounds by cytochrome P450 using deuterium isotope effects. J. Med. Chem. 34, 2871–2876 [DOI] [PubMed] [Google Scholar]
- 7. Meyer M. R., Peters F. T., Maurer H. H. (2008) The role of human hepatic cytochrome P450 isozymes in the metabolism of racemic 3,4-methylenedioxy-methamphetamine and its enantiomers. Drug Metab. Dispos. 36, 2345–2354 [DOI] [PubMed] [Google Scholar]
- 8. Meyer M. R., Peters F. T., Maurer H. H. (2009) Investigations on the human hepatic cytochrome P450 isozymes involved in the metabolism of 3,4-methylenedioxy-amphetamine (MDA) and benzodioxolyl-butanamine (BDB) enantiomers. Toxicol. Lett. 190, 54–60 [DOI] [PubMed] [Google Scholar]
- 9. Liu Y., Hao H., Xie H., Lv H., Liu C., Wang G. (2009) Oxidative demethylenation and subsequent glucuronidation are the major metabolic pathways of berberine in rats. J. Pharm. Sci. 98, 4391–4401 [DOI] [PubMed] [Google Scholar]
- 10. Paul L. D., Springer D., Staack R. F., Kraemer T., Maurer H. H. (2004) Cytochrome P450 isoenzymes involved in rat liver microsomal metabolism of californine and protopine. Eur. J. Pharmacol. 485, 69–79 [DOI] [PubMed] [Google Scholar]
- 11. Desgagné-Penix I., Facchini P. J. (2012) Systematic silencing of benzylisoquinoline alkaloid biosynthetic genes reveals the major route to papaverine in opium poppy. Plant J. 72, 331–344 [DOI] [PubMed] [Google Scholar]
- 12. Liscombe D. K., Facchini P. J. (2007) Molecular cloning and characterization of tetrahydroprotoberberine cis-N-methyltransferase, an enzyme involved in alkaloid biosynthesis in opium poppy. J. Biol. Chem. 282, 14741–14751 [DOI] [PubMed] [Google Scholar]
- 13. Hagel J. M., Beaudoin G. A., Fossati E., Ekins A., Martin V. J., Facchini P. J. (2012) Characterization of a flavoprotein oxidase from opium poppy catalyzing the final steps in sanguinarine and papaverine biosynthesis. J. Biol. Chem. 287, 42972–42983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dang T. T., Onoyovwi A., Farrow S. C., Facchini P. J. (2012) Biochemical genomics for gene discovery in benzylisoquinoline alkaloid biosynthesis in opium poppy and related species. Methods Enzymol. 515, 231–266 [DOI] [PubMed] [Google Scholar]
- 15. Desgagné-Penix I., Farrow S. C., Cram D., Nowak J., Facchini P. J. (2012) Integration of deep transcript and targeted metabolite profiles for eight cultivars of opium poppy. Plant Mol. Biol. 79, 295–313 [DOI] [PubMed] [Google Scholar]
- 16. Chenna R. (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31, 3497–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schmidt J., Raith K., Boettcher C., Zenk M. H. (2005) Analysis of benzylisoquinoline-type alkaloids by electrospray tandem mass spectrometry and atmospheric pressure photoionization. Eur. J. Mass Spectrom. 11, 325–333 [DOI] [PubMed] [Google Scholar]
- 18. Schmidt J., Boettcher C., Kuhnt C., Kutchan T. M., Zenk M. H. (2007) Poppy alkaloid profiling by electrospray tandem mass spectrometry and electrospray FT-ICR mass spectrometry after [ring-13C6]tyramine feeding. Phytochemistry 68, 189–202 [DOI] [PubMed] [Google Scholar]
- 19. Farrow S. C., Hagel J. M., Facchini P. J. (2012) Transcript and metabolite profiling in cell cultures of 18 plant species that produce benzylisoquinoline alkaloids. Phytochemistry 77, 79–88 [DOI] [PubMed] [Google Scholar]
- 20. Meisel L., Fonseca B., González S., Baeza-Yates R., Cambiazo V., Campos R., González M., Orellana A., Retamales J., Silva H. (2005) A rapid and efficient method for purifying high quality total RNA from peaches (Prunus persica) for functional genomics analyses. Biol. Res. 38, 83–88 [DOI] [PubMed] [Google Scholar]
- 21. Martín-Hernández A. M., Baulcombe D. C. (2008) Tobacco rattle virus 16-kilodalton protein encodes a suppressor of RNA silencing that allows transient viral entry in meristems. J. Virol. 82, 4064–4071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wijekoon C. P., Facchini P. J. (2012) Systematic knockdown of morphine pathway enzymes in opium poppy using virus-induced gene silencing. Plant J. 69, 1052–1063 [DOI] [PubMed] [Google Scholar]
- 23. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real time quantitative PCR and the 2−ΔΔCt method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 24. Dolejš L., Hanuš V. (1967) Mass spectrometry of rhoeadine type alkaloids. Tetrahedron 23, 2997–3005 [Google Scholar]
- 25. Tiainen P., Myllyharju J., Koivunen P. (2005) Characterization of a second Arabidopsis thaliana prolyl 4-hydroxylase with a distinct substrate specificity. J. Biol. Chem. 280, 1142–1148 [DOI] [PubMed] [Google Scholar]
- 26. Tucker G. T., Lennard M. S., Ellis S. W., Woods H.F., Cho A. K., Lin L. Y., Hiratsuka A., Schmitz D. A, Chu T. Y. (1994) The demethylenation of methylenedioxymethamphetamine (“ecstasy”) by debrisoquine hydroxylase (CYP2D6). Biochem. Pharmacol. 47, 1151–1156 [DOI] [PubMed] [Google Scholar]
- 27. Purpero V., Moran G. R. (2007) The diverse and pervasive chemistries of the α-keto acid dependent enzymes. J. Biol. Inorg. Chem. 12, 587–601 [DOI] [PubMed] [Google Scholar]
- 28. Loenarz C., Schofield C. J. (2008) Expanding chemical biology of 2-oxoglutarate oxygenases. Nat. Chem. Biol. 4, 152–156 [DOI] [PubMed] [Google Scholar]
- 29. Lin L. Y., Kumagai Y., Cho A. K. (1992) Enzymatic and chemical demethylenation of (methylenedioxy)amphetamine and (methylenedioxy)methamphetamine by rat brain microsomes. Chem. Res. Toxicol. 5, 401–406 [DOI] [PubMed] [Google Scholar]
- 30. Winzer T., Gazda V., He Z., Kaminski F., Kern M., Larson T. R., Li Y., Meade F., Teodor R., Vaistij F. E., Walker C., Bowser T. A., Graham I. A. (2012) A Papaver somniferum 10-gene cluster for synthesis of the anticancer alkaloid noscapine. Science 336, 1704–1708 [DOI] [PubMed] [Google Scholar]
- 31. Zulak K. G., Cornish A., Daskalchuk T. E., Deyholos M. K., Goodenowe D. B., Gordon P. M., Klassen D., Pelcher L. E., Sensen C. W., Facchini P. J. (2007) Gene transcript and metabolite profiling of elicitor-induced opium poppy cell cultures reveals the coordinate regulation of primary and secondary metabolism. Planta 225, 1085–1106 [DOI] [PubMed] [Google Scholar]
- 32. Decker G., Wanner G., Zenk M. H., Lottspeich F. (2000) Characterization of proteins in latex of opium poppy (Papaver somniferum) using two-dimensional gel electrophoresis and microsequencing. Electrophoresis 21, 3500–3516 [DOI] [PubMed] [Google Scholar]
- 33. Liu C.-J., Dixon R. A. (2001) Elicitor-induced association of isoflavone O-methyltransferase with endomembranes prevents the formation and 7-O-methylation of daidzein during isoflavonoid phytoalexin biosynthesis. Plant Cell 13, 2643–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Beaudoin G. A., Facchini P. J. (2013) Isolation and characterization of a cDNA encoding (S)-cis-N-methylstylopine 14-hydroxylase from opium poppy, a key enzyme in sanguinarine biosynthesis. Biochem. Biophys. Res. Comm. 431, 597–603 [DOI] [PubMed] [Google Scholar]
- 35. Ziegler J., Brandt W., Geissler R., Facchini P. J. (2009) Removal of substrate inhibition and increase in maximal velocity in the short chain dehydrogenase/reductase salutaridine reductase involved in morphine biosynthesis. J. Biol. Chem. 284, 26758–26767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Manoj K. M., Baburaj A., Ephraim B., Pappachan F., Maviliparambathu P. P., Vijayan U. K., Narayanan S. V., Periasamy K., George E. A., Mathew L. T. (2010) Explaining the atypical reaction profiles of heme enzymes with a novel mechanistic hypothesis and kinetic treatment. PLoS One 5, e10601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tracy T. S. (2006) Atypical cytochrome P450 kinetics: implications for drug discovery. Drugs R&D 7, 349–363 [DOI] [PubMed] [Google Scholar]
- 38. Ronsch H. (1977) Biosynthesis of alpinigenine by way of tetrahydroprotoberberine and protopine intermediates. Phytochemistry 16, 691–698 [Google Scholar]
- 39. Montgomery C. T., Cassels B. K., Shamma M. (1983) The rhoeadine alkaloids. J. Nat. Prod. 46, 441–453 [Google Scholar]
- 40. Tani C., Tagahara K. (1977) Studies on the alkaloids of Papaveraceous plants. XXVIII. The biosynthesis of rhoeadine. J. Pharm. Soc. Jpn. 97, 93–102 [PubMed] [Google Scholar]
- 41. Ikezawa N., Iwasa K., Sato F. (2007) Molecular cloning and characterization of methylenedioxy bridge-forming enzymes involved in stylopine biosynthesis in Eschscholzia californica. FEBS J. 274, 1019–1035 [DOI] [PubMed] [Google Scholar]
- 42. Díaz Chávez M. L., Rolf M., Gesell A., Kutchan T. M. (2011) Characterization of two methylenedioxy bridge-forming cytochrome P450-dependent enzymes of alkaloid formation in the Mexican prickly poppy Argemone mexicana. Arch. Biochem. Biophys. 507, 186–193 [DOI] [PubMed] [Google Scholar]
- 43. Morishige T., Dubouzet E., Choi K. B., Yazaki K., Sato F. (2002) Molecular cloning of columbamine O-methyltransferase from cultured Coptis japonica cells. Eur. J. Biochem. 269, 5659–5667 [DOI] [PubMed] [Google Scholar]
- 44. Dang T. T., Facchini P. J. (2012) Characterization of three O-methyltransferases involved in noscapine biosynthesis in opium poppy. Plant Physiol. 159, 618–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ibragimova M. U., Israilov I. A., Yunusov M. S., Yunusov S. Y. (1974) Alkaloids of Fumaria vaillantii structure of vaillantine. Chem. Nat. Compd. 10, 481–482 [Google Scholar]
- 46. Bahadur S., Shukla A. K. (1983) Studies on native medicinal plants, I. the quaternary alkaloids of Thalictrum javanicum. J. Nat. Prod. 46, 454–457 [Google Scholar]
- 47. Lassakaya O. E., Tolkachev O. N. (1978) Quaternary benzophenanthridine alkaloids 9,10-demethylene derivatives of sanguinarine. Chem. Nat. Comp. 44, 650–652 [Google Scholar]
- 48. Kametani T. (1968) The Chemistry of the Isoquinoline Alkaloids. Elsevier Science Publishers B.V., Amsterdam [Google Scholar]
- 49. Kametani T. (1974) The Chemistry of the Isoquinoline Alkaloids, Vol. 2, The Sendai Institute of Heterocyclic Chemistry, Japan [Google Scholar]
- 50. Runguphan W., Glenn W. S., O'Connor S. E. (2012) Redesign of a dioxygenase in morphine biosynthesis. Chem. Biol. 19, 674–678 [DOI] [PubMed] [Google Scholar]