

Background: STIM1 and Orai1, reconstituting a main cellular Ca2+ entry pathway, interact via their cytosolic strands.
Results: The extended transmembrane Orai1 N-terminal (ETON) region combines binding interface and gate for Orai1 activation by STIM1.
Conclusion: Several “hot spot” residues in the ETON region mediate STIM1 interaction, enabling conformational reorientation of the gate.
Significance: Identification of critical residues for protein-protein interaction are fundamental to therapeutic drug development.
Keywords: Calcium Channels, Calcium Intracellular Release, Calcium Signaling, Electrophysiology, Ion Channels
Abstract
STIM1 and Orai1 represent the two molecular key components of the Ca2+ release-activated Ca2+ channels. Their activation involves STIM1 C terminus coupling to both the N terminus and the C terminus of Orai. Here we focused on the extended transmembrane Orai1 N-terminal (ETON, aa73–90) region, conserved among the Orai family forming an elongated helix of TM1 as recently shown by x-ray crystallography. To identify “hot spot” residues in the ETON binding interface for STIM1 interaction, numerous Orai1 constructs with N-terminal truncations or point mutations within the ETON region were generated. N-terminal truncations of the first four residues of the ETON region or beyond completely abolished STIM1-dependent Orai1 function. Loss of Orai1 function resulted from neither an impairment of plasma membrane targeting nor pore damage, but from a disruption of STIM1 interaction. In a complementary approach, we monitored STIM1-Orai interaction via Orai1 V102A by determining restored Ca2+ selectivity as a consequence of STIM1 coupling. Orai1 N-terminal truncations that led to a loss of function consistently failed to restore Ca2+ selectivity of Orai1 V102A in the presence of STIM1, demonstrating impairment of STIM1 binding. Hence, the major portion of the ETON region (aa76–90) is essential for STIM1 binding and Orai1 activation. Mutagenesis within the ETON region revealed several hydrophobic and basic hot spot residues that appear to control STIM1 coupling to Orai1 in a concerted manner. Moreover, we identified two basic residues, which protrude into the elongated pore to redound to Orai1 gating. We suggest that several hot spot residues in the ETON region contribute in aggregate to the binding of STIM1, which in turn is coupled to a conformational reorientation of the gate.
Introduction
The CRAC4 channel signaling machinery consists of two key components, STIM1 (1, 2) and Orai (3–5). STIM1 acts as an endoplasmic reticulum-located Ca2+ sensor and mediates CRAC channel activation (1, 2, 6). After endoplasmic reticulum Ca2+ store depletion, STIM1 multimerizes and redistributes into punctae close to the plasma membrane (7, 8). It undergoes a switch to an extended conformation, thereby coupling to the CRAC channel Orai (9, 10). Finally, the coupling of STIM1 to Orai culminates in CRAC channel activation, which allows Ca2+ influx into the cell (11–14).
Recently, Hou et al. (15) have published the crystal structure of Drosophila Orai. It exhibits a hexameric assembly of Orai subunits with the ion pore located in the center, which is surrounded by the transmembrane domains. Thereby the first transmembrane domains form an inner ring around the ion pore, the second and the third ones form a middle ring, and the fourth transmembrane domains form the outer ring (15). Ca2+ enters the cell at a 6 Å narrow opening: the selectivity filter, which is composed of the glutamate Glu-106 in human Orai1 (16). Toward the cytoplasmic side, the pore opens to a wider cavity including hydrophobic side chains such as valine, phenylalanine, and lysine: for example, Val-102. The mutation of Val-102 to an alanine or a cysteine profoundly alters the selectivity of the pore and leads to constitutively active nonselective currents (17). Upon STIM1 binding, Orai1 V102A regains Ca2+ selectivity comparable with wild-type Orai1 (17). The selectivity filter and the hydrophobic cavity are followed by a flexible glycine hinge (Gly-98) (18), which may enable flexion of the upstream pore-lining region to reduce the impedance of Ca2+ flow after passing the selectivity filter (16).
Strikingly, this part of the cytosolic N-terminal strand upstream of the first transmembrane helix (TM1) forms a helical (19), extended transmembrane Orai1 N-terminal (ETON) region that comprises the N-terminal residues aa73–90, which are fully conserved among the three human homologues of Orai proteins and protrudes about 20 Å into the cytosol (16). The TM1 helix together with the ETON region contains three positively charged residues Arg-91, Lys-87, and Arg-83, which directly line the pore and thus have been supposed to form an electrostatic barrier impeding Ca2+ flow when the channel is in the closed state (16). The arginine Arg-91 inhibits store-operated current activation upon its mutation to a hydrophobic residue (20, 21). This barrier of the three positively charged residues must be released to let Ca2+ pass into the cell, which may be accomplished by an interaction of STIM1 with the conserved ETON regions forming the elongated pore (16). The CRAC-activating domain (CAD), a small Orai-activating STIM1 C-terminal fragment, has already been shown to interact with an N-terminal fragment (73–90) of Orai1 (22), underlining its relevance as the second major interaction site besides Orai1 C terminus (11, 12, 23). Orai1 is probably gated by a STIM1 binding to bridge the cytosolic TM1 and TM4 extended helices, thereby applying a force at the helical TM1 extension to form and stabilize the open pore state (16).
Another positively charged residue near the membrane, Lys-85 (24, 25), located on the pore-averted side of the helical TM1 extension, has been reported to abolish store-operated activation upon a K85E mutation due to a defect in gating together with a weaker STIM1 binding (24, 25).
In this study, we performed a systematic screen along the conserved ETON region to determine potential “hot spot” (26–28) residues in the binding interface with STIM1. A combined approach based on Orai1 N-terminal truncations and point mutations revealed that almost the whole ETON region provides an elongated low affinity binding interface for the interaction with STIM1. It additionally contains electrostatic gating elements that fine-tune and shape the elongated pore for controlled STIM1-dependent Ca2+ entry.
EXPERIMENTAL PROCEDURES
Molecular Biology
For N-terminal fluorescence labeling of human Orai1 (Orai1; accession number NM_032790, provided by the A. Rao laboratory), the constructs were cloned into the pEYFP-C1 (Clontech) expression vector via KpnI and XbaI restriction sites. For the alanine scanning mutagenesis, pEYFP-C1-Orai1 served as template. Site-directed mutagenesis (pEYFP-Orai1 S75W, S75A/W76A, W76A, W76P, W76S, W76E, W76R, L74S/W76S, L74R/W76R, L74E/W76E, R77A/K78A, R83A/K87A, K85E, R83E/K87E, N1–74 S75A/W76A, ΔN1–74 R83A/K87A, ΔN1–76 R83A/K87A) was performed using the QuikChangeTM XL site-directed mutagenesis kit (Stratagene). pEYFP-Orai1 N-terminal deletion mutants (Orai1 ΔN1–38, ΔN1–47, ΔN1–72, ΔN1–74, ΔN1–75, ΔN1–76, ΔN1–77, ΔN1–78, ΔN1–80, ΔN1–82, ΔN1–85, ΔN1–87, DN1–88, ΔN1–89) were amplified via PCR including an N-terminal KpnI and a C-terminal XbaI restriction site for cloning into the pEYFP-C1 vector. V102A mutants were accomplished analogously (using the QuikChangeTM XL site-directed mutagenesis kit (Stratagene) with the corresponding point mutants serving as template.
Human STIM1 (STIM1; accession number: NM_003156, provided by the T. Meyers laboratory) was subcloned from pcDNA3.1/V5-His TOPO via KpnI and ApaI restriction sites into a custom made mCherry vector (peGFPN-3 as backbone). pECFP-C1 STIM1 C terminus (aa233–685 WT and L251S) was used as template for the generation of pECFP-OASF (WT and L251S) by introducing a stop codon at position 475 (aa233–474) using the QuikChange XL site-directed mutagenesis kit (Stratagene). pEYFP-C1 STIM1 C terminus (aa233–685 WT and L251S) was used as template for the generation of pEYFP-OASF (WT and L251S) by introducing a stop codon at position 475. pEYFP-C1 STIM1 C terminus (aa233–685 WT) was used as template for the generation of pEYFP-233–485 by introducing a stop codon at position 486. All point mutations were performed using the QuikChange XL site-directed mutagenesis kit (Stratagene). STIM1 fragment 344–449 was amplified via PCR including an N-terminal KpnI and a C-terminal XbaI restriction site for cloning into the pECFP-C1 vector. All clones were confirmed by sequence analysis.
Biotinylation
Reagents
EZ-Link Sulfo-NHS-LC-biotin and high capacity streptavidin-Agarose resin were purchased from Pearce; anti-Orai1 and anti-β actin antibodies were from Sigma-Aldrich; anti-rabbit IgG peroxidase-linked was from Amersham Biosciences.
Biotinylation of Cell Surface Membrane Proteins
Cell biotinylation was performed as described previously (29). Briefly, HEK293 cells were transfected with YFP-Orai1 WT or different YFP-Orai1 mutants. After 24–72 h, cell surface proteins were incubated for 1h at 4 °C using EZ-Link sulfo-NHS-LC-biotin (0.5 mg/ml; Pierce). After incubation, 100 mm Tris was added to stop the reaction. Cells were washed twice with PBS to remove excess biotinylation agent and lysed with lysis buffer, pH 8.0, containing 100 mm NaCl, 20 mm Tris, 2 mm EDTA, 10% glycerol, 0.5% Nonidet P40 and supplemented by 20 μl/ml protease inhibitor mixture (Roche Applied Science). Lysed samples were centrifuged at 14,000 × g for 15 min. Finally, biotinylated proteins in the supernatant were precipitated using high capacity streptavidin-Agarose resin (Pearce) overnight at 4 °C on a rocking platform. The samples were resolved by 12% SDS-PAGE, and protein detection was achieved using the anti-Orai1 antibody diluted 1:2000 in PBS-Tween-20 (PBST). The primary antibody was removed, and blots were washed six times for 5 min each with PBST. To detect the primary antibody, blots were incubated for 1 h with horseradish peroxidase-conjugated rabbit anti-mouse IgG antibody diluted 1:5000 in PBST and then with enhanced chemiluminescence reagents for 2 min. Blots were then exposed to photographic films. The density of bands on the film was measured using scanning densitometry. Anti-β-actin (A5441; Sigma) was used to normalize protein expression.
Electrophysiology
Electrophysiological recordings comparing characteristics of 2–3 constructs were carried out in paired comparison on the same day. Expression patterns and levels of the various constructs were carefully monitored by confocal fluorescence microscopy and were not significantly changed by the introduced mutations. Electrophysiological experiments were performed at 20–24 °C, using the patch clamp technique in the whole-cell recording configuration. For STIM1/Orai as well as STIM1 C terminus/Orai current measurements, voltage ramps were usually applied every 5 s from a holding potential of 0 mV, covering a range of −90 to +90 mV over 1 s. The internal pipette solution for passive store depletion contained (in mm) 3.5 MgCl2, 145 cesium methanesulfonate, 8 NaCl, 10 HEPES, 20 EGTA, pH 7.2. Extracellular solution consisted of (in mm) 145 NaCl, 5 CsCl, 1 MgCl2, 10 HEPES, 10 glucose, 10 CaCl2, pH 7.4.
Confocal Fluorescence Microscopy
Confocal microscopy for co-localization experiments was performed similarly as in Ref. 30. In brief, a QLC100 real-time confocal system (VisiTech International) was used for recording fluorescence images connected to two Photometrics CoolSNAPHQ monochrome cameras (Roper Scientific) and a dual port adapter (dichroic, 505lp; cyan emission filter, 485/30; yellow emission filter, 535/50; Chroma Technology Corp.). This system was attached to an Axiovert 200M microscope (Zeiss) in conjunction with an argon ion multiwavelength (457, 488, 514 nm) laser (Spectra Physics). The wavelengths were selected by an acousto-optical tunable filter (VisiTech International). MetaMorph 5.0 software (Universal Imaging Corp.) was used to acquire images and to control the confocal system. Illumination times for CFP and YFP images that were recorded with a minimum delay consecutively were about 900 ms.
Thermophoresis Measurements
The thermophoresis experiments were carried out with the monolith NT.115 (Nanotemper). We employed the STIM1 233–485 C-terminal fragment as a surrogate for full-length STIM1 as it expressed much better in HEK293 cells than the shorter STIM1-OASF. In brief, YFP-STIM1 233–485 transfected HEK293 cells were washed 24 h after transfection with PBS and pelleted at 1500 × g for 10 min. The supernatant was discarded, and the pellet was resuspended in 350 μl of DMEM. The cells were lysed by adding 35 μl of lysis buffer (20 mm TRIS (pH 7.4), 1% Nonidet P40, 150 mm NaCl, and protease inhibitor) for 30 min and afterward centrifuged at 13,000 × g for 20 min. The supernatant containing YFP-STIM1 233–485 was stored at 4 °C until analysis. Orai1 peptides (aa70–91, aa77–91, aa70–91 L74E/W76E) were purchased from peptide 2.0. Up to 16 samples of different dilutions were prepared (concentration of YFP-STIM1 233–485 was kept constant, concentration of the Orai1 peptide was varied), and after an incubation time of 5 min, the reaction was transferred into glass capillaries and placed on the sample tray. The instrument recognized automatically the capillaries, and upon measuring the thermophoresis signal, the dissociation coefficient was calculated.
RESULTS
STIM1-dependent Activation of Orai1 Requires Almost the Whole ETON Region
The ETON region (aa73–90, Fig. 1, a and b), comprising the extended TM1 Orai1 N-terminal helix, is essential for store-operated CRAC current activation via STIM1 (11, 12, 22–24). For a thorough evaluation of the impact of this ETON region on STIM1 binding and Orai1 activation, we initially generated several Orai1 mutants with increasing N-terminal deletions in an attempt to uncover the minimal stretch of the ETON region that retains store-operated activation. The function of these Orai1 truncation mutants co-expressed in HEK293 cells with STIM1 was characterized by employing the whole-cell patch clamp technique. Orai1 ΔN1–38 partially and Orai1 ΔN1–47 fully lacking the arginine-/proline-rich region were activated via STIM1 in a comparable manner as wild-type Orai1 (supplemental Fig. 1a). Orai1 ΔN1–72 and Orai1 ΔN1–74 still displayed STIM1-dependent current activation, which was similar to that of the Orai1 ΔN1–73 mutant (31), although slightly reduced when compared with Orai1 wild-type (Fig. 1, c and e). Deletion of a further residue leading to Orai1 ΔN1–75 retained store-operated activation, yet further reduced current density (Fig. 1, c and e). However, the following Trp-76 (Orai1 ΔN1–76) or consecutive residues (Arg-77/Lys-78/Y80/Ser-82/Lys-85/Lys-87), when deleted, completely abolished STIM1-dependent CRAC current activation (Fig. 1, d and e). In line with this finding, STIM1 C-terminal fragments such as STIM1 344–449 or OASF (STIM1 233–474), both displaying an increased coupling to Orai1 proteins, thereby enhancing currents 2–3-fold (32), also failed to activate Orai1 ΔN1–76 and Orai1 ΔN1–78 (supplemental Fig. 1, b and c).
FIGURE 1.

N-terminal truncations including Trp-76 of the ETON region or beyond abolish STIM1-dependent Orai1 function. a, amino acid sequence of Orai1 N terminus displaying those residues (indicated by scissors) in the conserved ETON region (yellow area) up to which increasing N-truncations have been performed. b, schematic representation of two TM1 domains + ETON regions highlighting those residues up to which N-truncations have been performed (blue) in the ETON region. c, time course of whole cell inward currents at −74 mV activated by passive store depletion of HEK 293 cells co-expressing CFP-STIM1 and YFP-Orai1 ΔN1–72, ΔN1–74 and ΔN1–75 in comparison with wild-type YFP-Orai1. pF, picofarads. d, time course of whole cell inward currents at −74 mV activated by passive store depletion of HEK293 cells co-expressing CFP-STIM1 and YFP-Orai1 ΔN1–76 and ΔN1–78 in comparison with wild-type YFP-Orai1. e, block diagram comparing maximal current densities of Orai1 N-truncation mutants (ΔN1–38, -47, -72, -74, -75, -76, -77, -78, -80, -82, -85, -87) with that of wild-type Orai1. Error bars indicate S.E. f, Orai1 plasma membrane expression in HEK293 cells expressing Orai1 WT, Orai1 ΔN1–74, Orai1 ΔN1–76, or Orai1 ΔN1–78 determined by biotinylation as described under “Experimental Procedures” (n = 5; *, p < 0.05). Error bars indicate S.E. n.s., not significant. WB, Western blot.; ab, antibody.
To exclude impaired plasma membrane targeting of the respective Orai1 deletion mutants as the cause of loss of function, we estimated the amount targeted to the plasma membrane by biotinylation experiments. These studies revealed similar plasma membrane expression levels for YFP-labeled Orai1, Orai1 ΔN1–76, and Orai1 ΔN1–78 (Fig. 1f) in line with similar plasma membrane fluorescence intensities (data not shown).
Hence, based on these N-terminal deletion experiments, most (aa76–90) of the ETON region (aa73–90) is essential for STIM1-dependent Orai1 activation. As the loss of function is not caused by impairment of plasma membrane expression, these truncations apparently affected either STIM1 coupling or Orai1 gating.
Orai1 V102A Currents Enable Detection of STIM1 Interaction by a Rightward Shift in the Reversal Potential
In a complementary approach, we utilized the recently reported constitutively active Orai1 mutant V102A/V102C (17) to examine the impact of the N-terminal deletions on channel function and coupling to STIM1. Specifically, the V102A/V102C mutant is able to gate in the absence of STIM1, yielding constitutively active but nonselective currents, which regain selectivity upon coupling to STIM1 (supplemental Fig. 2, a–e) (17). The Orai1 mutant V102A/L273S or V102C/L276D (17) with a disrupted C-terminal binding site for STIM1 failed to re-establish Ca2+ selectivity as judged from a lack of rightward shift in the reversal potential in the presence of STIM1 (supplemental Fig. 2, c–e) in line with the predominant role of Orai1 C terminus in the coupling to STIM1. As interactions of both the C termini and the N termini of Orai1 with STIM1 are required for a functional channel (12, 22, 24, 33), we hypothesize that the shift in the reversal potential is induced by STIM1 binding to Orai1 N-terminal regions, as long as the Orai1 C terminus is intact. Thus, we utilized the potential rightward shift in the reversal potential as a read-out parameter for an N-terminal interaction of Orai1 with STIM1.
N-terminal deletion mutants up to residues 38, 72, and 74, which retained STIM1-dependent Orai1 currents when introduced into the Orai1 V102A core, yielded constitutively active currents that were nonselective in the absence of STIM1 and Ca2+-selective in its presence. This was clearly evident from the rightward shift in the reversal potential similar to wild-type Orai1 (supplemental Fig. 2, f–j). Thus, the N-terminal strand of Orai1 including residue 74 was dispensable for STIM1-mediated high Ca2+ selectivity of Orai1 in line with the intact ETON region coupling to STIM1.
When introduced into the Orai1 V102A core, the substantially less active Orai1 ΔN1–75 as well as the other nonfunctional N-truncation mutants Orai1 ΔN1–76 and ΔN1–78 (Fig. 1, c and d) displayed constitutive, nonselective currents with a reversal potential between +10 mV and +20 mV (Fig. 2, a and c), suggesting that these N-truncations did not severely damage the pore. This was also in line with the preserved 2-aminoethoxydiphenyl borate-evoked currents obtained with the nonfunctional truncation mutants (supplemental Fig. 3, a–d). Although the Orai1 ΔN1–75 V102A channels when co-expressed with STIM1 displayed a significant rightward shift in the reversal potential of constitutive currents, the previously (see Fig. 1) STIM1 nonresponsive Orai1 N-truncation mutants ΔN1–76 to ΔN1–82 with the V102A mutation lacked this shift in the reversal potential when co-expressed with STIM1 (Fig. 2, b and c and supplemental Fig. 4). Deletion of further residues up to aa85 resulted in smaller but still constitutively active, nonselective currents with a reversal potential around 0 mV independent of the presence or absence of STIM1 (supplemental Fig. 4). Truncation up to aa87 or of the whole NT resulted in almost nonfunctional channels (supplemental Fig. 4).
FIGURE 2.
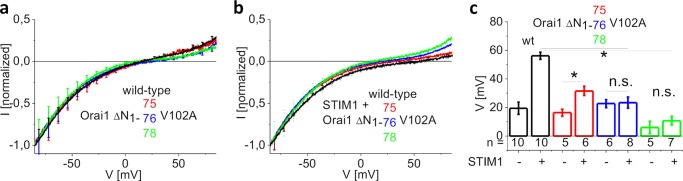
Nonfunctional Orai1 N-truncation mutants introduced into the Orai1 V102A core display no shift in reversal potential in the presence of STIM1. a, I/V relationships of normalized Orai1 V102A currents in comparison with those of Orai1 ΔN1–75 V102A, Orai1 ΔN1–76 V102A, and Orai1 ΔN1–78 V102A. b, analogue to panel a in the presence of STIM1. c, block diagram displaying reversal potentials of Orai1 V102A, Orai1 ΔN1–75 V102A, Orai1 ΔN1–76 V102A, and Orai1 ΔN1–78 V102A currents in the absence and presence of STIM1 (*, p < 0.05). Error bars indicate S.E. n.s., not significant.
In summary, these experiments established the inducible Ca2+ selectivity for Orai1 V102A N-terminal deletion mutants as a read-out parameter for detection of STIM1 interaction. The results obtained were perfectly in line with the previous experiments shown in Fig. 1 in that residues around aa76 of the ETON region were essential in contributing to the Orai1 N-terminal interaction with STIM1.
STIM1 Interaction with the Nonfunctional Orai1 N-terminal Deletion Mutants Is Significantly Reduced
In a further approach, we employed co-localization experiments to directly visualize a potentially reduced coupling of the nonfunctional truncation mutants Orai1 ΔN1–76 and Orai1 ΔN1–78 with the co-expressed STIM1 OASF (aa234–474) as well as the OASF L251S fragment (Fig. 3, a and b). Initially, we employed OASF L251S, which already provides the active, extended conformation (9, 10). Co-expression of OASF L251S with wild-type Orai1 resulted in almost 90% co-localization, leaving 10% OASF L251S in the cytosol as judged from cell density profiles (Fig. 3a). The cytosolic fraction was increased to ∼20% when co-expressed with Orai1 ΔN1–74, compatible with the reduced current density in comparison with wild-type Orai1. The nonfunctional Orai1 ΔN1–76 and the Orai1 ΔN1–78 displayed ∼50% attenuation in co-localization with OASF L251S, similar to Orai1 ΔN1–88 where almost the whole N terminus was deleted. An Orai1 mutant lacking the whole C terminus (Orai1 ΔC-term) exhibited a complete loss of co-localization with OASF L251S as evident from the density profile and confirmed the dominant role of Orai1 C terminus in the coupling process (12). Next, we utilized the wild-type OASF, which requires interaction with Orai1 to adopt the extended conformation (9). The coupling of OASF to Orai1 ΔN1–74 was dramatically reduced with about 80% determined in the cytosol, whereas Orai1 ΔN1–76 and Orai1 ΔN1–78 completely lacked co-localization (Fig. 3b) similar to Orai1 ΔN1–88 with almost the whole N terminus missing (data not shown). Hence, OASF, when compared with OASF L251S, was particularly sensitive to Orai1 N-terminal deletions in that it apparently required interaction with both the N-terminal strands as well as the C-terminal strands of Orai1. Therefore, the OASF fragment represented another sensitive tool for detection of hot spot residues (see next paragraph) in the ETON region binding interface with STIM1.
FIGURE 3.
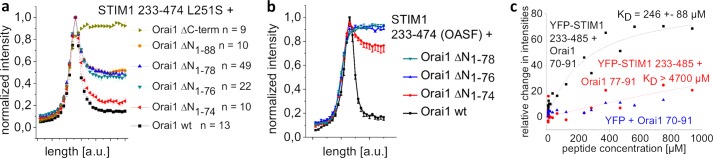
Nonfunctional Orai1 N-truncation mutants display a significant reduction in STIM1 binding. a, intensity plots representing the localization of STIM1 233–474 L251S across the cell when co-expressed with Orai1 ΔN1–74, Orai1 ΔN1–76, Orai1 ΔN1–78, Orai1 ΔN1–88, and Orai1 ΔC-term when compared with wild-type Orai1. a. u., arbitrary units. b, intensity plots representing the localization of STIM1 233–474 (OASF) across the cell when co-expressed with Orai1 ΔN1–74, Orai1 ΔN1–76, and Orai1 ΔN1–78 when compared with wild-type Orai1. c, binding curve fitted to a one-site binding model for YFP-STIM1 233–485 from a HEK293 cell lysate together with Orai1 NT(aa70–91) or Orai1 NT(aa77–91) in comparison with YFP from HEK293 cell lysate together with Orai1 NT(aa70–91), obtained from thermophoresis experiments.
For a more quantitative analysis, we utilized thermophoresis measurements to estimate the dissociation constant KD of the slightly extended OASF fragment YFP-STIM1 233–485 and the Orai1 N-terminal peptide aa70–91 containing the whole ETON region or the peptide aa77–91 corresponding to Orai1 ΔN1–76, which previously displayed a reduced interaction with STIM1. In accordance to both the functional (Figs. 1 and 2) and the co-localization (Fig. 3, a and b) experiments, we estimated (Fig. 3c) a rather weak KD of 246 ± 88 μm for the interaction between STIM1 233–485 and the ETON region (Orai1 aa70–91), whereas the truncated ETON segment (Orai1 aa77–91) exhibited no saturating binding curve (KD>4700 μm). In this behavior, the latter resembled that of free YFP with Orai1 aa70–91, corresponding to the negative control (Fig. 3c).
In summary, complete loss of STIM1-dependent Orai1 activation with progressive N-terminal deletions, including residue 76 or beyond, arises from a lack or an improper STIM1 binding to the truncated ETON region. In the following, we focused on characterizing hot spot residues in the ETON region that constitutes the binding interface with STIM1 for Orai1 current activation.
The Hydrophobic Residues Leu-74 and Trp-76 in the ETON Region Contribute in a Concerted Manner to Orai1-STIM1 Coupling
The serial N-terminal deletions examined above already revealed that amino acids downstream to Trp-76 were essential for STIM1 binding to Orai1 N terminus. Hence, we initially focused on that residue as a potential hot spot (34) within the binding interface of the ETON region, essential for STIM1 binding to and STIM1-mediated gating of Orai1 channels. However, several single point mutations of Trp-76 tested (W76A/W76S/W76R/W76E; supplemental Fig. 5a), except W76P, did not exert a substantial effect on STIM1-dependent current activation as drastic as was found with the nonfunctional Orai1 ΔN1–76. The loss of function upon proline substitution in the N terminus of Orai1 W76P, which dramatically interfered with its function (supplemental Fig. 5, b and c), probably by generating a kink in the ETON region, might imply that indeed the region around Trp-76 but not the residue alone was indispensable for STIM1-dependent Orai1 activation.
Therefore, we focused next on double point mutations around position 76 based on our observation that Orai1 ΔN1–74 in contrast to Orai1 ΔN1–76 still retained STIM1-dependent function. Among several mutants, which were summarized in supplemental Fig. 6, we identified a double mutation of the two hydrophobic residues Leu-74 and Trp-76 that impaired channel function. Three mutants, Orai1 L74E/W76E, Orai1 L74R/W76R, and Orai1 L74S/W76S (Fig. 4, a and b), were generated by switching the hydrophobic amino acids to charged (Glu and Arg) or polar (Ser) residues. All three mutants displayed a significant reduction in STIM1-dependent activation (Fig. 4c), which did not result from disrupted plasma membrane targeting as evident from biotinylation experiments exemplarily shown for Orai1 L74S/W76S (Fig. 4d). Introduction of these mutations in the Orai1 V102A core resulted in constitutive, nonselective currents in the absence of STIM1, which displayed slight, yet nonsignificant rightward shifts of the reversal potential in the presence of STIM1 (Fig. 4, e and f). Furthermore, co-expression with OASF revealed a significant reduction in its co-localization with all three Orai1 L74E/L74R/L74S and W76E/W76R/W76S mutants (Fig. 4g). Accordingly, thermophoresis measurements of YFP-STIM1 233–485 and the Orai1 aa70–91 L74E/W76E N-terminal peptide revealed an about 1.7-fold increase in KD to 406 ± 68 μm when compared with the wild-type Orai1 N-terminal peptide. Nevertheless, the previously determined KD with the shorter 77–91 N-terminal peptide was at least 10-fold higher, suggesting additional residues in the ETON region between 70 and 76 that also contribute to the interaction with STIM1 (Fig. 4h). In summary, Leu-74 and Trp-76 were identified as hydrophobic hot spot residues within the ETON region that contribute in a concerted manner to the binding interface with STIM1.
FIGURE 4.

The hydrophobic residues Leu-74 and Trp-76 of the ETON region contribute in a concerted manner to the coupling with STIM1. a, amino acid sequence of Orai1 N terminus displaying Leu-74 and Trp-76 in red in the conserved ETON region (yellow area) b, schematic representation of two TM1 domains + ETON regions highlighting Leu-74 and Trp-76 (blue) in the ETON region. c, time course of whole cell inward currents at −74 mV activated by passive store depletion of HEK 293 cells co-expressing CFP-STIM1 and YFP-Orai1 L74S/W76S, YFP-Orai1 L74E/W76E, and YFP-Orai1 L74R/W76R in comparison with wild-type YFP-Orai1. pF, picofarads. d, Orai1 plasma membrane expression in HEK293 cells, expressing Orai1 WT in comparison with Orai1 L74S/W76S determined by biotinylation as described under “Experimental Procedures” (n = 5; *, p < 0.05). Error bars indicate S.E. e, I/V relationship of normalized Orai1 V102A currents in comparison with that of Orai1 L74E/W76E/V102A, shown as example. Error bars indicate S.E. n.s., not significant. WB, Western blot; ab, antibody. f, block diagram displaying reversal potentials of Orai1 V102A, Orai1 L74S/W76S/V102A (SS), Orai1 L74E/W76E/V102A (EE), and Orai1 L74R/W76R/V102A (RR) in the absence and presence of STIM1 (*, p < 0.05). Error bars indicate S.E. g, intensity plots representing the localization of STIM1 233–474 (OASF) across the cell when co-expressed with Orai1 L74S/W76S, Orai1 L74E/W76E, and Orai1 L74R/W76R when compared with wild-type Orai1. a. u., arbitrary units. h, binding curve fitted to a one-site binding model for YFP-STIM1 233–485 from a HEK293 cell lysate together with Orai1 NT(aa70–91) or Orai1 NT(aa70–91 L74E/W76E) in comparison with YFP from HEK293 cell lysate together with Orai1 NT(aa70–91), obtained from thermophoresis measurements.
The Positively Charged Arg-77 and Lys-78 in the ETON Region Also Contribute to STIM1 Binding
We focused next on the potential role of the positively charged residues in the ETON region downstream to Trp-76 as they are also frequently found in protein-protein interfaces (35). Single point mutations of positively charged residues (Orai1 R77E, Orai1 K78E, Orai1 R83A, Orai1 K87A) did not severely impair STIM1-dependent current activation when compared with wild-type Orai1 (data not shown) (25). Only the single point mutant Orai1 K85E exhibited loss of function due to impaired gating together with a reduced STIM1 binding (supplemental Fig. 7) in line with Refs. 24 and 25.
Next, we constructed double point mutations and focused initially on residues Arg-77/Lys-78 adjacent to position 76 (Fig. 5, a and b). The Orai1 R77A/K78A double mutant displayed ∼50% reduced STIM1-dependent current densities (Fig. 5c), the attenuation of which was not caused by a diminished plasma membrane targeting (Fig. 5d). Introduction of this double R77A/K78A mutation into the Orai1 V102A core resulted in the expected nonselective, constitutive currents, which still exhibited a significant rightward shift of the reversal potential in the presence of STIM1 but not to the extent of the Orai1 V102A (Fig. 5, e and f). In accordance, co-localization of the Orai1 R77A/K78A mutant with OASF was partially reduced (Fig. 5g). Hence, we suggest that the positively charged Arg-77 and Lys-78 residues in the ETON region also contribute in a concerted manner to the STIM1 binding interface, but in a less dominant role as found for the hydrophobic Leu-74 and Trp-76.
FIGURE 5.

The positively charged residues Arg-77 and Lys-78 in the ETON region also contribute to STIM1 binding. a, amino acid sequence of Orai1 N terminus displaying Arg-77/Lys-78 in red in the ETON region (yellow area). b, schematic representation of two TM1 domains + ETON regions highlighting Arg-77 and Lys-78 (blue) in the ETON region. c, time course of whole cell inward currents at −74 mV activated by passive store depletion of HEK 293 cells co-expressing CFP-STIM1 and YFP-Orai1 R77A/K78A in comparison with wild-type YFP-Orai1. pF, picofarads. d, Orai1 plasma membrane expression in HEK293 cells, expressing Orai1 in comparison with Orai1 R77A/K78A as determined by biotinylation as described under “Experimental Procedures” (n = 5; *, p < 0.05). Error bars indicate S.E. n.s., not significant. WB, Western blot; ab, antibody. e, I/V relationships of normalized Orai1 R77A/K78A/V102A currents in the absence and presence of STIM1. Error bars indicate S.E. f, block diagram displaying reversal potentials of Orai1 V102A and Orai1 R77A/K78A/V102A in the absence and presence of STIM1 (*, p < 0.05). Error bars indicate S.E. g, intensity plots representing the localization of STIM1 233–474 (OASF) across the cell when co-expressed with Orai1 R77A/K78A when compared with wild-type Orai1. Error bars indicate S.E.
The Positively Charged Arg-83 and Lys-87 Contribute to the STIM1 Binding Interface in a Concerted Manner, and as Pore-lining Residues, They Provide Gating Function to the ETON Region
Next, double point mutation of the pore-lining positively charged residues Arg-83 and Lys-87 (Fig. 6, a and b) to alanines (Orai1 R83A/K87A) resulted in a substantial reduction of STIM1-dependent current activation (Fig. 6c). The reduced current density of Orai1 R83A/K87A was not caused by an impaired plasma membrane targeting as determined by biotinylation experiments (Fig. 6d). Co-localization with OASF was, however, reduced as evident from density profiles (Fig. 6e), suggesting a partially reduced interaction with STIM1. Strikingly, the introduction of these mutations into the Orai1 V102A core yielded a channel with novel characteristics (Fig. 6, f–h) in that the Orai1 R83A/K87A/V102A mutant displayed complete loss of function (Fig. 6, f and h). Hence, the concerted alanine substitutions at positions 83 and 87 apparently altered the geometry of the elongated pore in such a way that Ca2+ permeation was inhibited. In support of this, 2-aminoethoxydiphenyl borate failed to evoke currents from the double Orai1 R83A/K87A mutant compatible with a severe defect in the pore (supplemental Fig. 3g). It is tempting to speculate that the positive charges of the pore-lining residues Arg-83 and Lys-87 in the ETON region (15) contribute to the stabilization of the elongated pore, the alanine mutation of which leads to pore collapse.
FIGURE 6.

The positively charged Arg-83 and Lys-87 residues in the ETON region contribute to both STIM1 binding and the gate. a, amino acid sequence of Orai1 N terminus displaying Arg-83/Lys-87 in red in the ETON region (yellow area). b, schematic representation of two TM1 domains + ETON regions highlighting the positively charged residues Arg-83 and Lys-87 (blue) in the ETON region. c, time course of whole cell inward currents at −74 mV activated by passive store depletion of HEK 293 cells co-expressing CFP-STIM1 and YFP-Orai1 R83A/K87A in comparison with wild-type YFP-Orai1. d, plasma membrane expression in HEK293 cells expressing Orai1 WT in comparison with Orai1 R83A/K87A determined by biotinylation as described under “Experimental Procedures” (n = 5; *, p < 0.05). Error bars indicate S.E. n.s., not significant. WB, Western blot; ab, antibody. e, intensity plots representing the localization of STIM1 233–474 (OASF) across the cell when co-expressed with Orai1 R83A/K87A when compared with wild-type Orai1. Error bars indicate S.E. f, time course of whole cell inward currents at −74 mV activated by passive store depletion of HEK 293 cells co-expressing CFP-STIM1 and YFP-Orai1 R83A/K87A/V102A in the absence and presence of STIM1. g, I/V relationships of normalized Orai1 R83A/K87A/V102A currents in the absence and presence of STIM1. h, block diagram displaying reversal potentials of Orai1 V102A and Orai1 R83A/K87A/V102A in the absence and presence of STIM1 (*, p < 0.05). Error bars indicate S.E. i, time course of whole cell inward currents at −74 mV activated by passive store depletion of HEK 293 cells co-expressing CFP-STIM1 and YFP-Orai1 ΔN1–72 R83A/K87A/V102A, YFP-Orai1 ΔN1–74 R83A/K87A/V102A, and YFP-Orai1 ΔN1–76 R83A/K87A/V102A in comparison with YFP-Orai1 R83A/K87A/V102A (ctrl).
An overall disturbance of channel function could be excluded as Orai1 R83A/K87A/V102A was still activated by STIM1, leading to inward-rectifying Ca2+ currents that showed a rightward shift of the reversal potential similar to STIM1+Orai1 V102A (Fig. 6, f–h). Thus, interaction with STIM1 was still attainable with the R83A/K87A mutations in the ETON region, although the STIM1-dependent currents activated at a slower rate and to a lower extent than those obtained with Orai1 V102A.
As the activity of the Orai1 R83A/K87A/V102A mutant was strictly STIM1-dependent, we utilized it to confirm the impact of N-terminal truncations presented in Fig. 1. Consistently, Orai1 ΔN1–72 R83A/K87A/V102A and Orai1 ΔN1–74 R83A/K87A/V102A regained function in the presence of STIM1, whereas Orai1 ΔN1–76 R83A/K87A/V102A failed to activate, independent of the presence or absence of STIM1 (Fig. 6i). In summary, our results suggest an additional role of the pore-lining positively charged Arg-83 and Lys-87 residues in providing a gating function to the ETON region that, in conjunction as a binding interface for STIM1, fine-tunes pore width for controlled STIM1-dependent Ca2+ entry.
DISCUSSION
The gating of Orai1 channels requires both a robust STIM1 coupling to the Orai1 C terminus and a weaker one to the Orai1 N terminus, respectively (12, 22). The Orai1 N-terminal interaction is mediated via the conserved ETON region as demonstrated by systematic N-truncations as well as point mutations. Our study revealed multiple hot spots along the ETON region that contribute in a concerted manner to the STIM1 binding interface. In particular, the hydrophobic residues Leu-74 and Trp-76 clustered at the beginning of the ETON region are molecular determinants in the coupling to STIM1, the mutation of which decreased affinity to and almost abolished Orai1 activation via STIM1. The positively charged pore-lining residues Arg-83 and Lys-87 additionally function as a gate that likely shapes the exit at the cytosolic side by electrostatic tuning of the width and barrier of the elongated pore. Hence, the ETON region combines both binding interface and gate for the activation of Orai1 via STIM1, in line with the recently published crystal structure (15).
The relevance of the conserved ETON region (aa73–90) in triggering STIM1-mediated Orai1 activation has already been reported by several studies (22, 31, 33, 36), in that N-terminal truncations up to aa 74 maintain a functional channel, whereas a deletion up to aa 80 (33) or of the whole N terminus of Orai1 abolishes STIM1-dependent activation. Consistently, deletion of aa 74–85 (24) leads to nonfunctional Orai1 channels. In this study, we initially focused on characterizing the minimal domain of the ETON region that is essential for retaining STIM1-dependent activation of Orai1 currents. The minimal stretch, based on progressive N-terminal Orai1 truncations, included residues 76–90 of the ETON region, which still allowed at least partial STIM1-dependent activation. Residues upstream of 76, i.e. 72–75, also contributed to STIM1 binding as their deletion progressively reduced current densities, but not as pronounced as with deletion of the first 75 amino acids. Additional deletion of residue Trp-76 completely abolished STIM1-dependent Orai1 function. Park et al. (22) have reported weak binding of the CAD to the Orai1 N-terminal fragments aa1–91 and aa48–91 but not to Orai1 1–70 in support of STIM1 interaction with the conserved ETON region. Consistent with the proposed weak interaction of the ETON region, a peptide fragment aa70–91 and STIM1 C-terminal fragment interacted with a KD of ∼250 μm as determined by thermophoresis experiments compatible with a weak, transient interaction (37, 38). The KD shifted drastically above 4 mm with a shortened ETON region comprising aa77–91. Hence, this result, in line with the N-truncation Orai1 mutants, suggests Trp-76 together with residues 72–75 as a hot spot within the binding interface of the ETON region for interaction with STIM1. Thus, STIM1 requires almost the whole ETON region for STIM1-dependent Orai1 activation.
To further pinpoint hot spot residues mediating STIM1 binding, we carried out scanning mutagenesis of selected hydrophobic as well as positively charged residues in the ETON region as those amino acids are frequently found in protein-protein interfaces (35). Single point mutations of Trp-76 (except of a proline) within the full-length Orai1 failed to drastically impair STIM1-dependent Orai1 function. However, the double mutation of Leu-74 together with Trp-76 to more polar residues substantially reduced Orai1 activation consistent with a significant decrease in co-localization and affinity of STIM1 C-terminal fragments. Thus, Leu-74 and Trp-76 represent hydrophobic hot spot residues within the ETON region contributing in a concerted manner to the Orai1 N-terminal coupling with STIM1. The single W76P substitution likely introduced a break in the ETON helical region that severely interfered with the overall orientation of the STIM1 binding interface.
In addition to these hydrophobic residues, we further characterized positively charged residues along the ETON region that contributed to the interaction with STIM1. Except for the single amino acid mutant Orai1 K85E (24, 25), which has already been reported to abolish Orai1 channel function, we identified double mutations of positively charged residues, i.e. Arg77/Lys-78 and Arg-83/Lys-87, along the ETON region that diminished both current activation and STIM1 co-localization. Although Arg-77 and Lys-78 are located on the pore-averted side of the helical ETON region, Arg-83 and Lys-87 represent pore-lining residues close to the membrane as derived from the recent Orai crystal structure (15). Therefore, the latter two residues together with Arg-91 have been recently hypothesized to form a potential gate of the Orai1 channel (16). Utilization of the constitutively active Orai1 V102A mutant indeed enabled us to reveal a gating function of Arg-83 and Lys-87 as their mutation to alanines abolished Orai1 Arg-83/Lys-87 V102A channel function that was restored by the presence of STIM1. We (this work) and others (24) employed the Orai1 V102A mutant as an alternative and sensitive tool to monitor N-terminal interactions with STIM1 as monitored by restored Ca2+ selectivity as long as STIM1 coupling occurred. The striking loss of function of the generally constitutively active Orai1 V102A mutant upon alanine mutations of Arg-83 and Lys-87 suggested these residues as gating elements that provide both electrostatic stabilization and barrier to the pore jointly formed by the ETON regions in the closed conformation of the Orai1 channel complex. The presence of STIM1 restored function of this R83A/K87A Orai1 V102A mutant probably by tuning the geometry of the pore for Ca2+ permeation. In accordance, charge swap in the R83E/K87E Orai1 V102A mutant resulted in a constitutively active channel, supporting the concept of electrostatic pore stabilization (supplemental Fig. 8).
In summary, almost the whole ETON region functions as binding interface for Orai1 interaction with STIM1 and additionally provides electrostatic gating elements to fine-tune the shape of the elongated pore. STIM1 binding onto the ETON region occurs with a weak affinity in the μm range (37, 38) and requires at least the N-terminal strand starting with residue 76. The residues Leu-74 and Trp-76 acting in a concerted manner represent a hydrophobic hot spot interface for the Orai1 N-terminal interaction with STIM1 that is additionally affected by positively charged residues within the ETON region. Besides Lys-85 (24, 25), we identified the two pore-lining residues Arg-83 and Lys-87 that exert a gating function both by electrostatic stabilization and by barrier to the elongated pore of the closed channel. The identification of the hot spot residues may stimulate the search for small molecule Orai1 inhibitors enabling targeted disruption of STIM1 interaction with the ETON region.
Acknowledgments
We thank S. Buchegger for excellent technical assistance. We thank Robert Tampé and Volker Gatterdam (Institute of Biochemistry, Goethe-University, Frankfurt, Germany) as well as Harald Schwalbe and Krishna Saxena (Institute of Organic Chemistry and Chemical Biology, Goethe-University) for the opportunity to do the thermophoresis measurements in their laboratory as well as their support and advice.
This work was supported by the Austrian Science Fund (FWF Project P25210 to I. D., FWF Project P22747 to R. S., FWF Project P21925 to K. G., and FWF Project P25172 to C. R.) and the Austrian Research Promotion Agency (FFG, Austrian Nanoscience Initiative VO104-08-BI Project 819703 NSI-NABIOS to H. J. G).
This article was selected as a Paper of the Week.

This article contains supplemental Figs. 1–8.
- CRAC
- calcium release-activated calcium
- STIM1
- stromal interaction molecule 1
- ETON
- extended transmembrane Orai1 N-terminal
- aa
- amino acid(s)
- NT
- N terminus
- TM1
- transmembrane helix 1.
REFERENCES
- 1. Roos J., DiGregorio P. J., Yeromin A. V., Ohlsen K., Lioudyno M., Zhang S., Safrina O., Kozak J. A., Wagner S. L., Cahalan M. D., Veliçelebi G., Stauderman K. A. (2005) STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 169, 435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang S. L., Yu Y., Roos J., Kozak J. A., Deerinck T. J., Ellisman M. H., Stauderman K. A., Cahalan M. D. (2005) STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Prakriya M., Feske S., Gwack Y., Srikanth S., Rao A., Hogan P. G. (2006) Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233 [DOI] [PubMed] [Google Scholar]
- 4. Vig M., Beck A., Billingsley J. M., Lis A., Parvez S., Peinelt C., Koomoa D. L., Soboloff J., Gill D. L., Fleig A., Kinet J. P., Penner R. (2006) CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr. Biol. 16, 2073–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vig M., Peinelt C., Beck A., Koomoa D. L., Rabah D., Koblan-Huberson M., Kraft S., Turner H., Fleig A., Penner R., Kinet J. P. (2006) CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liou J., Kim M. L., Heo W. D., Jones J. T., Myers J. W., Ferrell J. E., Jr., Meyer T. (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Luik R. M., Wu M. M., Buchanan J., Lewis R. S. (2006) The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 174, 815–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luik R. M., Wang B., Prakriya M., Wu M. M., Lewis R. S. (2008) Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature 454, 538–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Muik M., Fahrner M., Schindl R., Stathopulos P., Frischauf I., Derler I., Plenk P., Lackner B., Groschner K., Ikura M., Romanin C. (2011) STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J. 30, 1678–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Korzeniowski M. K., Manjarrés I. M., Varnai P., Balla T. (2010) Activation of STIM1-Orai1 involves an intramolecular switching mechanism. Sci. Signal. 3, ra82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frischauf I., Schindl R., Derler I., Bergsmann J., Fahrner M., Romanin C. (2008) The STIM/Orai coupling machinery. Channels 2, 261–268 [DOI] [PubMed] [Google Scholar]
- 12. Muik M., Frischauf I., Derler I., Fahrner M., Bergsmann J., Eder P., Schindl R., Hesch C., Polzinger B., Fritsch R., Kahr H., Madl J., Gruber H., Groschner K., Romanin C. (2008) Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J. Biol. Chem. 283, 8014–8022 [DOI] [PubMed] [Google Scholar]
- 13. Navarro-Borelly L., Somasundaram A., Yamashita M., Ren D., Miller R. J., Prakriya M. (2008) STIM1-Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J. Physiol. 586, 5383–5401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Soboloff J., Spassova M. A., Tang X. D., Hewavitharana T., Xu W., Gill D. L. (2006) Orai1 and STIM reconstitute store-operated calcium channel function. J. Biol. Chem. 281, 20661–20665 [DOI] [PubMed] [Google Scholar]
- 15. Hou X., Pedi L., Diver M. M., Long S. B. (2012) Crystal structure of the calcium release-activated calcium channel Orai. Science 338, 1308–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rothberg B. S., Wang Y., Gill D. L. (2013) Orai channel pore properties and gating by STIM: implications from the Orai crystal structure. Sci. Signal. 6, pe9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McNally B. A., Somasundaram A., Yamashita M., Prakriya M. (2012) Gated regulation of CRAC channel ion selectivity by STIM1. Nature 482, 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang S. L., Yeromin A. V., Hu J., Amcheslavsky A., Zheng H., Cahalan M. D. (2011) Mutations in Orai1 transmembrane segment 1 cause STIM1-independent activation of Orai1 channels at glycine 98 and channel closure at arginine 91. Proc. Natl. Acad. Sci. U.S.A. 108, 17838–17843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu Y., Zheng X., Mueller G. A., Sobhany M., DeRose E. F., Zhang Y., London R. E., Birnbaumer L. (2012) Crystal structure of calmodulin binding domain of orai1 in complex with Ca2+ calmodulin displays a unique binding mode. J. Biol. Chem. 287, 43030–43041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Derler I., Fahrner M., Carugo O., Muik M., Bergsmann J., Schindl R., Frischauf I., Eshaghi S., Romanin C. (2009) Increased hydrophobicity at the N terminus/membrane interface impairs gating of the severe combined immunodeficiency-related ORAI1 mutant. J. Biol. Chem. 284, 15903–15915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feske S., Gwack Y., Prakriya M., Srikanth S., Puppel S. H., Tanasa B., Hogan P. G., Lewis R. S., Daly M., Rao A. (2006) A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185 [DOI] [PubMed] [Google Scholar]
- 22. Park C. Y., Hoover P. J., Mullins F. M., Bachhawat P., Covington E. D., Raunser S., Walz T., Garcia K. C., Dolmetsch R. E., Lewis R. S. (2009) STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Frischauf I., Muik M., Derler I., Bergsmann J., Fahrner M., Schindl R., Groschner K., Romanin C. (2009) Molecular determinants of the coupling between STIM1 and Orai channels: differential activation of Orai1–3 channels by a STIM1 coiled-coil mutant. J. Biol. Chem. 284, 21696–21706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McNally B. A., Somasundaram A., Jairaman A., Yamashita M., Prakriya M. (2013) The C- and N-terminal STIM1 binding sites on Orai1 are required for both trapping and gating CRAC channels. J. Physiol. 591, 2833–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lis A., Zierler S., Peinelt C., Fleig A., Penner R. (2010) A single lysine in the N-terminal region of store-operated channels is critical for STIM1-mediated gating. J. Gen. Physiol. 136, 673–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Keskin O., Ma B., Rogale K., Gunasekaran K., Nussinov R. (2005) Protein-protein interactions: organization, cooperativity, and mapping in a bottom-up Systems Biology approach. Phys. Biol. 2, S24–S35 [DOI] [PubMed] [Google Scholar]
- 27. Moreira I. S., Martins J. M., Ramos R. M., Fernandes P. A., Ramos M. J. (2013) Understanding the importance of the aromatic amino-acid residues as hot-spots. Biochim. Biophys. Acta 1834, 404–414 [DOI] [PubMed] [Google Scholar]
- 28. Raj M., Bullock B. N., Arora P. S. (2013) Plucking the high hanging fruit: A systematic approach for targeting protein-protein interactions. Bioorg. Med. Chem. 21, 4051–4057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schindl R., Fritsch R., Jardin I., Frischauf I., Kahr H., Muik M., Riedl M. C., Groschner K., Romanin C. (2012) Canonical transient receptor potential (TRPC) 1 acts as a negative regulator for vanilloid TRPV6 mediated Ca2+ influx. J. Biol. Chem. 287, 35612–35620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Singh A., Hamedinger D., Hoda J. C., Gebhart M., Koschak A., Romanin C., Striessnig J. (2006) C-terminal modulator controls Ca2+-dependent gating of Cav1.4 L-type Ca2+ channels. Nat. Neurosci. 9, 1108–1116 [DOI] [PubMed] [Google Scholar]
- 31. Li Z., Lu J., Xu P., Xie X., Chen L., Xu T. (2007) Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J. Biol. Chem. 282, 29448–29456 [DOI] [PubMed] [Google Scholar]
- 32. Muik M., Fahrner M., Derler I., Schindl R., Bergsmann J., Frischauf I., Groschner K., Romanin C. (2009) A cytosolic homomerization and a modulatory domain within STIM1 C terminus determine coupling to ORAI1 channels. J. Biol. Chem. 284, 8421–8426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zheng H., Zhou M. H., Hu C., Kuo E., Peng X., Hu J., Kuo L., Zhang S. L. (2013) Differential roles of the C and N termini of Orai1 protein in interacting with stromal interaction molecule 1 (STIM1) for Ca2+ release-activated Ca2+ (CRAC) channel activation. J. Biol. Chem. 288, 11263–11272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fahrner M., Muik M., Derler I., Schindl R., Fritsch R., Frischauf I., Romanin C. (2009) Mechanistic view on domains mediating STIM1-Orai coupling. Immunol. Rev. 231, 99–112 [DOI] [PubMed] [Google Scholar]
- 35. Keskin O., Ma B., Nussinov R. (2005) Hot regions in protein–protein interactions: the organization and contribution of structurally conserved hot spot residues. J. Mol. Biol. 345, 1281–1294 [DOI] [PubMed] [Google Scholar]
- 36. Bergsmann J., Derler I., Muik M., Frischauf I., Fahrner M., Pollheimer P., Schwarzinger C., Gruber H. J., Groschner K., Romanin C. (2011) Molecular determinants within N terminus of Orai3 protein that control channel activation and gating. J. Biol. Chem. 286, 31565–31575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perkins J. R., Diboun I., Dessailly B. H., Lees J. G., Orengo C. (2010) Transient protein-protein interactions: structural, functional, and network properties. Structure 18, 1233–1243 [DOI] [PubMed] [Google Scholar]
- 38. Ozbabacan S. E., Engin H. B., Gursoy A., Keskin O. (2011) Transient protein-protein interactions. Protein Eng. Des. Sel. 24, 635–648 [DOI] [PubMed] [Google Scholar]