

Background: The mechanisms initiating protein acylation in mitochondria are unknown.
Results: The pH and acyl-CoA concentrations of the mitochondrial matrix are sufficient to cause protein lysine acetylation and succinylation.
Conclusion: Protein acylation in mitochondria may be a nonenzymatic event facilitated by the alkaline pH and high acyl-CoA concentrations.
Significance: The mitochondrial deacylases SIRT3 and SIRT5 may have evolved to regulate nonenzymatic protein acylation.
Keywords: Acetyl Coenzyme A, Metabolic Diseases, Metabolic Regulation, Mitochondrial Metabolism, pH Regulation, Sirtuins, Acetylation, Nonenzymatic, Succinyl Coenzyme A, Succinylation
Abstract
Alterations in mitochondrial protein acetylation are implicated in the pathophysiology of diabetes, the metabolic syndrome, mitochondrial disorders, and cancer. However, a viable mechanism responsible for the widespread acetylation in mitochondria remains unknown. Here, we demonstrate that the physiologic pH and acyl-CoA concentrations of the mitochondrial matrix are sufficient to cause dose- and time-dependent, but enzyme-independent acetylation and succinylation of mitochondrial and nonmitochondrial proteins in vitro. These data suggest that protein acylation in mitochondria may be a chemical event facilitated by the alkaline pH and high concentrations of reactive acyl-CoAs present in the mitochondrial matrix. Although these results do not exclude the possibility of enzyme-mediated protein acylation in mitochondria, they demonstrate that such a mechanism may not be required in its unique chemical environment. These findings may have implications for the evolutionary roles that the mitochondria-localized SIRT3 deacetylase and SIRT5 desuccinylase have in the maintenance of metabolic health.
Introduction
Reversible lysine acetylation is a post-translational mechanism that regulates diverse cellular processes in prokaryotes and eukaryotes (1–3). In the mammalian nucleus and cytosol, protein acetylation is regulated via the opposing actions of lysine acetyltransferases and lysine deacetylases. In contrast, mitochondria contain one known enzyme with deacetylase activity, the NAD+-dependent sirtuin family member SIRT3. SIRT3 globally deacetylates mitochondrial proteins, 65% of which are acetylated at nearly 2,200 sites (4). Focused studies indicate that excess mitochondrial acetylation is detrimental to multiple processes of oxidative and intermediary mitochondrial metabolism and is implicated in the pathophysiology of diabetes, the metabolic syndrome, cardiac hypertrophy, mitochondrial dysfunction, and cancer (4–11). Despite the pervasive nature of acetylation in mitochondria and its implications for human disease, a viable mechanism responsible for initiating protein acetylation in this organelle remains unknown. Similarly, protein lysine succinylation was recently identified in mitochondria, but the mechanism initiating protein succinylation is unknown (12).
Consistent with rapid kinetics characteristic of enzymes, histone acetyltransferase and histone deacetylase-mediated changes in global histone acetylation can occur over a period of minutes (13, 14). In contrast, a review of the published experimental conditions in which mitochondrial protein hyperacetylation is observed (see Table 1) indicates that this phenotype develops gradually over a longer time period, e.g. days or weeks, suggesting a different mechanism responsible for protein acetylation in mitochondria.
TABLE 1.
Review of published experimental conditions in which mitochondrial protein hyperacetylation is observed
| Experimental condition | Tissue showing mitochondrial protein hyperacetylation | Reported time to mitochondrial protein hyperacetylation after onset of experimental condition | Reference |
|---|---|---|---|
| Fasting | Liver | 12–48 h | 34 |
| Caloric restriction | Liver | 16–18 weeks, possibly earlier | 36 |
| Chronic alcohol consumption | Liver | 3–6 weeks | 35 |
| High fat diet | Liver | 13 weeks, possibly earlier | 9 |
| SIRT3 KO | Liver, brown adipose, skeletal muscle | 12–13 weeks, possibly earlier | 8, 9, 28 |
| Frataxin conditional KO | Heart | 3.5 weeks, NSE-Cre | 7 |
| Hint2 KO | Liver | 20 weeks, possibly earlier | 11 |
The physiologic conditions of the mitochondrial matrix are distinct from those of any other cellular compartment (Fig. 1A). As the primary site of cellular carbon utilization, mitochondria generate between 0.1 and 1.5 mm steady-state concentrations of acetyl-CoA, which is 3–50 times higher than that of the cytosol and nucleus according to current estimates in yeast (15–17). Additionally, due to the extrusion of H+ ions across the inner mitochondrial membrane to generate a proton-motive force, the pH of the mitochondrial matrix is maintained at 7.9–8.0, which corresponds to a 6.3-fold greater hydroxide (OH−) ion concentration than the cytosol and nucleus, which maintain a pH of 7.2 (18).
FIGURE 1.
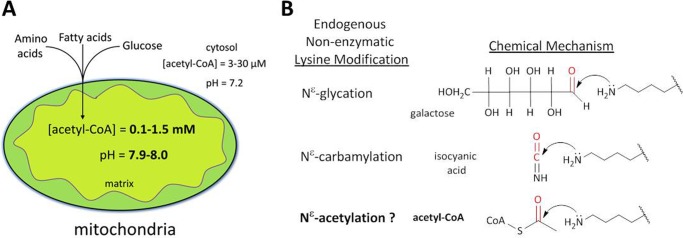
A, schematic highlighting the unique chemical environment of the mitochondrial matrix. Both the pH and the acetyl-CoA concentration of the mitochondrial matrix are considerably higher than the cytosol and nucleus (pH 8.0 versus pH 7.2, and 0.1–1.5 mm versus 3–30 μm. B, nonenzymatic acetylation is mechanistically analogous to other endogenous nonenzymatic lysine modifications such as glycation and carbamylation. In each example, the mechanism involves a primary amine performing a nucleophilic attack on the carbonyl carbon of the endogenous metabolite.
Review of evidence from kinetic characterization of acetyltransferases suggests that nonenzymatic acetylation may occur in the unique conditions of the mitochondrial matrix. Members of the GCN5-related N-acetyltransferase (GNAT) superfamily of enzymes employ a general base catalyst to deprotonate the positively charged ϵ-amino group of a lysine residue on the accepting protein, which is then primed for a nucleophilic attack on the carbonyl carbon of the acetyl group in acetyl-CoA (19, 20). This displaces the thioester bond, leaving free coenzyme A and an Nϵ-acetyl-lysine. Predictably, mutating the catalytic base of GCN5 abolishes acetyl transfer, but more importantly, the rate of acetyl transfer to histone substrate can be nearly completely recovered in the absence of enzyme merely by increasing the pH of the reaction buffer (19). Thus, chemical (nonenzymatic) acetylation of lysine residues is energetically favorable provided that acetyl-CoA is present and the pH is alkaline, conditions that are both satisfied by the chemical milieu of the mitochondrial matrix (5, 21). Furthermore, nonenzymatic lysine acetylation shares an analogous chemical mechanism with other endogenous nonenzymatic lysine modifications including glycation and carbamylation (Fig. 1B).
Here, we demonstrate that the minimal chemical conditions of the mitochondrial matrix are sufficient to cause nonenzymatic protein lysine acetylation. Furthermore, we broaden these findings by demonstrating that this distinct chemical environment also promotes nonenzymatic protein succinylation by succinyl-CoA, suggesting that this post-translational mechanism may extend to several short-chain acyl-CoA intermediates present within mitochondria. To our knowledge, this is the first study to systematically demonstrate that the chemical conditions of the mitochondrial matrix are sufficient to cause nonenzymatic acylation of proteins, which provides a viable mechanism for the pervasive protein acylation in this organelle and, perhaps, an evolutionary basis for the mitochondria-localized SIRT3 deacetylase and SIRT5 desuccinylase.
EXPERIMENTAL PROCEDURES
Mitochondrial Isolation
Fresh tissue (100–200 mg) from wild-type mice on an inbred mixed C57BL/6J x 129/Sv-ter background was dissected and immediately immersed in ice-cold mitochondrial isolation buffer (220 mm mannitol, 70 mm sucrose, 30 mm Tris-Cl (pH 7.4), 0.5 mm EGTA, and 0.1% BSA) supplemented with an EDTA-free protease inhibitor mixture (Roche Applied Science). Tissue was briefly minced with a razor blade and then homogenized in 1:5 w/v mitochondrial isolation buffer with a 2-ml hand Dounce homogenizer (liver = 10 passes). Mitochondria were then isolated using a standard differential centrifugation method as described previously (22) and then resuspended and frozen in nondenaturing buffer containing 50 mm Tris-Cl (pH 8.0 at 37 °C), 150 mm NaCl or 50 mm HEPES (pH 8.0), 150 mm NaCl at 1.50 μg/μl. For experiments, mitochondria were thawed on ice, sonicated twice for 15 s each on ice, and then cleared by centrifugation at 12,000 × g for 10 min. Supernatants containing soluble mitochondrial protein were used for in vitro assays described below. Protein concentration was determined via the BCA method.
In Vitro Incubation of Mitochondrial Protein and BSA with Acetyl-CoA, Succinyl-CoA, and Western Blotting
A 48 mm solution of acetyl-CoA (Sigma) and a 100 mm solution of sodium acetate were each made in 50 mm Tris-Cl, 150 mm NaCl, and adjusted to pH 8.0 at 37 °C. Serial dilutions were made using 50 mm Tris-Cl (pH 8.0 at 37 °C), 150 mm NaCl as the diluent. Twenty microliters of the appropriate dilution was added to 20 μl of soluble mitochondrial protein (1.5 μg/μl) in 50 mm Tris-Cl (pH 8.0 at 37 °C), 150 mm NaCl to achieve the desired concentration of acetyl-CoA or acetate. Total reaction volumes of 40 μl were incubated for 6 h at 37 °C at 400 rpm in an Eppendorf Thermomixer unless otherwise indicated. Reaction tubes were briefly centrifuged during the incubation to minimize condensation. For denatured protein experiments, soluble liver mitochondrial protein was heated at 95 °C for 10 min and cooled on ice prior to the addition of acetyl-CoA. To test for competitive inhibition by CoA, a 12 mm solution of CoA (Sigma) was made in 50 mm Tris-Cl, 150 mm NaCl, and adjusted to pH 8.0 at 37 °C. Dilutions of this CoA solution and acetyl-CoA solutions were added to 30 μg of native mitochondrial protein in 50 mm Tris-Cl (pH 8.0 at 37 °C), 150 mm NaCl to achieve the desired concentrations in a final volume of 40 μl and then incubated for 6 h at 37 °C and 400 rpm in an Eppendorf Thermomixer. For BSA experiments, 2 μg/μl solutions of nonacetylated, fatty acid-free BSA were made in 50 mm Tris-Cl, 150 mm NaCl and adjusted to pH 6.0, 7.0, or 8.0 at 37 °C. Twenty microliters of each BSA solution was added to 20 μl of a 3 mm solution of acetyl-CoA or 1 mm succinyl-CoA adjusted to the same pH for final conditions of 40 μl total volume, 1.5 mm acetyl-CoA or 0.5 mm succinyl-CoA, and 1 μg/μl BSA in 50 mm Tris-Cl, 150 mm NaCl at pH 6, 7, or 8. BSA reactions were incubated for 9 h as described earlier, or for 6 h for succinyl-CoA. Following the incubations, loading buffer was added, and the samples were separated by SDS-PAGE, transferred to nitrocellulose membrane, blocked, and probed with a polyclonal acetyl-lysine antibody (Cell Signaling) or a polyclonal succinyl-lysine antibody (PTM Biolabs, Inc.) and monoclonal antibodies against succinate dehydrogenase iron-sulfur subunit (SDHB) and Complex III Rieske protein (UQCRFS1)(Mitosciences/Abcam). Signal was visualized with HRP-conjugated polyclonal goat-anti-rabbit or goat-anti-mouse secondary antibodies and SuperSignal Pico West chemiluminescent substrate (Pierce) with exposure to x-ray film. The pH was adjusted with an Oakton 1100 series digital pH and temperature meter or Corning pH meter 240 electrode calibrated to the same set of Fisher Scientific buffer solution standards.
Immunoprecipitation
Mitochondrial protein extracts were prepared as described above, and 120 μg per sample was incubated with or without 1.5 mm acetyl-CoA at pH 8.0 and 37 °C for 4 h. After incubation, the samples were diluted 1:10 in 50 mm HEPES (pH 7.1), with 150 mm NaCl, and 25 μl of acetyl-lysine antibody-conjugated agarose beads (ImmuneChem) was added to each sample followed by incubation for 18 h at 4 °C with gentle rocking. Beads were centrifuged at 400 × g for 1 min at 4 °C, and the supernatant was decanted. The beads were washed three times for 5 min in 250 μl of 50 mm Tris-Cl (pH 7.25), 150 mm NaCl, 1 mm EDTA, 0.1% Nonidet P-40, and then boiled in loading buffer prior to SDS-PAGE.
In Vitro SIRT3 Deacetylation Assay
Soluble liver mitochondrial protein (30 μg) was incubated in the following final reaction conditions: 48.4 mm Tris-Cl (pH 8.0 at 30 °C), 162 mm NaCl, 5.7% glycerol, 1.1 mm MgCl2, 5.7 mm NAD+, ± 7.2 mm acetyl-CoA, ± 3 μg of GST-hSIRT3, ± 10 mm nicotinamide in a total volume of 70 μl. Reactions were incubated for 4 h at 30 °C in an Eppendorf Thermomixer set to 400 rpm. Reactions were briefly centrifuged at 2 h to minimize reaction condensation. Reactions were stopped with the addition of loading buffer and resolved with a 12% SDS-PAGE followed by immunoblotting with acetyl-lysine and Complex III Rieske protein antibodies as described earlier and a polyclonal SIRT3 antibody (Cell Signaling).
Acetyl-lysine Sequence Motif Analysis Using iceLogo
The flanking sequence data for acetyl sites was manually retrieved from UniProt. Using supplemental data from Hebert et al. (4), the 40 sites with evidence of mitochondrial localization undergoing the greatest statistically significant -fold changes (p < 0.05, Welch's t test with Storey correction) in acetylation in response to SIRT3 deletion (SIRT3 KO-Control Diet/WT-Control Diet) were compared with 40 sites with evidence of mitochondrial localization undergoing smaller statistically significant -fold changes (Welch's t test with Storey correction) in acetylation in response to SIRT3 deletion. Flanking sequence data were uploaded to iceLogo, and graphs were generated in filled logo mode.
PROPKA Analysis
Crystal structure data for the pig mitochondrial proteins malate dehydrogenase 2, succinate CoA ligase, citrate synthase, aconitase 2, and succinate dehydrogenase (Protein Data Bank IDs: 1MLD, 2FP4, 3ENJ, 1B0J, and 1ZOY) representing 74 distinct acetylation sites (as described in Ref. 4) were uploaded into the PROPKA 3.1 web interface (23, 24). The corresponding lysine residues for the homologous mouse proteins were determined through alignment in UniProt.
Image Analysis
Integrated densitometries of Western blot exposures were calculated using ImageJ. Schematics and figures were generated using ChemBioDraw Ultra 12.0 and Microsoft PowerPoint.
RESULTS
Nonenzymatic Acetylation in the Chemical Conditions of the Mitochondrial Matrix
To test the hypothesis that mitochondrial Nϵ-acetylation occurs chemically, we incubated soluble liver mitochondrial proteins in nondenaturing buffer with varying concentrations of acetate or acetyl-CoA at pH 8.0, the pH of the mitochondrial matrix. After incubating mitochondrial proteins in 50 mm acetate for 5 h, there was no change in protein acetylation (data not shown). In contrast, incubating mitochondrial proteins with low mm concentrations of acetyl-CoA generated dose- and time-dependent increases in lysine acetylation (Fig. 2, A–E). We reasoned that if a mitochondria-localized enzyme were responsible for this acetylation, then denaturing the mitochondrial extracts prior to incubating with acetyl-CoA would eliminate the acetyltransferase activity. In contrast, if the observed increase in acetylation were nonenzymatic, then protein denaturation might promote acetylation by increasing the number of solvent-accessible lysines available to react with acetyl-CoA. Strikingly, and consistent with the latter hypothesis, our data show that heat-induced denaturation of the mitochondrial extracts resulted in a dose- and time-dependent increase in lysine acetylation, which was substantially greater relative to the native (unheated) state (Fig. 2, A–E). Incubating native and denatured cardiac mitochondrial protein with acetyl-CoA generated identical results (data not shown). The increase in acetylation was not caused by the denaturation process as evinced by loss of acetylation while incubating denatured protein under acidic conditions (Fig. 2F). The observed lysine acetylation was equally robust in non-amine-containing buffers, was unaffected by buffer concentration, and was directly related to protein content (data not shown).
FIGURE 2.

Nonenzymatic Nϵ-acetylation in the chemical conditions of the mitochondrial matrix. A, acetyl-CoA concentration-dependent increases in mitochondrial protein acetylation under native and denatured conditions. The mouse liver mitochondrial extracts (30 μg) were incubated over a range of acetyl-CoA concentrations previously determined to occur in mitochondria (15, 16) (0.1–1.5 mm, and 1 μl of matrix volume/mg of mitochondrial protein) for 6 h at 37 °C with gentle mixing. For denatured samples, mitochondrial extracts (30 μg) were heated at 95 °C for 10 min prior to the addition of acetyl-CoA. IB, immunoblot; ac-lysine, acetyl-lysine. B, quantification of densitometry data in A normalized to Complex III. The control reaction with the smaller arbitrary value was set to zero, and the remaining data points were transformed accordingly. AU, arbitrary units. C, time-dependent incorporation of acetyl groups into native and denatured mitochondrial protein lysine residues. D, quantification of the densitometry data in C normalized to Complex III. E, time-dependent incorporation of acetyl groups into native mitochondrial protein over a broader range of time points. F, acetylation is not caused by the denaturation process and is pH-dependent. In A, C, E, and F, the nitrocellulose membrane used for immunoblotting was stained with the nonspecific protein marker Ponceau S to show equal loading as indicated.
Although the increase in Nϵ-acetylation in the denatured state showed that robust acetyl transfer can occur in the absence of a functional enzyme in these conditions, it did not exclude the possibility of enzyme-mediated acetylation in the native state. Acetyltransferases in the GCN5 and MYST families are competitively inhibited by their product, coenzyme A, with inhibition constants in the low micromolar range (14, 25). Protein acetyltransferases outside the GCN5 and MYST families are predicted to undergo competitive inhibition because of their structurally conserved CoA-binding domains (20). Thus, if a protein acetyltransferase were responsible for acetyl transfer in the native state, it would likely be competitively inhibited in the presence of CoA. As shown in Fig. 3A, increasing the concentration of CoA to double that of acetyl-CoA had no effect on the protein acetyl transfer reaction. This result suggests that acetyl transfer under native protein conditions is not competitively inhibited by CoA, and most likely occurs independently of CoA binding.
FIGURE 3.
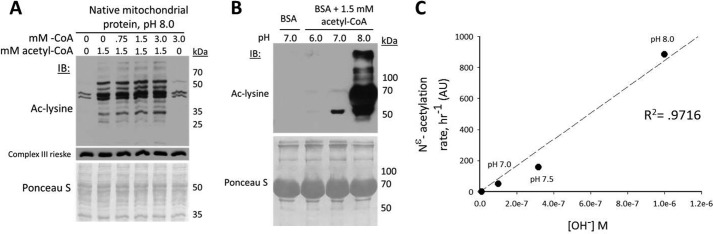
The acetyl transfer mechanism is not competitively inhibited by coenzyme A, and its rate is directly proportional to hydroxide ion concentration. A, CoA does not competitively inhibit the observed acetyl transfer reaction. Mitochondrial extracts were prepared from liver. IB, immunoblot; ac-lysine, acetyl-lysine. B, the physiologic pH (8.0) and acetyl-CoA concentration (1.5 mm) of the mitochondrial matrix are sufficient to induce widespread lysine acetylation of a nonmitochondrial protein (BSA) under native conditions. C, lysine acetylation rate is directly proportional to the concentration of hydroxide ions, consistent with specific base catalysis. AU, arbitrary units.
If protein acetylation in mitochondria is nonenzymatic, then the minimal chemical conditions of the mitochondrial matrix (pH 8.0, 0.1–1.5 mm acetyl-CoA) should also induce acetylation of a nonmitochondrial protein. Incubating nonacetylated BSA for 9 h in 1.5 mm acetyl-CoA at pH 6 or 7 resulted in no, or minimal increases in lysine acetylation. In contrast, incubating BSA at pH 8.0 induced a striking increase in lysine acetylation (Fig. 3B), which was further increased at higher pH and directly proportional to the concentration of hydroxide ions (Fig. 3C).
Nonenzymatic Succinylation in the Chemical Conditions of the Mitochondrial Matrix
Recent studies have identified lysine succinylation on mitochondrial and bacterial proteins, but a mechanism responsible for succinyl transfer was not identified (12, 26, 27). Using a validated succinyl-lysine antibody, we confirmed via Western blot that there are many succinylated proteins in mouse liver mitochondrial extracts (Fig. 4A). Like acetyl-CoA, succinyl-CoA is an inherently reactive short-chain CoA thioester that maintains steady-state concentrations in the millimolar range (0.1–0.6 mm) in the mitochondrial matrix (Fig. 4B) (15). Accordingly, we hypothesized that incubating protein with mitochondrial concentrations of succinyl-CoA at physiological pH would initiate nonenzymatic succinylation. Consistently, incubating nonacetylated BSA with 0.5 mm succinyl-CoA caused a pH-dependent increase in lysine succinylation (Fig. 4C). To our knowledge, this is the first study reporting nonenzymatic protein succinylation by succinyl-CoA at physiological pH. These results demonstrated that the pH and acyl-CoA concentrations of the mitochondrial matrix are sufficient to induce widespread and enzyme-independent protein lysine acylation. The specific base-catalyzed nucleophilic acyl substitution mechanism predicted for these reactions is summarized in Fig. 5.
FIGURE 4.
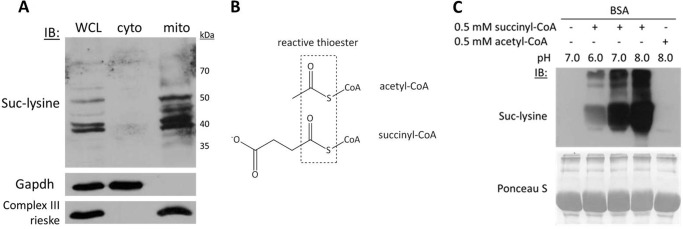
Protein succinylation is abundant within mitochondria and also occurs nonenzymatically at physiological pH. A, fractionation of mouse liver tissue into whole cell lysate (WCL), cytoplasm (cyto), and mitochondrial (mito) fractions followed by Western blotting with an anti-succinyl-lysine (Suc-lysine) antibody. Gapdh and respiratory Complex III Rieske protein antibodies were used to confirm cytoplasmic and mitochondrial subfractions, respectively. IB, immunoblot. B, acetyl-CoA and succinyl-CoA are both inherently reactive CoA thioesters. C, the physiologic pH (8.0) and succinyl-CoA concentration (0.1–0.6 mm) (15) of the mitochondrial matrix are sufficient to induce lysine succinylation of protein under native conditions.
FIGURE 5.
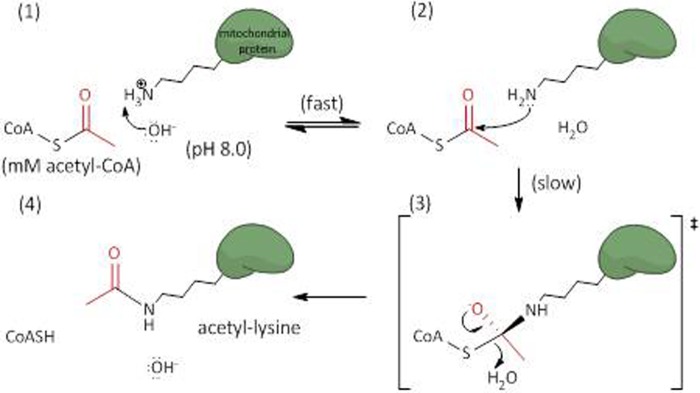
Schematic illustrating the specific base catalyzed acyl transfer mechanism. The alkaline environment of the mitochondrial matrix reflects an increased concentration of hydroxide ions (OH−) free to abstract protons from accessible lysine residues (panel 1). The deprotonated lysine can then perform a nucleophilic attack on the terminal carbonyl carbon of the acyl-CoA (panel 2). A putative tetrahedral intermediate is formed (panel 3), which then collapses, displacing the thioester bond and leaving an acylated (acetylated) lysine, CoASH, and hydroxide (panel 4). This mechanism was adapted from that of the GCN5 acetyltransferase, which employs a general base catalyst to deprotonate lysine residues and initiate acetyl transfer (19).
The SIRT3 Deacetylase Reverses Nonenzymatic Acetylation of Mitochondrial Proteins
The NAD+-dependent deacetylase SIRT3 regulates global acetylation of mitochondrial proteins (4, 28). Therefore, we hypothesized that SIRT3 target proteins would become hyperacetylated in the presence of acetyl-CoA at pH 8 and that recombinant SIRT3 would mitigate the acetyl-CoA-induced increase in mitochondrial protein acetylation. Immunoprecipitation and Western blotting of mitochondrial proteins after incubation with acetyl-CoA showed that the SIRT3 targets long-chain acyl-CoA dehydrogenase (LCAD) and NDUFA9 (5, 29) were hyperacetylated (Fig. 6A). Furthermore, the addition of recombinant SIRT3 in the presence of NAD+ markedly reduced the acetyl-CoA-induced increase in Nϵ-acetylation, an effect that was attenuated upon the addition of the sirtuin inhibitor, nicotinamide (Fig. 6B).
FIGURE 6.

The SIRT3 deacetylase attenuates acetyl-CoA-induced acetylation of mitochondrial proteins. A, acetylation of SIRT3 target proteins in the presence of acetyl-CoA at pH 8.0. IP, immunoprecipitation; IB, immunoblot; Ac-lysine, acetyl-lysine; LCAD, long-chain acyl-CoA dehydrogenase. B, recombinant SIRT3 reverses acetyl-CoA-induced acetylation of mitochondrial proteins at pH 8.0. A SIRT3 antibody was used to demonstrate the presence of recombinant (59 kDa) and endogenous (28 kDa) SIRT3. Mitochondrial extracts were prepared from liver. NAD+, nicotinamide adenine dinucleotide; NAM, nicotinamide (Sirtuin inhibitor); hSIRT3, human SIRT3.
Informatics Analysis of Protein Microenvironment and Acetylation
Although the observed acetylation mechanism was generally pH-dependent, our data indicated that certain mitochondrial proteins and/or lysine residues within those proteins will more readily undergo nonenzymatic acetylation than others (Figs. 2, 3, and 6). This observation suggests that the tendency of a particular lysine to become acetylated at pH 8.0 may be facilitated by additional factors of the protein microenvironment such as local sequence features, hydrophobicity, or Coulombic interactions with proximal residues. We used a bioinformatics approach to test this hypothesis.
Hebert et al. (4) had demonstrated that proximal positive charges are overrepresented among mitochondrial acetyl-lysine sequence motifs. Using data from the study, our further analysis indicated that there are significantly (p < 0.004) more proximal positively charged residues flanking the acetyl sites undergoing the greatest increases (-fold change 19.4–99.0) in acetylation in response to SIRT3 deletion when compared with those sites undergoing smaller increases in acetylation (-fold change 2.00–2.14) (Fig. 7, A and B). This finding suggests that these sites represent better substrates of SIRT3 due to its negatively charged peptide-binding pocket and, therefore, the acetylation levels at these sites are minimized when SIRT3 is present, as was suggested previously (4, 30). The increase in flanking positive charges among these sites may also suggest that the proximal positive charges are lowering the acid dissociation constant, or pKa, of the target lysines, thereby promoting their nucleophilic character at physiological pH and, consequently, the large increase in acetylation when SIRT3 is absent (31). However, when this hypothesis was tested by plotting acetyl-lysine -fold change in response to SIRT3 deletion against predicted pKa values calculated from crystal structure data of five mitochondrial enzymes using PROPKA 3.1 (23, 24), there was no correlation (Fig. 7C). This suggests that other factors affect SIRT3-regulated lysine acetylation, such as protein conformational flexibility, substrate binding dynamics, or transient stabilization of acetyl-CoA with protein structure, which are not accounted for in this model.
FIGURE 7.

Bioinformatics analysis of protein microenvironment and acetylation. A, acetyl sites undergoing the greatest increases in acetylation in response to SIRT3 deletion have significantly more proximal positively charged residues than sites undergoing smaller increases in acetylation (Wilcoxon rank sum test). B, iceLogo analysis of site-specific amino acid abundance between the two sequence sets in A. Amino acids with greater frequency at a specific site appear at the top, and those with lower frequency appear at the bottom. Positively charged residues are highlighted in blue, and negatively charged residues are highlighted in red. C, scatter plot of the predicted lysine ϵ-amino group acid dissociation constants calculated using structural data from five mitochondrial proteins encompassing 74 acetylation sites and their corresponding acetyl-lysine -fold changes in response to SIRT3 deletion as detailed in Hebert et al. (4). PROPKA 3.1 pKa prediction accounts for hydrogen bonding, desolvation effects, and Coulombic interactions on both internal and surface residues, but does not account for conformational flexibility (23).
DISCUSSION
Alterations in mitochondrial protein acetylation or an inability to dispose of excess mitochondrial acetyl-CoA are implicated in the pathophysiology of diabetes, the metabolic syndrome, cancer, cardiac hypertrophy, and mitochondrial disorders such as Friedreich ataxia (6–10). Despite ongoing characterization of the mechanisms underlying protein deacetylation in mitochondria, a viable mechanism responsible for the extensive acetylation in this organelle has remained elusive. Our data demonstrate that the high physiologic pH and acyl-CoA concentrations present in the mitochondrial matrix are sufficient to cause widespread lysine acetylation and succinylation of mitochondrial and nonmitochondrial proteins in an enzyme-independent manner.
It is important to acknowledge that these studies were performed in vitro. However, a nonenzymatic reaction is one that, by definition, cannot be proven in vivo and must be removed from its cellular context and demonstrated in the absence of interfering enzymes. This is the standard by which other intracellular nonenzymatic lysine modifications caused by endogenous metabolites, such as Nϵ-glycation and Nϵ-carbamylation, have been proven (32, 33). These reactions are now widely accepted as nonenzymatic because the modifications are detected on proteins under normal physiological conditions and increases in either modification are closely associated with metabolic states that increase the endogenous modifying metabolites (e.g. reducing sugars and isocyanic acid in diabetes and uremia, respectively). Similarly, our data predict that physiological and pathological metabolic states causing prolonged increases in mitochondrial acetyl-CoA utilization would also promote hyperacetylation of mitochondrial proteins. Consistent with this prediction are data showing that prolonged fasting, caloric restriction, high fat diets, and chronic alcohol consumption all cause hyperacetylation of hepatic mitochondrial proteins (Table 1) (4, 9, 34–36). Moreover, all are also associated with increased ketone body production, which is stimulated, in part, by excess acetyl-CoA in hepatic mitochondria (37, 38). These findings strongly suggest that increases in the mitochondrial pool of acetyl-CoA during extended metabolic stress are a primary factor underlying increased protein acetylation in this organelle.
Loss of protein deacetylation capability in mitochondria via genetic ablation of SIRT3 has almost universally harmful and disease-causing consequences. These include increased reactive oxygen species and a predisposition to diabetes, the metabolic syndrome, cardiac hypertrophy, hearing loss, and cancer (6, 8–10, 39). Thus, all evidence to date would suggest that unopposed mitochondrial acetylation is detrimental to mitochondrial function and metabolic homeostasis, which stands in contrast to nuclear histone acetylation that serves as an important gene regulation mechanism for which protein acetyltransferases have been identified. Nevertheless, these studies do not preclude the existence of a mitochondrial acetyltransferase or succinyltransferase. Indeed, a GCN5 acetyltransferase homolog previously implicated in the formation of lysosome-related organelles (Bloc1s1 or GCN5L1) can localize to mitochondria (40). Rather, these data question what physiologic role a mitochondrial acetyltransferase would play considering that unopposed acetylation compromises numerous metabolic processes and the chemical environment of the mitochondrial matrix is sufficient to initiate nonenzymatic lysine acetylation that is targeted by the SIRT3 deacetylase. Further kinetic and molecular characterizations of GCN5L1 or other potential candidates and their roles in the varied conditions associated with mitochondrial protein hyperacetylation are needed.
Interestingly, a recent study highlighted the role of acetyl phosphate metabolism in regulating global shifts in protein acetylation in Escherichia coli (41). The authors concluded that acetyl phosphate likely influences global protein acetylation via nonenzymatic mechanisms. About 10% of the acetylation events reported as nonenzymatic were also regulated by the sirtuin CobB, suggesting an evolutionary role for this sirtuin in the regulation of nonenzymatic acetylation in bacteria. Our data complement these recent findings by suggesting that mammalian mitochondria, the symbiotic descendants of bacteria, also undergo nonenzymatic lysine acetylation and succinylation, which are likely mediated by variation in acetyl-CoA and succinyl-CoA metabolism. Although acetyl phosphate is not known to participate in eukaryotic metabolism, the acyl phosphate 1,3-bisphosphoglycerate is generated in prokaryotes and eukaryotes during glycolysis and, interestingly, was recently demonstrated to nonenzymatically modify several glycolytic enzymes (42). The similarity between these studies and ours is that, of the carbonyl-containing functional groups commonly found in biological systems, acyl phosphates and thioesters are the two most reactive toward nucleophilic acyl substitution (43). This fact suggests that diverse organisms may have conserved mechanisms for coping with the inherent reactivity of these metabolites toward endogenous proteins, which has broad implications for our understanding and treatment of metabolic disease.
Taken together with previous findings showing that many mitochondrial acetylation sites do not affect enzyme activity (29, 44–46), our findings support the hypothesis that protein lysine acetylation in mitochondria is, at least in part, a slow chemical consequence of mitochondrial metabolism facilitated by locally high concentrations of reactive acetyl-CoA and the uniquely alkaline environment of the mitochondrial matrix. Furthermore, our data indicate that another abundant mitochondrial metabolite, succinyl-CoA, promotes nonenzymatic protein lysine succinylation in the chemical conditions of the mitochondrial matrix (Fig. 4). These findings further support the notion that in addition to their important roles as metabolic intermediates, excess short-chain acyl-CoA thioesters may also act as nonenzymatic chemical donors, a hypothesis first proposed in 1982 by Kirshenbaum (47). Importantly, although mitochondria-localized SIRT5 is a weak deacetylase, it has efficient lysine desuccinylase activity in vitro and was recently shown to regulate various metabolic pathways via desuccinylation (12, 27). Thus, SIRT3 and SIRT5 may have evolved to exploit the fasting-induced rise in mitochondrial NAD+ (48), thereby regulating the chemical acylation of proteins caused by a concurrent increase in mitochondrial acyl-CoA utilization. Consistent with a recent review (5), we propose that SIRT3 is a quality control deacetylase that protects mitochondrial enzymes and proteins from chemical acetylation-induced impairment, especially during metabolic states predicted to increase the mitochondrial acetyl-CoA pool such as fasting, caloric restriction, or high fat, ketogenic diets (4, 16, 37). More broadly, the findings that acetylation is a ubiquitous protein modification (2, 4, 49), acetyl-CoA-induced protein acetylation is energetically favorable at physiological pH (19) (Fig. 3, B and C), and altering intracellular pH causes corresponding changes in protein acetylation (50) raise the intriguing possibility that an unexpected number of cellular acylation events may be chemical in nature and that certain deacetylases/deacylases may serve to regulate the effects of unavoidable chemical acylation of cellular proteins (51).
Acknowledgments
We thank Dr. W. Zhang, K. Koehler, C. Babbey, T. Renkens, and members of the Payne laboratory for discussion and comments.
Footnotes
This work was supported, in whole or in part, by National Institutes of Health Grant 1P01HL085098 (to R. M. P.). This work was also supported by American Heart Association Grant in Aid 0855646G (to R. M. P.) and American Heart Association Fellowship 11PRE7290079 (to G. R. W.).
This article was selected as a Paper of the Week.
REFERENCES
- 1. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 2. Strahl B. D., Allis C. D. (2000) The language of covalent histone modifications. Nature 403, 41–45 [DOI] [PubMed] [Google Scholar]
- 3. Starai V. J., Celic I., Cole R. N., Boeke J. D., Escalante-Semerena J. C. (2002) Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298, 2390–2392 [DOI] [PubMed] [Google Scholar]
- 4. Hebert A. S., Dittenhafer-Reed K. E., Yu W., Bailey D. J., Selen E. S., Boersma M. D., Carson J. J., Tonelli M., Balloon A. J., Higbee A. J., Westphall M. S., Pagliarini D. J., Prolla T. A., Assadi-Porter F., Roy S., Denu J. M., Coon J. J. (2013) Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 49, 186–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Newman J. C., He W., Verdin E. (2012) Mitochondrial protein acylation and intermediary metabolism: regulation by sirtuins and implications for metabolic disease. J. Biol. Chem. 287, 42436–42443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Finley L. W., Carracedo A., Lee J., Souza A., Egia A., Zhang J., Teruya-Feldstein J., Moreira P. I., Cardoso S. M., Clish C. B., Pandolfi P. P., Haigis M. C. (2011) SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell 19, 416–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wagner G. R., Pride P. M., Babbey C. M., Payne R. M. (2012) Friedreich's ataxia reveals a mechanism for coordinate regulation of oxidative metabolism via feedback inhibition of the SIRT3 deacetylase. Hum. Mol. Genet. 21, 2688–2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jing E., Emanuelli B., Hirschey M. D., Boucher J., Lee K. Y., Lombard D., Verdin E. M., Kahn C. R. (2011) Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. U.S.A. 108, 14608–14613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hirschey M. D., Shimazu T., Jing E., Grueter C. A., Collins A. M., Aouizerat B., Stančáková A., Goetzman E., Lam M. M., Schwer B., Stevens R. D., Muehlbauer M. J., Kakar S., Bass N. M., Kuusisto J., Laakso M., Alt F. W., Newgard C. B., Farese R. V., Jr., Kahn C. R., Verdin E. (2011) SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 44, 177–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hafner A. V., Dai J., Gomes A. P., Xiao C. Y., Palmeira C. M., Rosenzweig A., Sinclair D. A. (2010) Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2, 914–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Martin J., Maurhofer O., Bellance N., Benard G., Graber F., Hahn D., Galinier A., Hora C., Gupta A., Ferrand G., Hoppeler H., Rossignol R., Dufour J. F., St-Pierre M. V. (2013) Disruption of the histidine triad nucleotide-binding Hint2 gene in mice affects glycemic control and mitochondrial function. Hepatology 57, 2037–2048 [DOI] [PubMed] [Google Scholar]
- 12. Du J., Zhou Y., Su X., Yu J. J., Khan S., Jiang H., Kim J., Woo J., Kim J. H., Choi B. H., He B., Chen W., Zhang S., Cerione R. A., Auwerx J., Hao Q., Lin H. (2011) Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Katan-Khaykovich Y., Struhl K. (2002) Dynamics of global histone acetylation and deacetylation in vivo: rapid restoration of normal histone acetylation status upon removal of activators and repressors. Genes Dev. 16, 743–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tanner K. G., Langer M. R., Kim Y., Denu J. M. (2000) Kinetic mechanism of the histone acetyltransferase GCN5 from yeast. J. Biol. Chem. 275, 22048–22055 [DOI] [PubMed] [Google Scholar]
- 15. Hansford R. G., Johnson R. N. (1975) The steady state concentrations of coenzyme A-SH and coenzyme A thioester, citrate, and isocitrate during tricarboxylate cycle oxidations in rabbit heart mitochondria. J. Biol. Chem. 250, 8361–8375 [PubMed] [Google Scholar]
- 16. Garland P. B., Shepherd D., Yates D. W. (1965) Steady-state concentrations of coenzyme A, acetyl-coenzyme A and long-chain fatty acyl-coenzyme A in rat-liver mitochondria oxidizing palmitate. Biochem. J. 97, 587–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cai L., Sutter B. M., Li B., Tu B. P. (2011) Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol. Cell 42, 426–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Casey J. R., Grinstein S., Orlowski J. (2010) Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 11, 50–61 [DOI] [PubMed] [Google Scholar]
- 19. Tanner K. G., Trievel R. C., Kuo M. H., Howard R. M., Berger S. L., Allis C. D., Marmorstein R., Denu J. M. (1999) Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J. Biol. Chem. 274, 18157–18160 [DOI] [PubMed] [Google Scholar]
- 20. Marmorstein R. (2001) Structure and function of histone acetyltransferases. Cell. Mol. Life Sci. 58, 693–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Baddiley J., Kekwick R. A., Thain E. M. (1952) A new method for acetylating proteins. Nature 170, 968–970 [DOI] [PubMed] [Google Scholar]
- 22. MacKenzie J. A., Payne R. M. (2004) Ribosomes specifically bind to mammalian mitochondria via protease-sensitive proteins on the outer membrane. J. Biol. Chem. 279, 9803–9810 [DOI] [PubMed] [Google Scholar]
- 23. Sondergaard C. R., Olsson M. H. M., Rostkowski M., Jensen J. H. (2011) Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem Theory Comput. 7, 2284–2295 [DOI] [PubMed] [Google Scholar]
- 24. Stanton C. L., Houk K. N. (2008) Benchmarking pKa prediction methods for residues in proteins. J. Chem. Theory Comput. 4, 951–966 [DOI] [PubMed] [Google Scholar]
- 25. Albaugh B. N., Arnold K. M., Denu J. M. (2011) KAT(ching) metabolism by the tail: insight into the links between lysine acetyltransferases and metabolism. Chembiochem 12, 290–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Z., Tan M., Xie Z., Dai L., Chen Y., Zhao Y. (2011) Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 7, 58–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Park J., Chen Y., Tishkoff D. X., Peng C., Tan M., Dai L., Xie Z., Zhang Y., Zwaans B. M., Skinner M. E., Lombard D. B., Zhao Y. (2013) SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 50, 919–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lombard D. B., Alt F. W., Cheng H. L., Bunkenborg J., Streeper R. S., Mostoslavsky R., Kim J., Yancopoulos G., Valenzuela D., Murphy A., Yang Y., Chen Y., Hirschey M. D., Bronson R. T., Haigis M., Guarente L. P., Farese R. V., Jr., Weissman S., Verdin E., Schwer B. (2007) Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 27, 8807–8814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hirschey M. D., Shimazu T., Goetzman E., Jing E., Schwer B., Lombard D. B., Grueter C. A., Harris C., Biddinger S., Ilkayeva O. R., Stevens R. D., Li Y., Saha A. K., Ruderman N. B., Bain J. R., Newgard C. B., Farese R. V., Jr., Alt F. W., Kahn C. R., Verdin E. (2010) SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith B. C., Settles B., Hallows W. C., Craven M. W., Denu J. M. (2011) SIRT3 substrate specificity determined by peptide arrays and machine learning. ACS Chem. Biol. 6, 146–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Harris T. K., Turner G. J. (2002) Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life 53, 85–98 [DOI] [PubMed] [Google Scholar]
- 32. Stark G. R. (1965) Reactions of cyanate with functional groups of proteins. 3. Reactions with amino and carboxyl groups. Biochemistry 4, 1030–1036 [DOI] [PubMed] [Google Scholar]
- 33. Maillard L. C. (1912) Action of amino acids on sugars. Formation of melanoidins in a methodical way. Compt. Rend. 154, 66 [Google Scholar]
- 34. Kim S. C., Sprung R., Chen Y., Xu Y., Ball H., Pei J., Cheng T., Kho Y., Xiao H., Xiao L., Grishin N. V., White M., Yang X. J., Zhao Y. (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618 [DOI] [PubMed] [Google Scholar]
- 35. Fritz K. S., Galligan J. J., Smathers R. L., Roede J. R., Shearn C. T., Reigan P., Petersen D. R. (2011) 4-Hydroxynonenal inhibits SIRT3 via thiol-specific modification. Chem. Res. Toxicol. 24, 651–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schwer B., Eckersdorff M., Li Y., Silva J. C., Fermin D., Kurtev M. V., Giallourakis C., Comb M. J., Alt F. W., Lombard D. B. (2009) Calorie restriction alters mitochondrial protein acetylation. Aging Cell 8, 604–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McGarry J. D., Foster D. W. (1980) Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem. 49, 395–420 [DOI] [PubMed] [Google Scholar]
- 38. Jenkins D. W., Eckle R. E., Craig J. W. (1971) Alcoholic ketoacidosis. JAMA 217, 177–183 [PubMed] [Google Scholar]
- 39. Someya S., Yu W., Hallows W. C., Xu J., Vann J. M., Leeuwenburgh C., Tanokura M., Denu J. M., Prolla T. A. (2010) Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Scott I., Webster B. R., Li J. H., Sack M. N. (2012) Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem. J. 443, 655–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weinert B. T., Iesmantavicius V., Wagner S. A., Schölz C., Gummesson B., Beli P., Nyström T., Choudhary C. (2013) Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol. Cell 51, 265–272 [DOI] [PubMed] [Google Scholar]
- 42. Moellering R. E., Cravatt B. F. (2013) Functional lysine modification by an intrinsically reactive primary glycolytic metabolite. Science 341, 549–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McMurry J., Begley T. P. (2005) The Organic Chemistry of Biological Pathways, pp. 26–28, Roberts and Co. Publishers, Englewood, CO [Google Scholar]
- 44. Shimazu T., Hirschey M. D., Hua L., Dittenhafer-Reed K. E., Schwer B., Lombard D. B., Li Y., Bunkenborg J., Alt F. W., Denu J. M., Jacobson M. P., Verdin E. (2010) SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 12, 654–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yu W., Dittenhafer-Reed K. E., Denu J. M. (2012) SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 287, 14078–14086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yu W., Lin Y., Yao J., Huang W., Lei Q., Xiong Y., Zhao S., Guan K.-L. (2009) Lysine 88 acetylation negatively regulates ornithine carbamoyltransferase activity in response to nutrient signals. J. Biol. Chem. 284, 13669–13675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kirschenbaum D. M. (1982) Nonenzymic protein modification: a general phenomenon? Med. Hypotheses 8, 491–493 [DOI] [PubMed] [Google Scholar]
- 48. Yang H., Yang T., Baur J. A., Perez E., Matsui T., Carmona J. J., Lamming D. W., Souza-Pinto N. C., Bohr V. A., Rosenzweig A., de Cabo R., Sauve A. A., Sinclair D. A. (2007) Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130, 1095–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., Yao Y., Ning Z. B., Zeng R., Xiong Y., Guan K. L., Zhao S., Zhao G. P. (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McBrian M. A., Behbahan I. S., Ferrari R., Su T., Huang T. W., Li K., Hong C. S., Christofk H. R., Vogelauer M., Seligson D. B., Kurdistani S. K. (2013) Histone acetylation regulates intracellular pH. Mol. Cell 49, 310–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tawfik D. S. (2010) Messy biology and the origins of evolutionary innovations. Nat. Chem. Biol. 6, 692–696 [DOI] [PubMed] [Google Scholar]