

Background: PsbO is an intrinsically disordered subunit of photosystem II.
Results: Temperature-sensitive PsbO dynamics are identified by reaction-induced Fourier transform infrared spectroscopy and the removal and reconstitution of PsbO.
Conclusion: PsbO serves as an organizational template and undergoes flash-induced hydrogen-bonding changes, coupled with the catalytic cycle of water oxidation.
Significance: PsbO samples a rough conformational landscape when bound to its target, the photosystem II reaction center.
Keywords: Intrinsically Disordered Proteins, Metalloproteins, Photosynthesis, Protein Dynamics, Protein-Protein Interactions, Conformational Landscape
Abstract
Photosystem II (PSII) is a membrane-bound enzyme that utilizes solar energy to catalyze the photooxidation of water. Molecular oxygen is evolved after four sequential light-driven oxidation reactions at the Mn4CaO5 oxygen-evolving complex, producing five sequentially oxidized states, Sn. PSII is composed of 17 membrane-spanning subunits and three extrinsic subunits, PsbP, PsbQ, and PsbO. PsbO is intrinsically disordered and plays a role in facilitation of the water oxidizing cycle. Native PsbO can be removed and substituted with recombinant PsbO, thereby restoring steady-state activity. In this report, we used reaction-induced Fourier transform infrared spectroscopy to obtain information concerning the role of PsbP, PsbQ, and PsbO during the S state cycle. Light-minus-dark difference spectra were acquired, monitoring structural changes associated with each accessible flash-induced S state transition in a highly purified plant PSII preparation (Triton X-100, octylthioglucoside). A comparison of S2 minus S1 spectra revealed that removal of PsbP and PsbQ had no significant effect on the data, whereas amide frequency and intensity changes were associated with PsbO removal. These data suggest that PsbO acts as an organizational template for the PSII reaction center. To identify any coupled conformational changes arising directly from PsbO, global 13C-PsbO isotope editing was employed. The reaction-induced Fourier transform infrared spectra of accessible S states provide evidence that PsbO spectral contributions are temperature (263 and 277 K) and S state dependent. These experiments show that PsbO undergoes catalytically relevant structural dynamics, which are coupled over long distance to hydrogen-bonding changes at the Mn4CaO5 cluster.
Introduction
Although much attention is focused on lowest energy protein structures, as revealed by high resolution x-ray crystallography, enzymes sample an ensemble of conformational substates, called the conformational landscape. The primary sequence encodes not only information about static structure but also the dynamic range. These factors determine the function and the adaptability of the protein (see, for example, Refs. 1–3 for reviews, and references therein). Natively unfolded or intrinsically disordered proteins (IDPs)2 are a limiting case in which a wide array of function-related structural organizations is readily detectable. This malleability has been postulated to play an important role in an IDP binding to its target protein (reviewed in Refs. 4 and 5). Binding is often observed to result in a decrease in disorder, either through a conformational selection or induced fit mechanism, although exceptions have been noted (see Ref. 5 and references therein).
Photosystem II (PSII) is a complex membrane protein consisting both of integral, membrane-spanning subunits and extrinsic subunits. A monomeric unit of PSII consists of at least 20 distinct protein subunits, which are composed of 17 integral subunits and 3 extrinsic polypeptides, PsbP, PsbQ, and PsbO (Fig. 1A) (6, 7). The hydrophobic intrinsic subunits, which bind most of the redox-active cofactors, are D1, D2, CP43, and CP47. The light-induced electron transfer pathway in the reaction center involves the dimeric chlorophyll (Chl) donor, P680, accessory chlorophyll molecules, quinone acceptors, and a tyrosine residue, YZ, which is Tyr-161 of the D1 polypeptide (reviewed in Refs. 8 and 9).
FIGURE 1.
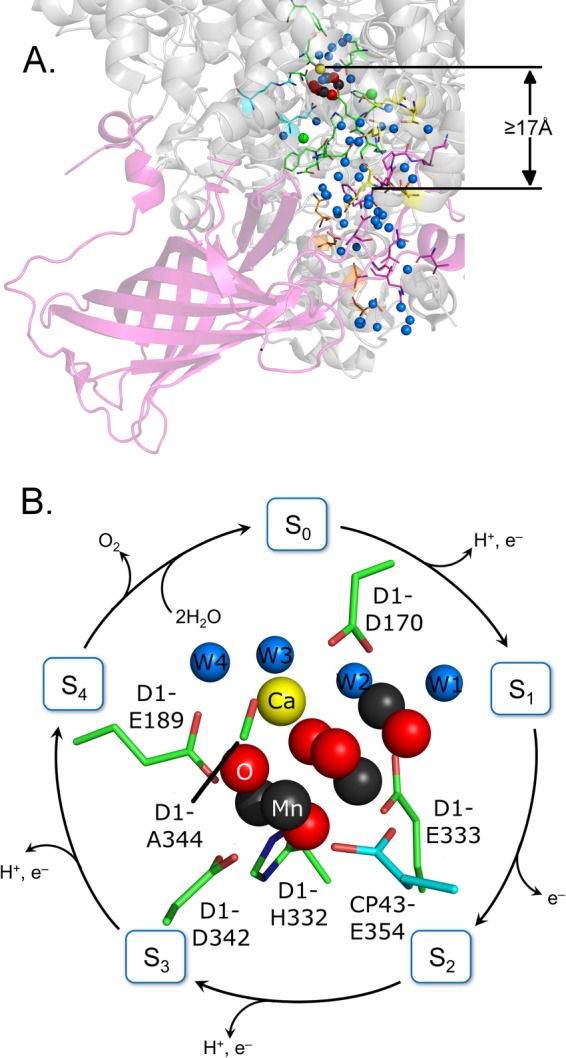
Diagram of PsbO (A) from the 1.9-Å cyanobacterial PSII crystal structure (PDB 3ARC) (7). PsbO is oriented to illustrate its relationship to the hydrogen-bonded water network (blue spheres) that extends from the Mn4CaO5 (dark gray, yellow, and red spheres) oxygen-evolving complex to the lumen. Amino acids of the CP43 (cyan sticks), D1 (green sticks), D2 (yellow sticks), PsbU (orange sticks), and PsbO (pink) subunits are color coded. The distance from the OEC calcium to the nearest PsbO residue is at least 17 Å. B, shows a model for flash-dependent photosynthetic water oxidation. A Mn4CaO5 structure, including metal ligands, is shown in the inset (PDB 3ARC). For the FT-IR experiments, presented here, a single 532-nm flash followed by 20-min dark adaptation synchronizes PSII in the S1 state. Subsequent actinic flashes promote centers through the S state cycle. Molecular oxygen is evolved on the third flash (S3 to S0 transition).
The oxygen-evolving complex (OEC) is a Mn4CaO5 cluster (Fig. 1B, inset). Four sequential photooxidations (Fig. 1B) of the OEC are required to produce molecular oxygen (10). Oxygen release from the OEC fluctuates with period four. The OEC cycles through five sequentially oxidized states, called the Sn states. A single flash given to a dark-adapted sample (S1 state) generates the S2 state (Fig. 1B), which corresponds to the oxidation of Mn(III) to Mn(IV) (11). Subsequent flashes advance the OEC to higher oxidation states and higher S states, and oxygen is evolved on the transition from S3 to S0. S4 is an unstable state, which is produced by flash excitation of S3 and converts to S0 in the dark. Calcium and chloride play important, but not completely understood, roles in the S state cycle. Flash excitation can be coupled with spectroscopic techniques to identify structural changes associated with each S state transition (for examples, see Refs. 12–15).
In addition to intrinsic subunits, PSII also contains extrinsic subunits (16, 17). For example, in plants, the three extrinsic lumenal subunits (PsbO, PsbP, and PsbQ) are necessary to maintain high steady-state rates of oxygen evolution activity (17). One proposed role of the extrinsic polypeptides, PsbP and PsbQ, is in the retention of the OEC calcium and chloride ions (18). PsbO also plays a role in retention of calcium and chloride and accelerates the steady-state rate of oxygen evolution (reviewed in Ref. 16), although a low level of activity is observed in its absence (19). The PsbO subunit is natively unfolded in solution (20), and cyanobacterial PsbO is known to form a β barrel (Fig. 1A) when bound to the PSII reaction center (7).
An extensive set of mutagenesis studies has been performed on plant and cyanobacterial PsbO (for examples, see Ref. 16 and 21–47). Experiments have also investigated the roles of PsbO in green algae (48) and the two PsbO subunits, PsbO1 and PsbO2, in Arabidopsis thaliana (49–59). These studies and chemical cross-linking (see Refs. 60 and 61, for examples) have identified domains that are important in structure, function, and assembly. Although one copy of PsbO is bound per reaction center in thermophilic cyanobacteria (6, 7), plant PSII binds two copies of PsbO (19, 23, 25, 32, 41, 50, 62–64).
Previous work showed that PsbO stabilizes the metal cluster and acts as a catalyst for the water splitting reactions (reviewed in Ref. 16 and 17). Functional roles for PsbO carboxylic acid and amide side chains were proposed, based on studies using reaction-induced FT-IR spectroscopy to study one transition, the S1 to S2 transition (65, 66). These experiments suggested that amino acid side chains on the PsbO subunit are involved in proton transfer reactions on the S1 to S2 transition at 200 K (65, 66). Interestingly, the description of PsbO structural changes were altered with temperature. These data suggest that complex PsbO dynamics accompany OEC oxidation.
To describe the role of PsbO in the PSII conformational landscape during other parts of the S state cycle, here we examine reaction-induced Fourier transform infrared (FT-IR) difference spectra for each accessible S state transition (Fig. 1B). Reaction-induced FT-IR spectroscopy has been used to study catalysis-induced conformational changes in other proteins (reviewed by Ref. 67). In application to S state cycling events, this technique was described originally in Refs. 13 and 14. The PSII sample is synchronized in the S1 state, and laser flashes are employed to advance the Mn4CaO5 cluster sequentially to the S2, S3, and S0 states. Difference FT-IR spectra are constructed for each flash-induced transition. For example, in a S2 minus S1 spectrum, unique vibrational bands of the S2 state are positive and unique vibrational bands of the S1 state are negative. Frequencies and intensities in these spectra have been reported to oscillate with period four. On the time scale employed, this technique reflects long-lived structural dynamics on the donor side of PSII (68, 69).
This reaction-induced FT-IR technique has been used recently (70, 71) to study the S state cycle in a plant PSII preparation, isolated with octylthioglucoside (72) and Triton X-100 (73). This preparation provides an advantage for spectroscopic studies in that it is highly resolved and stable. The PsbP, PsbQ, and PsbO extrinsic subunits have been reported to be retained by this preparation as isolated (72), but PsbP and PsbQ can be removed selectively by NaCl washes (74), and then PsbO can be removed by urea (75) or CaCl2 (76) washing.
In this study, these PSII purification methods and reaction-induced FT-IR spectroscopy were used to identify extrinsic subunit influence on the PSII conformational landscape. These results provide evidence that the IDP, PsbO, provides a template for organizing its target, the PSII reaction center. To identify structural changes arising directly from PsbO, spinach PsbO was recombinantly expressed and 13C-labeled, as described previously (66, 77). The 12C (natural abundance) and globally 13C-labeled recombinant proteins bind and restore steady-state oxygen activity (66, 77). This isotope-editing approach detects structural changes only in PsbO, with no spectral contribution from the rest of the PSII reaction center. These approaches provide evidence that PsbO undergoes temperature-dependent structural dynamics, which are coupled with the S state cycle.
EXPERIMENTAL PROCEDURES
Isolation of PSII
PSII-enriched thylakoid membranes were isolated from market spinach, as described previously (73). Membranes were solubilized (72) with n-octyl-β-d-thioglucoside (0.4%, pH 6.0), followed by treatment with 10 mm MgCl2 to remove light-harvesting complexes (LHCII, CP24, CP26, and CP29). Oxygen evolution activity of isolated PSII samples was measured (78). Samples were suspended in 400 mm sucrose, 50 mm MES-NaOH (pH 6.0), 60 mm NaCl, and 20 mm CaCl2. The electron acceptors, recrystallized 2,6-dichloro-1,4-benzoquinone (500 μm) and potassium ferricyanide (1 mm), were used and prepared immediately prior to the experiment. Steady-state oxygen evolution rates for PSII samples as isolated were 1100 ± 80 μmol of O2 (mg of Chl-h)−1 (see Table 1) using 2,6-dichloro-1,4-benzoquinone and potassium ferricyanide as acceptors at 25 °C. The use of 7 mm potassium ferricyanide alone, as in the FT-IR experiments, gave a steady-state oxygen evolution rate of 250 ± 45 μmol of O2 (mg of Chl-h)−1.
TABLE 1.
Steady-state O2 evolution activity of PSII samples
| PSII sample | −Ca2+a,b | +Ca2+a,c | % NaCl-PSIId |
|---|---|---|---|
| Untreated | 1100 ± 80 | 1300 ± 20 | |
| NaCl-PSII | 690 ± 20 | 970 ± 80 | 100 |
| Urea-PSII | 220 ± 10 | 580 ± 65 | 60 |
| Native-PSII | 560 ± 17 | 830 ± 18 | 85 |
| 12C-PSII | 410 ± 8 | 800 ± 74 | 82 |
| 13C-PSII | 500 ± 19 | 770 ± 41 | 79 |
a Steady-state oxygen evolution activity (μmol of O2 (mg of Chl-h)−1).
b The assay buffer used was 400 mm sucrose, 50 mm MES-NaOH (pH 6.0), and 100 mm NaCl.
c The assay buffer contained 400 mm sucrose, 50 mm MES-NaOH (pH 6.0), 60 mm NaCl, and 20 mm CaCl2.
d Percent activity (+Ca2+) relative to NaCl-PSII samples.
Depletion of Extrinsic Subunits
PSII samples were incubated in high ionic strength buffer (400 mm sucrose, 50 mm MES-NaOH (pH 6.0), and 2 m NaCl) to extract the extrinsic polypeptides PsbP and PsbQ (NaCl-PSII) (77, 79, 80). To remove PsbO, NaCl-PSII was incubated in buffer containing 400 mm sucrose, 50 mm MES-NaOH (pH 6.0), 2.6 m urea, and 200 mm NaCl (Urea-PSII) (77, 79, 80). As an alternative method for PsbO removal, NaCl-PSII was incubated in buffer containing 400 mm sucrose, 50 mm MES-NaOH (pH 6.0), and 1 m CaCl2 (CaCl2-PSII) (79).
Estimation of PsbO Content
Neville SDS-PAGE analysis of PSII samples (Fig. 2A) was performed as described previously (81). For Triton X-100-isolated PSII membranes (BBY), 5 μg of chlorophyll was loaded per lane and for n-octyl-β-d-thioglucoside-isolated PSII, 1 μg of chlorophyll was loaded per lane. Gels were stained with 0.05% Coomassie Brilliant Blue R (Coomassie stain) to detect total protein. To determine the content of PsbO, Western analysis was performed using an anti-PsbO antibody raised in rabbits (82). Unstained gels were blotted onto a polyvinylidene fluoride (PVDF) membrane. The PVDF membrane was subjected to sequential incubations with the primary anti-PsbO antibody (82) and then with a secondary anti-rabbit protein A-alkaline phosphatase conjugate (Calbiochem, San Diego, CA). The Western blot (Fig. 2B) was developed with a 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (BCIP/NBT) liquid substrate (Thermo Scientific, Waltham, MA). Densitometry was used to quantitate the amount of PsbO in samples used for FT-IR spectroscopy. A Western blot containing a standard curve of NaCl-PSII samples (data not shown) was employed (as performed previously in Ref. 79). Comparison of the integrated areas showed that urea or CaCl2 treatments removed 82 and 92% of native PsbO, respectively. Reconstitution with 12C- or 13C-PsbO to urea-PSII gave quantitative (120%) rebinding. This analysis is consistent with previous results for a different PSII preparation (24, 79).
FIGURE 2.

Urea-SDS-PAGE (81) (A) and Western blot (B) of PSII samples used for FT-IR experiments. The lanes represent: 1) PSII membranes (BBY), isolated by Triton X-100; 2) PSII, isolated by n-octyl-β-d-thioglucoside and Triton X-100; 3) NaCl-washed PSII (PsbP/PsbQ-depleted); 4) urea-washed PSII (PsbP/PsbQ and PsbO-depleted); 5) PSII reconstituted with 12C-PsbO; 6) PSII reconstituted with 13C-PsbO; 7) molecular weight markers (labels in kDa on the right); 8) 12C-PsbO; and 9) 13C-PsbO. Sample treatments are described in detail under “Experimental Procedures.” The amount of PsbO remaining in the urea-PSII sample is ∼20% (see “Experimental Procedures” for details). In 12C-PSII and 13C-PSII samples, a slight difference in migration for recombinant PsbO was observed, as expected (24). The amount of 12C/13C-PsbO in lanes 8 and 9 corresponds to 2 mol of PsbO/mol of PSII. Along the left, the electrophoretic migration of PsbO, PsbP, and PsbQ subunits is labeled. A Western blot analysis with anti-PsbO antibody (see “Experimental Procedures” for details) is shown in B.
Isolation and Purification of PsbO
Recombinant PsbO was expressed in and purified from Escherichia coli BL21(DE3)/pLysS cells containing the PsbO expression plasmid (24, 77, 80). The recombinant protein is different in two respects from native, spinach PsbO. The recombinant protein has an N-terminal methionine and also has a Val-235 to Ala mutation, which has been reported to arise spontaneously from propagation in E. coli (24). These changes have no influence on PsbO binding at room temperature or on reconstituted steady-state activity (24, 25, 65, 77). Global 13C-enrichment of recombinant PsbO was accomplished with [13C]glucose and minimal media (66, 77). This protocol incorporates the 13C-isotope at all PsbO carbons with at least 90% labeling, as assessed from gas chromatography/mass spectrometry analysis on acid-hydrolyzed PsbO samples (77).
Recombinant and native PsbO were purified by a method previously described (20). Isolated PsbO samples were dialyzed (24) into 400 mm sucrose, 50 mm MES-NaOH (pH 6.0), 60 mm NaCl, and 20 mm CaCl2. Samples were frozen and stored at −70 °C.
Sequencing
Nucleic acid sequencing (Eurofins MWG Operon) of the recombinant PsbO-containing plasmid confirmed the presence of the N-terminal methionine and the V235A mutation. N-terminal protein sequencing (University of Minnesota, BioMedical Genomics Center) of recombinant PsbO confirmed the presence of an N-terminal methionine residue. Electrospray ionization (ESI)-mass spectrometry (Georgia Institute of Technology, BioAnalytical Mass Spectrometry Facilities) determined a molecular mass of 26,634 Da, consistent with N-terminal methionine residue and V235A mutation in recombinant PsbO, and the expected molecular mass of 26,530 Da for native, spinach PsbO.
PsbO Reconstitution
Native, 12C-PsbO, or 13C-PsbO was reconstituted to urea-PSII (2 μm) in 400 mm sucrose, 50 mm MES-NaOH (pH 6.0), 60 mm NaCl, and 20 mm CaCl2 supplemented with 0.33 mg ml−1 of bovine serum albumin (Fraction V). The PsbO concentration during reconstitution was 10 μm, corresponding to 5 mol of PsbO/mol of PSII reaction center. The PsbO concentration was estimated using the extinction coefficient (16 mm−1 cm−1 at 276 nm; see Ref. 19). Rebinding of PsbO was carried out at room temperature, in the dark, for 1 h. Oxygen evolution assays were performed to assess the efficiency of PsbO reconstitution, as described in Table 1. Nonspecifically bound PsbO was removed by centrifugation (12,000 × g, 4 °C, 10 min). PsbO-reconstituted (12C/13C-PSII or native-PSII) samples were resuspended in the appropriate buffer and analyzed by urea-SDS-PAGE (see Fig. 2).
13C-PsbO Labeling and Rebinding Assessed by FT-IR Absorption Spectroscopy
FT-IR absorption spectra were collected from 12C-PSII and 13C-PSII samples. An FT-IR difference (12C-PSII minus 13C-PSII) spectrum was generated (Fig. 3A). As expected, the isotope shifts from reconstituted PsbO, bound to PSII (Fig. 3A), were comparable with the isotope shifts observed in PsbO in solution (Fig. 3B). The magnitude of the amide I and II isotope shifts was consistent with the expected downshifts for the 13C-isotopologue (65, 66).
FIGURE 3.
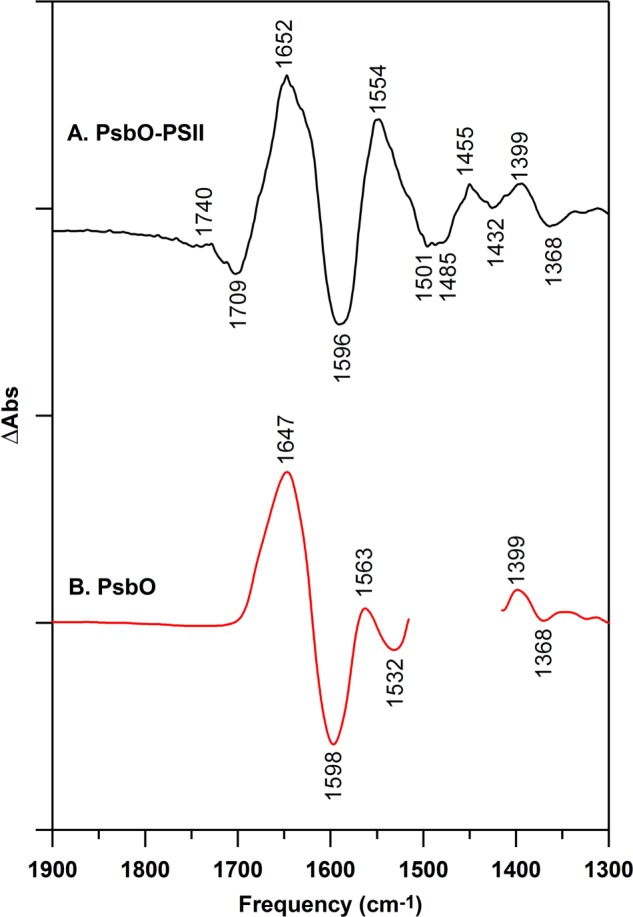
Isotope shifts caused by global 13C labeling of PsbO, as assessed from FT-IR absorbance spectra. Difference spectra were constructed to measure the isotope-induced downshift. In A, data were derived from 12C/13C-PsbO bound to PSII (12C- minus 13C-PSII; black), and in B, data were derived from PsbO in D2O buffer (12C- minus 13C-PsbO; red). In A, samples were prepared in 400 mm sucrose, 50 mm MES (pH 6.0), 60 mm NaCl, and 20 mm CaCl2, as described under “Experimental Procedures” for reaction-induced FT-IR spectroscopy. In B, PsbO (red) samples were prepared at 100 μm in 50 mm sucrose, 50 mm MES (pD 6.0), 60 mm NaCl, and 20 mm CaCl2. The 1450 cm−1 region for isolated PsbO (red) has overlapping H-O-D contributions and was removed for clarity. The y axis tick marks represent 5 × 10−2 absorbance units.
Reaction-induced FT-IR Spectroscopy
Reaction-induced FT-IR spectroscopy was performed at 263 and 277 K, as previously described (70, 71). Samples (PSII, NaCl-PSII, urea-PSII, CaCl2-PSII, native-PSII, 12C-PSII, or 13C-PSII) for FT-IR experiments were suspended in 400 mm sucrose, 50 mm MES-NaOH (pH 6.0), 60 mm NaCl, and 20 mm CaCl2 by three rounds of centrifugation (50,000 × g, 4 °C, 15 min) and resuspension and frozen (−70 °C) until use. Samples were thawed and potassium ferricyanide (7 mm) was added, and the sample was centrifuged (50,000 × g, 4 °C, 15 min) to produce a pellet. The sample was spread on a calcium fluoride window and concentrated briefly under N2 gas to give an O-H stretching (3370 cm−1) to amide II (1550 cm−1) absorbance ratio greater than 3. Samples were covered with a second CaF2 window and the edges were sealed with high vacuum grease and parafilm to prevent dehydration. Acquisition parameters for FT-IR spectroscopy were as follows: 8 cm−1 spectral resolution; 60 kHz mirror speed; four levels of zero filling; Happ-Genzel apodization; and Mertz phase correction. Samples were preflashed with a single saturating 532 nm laser flash (40 mJ cm−2), followed by 20 min dark adaptation to synchronize PSII centers in the S1 state. Dark-adapted PSII samples were then given four more (actinic) flashes. Each single actinic flash was followed by 15 s of data acquisition for each accessible S state. Therefore the flash frequency is one flash per 15 s. The reaction cycle was conducted once for each sample, so that each sample received only 5 total laser flashes. Reaction-induced difference spectra were constructed by the ratio of single-channel data, which were collected before and after each actinic flash. Rapid scan data were normalized to an amide II intensity of 0.5 absorbance units to correct for small differences in the path length (6 μm) and concentration between samples.
The Steady-state Rate of Ferricyanide Reduction at 263 and 277 K and Estimate of Flash Yield of Electron Transfer in FT-IR Samples
Experiments were conducted to quantitate the light-induced, steady-state rate of electron transfer to 7 mm potassium ferricyanide in PSII FT-IR samples at 263 and 277 K. PSII FT-IR samples were illuminated with a 633 nm HeNe (Melles-Griot, Carlsbad, CA) laser (2 milliwatt). The concentration of ferrocyanide, produced under 90 s of continuous illumination, was assessed from the amplitude of the 2038 cm−1 band in a 633-nm light-induced difference spectrum and a standard curve of potassium ferrocyanide. The standard curve was derived by addition of a known concentration of ferrocyanide to PSII and measurement of the 2038 cm−1 band in the dark in the FT-IR sample cell. Control experiments showed that no detectable ferrocyanide was produced by illumination of the ferricyanide-containing buffer (no PSII) and that no detectable ferrocyanide was produced in ferricyanide-containing PSII without 633 nm illumination. The FT-IR sample volume was estimated as 40 μl, the samples contained 0.3 mg of Chl (antenna size 55 Chl/reaction center), and the 633 nm difference spectra were corrected for any small difference in amide II absorbance. The derived electron transfer rates were 1.1 ± 0.3 μmol of electrons (mg of Chl-h)−1 or 55 mol of electrons (mol of PSII reaction center-h)−1 at 263 K and 1.2 ± 0.5 μmol of electrons (mg of Chl-h)−1 or 61 mol of electrons (mol of PSII reaction-h)−1 at 277 K. Using the same procedure, the yield of ferrocyanide produced by the first actinic 532 nm flash at 277 K was ∼1 mol of electron per mol of PSII reaction center, confirming the validity of the procedure. This experiment supports the conclusion that there is no difference in acceptor side electron transfer when the two temperatures are compared. Note that the electron transfer rate under continuous illumination is slower than the rate predicted by the oxygen evolution assay (see above). This difference is attributable to the rapid mixing and dilute sample conditions in the oxygen assay, as compared with the concentrated FT-IR samples.
RESULTS
The steady-state oxygen evolution rates of PSII preparations used in this study are presented in Table 1. Removal of extrinsic polypeptides was performed by washing with 2 m NaCl (PsbP, PsbQ depleted, NaCl-PSII) and then either 2.6 m urea (PsbO, PsbP, and PsbQ depleted, urea-PSII) or 1 m CaCl2 (PsbO, PsbP, and PsbQ depleted, CaCl2-PSII). Removal of the PsbP and PsbQ subunits was confirmed by SDS-PAGE analysis of NaCl-PSII (Fig. 2, lane 3), compared with untreated PSII (Fig. 2, lane 2). The oxygen evolution activity of NaCl-PSII was dependent on exogenous calcium and chloride, as expected (see Table 1 and Ref. 83). Treatment with 2.6 m urea removed ∼80% of natively bound PsbO (urea-PSII, Fig. 2, lane 4, and also see Refs. 77 and 79)) and significantly decreased steady-state rates of oxygen evolution (Table 1). CaCl2 treatment also decreased oxygen evolution and removed 90% of PsbO (see “Experimental Procedures” for details).
Urea-PSII was reconstituted with native PsbO (generating Native-PSII, data not shown) or recombinant PsbO (12C-PSII and 13C-PSII, ≥90% enriched) (Fig. 2, lanes 5 and 6), which restore oxygen evolution activity to ∼80% of the control, NaCl-PSII (Table 1). Previous analysis (77) determined that the extent of 13C labeling is greater than 90%. SDS-PAGE and Western blot analyses (Fig. 2, lanes 5 and 6) confirmed that 12C-PsbO and 13C-PsbO were bound to PSII. Both 12C- and 13C-PsbO were quantitatively reconstituted, as assessed from the Western blot (Fig. 2, lanes 8 and 9, see “Experimental Procedures” for details).
These results are consistent with previous studies of other spinach PSII preparations (20, 64, 66, 72, 77, 79, 80). The V235A mutation employed here does not affect the solution structure, binding, or activity at room temperature (24). The use of the V235A variant has the advantage that the migration of this protein on an SDS-PAGE gel is slightly different from the wild-type, allowing a ready verification of reconstitution, as shown above (Fig. 2, lanes 5 and 6).
To monitor the effects of the removal of the extrinsic polypeptides, we performed reaction-induced FT-IR spectroscopy (S2 minus S1) on NaCl-PSII (Fig. 4B), urea-PSII (Fig. 4C), native-PSII (Fig. 4E), and 12C-PSII (Fig. 4F). These spectra are dominated by contributions from the donor side of PSII. Double difference spectra (Fig. 5, A–E) were generated by subtraction of the S2 minus S1 difference spectra, associated with each treatment, from the control (untreated) PSII S2 minus S1 spectrum (Fig. 4A). Before construction of the double difference data, the spectra were scaled to reflect accurately the alterations induced by subunit removal. First, difference spectra were corrected for amide II absorption relative to an open beam background, reflecting any small differences in concentration and path length between samples. Second, spectra were then normalized to the intensity of the ferricyanide and ferrocyanide bands (2116 and 2038 cm−1, data not shown), which reflect any small changes in the amount of charge separation in the preparations. These methods have been used previously for PSII spectra (70, 71, 84) and give control double difference spectra (Fig. 5F) with flat baselines and no vibrational bands, as expected. Because the difference spectra are presented on the basis of total protein (amide II intensity), it was then necessary to use a correction factor to reflect the depletion of subunits. Each plant PSII reaction center is assumed to contain one copy of PsbP and PsbQ (6, 85). There is considerable evidence for two copies of PsbO (reviewed in Refs. 16 and 86) per plant PSII reaction center, and our SDS-PAGE and Western analyses provide evidence that the removal and reconstitution is quantitative (see Fig. 2 and “Experimental Procedures” for details). The scaling factors employed are based on this information and the molecular masses of PsbP (16.5 kDa), PsbQ (20 kDa) (17), PsbO (26.5 kDa) (87), and PSII (350 kDa) and are 90% for NaCl-PSII (PsbP/PsbQ depleted) and 74% for urea-PSII (PsbP/PsbQ and PsbO depleted).
FIGURE 4.
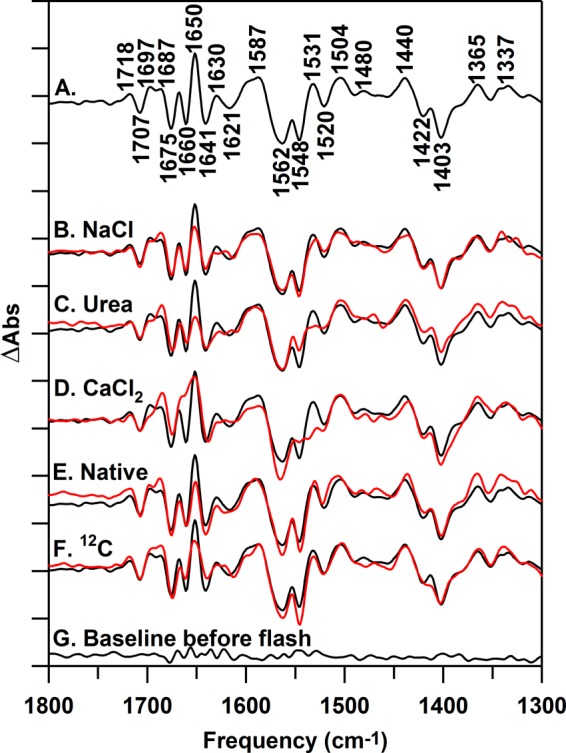
Reaction-induced FT-IR difference spectra, associated with the S1 to S2 transition, collected at 263 K. In A, the difference spectrum for untreated PSII is shown. The difference spectra in red are NaCl-PSII (B), urea-PSII (C), CaCl2-PSII (D), native-PSII (E), and 12C-PSII (F). The black spectrum in B–F is untreated PSII repeated from A. All samples were prepared at 15 μm PSII (0.75 mg of Chl ml−1) in 400 mm sucrose, 50 mm MES (pH 6.0), 60 mm NaCl, and 20 mm CaCl2 before pelleting. The spectrum in G is a representative (S1 minus S1) baseline recorded prior to illumination. Difference spectra were normalized as described in the text. Difference spectra were generated from 10 (A), 13 (B), 10 (C), 10 (D), 15 (E), 19 (F), and 10 (G) spectral averages. The y axis tick marks represent 2 × 10−4 absorbance units.
FIGURE 5.

Reaction-induced FT-IR double difference spectra, associated with the S1 to S2 transition, recorded at 263 K. The double difference spectra are (A) PSII minus NaCl-PSII, (B) PSII minus urea-PSII, (C) PSII minus CaCl2-PSII, (D) PSII minus native-PSII, and (E) PSII minus 12C-PSII. All samples were prepared at 15 μm PSII (0.75 mg of Chl ml−1) concentrations in 400 mm sucrose, 50 mm MES (pH 6.0), 60 mm NaCl, and 20 mm CaCl2 before pelleting. The spectrum in F is a control double difference spectrum, which was generated by subtraction of one-half of the data in A from the other half and division by the square root of two. Difference spectra, used to generate the double differences, are shown in Fig. 4 and were normalized before subtraction as described in the text. Double difference spectra were generated from 23 (A), 20 (B), 20 (C), 25 (D), 29 (E), and 10 (F) spectral averages. The y axis tick marks represent 2 × 10−4 absorbance units.
To establish the level of noise and baseline fluctuation in these data, we generated a control double difference spectrum. This control was produced using the relevant correction factors described above and by subtracting one-half of a data set from its other half. This control should exhibit no vibrational bands and is shown in Fig. 5F. In comparison, the CaCl2 spectrum (Fig. 5C) exhibits vibrational bands that are significant relative to the noise at −1682, −1661, −1547, and +1532 cm−1. These are bands typical of amide I (C = O stretch) and amide II (CN stretch/NH bending) bands in proteins. These bands have frequencies sensitive to secondary structure and hydrogen bonding (88). The urea-PSII spectrum also exhibited bands, significant relative to the noise (Fig. 5B). We conclude that removal of PsbO using either method results in a change in conformational sampling in the PSII reaction center during the S1 to S2 transition. Rebinding of native or recombinant protein reversed the majority of the effect of PsbO removal (Fig. 5, D and E). However, compared with the control (Fig. 5F), the PSII minus NaCl-PSII spectrum (Fig. 5A) exhibited no significant changes in the mid-infrared region.
This result supports the use of the recombinant protein in these spectroscopic studies and shows that PsbO removal has a significant effect on PSII structural transformations during the S1 to S2 transition. In agreement, previous EPR studies have concluded that although urea and CaCl2 treatments do not significantly alter hyperfine splittings in the S2 multiline signal, the intensity of the S2 g = 4.1 EPR signal is altered (89, 90) by PsbO removal. Note that, here, a subsequent flash to each of these preparations (S2 to S3 transition) gave the same spectral result in each type of preparation (data not shown).
To identify direct spectral contributions from PsbO in this PSII preparation, FT-IR difference spectra (Fig. 6) were recorded for 12C- (black) and 13C-PSII (red). Corresponding isotope-edited spectra were generated as 12C- minus 13C-PSII (from data in Fig. 6) and as a function of flash number at 263 K (Fig. 7, A–D). Oxygen evolution occurs on the first reaction cycle at this temperature, but the sample cannot carry out a second enzymatic cycle, under these flash conditions, possibly due to a limitation in water diffusion or a conformational gate (84). There is evidence that the steady-state rate of electron transfer under continuous illumination is slower in the FT-IR samples, when compared with a room temperature oxygen assay (“Experimental Procedures”). However, the flash-induced yield of ferrocyanide was ∼1 mol/mol of reaction center and the amplitudes of the ferrocyanide bands were similar for each S state transition (data not shown), arguing that there is no acceptor side limitation under these flash conditions.
FIGURE 6.

Reaction-induced FT-IR difference spectra of 12C-PSII (black) or 13C-PSII (red), recorded at 263 (A–E) and 277 (F–J) K. The difference spectra are associated with the, A and F, first (S2 minus S1); B and G, second (S3 minus S2); C and H, third (S0 minus S3); and D and I, fourth (S1 minus S0) flashes given to a dark-adapted sample. All samples were prepared at 15 μm PSII (0.75 mg of Chl ml−1) concentrations in 400 mm sucrose, 50 mm MES (pH 6.0), 60 mm NaCl, and 20 mm CaCl2 before pelleting. The spectra in E and J are representative (S1 minus S1) baselines recorded prior to illumination. Difference spectra were normalized as described in the text. Difference spectra were generated from 19 (A–E, black), 19 (A–E, red), 22 (F-I, black), and 25 (F–I, red) spectral averages. The y axis tick marks represent 2 × 10−4 absorbance units.
FIGURE 7.

Isotope-edited (12C- minus 13C-PSII) FT-IR spectra, recorded at 263 (A–E) and 277 (F–J) K. The data are associated with the first (S2 minus S1) (A and F), second (S3 minus S2) (B and G), third (S0 minus S3) (C and H), and fourth (S1 minus S0) (D and I) flashes given to a dark-adapted sample. All samples were prepared at 15 μm PSII (0.75 mg of Chl ml−1) concentrations in 400 mm sucrose, 50 mm MES (pH 6.0), 60 mm NaCl, and 20 mm CaCl2 before pelleting. The spectra in E and J are representative double difference spectra, which were generated by subtraction of one-half of the data in A and F from the other half and division by the square root of two. Difference spectra, used to generate the isotope-edited spectra, are shown in Fig. 6 and were normalized before subtraction as described in the text. Isotope-edited spectra were generated from 38 (A–D), 19 (E), 47 (F–I), and 22 (J) 22 spectral averages. The y axis tick marks represent 2 × 10−4 absorbance units.
Data were corrected as described above for path length, concentration, and charge separation differences. 12C and 13C reconstitutions were performed using the same PSII sample, so corrections for subunit composition were not necessary. Again, construction of control double difference spectra, by subtraction of one-half of a data set from the other, provides an estimate of the background and signal to noise of the measurement (Fig. 7, E and J).
The isotope-edited (12C- minus 13C-PSII) FT-IR spectrum associated with the S1 to S2 transition at 263 K is displayed in Fig. 7A. To appear in this double difference spectrum, the band must be altered in frequency and/or intensity by the photooxidation reaction and also be sensitive to 13C global labeling of PsbO. Thus, to be observed, the spectral contribution must originate from the PsbO subunit. For example, in the S2 minus S1 isotope-edited spectrum, the 12C-labeled S2 state will exhibit a positive band, and its 13C-labeled S2 isotopologue will exhibit a negative band. 12C/13C-labeled S1 bands will be reversed in sign.
To enhance the signal from just PsbO, on the background of the unlabeled PSII, spectra were acquired from just one reaction cycle on 19–25 FT-IR samples and then averaged. The resulting isotope-edited spectrum (Flash 1, Fig. 7A) displays bands at −1656, +1637, −1597, and −1549 cm−1, which are directly attributable to PsbO.
The 1656 and 1549 cm−1 bands have frequencies typical of the amide I and II vibrational modes in proteins (88). A frequency of 1656 cm−1 in H2O buffer is typical of an α helical or a disordered region of secondary structure (88). Cyanobacterial PsbO is primarily β sheet, with some disordered and a minority of α helical regions (7), so we attribute the negative 1656 cm−1 band to a disordered region of PsbO, which increases in structure and hydrogen bonding during transition from the S1 to S2 state. The resulting positive S2 band is not detected in Fig. 7A. This might occur if the PsbO β sheet is formed in the S2 state, because transition dipole coupling splits the amide band of β sheet into high and low frequency components, each with lower intensity (88). This would make the positive bands more difficult to detect.
The magnitude of the expected isotope shift for amide bands can be determined from FT-IR absorption spectra (Fig. 3A). The FT-IR absorption spectrum of 12C-PSII and 13C-PSII were subtracted (dark, no photolysis flash, 12C minus 13C). As shown, global 13C labeling is predicted to downshift both amide I (∼1650 cm−1) and amide II (1554 cm−1) bands by approximately ∼50 cm−1. Note that the isotope-shifted bands are less intense in Fig. 3A, compared with the 12C-labeled bands (77). Thus, we expect the signature of the amide isotope shift to be a band at ∼1650 and 1554 cm−1.
This comparison accounts for the −1656 and −1549 cm−1 bands in Fig. 7A, assigning them to the PsbO amide bond. To appear in the spectrum, the photooxidation reaction must change the frequency or amplitude of amide bands. Such a change is consistent with an alteration of PsbO amide hydrogen bonding during the S1 to S2 transition. This may correspond to an increase in order.
The frequency of +1637 cm−1 (Fig. 7A) is consistent with an assignment to a hydrogen-bonding change to an amide side chain of Gln or Asn (91). The band from the 13C-isotopologue may be detectable as −1597 cm−1 (Fig. 7A). Previously, a PsbO band at +1632 cm−1 was observed at 277 K in a PSII preparation isolated with lauryl maltoside. This band was also assigned to Gln/Asn, based on specific isotopic labeling (66). The amide I region of that spectrum was not reported, due to the lower stability of the baseline in that sample. Thus, a Gln/Asn assignment accounts for the +1637 cm−1 band described in this current study, but the band is observed at slightly lower temperature in this octylthioglucoside preparation, compared with a lauryl maltoside-isolated PSII preparation.
The observation of these PsbO amide bands supports the conclusion that PsbO has a responsive structure, which is altered during the S1 to S2 transition. Interestingly, the S2 to S3 (Flash 2, Fig. 7B) and S3 to S0 (Flash 3, Fig. 7C) transitions also generated spectra that exhibited amide bands, but with differences in frequency and sign. The S0 to S1 (Flash 4, Fig. 7D) spectrum was similar to the negative control, the control double difference spectrum in Fig. 7E. Thus, at 263 K, the effects of the S state transitions on PsbO conformation are distinguishable.
The effect of temperature on PsbO dynamics was tested by performing the isotope-editing experiment at 277 K. At this temperature, the yield of potassium ferrocyanide produced on each flash was similar and also similar to the flash-induced ferrocyanide yields at 263 K. The rate of ferrocyanide production under continuous illumination was also indistinguishable at 277 and 263 K.
Isotope-edited spectra for each accessible S state transition were constructed and are shown in Fig. 7, F–I. Interestingly, the 277 K spectra are distinct when compared with the 263 K data (Fig. 7, A–D). On the first flash (Fig. 7F), the spectra exhibited bands, some of which may be significant compared with the noise (Fig. 7J), throughout the mid-infrared region. On the second flash (Fig. 7G), the spectra were altered and exhibited significant bands at −1652, −1548, +1441, −1400, and −1356 cm−1. There was less intensity in the isotope-edited spectra on subsequent flashes (Fig. 7, H and I) at 277 K. Peptide backbone contributions can account for most of the observed frequencies in Fig. 7G (see Fig. 3A), with possible overlapping contributions from deprotonated carboxylate side chains (65, 66). Like amide frequencies, carboxylate frequencies are sensitive to changes in hydrogen bonding (92).
These data provide evidence that the most significant change in the PsbO structure occurs during the S2 to S3 transition at 277 K. Again, we attribute this change to an alteration in PsbO hydrogen bonding. Thus, reaction-induced FT-IR spectroscopy establishes that IDP PsbO exhibits temperature- and flash-dependent dynamics, which are linked with oxidation of the Mn4CaO5 cluster.
DISCUSSION
The understanding of how conformational selection contributes to enzymatic catalysis is still limited (3). Although static structures provide important information concerning the lowest energy conformer, proteins sample numerous conformational substates along the reaction coordinate (1–3). Descriptions of energy landscapes, which may facilitate enzymatic rate acceleration, are beginning to be developed using theoretical and spectroscopic (NMR (93, 94) and FT-IR (95)) approaches.
The extrinsic PSII subunits are important in biological water oxidation (16, 17, 75, 79, 83, 96). The PsbP and PsbQ extrinsic polypeptides have been shown to modulate the calcium and chloride requirements for efficient O2 evolution (17, 74, 79, 83, 97). PsbO also plays a critical role. The removal of PsbO had differential effects on the lifetimes and structural changes associated with the S state transitions. PsbO-depleted PSII samples exhibited longer-lived S2 and S3 states (in the dark) and delayed O2 release from the OEC (98). Alterations in S state lifetimes were observed in PsbO-deficient cyanobacteria (99) and in Arabidopsis (52), in which only one of the two isoforms of PsbO (PsbO-2) was expressed. Complete suppression of PsbP expression in Arabidopsis resulted in a highly unstable OEC in the mutants, which exhibited an increased S2 state lifetime (100). Depletion of PsbP and PsbQ slowed the rate of electron transfer 6–12-fold at the acceptor side of PSII; this observation was unaffected by removal of the PsbO subunit (101).
PsbO is an IDP (20). IDPs rely on their conformational promiscuity in solution to bind to a target or an array of target proteins (reviewed in Ref. 4 and 5). Previous analysis demonstrated that PsbO undergoes a secondary structural change, reducing the amount of disordered structure and increasing β sheet character (77), without altering the overall size or shape (102). Mutation of the lone tryptophan residue (W241F) in spinach PsbO resulted in defective binding to PSII and reduced steady-state oxygen evolution activity, but this effect could be eliminated by deletion of the six amino-terminal residues (40). Mutation at Arg-151, Arg-161, (38), Asp-157, (41), or Leu-245 (29) also resulted in reduced binding. It was suggested that these mutations increase the structural stability of isolated PsbO, which, in turn, interferes with the inherent flexibility required for PsbO binding. These previous mutational and spectroscopic studies concluded that PsbO flexibility is essential for effective PSII binding and restoration of high O2 evolution activity.
We show here that removal of PsbO either by urea or CaCl2 treatment alters the conformational landscape of the PSII reaction center. This was assessed by monitoring the structural changes associated with the S1 to S2 transition. Both urea- and CaCl2-treated preparations are known to form an S2 state. Previously, PsbO removal (with both urea and CaCl2) has been shown to alter the equilibrium between two EPR signals arising from the S2 state (for examples, see Ref. 86, 89, and 103). Our work provides evidence that this change in magnetic coupling may be caused by a conformational selection, which is mediated by PsbO binding to PSII. These experiments are consistent with the idea that PSII undergoes extensive dynamics on many time scales as previously proposed from analysis of transient infrared kinetics and FT-IR spectroscopy (68, 69) and that there is a distribution of active conformations. Interestingly, in our studies, removal of PsbP and PsbQ had no significant effect on the spectra associated with the S1 to S2 transition.
Reaction-induced FT-IR spectroscopy, isotopic labeling, and temperature were then used to probe the conformational dynamics of PsbO in all accessible S states. We hypothesized that, when bound to PSII, PsbO can sample a broad distribution of substates in a rough conformational landscape. This hypothesis was based on previous isotope-edited FT-IR experiments, conducted for the S1 to S2 transition, which showed surprising temperature dependence. Also, significant alterations were observed when data obtained under steady-state illumination were compared with spectra acquired on a long time scale after a saturating flash (65, 66).
To obtain more information, here, we monitored PsbO dynamics during the entire S state cycle (Fig. 1B) with a PSII preparation that exhibits enhanced signal to noise. Our results show that contributions from PsbO at 263 and 277 K are distinguishable and are most significant on the S2 to S3 transition at 277 K. This work provides evidence that PsbO participates in temperature-sensitive conformational dynamics, which are linked with the S state cycle. This conclusion provides a rationalization for the observation of broad PsbO isotope-edited spectra under illumination at 200 K (65). Under steady-state illumination, it is likely that many conformational substates of PsbO and PSII can be populated. The data suggest that PsbO retains its intrinsically disordered character, even when bound to its target, the PSII reaction center.
Previously, direct carboxylate deprotonation was inferred from 200 K data, but was not observed at 277 K in a lauryl maltoside-isolated PSII preparation (65, 66). Protonated and deprotonated carboxylates can be easily distinguished by FT-IR spectroscopy (67, 91). Protonation of the carboxylate side chain gives rise to C = O stretching bands in the 1720–1700 cm−1 region, whereas deprotonated carboxylates have delocalized asymmetric and symmetric CO stretching vibrations at ∼1560 and 1390 cm−1 (65). The reaction-induced FT-IR spectra presented here do not exhibit significant intensity in the 1770–1700 cm−1 region. Therefore, this work supports one of two conclusions: that any PsbO deprotonation event occurs in a small population of centers or that the deprotonation event is facile and therefore not observable at higher temperature under these conditions.
A previous FT-IR study concluded that no significant changes occurred in the mid-infrared region, associated with the S1 to S2 state transition, when PsbO was removed (104). However, spectral changes were attributed to removal of the PsbP subunit, but not the PsbQ or PsbO subunits. It was proposed that PsbP affects the peptide conformation around the OEC without alteration of the ligand structure. In our experiments, by contrast, PsbO depletion with urea or CaCl2 resulted in significant changes in the reaction-induced FT-IR spectra, whereas PsbP and PsbQ removal did not have a significant effect, relative to a negative control. In our studies, FT-IR experiments were conducted on a highly purified PSII preparation (72), which is devoid of light harvesting complexes (105). This PSII preparation exhibits high steady-state activity and stability. In addition, samples were subjected to the minimum number of flashes, including only one reaction cycle (4 flashes plus a preflash), and the amount of time between preparing the sample and running the experiment was strictly controlled to improve stability and reproducibility. Data were obtained from 10 to 25 different FT-IR samples and averaged to achieve the final signal to noise. Also, spectra were corrected in intensity for any small changes in protein concentration, path length, charge separation, and subunit content. By contrast, in Ref. 104, there may have been lower intrinsic activity and stability in the PSII preparation employed, and there was a resulting decrease in the signal to noise ratio. Also, in Ref. 104, there was a lower number of averages (3 to 5), a small number of samples employed (1 to 2), samples were frozen/thawed to conduct multiple reaction cycles, and there was no reported correction for protein concentration, path length, charge separation, or subunit composition.
PsbO is ∼17 Å from the Mn4CaO5 cluster (Fig. 1A). How are these conformational changes in PsbO linked with oxidation of the metal cluster? PSII contains a catalytically important hydrogen-bonded water network surrounding the OEC (70). This network (Fig. 1A) extends from the OEC to the lumen, including a number of residues in PsbO, and may play a functional role in proton transport (106–109). We propose that the hydrogen-bonding changes in PsbO are propagated through this network, when manganese is oxidized. Interestingly, the S2 to S3 transition has been proposed to be associated with a change in manganese-manganese and manganese-calcium bond lengths (12, 15). There are likely to be differences between cyanobacterial and plant PSII (see Ref. 16 for review). An example is the difference in binding stoichiometries of PsbO per PSII reaction center. However, the 1.9-Å cyanobacterial structure (7) serves as a reasonable starting point for our spectral interpretation. Previously, modeling of the PsbO structure has suggested that clusters of carboxylic acid side chains may be important in function (110). The FT-IR data, presented here, suggest that the amide bonds of PsbO are sensors for the functional, extended hydrogen-bonded water network in the OEC. Spectral differences observed at 263 K and 277 K may be due to inhibited diffusion of water or to a conformational gate at 263 K, under the conditions employed for flash excitation (84).
Studies of alcohol dehydrogenases have shown that long-range conformation selection is important in catalysis (111). The work presented here suggests that long-range conformational interactions may also play an important role in the PSII reaction center. Our studies provide additional insight into the role of a single subunit, PsbO, in navigating the conformational landscape, which is associated with photosynthetic water oxidation.
This work was supported by National Science Foundation Grant MCB 08-42246 (to B. A. B.)
- IDP
- intrinsically disordered protein
- FT-IR
- Fourier transform infrared
- OEC
- oxygen-evolving complex
- PSII
- photosystem II
- Chl
- chlorophyll.
REFERENCES
- 1. Henzler-Wildman K., Kern D. (2007) Dynamic personalities of proteins. Nature 450, 964–972 [DOI] [PubMed] [Google Scholar]
- 2. Baldwin A. J., Kay L. E. (2009) NMR spectroscopy brings invisible protein states into focus. Nat. Chem. Biol. 5, 808–814 [DOI] [PubMed] [Google Scholar]
- 3. Nashine V. C., Hammes-Schiffer S., Benkovic S. J. (2010) Coupled motions in enzyme catalysis. Curr. Opin. Chem. Biol. 14, 644–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fink A. L. (2005) Natively unfolded proteins. Curr. Opin. Struct. Biol. 15, 35–41 [DOI] [PubMed] [Google Scholar]
- 5. Tompa P. (2012) Intrinsically disordered proteins. A 10-year recap. Trends Biochem. Sci. 37, 509–516 [DOI] [PubMed] [Google Scholar]
- 6. Kashino Y., Lauber W. M., Carroll J. A., Wang Q., Whitmarsh J., Satoh K., Pakrasi H. B. (2002) Proteomic analysis of a highly active photosystem II preparation from the cyanobacterium Synechocystis sp. PCC 6803 reveals the presence of novel polypeptides. Biochemistry 41, 8004–8012 [DOI] [PubMed] [Google Scholar]
- 7. Umena Y., Kawakami K., Shen J.-R., Kamiya N. (2011) Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473, 55–60 [DOI] [PubMed] [Google Scholar]
- 8. Nelson N., Yocum C. F. (2006) Structure and function of photosystem I and II. Annu. Rev. Plant Biol. 57, 521–565 [DOI] [PubMed] [Google Scholar]
- 9. Barry B. A. (2011) Proton coupled electron transfer and redox active tyrosines in photosystem II. J. Photochem. Photobiol. B 104, 60–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Joliot P., Kok B. (1975) in Bioenergetics of Photosynthesis (Govindjee, ed) pp. 388–412, Academic Press, New York [Google Scholar]
- 11. Haumann M., Grabolle M., Neisius T., Dau H. (2002) The first room-temperature x-ray absorption spectra of higher oxidation states of the tetra-manganese complex of photosystem II. FEBS Lett. 512, 116–120 [DOI] [PubMed] [Google Scholar]
- 12. Liang W., Roelofs T. A., Cinco R. M., Rompel A., Latimer M. J., Yu W. O., Sauer K., Klein M. P., Yachandra V. K. (2000) Structural change of the Mn cluster during the S2→S3 state transition of the oxygen-evolving complex of photosystem II. Does it reflect the onset of water/substrate oxidation? Determination by Mn x-ray absorption spectroscopy. J. Am. Chem. Soc. 122, 3399–3412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hillier W., Babcock G. T. (2001) S-state dependent Fourier transform infrared difference spectra for photosystem II oxygen evolving complex. Biochemistry 40, 1503–1509 [DOI] [PubMed] [Google Scholar]
- 14. Noguchi T., Sugiura M. (2001) Flash-induced Fourier transform infrared detection of the structural changes during the S-state cycle of the oxygen-evolving complex in photosystem II. Biochemistry 40, 1497–1502 [DOI] [PubMed] [Google Scholar]
- 15. Haumann M., Müller C., Liebisch P., Iuzzolino L., Dittmer J., Grabolle M., Neisius T., Meyer-Klaucke W., Dau H. (2005) Structural and oxidation state changes of the photosystem II manganese complex in four transitions of the water oxidation cycle (S0 → S1, S1 → S2, S2 → S3, and S3,4 → S0) characterized by X-ray absorption spectroscopy at 20K and room temperatures. Biochemistry 44, 1894–1908 [DOI] [PubMed] [Google Scholar]
- 16. Popelkova H., Yocum C. F. (2011) PsbO, the manganese-stabilizing protein. Analysis of the structure-function relations that provide insights into its role in photosystem II. J. Photochem. Photobiol. B 104, 179–190 [DOI] [PubMed] [Google Scholar]
- 17. Bricker T. M., Roose J. L., Fagerlund R. D., Frankel L. K., Eaton-Rye J. J. (2012) The extrinsic proteins of photosystem II. Biochim. Biophys. Acta 1817, 121–142 [DOI] [PubMed] [Google Scholar]
- 18. Yocum C. F. (2008) The calcium and chloride requirements of the O2 evolving complex. Coord. Chem. Rev. 252, 296–305 [Google Scholar]
- 19. Xu Q., Bricker T. M. (1992) Structural organization of proteins on the oxidizing side of photosystem II. Two molecules of the 33-kDa manganese-stabilizing proteins per reaction center. J. Biol. Chem. 267, 25816–25821 [PubMed] [Google Scholar]
- 20. Lydakis-Simantiris N., Hutchison R. S., Betts S. D., Barry B. A., Yocum C. F. (1999) Manganese stabilizing protein of photosystem II is a thermostable natively unfolded polypeptide. Biochemistry 38, 404–414 [DOI] [PubMed] [Google Scholar]
- 21. Eaton-Rye J. J., Murata N. (1989) Evidence that the amino-terminus of the 33 kDa extrinsic protein is required for binding to the photosystem II complex. Biochim. Biophys. Acta 977, 219–226 [DOI] [PubMed] [Google Scholar]
- 22. Burnap R. L., Qian M., Shen J.-R., Inoue Y., Sherman L. A. (1994) Role of disulfide linkage and putative intermolecular binding residues in the stability and binding of the extrinsic manganese-stabilizing protein to the photosystem II reaction center. Biochemistry 33, 13712–13718 [DOI] [PubMed] [Google Scholar]
- 23. Seidler A. (1994) Introduction of a histidine tail at the N-terminus of a secretory protein expressed in Escherichia coli. Protein Eng. 7, 1277–1280 [DOI] [PubMed] [Google Scholar]
- 24. Betts S. D., Ross J. R., Pichersky E., Yocum C. F. (1996) Cold-sensitive assembly of a mutant manganese-stabilizing protein caused by a Val to Ala replacement. Biochemistry 35, 6302–6307 [DOI] [PubMed] [Google Scholar]
- 25. Betts S. D., Ross J. R., Pichersky E., Yocum C. F. (1997) Mutation Val235-Ala weakens biniding of the 33-kDa manganese stabilizing protein of photosystem II to one of two sites. Biochemistry 36, 4047–4053 [DOI] [PubMed] [Google Scholar]
- 26. Miura T., Shen J.-R., Takahashi S., Kamo M., Nakamura E., Ohta H., Kamei A., Inoue Y., Domae N., Takio K., Nakazato K., Inoue Y., Enami I. (1997) Identification of domains on the extrinsic 33-kDa protein possibly involved in electrostatic interaction with photosystem II complex by means of chemical modification. J. Biol. Chem. 272, 3788–3798 [DOI] [PubMed] [Google Scholar]
- 27. Qian M., Al-Khaldi S. F., Putnam-Evans C., Bricker T. M., Burnap R. L. (1997) Photoassembly of the photosystem II (Mn)4 cluster in site-directed mutants impaired in the binding of the manganese-stabilizing protein. Biochemistry 36, 15244–15252 [DOI] [PubMed] [Google Scholar]
- 28. Betts S. D., Lydakis-Simantiris N., Ross J. R., Yocum C. F. (1998) The carboxyl-terminal tripeptide of the manganese-stabilizing protein is required for quantitative assembly into photosystem II and for high rates of oxygen evolution activity. Biochemistry 37, 14230–14236 [DOI] [PubMed] [Google Scholar]
- 29. Lydakis-Simantiris N., Betts S. D., Yocum C. F. (1999) Leucine 245 is a critical residue for folding and function of the manganese stabilizing protein of photosystem II. Biochemistry 38, 15528–15535 [DOI] [PubMed] [Google Scholar]
- 30. Al-Khaldi S. F., Coker J., Shen J. R., Burnap R. L. (2000) Characterization of site-directed mutants in manganese-stabilizing protein (MSP) of Synechocystis sp. PCC6803 unable to grow photoautotrophically in the absence of cytochrome c-550. Plant Mol. Biol. 43, 33–41 [DOI] [PubMed] [Google Scholar]
- 31. Motoki A., Usui M., Shimazu T., Hirano M., Katoh S. (2002) A domain of the manganese-stabilizing protein from Synechococcus elongatus involved in functional binding to photosystem II. J. Biol. Chem. 277, 14747–14756 [DOI] [PubMed] [Google Scholar]
- 32. Popelkova H., Im M. M., D'Auria J., Betts S. D., Lydakis-Simantiris N., Yocum C. F. (2002) N-terminus of the photosystem II manganese stabilizing protein: Effects of sequence elongation and truncation. Biochemistry 41, 2702–2711 [DOI] [PubMed] [Google Scholar]
- 33. Popelkova H., Im M. M., Yocum C. F. (2002) N-terminal truncations of manganese stabilizing protein identify two amino acid sequences required for binding of the eukaryotic protein to Photosystem II and reveal the absence of one binding-related sequence in cyanobacteria. Biochemistry 41, 10038–10045 [DOI] [PubMed] [Google Scholar]
- 34. Popelkova H., Im M. M., Yocum C. F. (2003) Binding of manganese stabilizing protein to Photosystem II. Identification of essential N-terminal threonine residues and domains that prevent nonspecific binding. Biochemistry 42, 6193–6200 [DOI] [PubMed] [Google Scholar]
- 35. Popelkova H., Wyman A., Yocum C. (2003) Amino acid sequences and solution structures of manganese stabilizing protein that affect reconstitution of photosystem II activity. Photosyn. Res. 77, 21–34 [DOI] [PubMed] [Google Scholar]
- 36. Wyman A. J., Yocum C. F. (2005) Structure and activity of the photosystem II manganese-stabilizing protein: Role of the conserved disulfide bond. Photosyn. Res. 85, 359–372 [DOI] [PubMed] [Google Scholar]
- 37. Zhang F., Gao J., Weng J., Tan C., Ruan K., Xu C., Jiang D. (2005) Structural and functional differentiation of three groups of tyrosine residues by acetylation of N-acetylimidazole in manganese stabilizing protein. Biochemistry 44, 719–725 [DOI] [PubMed] [Google Scholar]
- 38. Popelkova H., Betts S. D., Lydakis-Symantiris N., Im M. M., Swenson E., Yocum C. F. (2006) Mutagenesis of basic residues R151 and R161 in manganese-stabilizing protein of photosystem II causes inefficient binding of chloride to the oxygen-evolving complex. Biochemistry 45, 3107–3115 [DOI] [PubMed] [Google Scholar]
- 39. Popelková H., Yocum C. F. (2007) Current status of the role of Cl− ion in the oxygen-evolving complex. Photosyn. Res. 93, 111–121 [DOI] [PubMed] [Google Scholar]
- 40. Wyman A. J., Popelkova H., Yocum C. F. (2008) Site-directed mutagenesis of conserved C-terminal tyrosine and tryptophan residues of PsbO, the photosystem II manganese-stabilizing protein, alters its activity and fluorescence properties. Biochemistry 47, 6490–6498 [DOI] [PubMed] [Google Scholar]
- 41. Popelkova H., Commet A., Yocum C. F. (2009) Asp157 is required for the function of PsbO, the photosystem II manganese-stabilizing protein. Biochemistry 48, 11920–11928 [DOI] [PubMed] [Google Scholar]
- 42. Popelkova H., Commet A., Yocum C. F. (2010) The double mutation ΔL6MW241F in PsbO, the photosystem II manganese stabilizing protein, yields insights into the evolution of its structure and function. FEBS Lett. 584, 4009–4014 [DOI] [PubMed] [Google Scholar]
- 43. Roose J. L., Yocum C. F., Popelkova H. (2010) Function of PsbO, the photosystem II manganese-stabilizing protein. Probing the role of aspartic acid 157. Biochemistry 49, 6042–6051 [DOI] [PubMed] [Google Scholar]
- 44. Popelkova H., Boswell N., Yocum C. (2011) Probing the topography of the photosystem II oxygen evolving complex: PsbO is required for efficient calcium protection of the manganese cluster against dark-inhibition by an artificial reductant. Photosyn. Res. 110, 111–121 [DOI] [PubMed] [Google Scholar]
- 45. Roose J. L., Yocum C. F., Popelkova H. (2011) Binding stoichiometry and affinity of the manganese-stabilizing protein affects redox reactions on the oxidizing side of photosystem II. Biochemistry 50, 5988–5998 [DOI] [PubMed] [Google Scholar]
- 46. Commet A., Boswell N., Yocum C. F., Popelka H. (2012) pH optimum of the photosystem II H2O oxidation reaction. Effects of PsbO, the manganese-stabilizing protein, Cl− retention, and deprotonation of a component required for O2 evolution activity. Biochemistry 51, 3808–3818 [DOI] [PubMed] [Google Scholar]
- 47. Popelka H., Yocum C. (2012) Probing the N-terminal sequence of spinach PsbO. Evidence that essential threonine residues bind to different functional sites in eukaryotic photosystem II. Photosyn. Res. 112, 117–128 [DOI] [PubMed] [Google Scholar]
- 48. Mayfield S. P., Rahire M., Frank G., Zuber H., Rochaix J. D. (1987) Expression of the nuclear gene encoding oxygen-evolving enhancer protein 2 is required for high levels of photosynthetic oxygen evolution in Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. U.S.A. 84, 749–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Murakami R., Ifuku K., Takabayashi A., Shikanai T., Endo T., Sato F. (2002) Characterization of an Arabidopsis thaliana mutant with impaired psbO, one of two genes encoding extrinsic 33-kDa proteins in photosystem II. FEBS Lett. 523, 138–142 [DOI] [PubMed] [Google Scholar]
- 50. Murakami R., Ifuku K., Takabayashi A., Shikanai T., Endo T., Sato F. (2005) Functional dissection of two Arabidopsis PsbO proteins. FEBS J. 272, 2165–2175 [DOI] [PubMed] [Google Scholar]
- 51. Yi X., McChargue M., Laborde S., Frankel L. K., Bricker T. M. (2005) The manganese-stabilizing protein is required for photosystem II assembly/stability and photoautotrophy in higher plants. J. Biol. Chem. 280, 16170–16174 [DOI] [PubMed] [Google Scholar]
- 52. Liu H., Frankel L. K., Bricker T. M. (2007) Functional analysis of photosystem II in a PsbO-1-deficient mutant in Arabidopsis thaliana. Biochemistry 46, 7607–7613 [DOI] [PubMed] [Google Scholar]
- 53. Lundin B., Hansson M., Schoefs B., Vener A. V., Spetea C. (2007) The Arabidopsis PsbO2 protein regulates dephosphorylation and turnover of the photosystem II reaction centre D1 protein. Plant J. 49, 528–539 [DOI] [PubMed] [Google Scholar]
- 54. Lundin B., Thuswaldner S., Shutova T., Eshaghi S., Samuelsson G., Barber J., Andersson B., Spetea C. (2007) Subsequent events to GTP binding by the plant PsbO protein. Structural changes, GTP hydrolysis and dissociation from the photosystem II complex. Biochim. Biophys. Acta 1767, 500–508 [DOI] [PubMed] [Google Scholar]
- 55. Bricker T. M., Frankel L. K. (2008) The psbO1 mutant of Arabidopsis cannot efficiently use calcium in support of oxygen evolution by photosystem II. J. Biol. Chem. 283, 29022–29027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lundin B., Nurmi M., Rojas-Stuetz M., Aro E.-M., Adamska I., Spetea C. (2008) Towards understanding the functional difference between the two PsbO isoforms in Arabidopsis thaliana. Insights from phenotypic analyses of psbO knockout mutants. Photosyn. Res. 98, 405–414 [DOI] [PubMed] [Google Scholar]
- 57. Yi X., Hargett S. R., Frankel L. K., Bricker T. M. (2008) The effects of simultaneous RNAi suppression of PsbO and PsbP protein expression in photosystem II of Arabidopsis. Photosyn. Res. 98, 439–448 [DOI] [PubMed] [Google Scholar]
- 58. Allahverdiyeva Y., Mamedov F., Holmström M., Nurmi M., Lundin B., Styring S., Spetea C., Aro E.-M. (2009) Comparison of the electron transport properties of the psbo1 and psbo2 mutants of Arabidopsis thaliana. Biochim. Biophys. Acta 1787, 1230–1237 [DOI] [PubMed] [Google Scholar]
- 59. Dwyer S. A., Chow W. S., Yamori W., Evans J. R., Kaines S., Badger M. R., von Caemmerer S. (2012) Antisense reductions in the PsbO protein of photosystem II leads to decreased quantum yield but similar maximal photosynthetic rates. J. Exp. Bot. 63, 4781–4795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Frankel L. K., Bricker T. M. (1995) Interaction of the 33-kDa extrinsic protein with photosystem II. Identification of domains on the 33-kDa protein that are shielded from NHS-biotinylation by photosystem II. Biochemistry 34, 7492–7497 [DOI] [PubMed] [Google Scholar]
- 61. Enami I., Kamo M., Ohta H., Takahashi S., Miura T., Kusayanagi M., Tanabe S., Kamei A., Motoki A., Hirano M., Tomo T., Satoh K. (1998) Intramolecular cross-linking of the extrinsic 33-kDa protein leads to loss of oxygen evolution but not its ability of binding to photosystem II and stabilization of the manganese cluster. J. Biol. Chem. 273, 4629–4634 [DOI] [PubMed] [Google Scholar]
- 62. Yamamoto Y., Nakayama S., Cohn C. L., Krogmann D. W. (1987) Highly efficient purification of the 33-, 24-, and 18-kDa proteins in spinach photosystem II by butanol/water phase partitioning and high-performance liquid chromatography. Arch. Biochem. Biophys. 255, 156–161 [DOI] [PubMed] [Google Scholar]
- 63. Leuschner C., Bricker T. M. (1996) Interaction of the 33 kDa extrinsic protein with photosystem II. Rebinding of the 33 kDa extrinsic protein to photosystem II membranes which contain four, two, or zero manganese per photosystem II reaction. Biochemistry 35, 4551–4557 [DOI] [PubMed] [Google Scholar]
- 64. Popelkova H., Commet A., Kuntzleman T., Yocum C. F. (2008) Inorganic cofactor stabilization and retention. The unique functions of the two psbO subunits of eukaryotic photosystem II. Biochemistry 47, 12593–12600 [DOI] [PubMed] [Google Scholar]
- 65. Hutchison R. S., Steenhuis J. J., Yocum C. F., Razeghifard M. R., Barry B. A. (1999) Deprotonation of the 33-kDa extrinsic manganese-stabilizing subunit accompanies photooxidation of manganese in photosystem II. J. Biol. Chem. 274, 31987–31995 [DOI] [PubMed] [Google Scholar]
- 66. Sachs R. K., Halverson K. M., Barry B. A. (2003) Specific isotopic labeling and photooxidation-linked structural changes in the manganese-stabilizing subunit of photosystem II. J. Biol. Chem. 278, 44222–44229 [DOI] [PubMed] [Google Scholar]
- 67. Kötting C., Gerwert K. (2005) Proteins in action monitored by time-resolved FTIR spectroscopy. ChemPhysChem 6, 881–888 [DOI] [PubMed] [Google Scholar]
- 68. Barry B. A., Cooper I. B., De Riso A., Brewer S. H., Vu D. M., Dyer R. B. (2006) Time-resolved vibrational spectroscopy detects protein-based intermediates in the photosynthetic oxygen-evolving cycle. Proc. Natl. Acad. Sci. U.S.A. 103, 7288–7291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. De Riso A., Jenson D. L., Barry B. A. (2006) Calcium exchange and structural changes during the photosynthetic oxygen evolving cycle. Biophys. J. 91, 1999–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Polander B. C., Barry B. A. (2012) A hydrogen-bonding network plays a catalytic role in photosynthetic oxygen evolution. Proc. Natl. Acad. Sci. U.S.A. 109, 6112–6117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Polander B. C., Barry B. A. (2013) Calcium and the hydrogen-bonded water network in the photosynthetic oxygen-evolving complex. J. Phys. Chem. Lett. 4, 786–791 [DOI] [PubMed] [Google Scholar]
- 72. Mishra R. K., Ghanotakis D. F. (1994) Selective extraction of CP 26 and CP 29 proteins without affecting the binding of the extrinsic proteins (33, 23 and 17 kDa) and the DCMU sensitivity of a photosystem II core complex. Photosyn. Res. 42, 37–42 [DOI] [PubMed] [Google Scholar]
- 73. Berthold D. A., Babcock G. T., Yocum C. F. (1981) A highly resolved, oxygen-evolving photosystem II preparation from spinach thylakoid membranes. FEBS Lett. 134, 231–234 [Google Scholar]
- 74. Ghanotakis D. F., Babcock G. T., Yocum C. F. (1985) On the role of water-soluble polypeptides (17, 23 kDa), calcium and chloride in photosynthetic oxygen evolution. FEBS Lett. 192, 1–33915890 [Google Scholar]
- 75. Miyao M., Murata M. (1984) Role of the 33-kDa polypeptide in preserving Mn in the photosynthetic oxygen-evolving system and its replacement by chloride ions. FEBS Lett. 170, 350–354 [Google Scholar]
- 76. Ono T., Inoue Y. (1984) Ca2+-dependent restoration of O2-evolving activity in CaCl2-washed PSII particles depleted of 33, 24, and 16 kDa proteins. FEBS Lett. 168, 281–286 [Google Scholar]
- 77. Hutchison R. S., Betts S. D., Yocum C. F., Barry B. A. (1998) Conformational changes in the extrinsic manganese stabilizing protein can occur upon binding to the photosystem II reaction center. An isotope editing and FT-IR study. Biochemistry 37, 5643–5653 [DOI] [PubMed] [Google Scholar]
- 78. Barry B. A. (1995) Tyrosyl radicals in photosystem II. Methods Enzymol. 258, 303–319 [DOI] [PubMed] [Google Scholar]
- 79. Bricker T. M. (1992) Oxygen evolution in the absence of the 33-kilodalton manganese-stabilizing protein. Biochemistry 31, 4623–4628 [DOI] [PubMed] [Google Scholar]
- 80. Betts S. D., Hachigian T. M., Pichersky E., Yocum C. F. (1994) Reconstitution of the spinach oxygen-evolving complex with recombinant Arabidopsis manganese-stabilizing protein. Plant Mol. Biol. 26, 117–130 [DOI] [PubMed] [Google Scholar]
- 81. Piccioni R., Bellemare G., Chua N. H. (1982) in Methods in Chloroplast Molecular Biology (Edlemann M., Hallick R. B., Chua N. H., eds) pp. 985–1014, Elsevier Biomedical, Amsterdam, The Netherlands [Google Scholar]
- 82. Noren G. H., Boerner R. J., Barry B. A. (1991) EPR characterization of an oxygen-evolving photosystem II preparation from the transformable cyanobacterium Synechocystis 6803. Biochemistry 30, 3943–3950 [DOI] [PubMed] [Google Scholar]
- 83. Miyao M., Murata M. (1984) Calcium ions can be substituted for the 24-kDa polypeptide in photosynthetic oxygen evolution. FEBS Lett. 168, 118–120 [Google Scholar]
- 84. Polander B. C., Barry B. A. (2013) Detection of an intermediary protonated water cluster in photosynthetic oxygen evolution. Proc. Natl. Acad. Sci. U.S.A. 110, 10634–10639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Thornton L. E., Ohkawa H., Roose J. L., Kashino Y., Keren N., Pakrasi H. B. (2004) Homologs of plant PsbP and PsbQ proteins are necessary for regulation of photosystem II activity in the cyanobacterium Synechocystis 6803. The Plant Cell 16, 2164–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bricker T. M., Frankel L. K. (1998) The structure and function of the 33 kDa extrinsic protein of photosystem II. A critical assessment. Photosynth. Res. 56, 157–173 [Google Scholar]
- 87. Seidler A. (1996) The extrinsic polypeptides of photosystem II. Biochim. Biophys. Acta 1277, 35–60 [DOI] [PubMed] [Google Scholar]
- 88. Surewicz W. K., Mantsch H. H., Chapman D. (1993) Determination of protein secondary structure by Fourier transform infrared spectroscopy. A critical assessment. Biochemistry 32, 389–394 [DOI] [PubMed] [Google Scholar]
- 89. Miller A.-F., De Paula J. C., Brudvig G. W. (1987) Formation of the S2 state and structure of the Mn complex in photosystem II lacking the extrinsic 33 kilodalton polypeptide. Photosynth. Res. 12, 205–218 [DOI] [PubMed] [Google Scholar]
- 90. Campbell K. A., Gregor W., Pham D. P., Peloquin J. M., Debus R. J., Britt R. D. (1998) The 23 and 17 kDa extrinsic proteins of photosystem II modulate the magnetic properties of the S1-state manganese cluster. Biochemistry 37, 5039–5045 [DOI] [PubMed] [Google Scholar]
- 91. Barth A. (2007) Infrared spectroscopy of proteins. Biochim. Biophys. Acta 1767, 1073–1101 [DOI] [PubMed] [Google Scholar]
- 92. Speranskiy K., Kurnikova M. (2004) Accurate theoretical prediction of vibrational frequencies in an inhomogeneous dynamic environment. A case study of a glutamate molecule in water solution and in a protein-bound form. J. Chem. Phys. 121, 1516–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Boehr D. D., McElheny D., Dyson H. J., Wright P. E. (2006) The dynamic energy landscape of dihydrofolate reductase catalysis. Science 313, 1638–1642 [DOI] [PubMed] [Google Scholar]
- 94. Masterson L. R., Shi L., Metcalfe E., Gao J., Taylor S. S., Veglia G. (2011) Dynamically committed, uncommitted, and quenched states encoded in protein kinase A revealed by NMR spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 108, 6969–6974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kapoor S., Triola G., Vetter I. R., Erlkamp M., Waldmann H., Winter R. (2012) Revealing conformational substates of lipidated N-Ras protein by pressure modulation. Proc. Natl. Acad. Sci. U.S.A. 109, 460–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bricker T. M., Frankel L. K. (2011) Auxiliary functions of the PsbO, PsbP, and PsbQ proteins of higher plant photosystem II. A critical analysis. J. Photochem. Photobiol. B 104, 165–178 [DOI] [PubMed] [Google Scholar]
- 97. Bricker T. M., Roose J. L., Zhang P., Frankel L. K. (2013) The PsbP family of proteins. Photosynth. Res., 10.1007/s11120-013-9820-7 [DOI] [PubMed] [Google Scholar]
- 98. Miyao M., Murata N., Lavorel J., Maison-Peteri B., Boussac A., Etienne A.-L. (1987) Effect of the 33-kDa protein on the S-state transitions in photosynthetic oxygen evolution. Biochim. Biophys. Acta 890, 151–159 [Google Scholar]
- 99. Burnap R. L., Shen J.-R., Jursinic P. A., Inoue Y., Sherman L. A. (1992) Oxygen yield and thermoluminiscence characteristics of a cyanobacterium lacking the manganese-stabilizing protein of photosystem II. Biochemistry 31, 7404–7410 [DOI] [PubMed] [Google Scholar]
- 100. Yi X., Hargett S. R., Frankel L. K., Bricker T. M. (2006) The PsbQ protein is required in Arabidopsis for photosystem II assembly/stability and photoautotrophy under low light conditions. J. Biol. Chem. 281, 26260–26267 [DOI] [PubMed] [Google Scholar]
- 101. Roose J. L., Frankel L. K., Bricker T. M. (2010) Documentation of significant electron transport defects on the reducing side of photosystem II upon removal of the PsbP and PsbQ extrinsic subunits. Biochemistry 49, 36–41 [DOI] [PubMed] [Google Scholar]
- 102. Svensson B., Tiede D. M., Nelson D. R., Barry B. A. (2004) Structural studies of the manganese stabilizing subunit in photosystem II. Biophys. J. 86, 1807–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Styring S., Miyao M., Rutherford A. W. (1987) Formation and flash-dependent oscillation of the S2-state multiline EPR signal in an oxygen-evolving photosystem-II preparation lacking the three extrinsic proteins in the oxygen-evolving system. Biochim. Biophys. Acta 890, 32–38 [Google Scholar]
- 104. Tomita M., Ifuku K., Sato F., Noguchi T. (2009) FTIR evidence that the PsbP extrinsic protein induces protein conformational changes around the oxygen-evolving Mn cluster in photosystem II. Biochemistry 48, 6318–6325 [DOI] [PubMed] [Google Scholar]
- 105. Ouellette A. J., Barry B. A. (2002) Tandem mass spectrometric identification of spinach photosystem II light-harvesting components. Photosyn. Res. 72, 159–173 [DOI] [PubMed] [Google Scholar]
- 106. Murray J. W., Barber J. (2007) Structural characteristics of channels and pathways in photosystem II including the identification of an oxygen channel. J. Struct. Biol. 159, 228–237 [DOI] [PubMed] [Google Scholar]
- 107. Freier E., Wolf S., Gerwert K. (2011) Proton transfer via a transient linear water-molecule chain in a membrane protein. Proc. Natl. Acad. Sci. U.S.A. 108, 11435–11439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Dau H., Zaharieva I., Haumann M. (2012) Recent developments in research on water oxidation by photosystem II. Curr. Opin. Chem. Biol. 16, 3–10 [DOI] [PubMed] [Google Scholar]
- 109. Bondar A.-N., Dau H. (2012) Extended protein/water H-bond networks in photosynthetic water oxidation. Biochim. Biophys. Acta 1817, 1177–1190 [DOI] [PubMed] [Google Scholar]
- 110. Shutova T., Klimov V. V., Andersson B., Samuelsson G. (2007) A cluster of carboxylic groups in PsbO protein is involved in proton transfer from the water oxidizing complex of photosystem II. Biochim. Biophys. Acta 1767, 434–440 [DOI] [PubMed] [Google Scholar]
- 111. Nagel Z. D., Cun S., Klinman J. P. (2013) Identification of a long-range protein network that modulates active site dynamics in extremophilic alcohol dehydrogenases. J. Biol. Chem. 288, 14087–14097 [DOI] [PMC free article] [PubMed] [Google Scholar]