

Background: FAD synthase is known to catalyze the biosynthesis of FAD in cytosol and mitochondria.
Results: The existence of a nuclear FAD synthase and a FAD-hydrolyzing activity is demonstrated.
Conclusion: A dynamic pool of FAD exists in the nucleus.
Significance: Nuclear, mitochondrial, and cytosolic FAD synthase pools constitute a flavin network involved in the regulation of cellular metabolism and epigenetic events.
Keywords: Confocal Microscopy, FAD, FMN, Nucleus, Rat, Riboflavin, FAD Synthase, FAD Hydrolysis, NUDIX Hydrolase
Abstract
FAD is a redox cofactor ensuring the activity of many flavoenzymes mainly located in mitochondria but also relevant for nuclear redox activities. The last enzyme in the metabolic pathway producing FAD is FAD synthase (EC 2.7.7.2), a protein known to be localized both in cytosol and in mitochondria. FAD degradation to riboflavin occurs via still poorly characterized enzymes, possibly belonging to the NUDIX hydrolase family. By confocal microscopy and immunoblotting experiments, we demonstrate here the existence of FAD synthase in the nucleus of different experimental rat models. HPLC experiments demonstrated that isolated rat liver nuclei contain ∼300 pmol of FAD·mg−1 protein, which was mainly protein-bound FAD. A mean FAD synthesis rate of 18.1 pmol·min−1·mg−1 protein was estimated by both HPLC and continuous coupled enzymatic spectrophotometric assays. Rat liver nuclei were also shown to be endowed with a FAD pyrophosphatase that hydrolyzes FAD with an optimum at alkaline pH and is significantly inhibited by adenylate-containing nucleotides. The coordinate activity of these FAD forming and degrading enzymes provides a potential mechanism by which a dynamic pool of flavin cofactor is created in the nucleus. These data, which significantly add to the biochemical comprehension of flavin metabolism and its subcellular compartmentation, may also provide the basis for a more detailed comprehension of the role of flavin homeostasis in biologically and clinically relevant epigenetic events.
Introduction
Riboflavin (Rf, vitamin B2)5 is the precursor of FMN and FAD, two cofactors that ensure the functionality of many enzymes, namely flavoenzymes, involved in fundamental biochemical processes, such as the oxidative metabolism of carbohydrates, amino acids, and fatty acids and the mitochondrial electron transport (1–3). A number of other regulatory processes crucial for cell life and death, among which are ROS production, antioxidant defense, protein folding, and chromatin remodeling, also depend on more than 100 different flavoproteins, 75% of which use FAD as cofactor. Although in the large majority of cases flavoenzymes are oxidoreductases, they can also catalyze non-redox reactions (i.e., transfer, synthesis, isomerization, and ligations) (for reviews see Refs. 4–7). Consistently, FAD-dependent enzyme deficiency and/or impairment of flavin homeostasis in humans and experimental animals have been linked to several diseases, such as cancer, cardiovascular diseases, anemia, abnormal fetal development, and neuromuscular and neurological disorders (for reviews see Ref. 8 and references therein).
Although bacteria, protists, fungi, plants, and some animals can synthesize Rf, mammals must obtain this water-soluble vitamin from food and, to a lesser extent, from intestinal microflora. Cells can take up Rf via specific translocators, three of which have been recently characterized in humans (9–12). Once in the cell, Rf is converted into the flavin cofactor FAD via the sequential action of riboflavin kinase (ATP:riboflavin 5′ phosphotransferase, EC 2.7.1.26), which transfers a phosphoryl group from ATP to Rf to form FMN, and FAD synthase, previously known as FAD synthetase (FADS; ATP:FMN adenylyltransferase, EC 2.7.7.2), the enzyme responsible for FMN adenylation to FAD. In mammals, riboflavin kinase and FADS are two distinct monofunctional enzymes that have been purified from rat tissues and biochemically characterized (13–15).
A controversial matter in FADS biochemistry is its subcellular localization. For many years it has been assumed that in eukaryotes FAD biosynthesis occurred only in the cytosol (13, 14, 16, 17). However, using cell fractionation procedures and enzymatic activity measurements, we have demonstrated the presence of FADS activity in mitochondria from rat liver (18, 19), Saccharomyces cerevisiae (20, 21) and Nicotiana tabacum Yellow Bright-2 (22). More recently, the existence of different natural FADS isoforms with distinct features regarding molecular mass and subcellular localization has been confirmed by biochemical and immunohistochemical approaches (23).
Our hypothesis was that different FADS isoforms with compartment-specific functions may exist in eukaryote stems from the cloning and functional characterization of two products of the human FADS gene (FLAD1), namely transcript variants 1 (hFADS1) and 2 (hFADS2) (24, 25). The two corresponding protein products were shown to differ for an extra sequence of 97 amino acids at the N terminus (present only in hFADS1) containing a putative mitochondrial targeting peptide, as predicted by bioinformatics. Accordingly, a subsequent paper conclusively demonstrated that, in humans, hFADS1 is mitochondrial, whereas hFADS2 is cytosolic (23).
Recently, two additional splicing variants for the FLAD1 gene have been deposited in the NCBI Entrez Gene database (gene identifier 80308), namely transcript variants 3 and 4, whose simultaneous presence with hFADS1 has been observed at the transcriptional level in the intestinal HT-29 cell line (8). The specific tissue distribution and subcellular localization of these isoforms are still uncharacterized and enigmatic. The possibility of other subcellular localizations, different from cytosol and mitochondria, has not been excluded by the available studies. In fact, in two recent papers, a plasma membrane localization in neuronal cells (26) and a nuclear localization, presumably associated to the nuclear flavoprotein lysine-specific demethylases in adipocytes (27), have been suggested for FADS.
In this paper, by confocal microscopy and immunoblotting approaches, we show for the first time the nuclear localization of FADS in different experimental rat models. The existence of FAD synthesizing and hydrolyzing activities involved in maintaining FAD homeostasis in isolated rat liver nuclei is also demonstrated.
EXPERIMENTAL PROCEDURES
Materials
All chemicals were of analytical or highest available grade and, unless otherwise stated, were obtained from Sigma-Aldrich. PVDF Hybond-P was from Amersham Biosciences GE Healthcare. The dye reagent for protein assay was from Bio-Rad. Anti-OxPhos Complex II 70-kDa subunit mouse monoclonal antibody (anti-succinate dehydrogenase) and Alexa Fluor-conjugated anti-rabbit or anti-mouse IgG secondary antibodies were from Molecular Probes, Inc. Mouse anti-Hsp60 monoclonal antibody was from Stressgen. Monoclonal mouse anti-β-actin and anti-lamin A/C antibodies were from Abcam. Mouse anti-tubulin monoclonal antibody was from Sigma-Aldrich. Peroxidase-conjugated anti-rabbit and anti-mouse IgG secondary antibodies were from Thermo Scientific. Solvents and salts used for HPLC were from J. T. Baker.
Cell Culture
BHK-21 cells were purchased from ATCC and cultured as described in Refs. 28 and 29. INS-1E cells, a clonal β-cell line derived from rat insulinoma (a generous gift from Prof. C. Wollheim, University of Genève), were grown in complete medium as described in Ref. 28. Cardiac myocytes and fibroblasts were prepared from ventricles of neonatal Wistar rats (0–2 days after birth) essentially as in Refs. 30 and 31. Astrocytes were prepared from primary cell cultures of neocortical tissues as described in Ref. 32. The cells were maintained in a humidified incubator at 37 °C in the presence of 5% CO2 until used.
Cell Lysate Preparation
Confluent cells seeded on plastic 6-well plates were washed twice with ice-cold PBS and harvested with lysis buffer (150 mm NaCl, 5 mm EDTA, 5 mm EGTA, 50 mm HEPES, 1% Triton X-100, 10 mm β-mercaptoethanol, 0.1 mm PMSF, 2 μg/ml aprotinin/leupeptin/pepstatin). Lysates were frozen at −20 °C for at least 2 h and thereafter thawed and centrifuged at 16,000 × g for 15 min. Pellet (Triton X-100-insoluble proteins) and supernatant (Triton X-100-soluble proteins) fractions were resuspended in loading buffer (50 mm Tris-HCl, 2% SDS, 10% glycerol, 100 mm DTT).
Cell Fractionation
Subcellular fractionation of neonatal rat cardiac fibroblasts was performed essentially as described in Ref. 33. Briefly, confluent 150-mm dishes were washed twice with ice-cold PBS and thereafter lysed with 2 ml of Buffer A (10 mm HEPES, pH 7.9, 10 mm KCl, 0.1 mm EDTA, 0.1 mm EGTA, 1 mm DTT, 0.5 mm PMSF, 2 μg/ml Aprotinin/Leupeptin/Pepstatin). The cell homogenate was then passed six times through a 30-gauge needle and centrifuged at 700 × g for 10 min to obtain the crude nuclear fraction. The crude nuclear pellet was resuspended in 200 μl of Buffer C (20 mm HEPES, pH 7.9, 0.4 mm KCl, 1 mm EDTA, 1 mm EGTA, 1 mm DTT) and left for 30 min on a rotating wheel. Soluble nuclear proteins were then separated from DNA and debris by centrifugation at 16,000 × g for 30 min. Cytosolic proteins were obtained by centrifugating the first cell homogenate supernatant at 21,000 × g for 2 h. The two subcellular protein fractions were finally resuspended in loading buffer (see above) and subjected to immunoblotting.
Preparation of Pure Nuclei from Rat Liver
Nuclei were isolated from the liver of male Wistar rats by differential centrifugation in sucrose gradient essentially as in Ref. 34. Briefly, rat liver was homogenized in ice-cold medium A (0.25 m sucrose, 3 mm CaCl2, 1 mm PMSF) through a Potter-Elvehjem homogenizer and centrifuged at 700 × g for 6 min at 4 °C. The pellet was then resuspended and manually homogenized in ice-cold medium B (0.34 m sucrose, 3 mm CaCl2, 1 mm PMSF) in a 1:4 ratio and centrifuged at 700 × g for 10 min at 4 °C. The obtained pellet was resuspended and manually homogenized in ice-cold medium B in a 1:3 ratio, and an equal volume of medium A was gently stratified under it. After centrifuging at 1500 × g for 15 min at 4 °C, the crude nuclei pellet was resuspended and manually homogenized in ice-cold medium C (2.2 m sucrose, 3 mm CaCl2, 1 mm PMSF) in a 1:3 ratio and then ultracentrifuged at 45,000 × g for 1 h at 4 °C. The white pellet containing the isolated nuclei, on the bottom of the tube, was finally resuspended in ice-cold Tris-Cl buffer (20 mm Tris-Cl, pH 7.5, 0.5 mm PMSF). Where indicated, isolated nuclei were solubilized by treatment with the non-ionic detergent Triton X-100 (0.1%) and subjected to immunoblotting or ultrafiltered by centrifugal filters (Microcon YM-10, Millipore) and utilized for the HPLC and enzymatic assays.
Immunoblotting
SDS-PAGE separated proteins were electrotransferred onto a PVDF membrane using a transblot semidry electrophoretic transfer cell (Sigma-Aldrich). The immobilized proteins were incubated overnight with a 3000-fold dilution of a polyclonal antiserum against hFADS, as in Ref. 23. Other antibodies were used to reveal and quantify protein markers, including a mouse monoclonal anti-β-actin antibody (1:10,000 dilution), a mouse monoclonal anti-tubulin antibody (1:2,000 dilution), a mouse monoclonal anti-lamin A/C antibody (1:500 dilution), a mouse monoclonal anti-succinate dehydrogenase antibody (1:2,000 dilution), and a rabbit polyclonal anti-rat dimethylglycine dehydrogenase antibody (1:1,000 dilution). The bound antibodies were visualized with the aid of secondary anti-rabbit or anti-mouse IgG antibodies conjugated with peroxidase (1:3,500 dilution). Quantitative evaluation of immunoreactive protein bands was carried by densitometric analysis using the ImageQuant 5.2 software (Molecular Dynamics).
Cell Immunofluorescence and Confocal Microscopy
Immunofluorescence experiments were performed essentially as described in Ref. 35. Briefly, cells seeded on glass coverslips at the desired confluence were fixed with 3.7% paraformaldehyde/PBS for 20 min and washed three times with PBS. After permeabilization with 0.1% Triton X-100 and blocking with 0.1% gelatin in PBS, the cells were incubated with the specific anti-FADS antiserum raised in rabbit (23) (1:300 dilution) or coincubated with a monoclonal anti-Hsp60 antibody (1:100 dilution) overnight at 4 °C. Coverslips were then washed three times with 0.1% gelatin in PBS and incubated at room temperature for 1 h with Alexa Fluor 488 or 568 secondary antibodies (1:1000 dilution). After one wash with PBS, nuclei were counterstained with Hoechst 33658 (2 μm). The coverslips were finally washed three times with ice-cold PBS, rapidly rinsed in distilled water, and then mounted on microscope slides with Mowiol® 4-88 mounting medium. Confocal images were captured with a Leica TCS SP5 confocal microscope (Leica Microsystems, Mannheim, Germany) using 40× (N.A. = 1.25) or 63× (N.A. = 1.32) oil immersion objectives, a 100-milliwatt argon laser (488 and 543 nm lines) and a 50-milliwatt diode (405 nm) as in Ref. 36. Confocal images were analyzed using the software Image J (37).
Quantitation of Flavins
Rf, FMN, and FAD content was measured in neutralized perchloric extracts by means of HPLC, essentially as described in Refs. 18–20, 22, and 38. Separation was achieved by means of a Gilson HPLC system including a model 306 pump and a model 307 pump equipped with a FP-2020 Plus Jasco fluorescence detector and a Unipoint system software. Quantitative determination of Rf, FMN, and FAD was carried out using a calibration curve made in each experiment by using standard solutions diluted in the extraction solution.
Measurements of FAD Synthesis Rate
Detergent-solubilized or ultrafiltered nuclei (0.1–0.2 mg) were preincubated at 37 °C in 100 μl of a medium consisting of 50 mm Tris-HCl (pH 7.5), 10 μm FMN, and 5 mm ATP; where indicated, either 5 mm MgCl2 or 1 mm EDTA was added. At the appropriate time, the reaction was stopped either by treatment with perchloric acid followed by neutralization or by boiling at 100 °C for 3 min followed by centrifugation at 14,000 × g for 4 min at 4 °C. In the former case, Rf, FMN, and FAD content was analyzed using HPLC (see above). In the latter case, newly synthesized FAD was measured spectrophotometrically by using the FAD-detecting system, as described in Refs. 18 and 20. Briefly, the amount of synthesized FAD was determined by revealing the reconstituted holo-d-amino acid oxidase activity derived from FAD binding to the apo-d-amino acid oxidase, using 25 mm d-alanine as substrate. NADH oxidation in the l-lactate dehydrogenase (LDH)-coupled reaction was followed spectrophotometrically at 340 nm, and the rate was calculated by measuring the slope of the tangent to the initial part of the experimental curve. This rate was proven to be proportional to FAD concentration. Calibration curves were obtained by using standard FAD solutions, and corrections were also made to account for the inhibition caused by FMN and ATP added to the reconstitution assay.
Measurements of FAD Hydrolysis Rate
Metabolism of externally added FAD was monitored in detergent solubilized rat liver nuclei by means of fluorimetric and HPLC measurements, essentially as in Refs. 38 and 39. In the case of fluorimetric measurements, flavin derivative emission spectra (excitation wavelength at 450 nm) and time drive measurements (excitation and emission wavelengths at 450 and 520 nm, respectively) were carried out at 37 °C in 50 mm Tris-HCl (pH 7.5), 5 mm MgCl2 by means of a LS50B PerkinElmer Life Sciences spectrofluorimeter. Flavin fluorescence emission spectra were corrected as in Ref. 40 by adding a few crystals of sodium dithionite to the nuclei suspension. When FAD hydrolysis was measured, the endogenous flavin fluorescence was not considered because it was found to be negligible when compared with that externally added to the subcellular suspension. In each experiment, FAD, FMN, and Rf fluorescence values were calibrated using standard flavin solutions. Their concentrations were spectrophotometrically calculated by using the ϵ450 values of 12.2 mm−1·cm−1 for FMN and Rf, and 11.3 mm−1·cm−1 for FAD. Under the experimental conditions used here, the FAD fluorescence constant (KFAD) was proven to be ∼10 times lower than those of FMN and Rf (KFMN/Rf), which did not differ significantly from each other (19, 41). Thus, the rate of FAD hydrolysis, i.e., the rate of FMN + Rf formation, expressed as nmol of FAD hydrolyzed·min−1·mg−1 protein and calculated from the rate of fluorescence intensity increase, was measured as the slope of the tangent to the initial part of the experimental curve by applying the following equation,
where ΔF is expressed in fluorescence arbitrary units, Vf is expressed in ml, ΔK = KFMN/Rf − KFAD is expressed as μm−1, Δt is expressed in min, and m is the mass of proteins in mg.
HPLC measurements were carried out on neutralized perchloric extracts of aliquots of the subcellular suspension (100 μl) taken at various times (see above). The kinetic parameters were calculated with the Grafit software (version 3.00; Erithacus Software), as in Refs. 22 and 38.
Other Assays
Protein concentration was assayed according to Bradford (42), using bovine serum albumin as standard. Lactate dehydrogenase (EC 1.1.1.27) activity was spectrophotometrically measured at 25 °C in 50 mm Tris-Cl (pH 7.5) by following the decrease in absorbance at 340 nm caused by NADH oxidation (ϵ340 = 6.2 mm−1 × cm−1). The reaction was started by adding rat homogenate or isolated nuclei (0.05–0.1 mg of protein) to the reaction mixture containing 1 mm pyruvate, 2.5 mg/ml rotenone, and 0.1 mm NADH, essentially as in Ref. 43.
Nicotinamide-mononucleotide adenylyltransferase activity (EC 2.7.7.1) was continuously revealed by means of a spectrofluorimetric enzymatic analysis carried out essentially as in Ref. 44. Rat homogenate or isolated nuclei (0.6 mg of protein each) were added to 50 mm Tris-HCl (pH 7.6) supplemented with 20 mm MgCl2 in the presence of the NAD+ detecting system consisting of 0.35% ethanol, 5 IU alcohol dehydrogenase (EC 1.1.1.1), and 35 mm semicarbazide. The reaction was started by adding 1 mm nicotinamide mononucleotide and 5 mm ATP. Under the experimental conditions described here, newly synthesized NAD+ was immediately reduced to NADH. The rate of NADH appearance, which reflects the rate of NAD+ synthesis, was fluorimetrically measured by means of a LS50B PerkinElmer Life Sciences spectrofluorimeter, with excitation and emission wavelengths set at 340 and 456 nm, respectively. The rate of fluorescence increase was obtained by measuring the slope of the tangent to the initial part of the experimental curve and expressed as nmol of NAD+ synthesized·min−1·mg−1 protein. In each experiment, the rate of NAD+ synthesis in the homogenates and nuclear samples was calibrated by adding a standard solution of NAD+ to the NAD+ detecting system.
RESULTS
Evidence for a Nuclear FADS
During a previous work (23) aimed at assessing the subcellular localization of the human isoforms hFADS1 and hFADS2, confocal microscopy analysis performed on BHK-21 cells subjected to overexpression experiments revealed an unexpected nuclear localization of the natural protein in most of the nontransfected cells (see Fig. 6 in Ref. 23). This observation was next confirmed by immunofluorescence experiments performed on the same cell line in control conditions (Fig. 1, A–C). Colocalization analysis performed with National Institutes of Health ImageJ software clearly showed a nuclear localization of FADS (white pixels in the merged images depicted in Fig. 1C). In parallel, to gain insight into the apparent molecular mass of the natural isoforms of FADS cross-reacting with the anti-FADS antiserum produced in our laboratory (23), immunoblotting analyses were carried out on BHK-21 cell lysates (Fig. 1D) obtained in the presence of Triton X-100 (1%).
FIGURE 6.

Chromatographic and enzymatic evidence of nuclear FAD synthesis. A, upper panel, typical HPLC chromatogram of neutralized perchloric acid extracts of isolated nuclei from rat liver showing flavin content (see “Experimental Procedures”). Lower panel, time course of FAD synthesis catalyzed by rat liver nuclei added to 2 μm FMN, 5 mm ATP, and 5 mm MgCl2 in 50 mm Tris-Cl (pH 7.5) at 37 °C, in the absence (●) or in the presence (○) of 50 μm HgCl2. B, the amount of FAD in ultrafiltered rat liver nuclei was enzymatically assayed by using the FAD detecting system (FADS D.S.), here schematized and described in greater detail under “Experimental Procedures.” Ultrafiltered nuclei (0.06 mg of protein) were incubated at 37 °C for 5 min in 100 μl of 50 mm Tris-HCl (pH 7.5) in the absence (dotted line, −FMN & ATP) or in the presence of the substrate pair 2 μm FMN and 1 mm ATP (straight lines). The effect of 5 mm MgCl2 on FAD synthesis was also evaluated.
FIGURE 1.
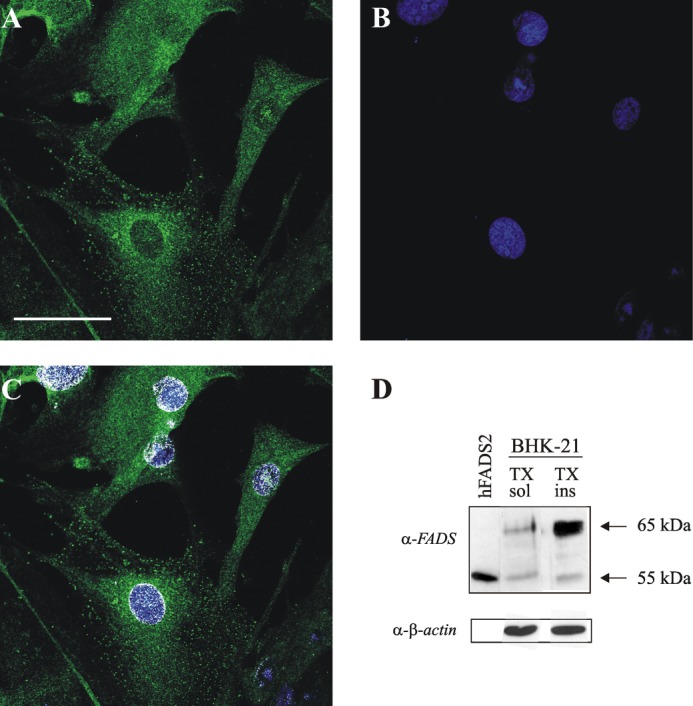
Fluorescence microscopy and immunoblotting evidence of FADS localization in BHK-21 cell nuclei. A, fixed and permeabilized BHK-21 cells were incubated with the polyclonal anti-FADS antiserum followed by incubation with an Alexa Fluor 488-conjugated anti-mouse antibody (green). B, nuclei were stained with Hoechst 33658 (blue, B). C, colocalization of FADS with the nuclear marker is depicted in white. Scale bar, 50 μm. D, immunoblotting of different anti-FADS-immunoreactive bands in BHK-21 cells. BHK-21 cells were lysed using 1% Triton X-100 as described under “Experimental Procedures.” Both the Triton X-100-soluble (TX sol) and Triton X-100-insoluble (TX ins) fractions were analyzed by immunoblotting with the anti-FADS antiserum. His6-hFADS2 (0.03 μg) was also loaded (hFADS2, first lane) as a control. The same PVDF membrane was tested with the anti-β-actin antibody. The arrows indicate the position of the main anti-FADS-immunoreactive bands.
In these experimental conditions, a major band migrating at ∼65 kDa accompanied by a 55-kDa immunoreactive band was evident, whereas other less abundant protein bands could also be observed. As visible from the representative blot reported in Fig. 1D, these bands were observed both in the soluble (supernatant) and insoluble (pellet, presumably DNA-containing) fractions of cell lysates. Notwithstanding the apparent enrichment of the 65-kDa band in the Triton X-100-insoluble pellet, if compared with the β-actin band (more than 75% released in the soluble fraction), the majority of FADS bands was shown to be released in the supernatant (5 ng/100 mg of protein), whereas its total amount in the pellet was less than 10%. Whether the different immunoreactive bands correspond to post-translationally modified forms or to different isoforms of the enzyme is currently unknown. FADS subcellular localization was next explored by confocal fluorescence microscopy on primary cultures of neonatal rat ventricular cardiomyocytes, a cell type in which the oxidative metabolism is expected to crucially depend on flavin cofactor supply (Fig. 2).
FIGURE 2.

Confocal evidence of mitochondrial and nuclear localization of FADS in primary cultures of neonatal rat ventricular cardiomyocytes. A, fixed and permeabilized neonatal rat cardiomyocytes were incubated with the polyclonal anti-FADS antiserum followed by incubation with an Alexa Fluor 568-conjugated anti-rabbit antibody (red). B, mitochondria were revealed with the monoclonal anti-Hsp60 antibody and an Alexa Fluor 488-conjugated anti-mouse antibody (green). C, nuclei were counterstained with Hoechst 33658 (blue). D and E, the colocalization of FADS with mitochondria (D) and nuclei (E) is depicted in white. Scale bar, 50 μm.
After fixation, permeabilization, and incubation with the anti-FADS-specific antiserum, the immunocomplexes were visualized with a secondary antibody conjugated with Alexa Fluor 568 (Fig. 2A). Mitochondria were marked by using a monoclonal antibody against the mitochondrial chaperonin Hsp60 followed by a secondary antibody conjugated with Alexa Fluor 488 (Fig. 2B). After nuclei counterstaining with Hoechst 33568 (Fig. 2C), the coverslips were analyzed by confocal fluorescence microscopy. Colocalization analysis clearly showed both a mitochondrial and a nuclear localization of FADS (white pixels in the merged images depicted in Fig. 2, D and E). The nuclear localization of FADS was further confirmed by confocal microscopy experiments performed on primary cultures of rat astrocytes (Fig. 3A) and insulin secreting INS1-E β cells (Fig. 3B), a model known for being particularly rich in FAD cofactors (45).
FIGURE 3.

Confocal evidence of a nuclear FADS localization in rat primary culture and cell lines. Fixed and permeabilized rat neonatal astrocytes (row A) and insulin secreting INS-1E β-cells (row B) were incubated with the polyclonal anti-FADS antiserum followed by incubation with an Alexa Fluor 568-conjugated anti-rabbit antibody (red). Nuclei were stained with Hoechst 33658 (blue). In the last panel of each row, the colocalization between FADS and the nuclear marker is depicted in white. Scale bar, 50 μm.
Also in these cell models, the distribution ratio of FADS between the pellet (insoluble fraction) and the supernatant (soluble fraction), as analyzed by immunoblotting, confirmed an enrichment of α-FADS cross-reactive bands (at 65 and 55 kDa) in the pellet (data not shown). During our survey of different rat cell models aimed at assessing the subcellular localization of FADS, a marked nuclear localization was observed in primary cultures of neonatal rat ventricular fibroblasts (Fig. 4, A–C).
FIGURE 4.
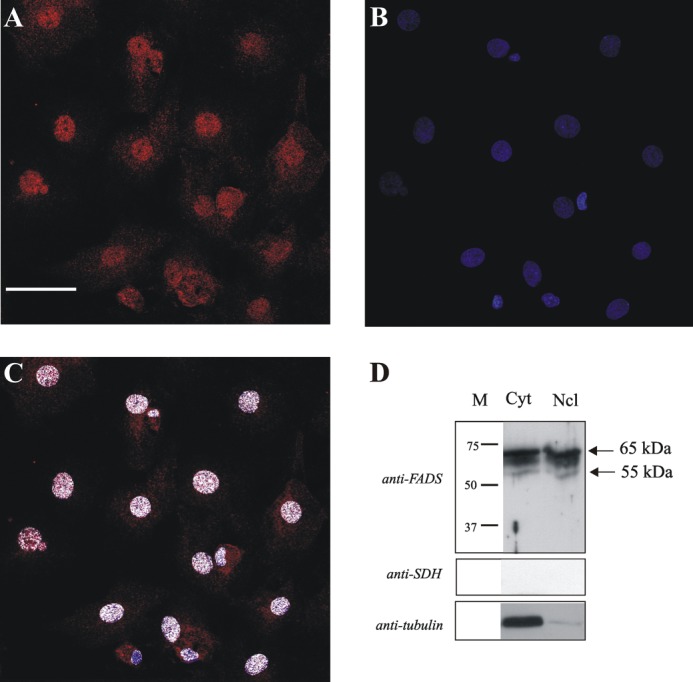
Confocal microscopy and immunoblotting evidence of FADS localization in the nucleus of neonatal rat ventricular fibroblasts. A, fixed and permeabilized neonatal rat ventricular fibroblasts were incubated with the polyclonal anti-FADS antiserum followed by incubation with an Alexa Fluor 568-conjugated anti-rabbit antibody (red). B, nuclei were stained with Hoechst 33658 (blue). C, the colocalizion of FADS with Hoechst is depicted in white. Scale bar, 50 μm. Subcellular fractions of cardiac fibroblasts were obtained as described under “Experimental Procedures.” D, The cytosolic (Cyt) and nuclear (Ncl) fractions were analyzed by immunoblotting with the anti-FADS antiserum. After stripping, the same PVDF membrane was tested with anti-tubulin and anti-succinate dehydrogenase antibodies (anti-SDH), used as cytosolic and mitochondrial markers, respectively. The arrows indicate the position of the main anti-FADS-immunoreactive bands.
Given the strong nuclear signal and the possibility of obtaining more material at a lower cost in respect to cardiac myocytes, we elected to perform a subcellular fractionation on this cell model. After the simplified subfractionation procedure described under “Experimental Procedures,” the cytosolic and the nuclear fractions were analyzed by immunoblotting.
Loading equal amounts of each fraction, again different anti-FADS cross-reactive bands were revealed (Fig. 4D). The migrating range was found to be 65–55 kDa in both the fractions. The absence of mitochondrial contamination in the nuclear fraction was assessed by following the distribution of the specific marker enzyme (succinate dehydrogenase flavoprotein subunit). Only a minimal cytosolic protein contamination was observed by using tubulin as cytosolic marker (Fig. 4D).
Pure Nuclei Isolated from Rat Liver Show a FAD Synthesizing Activity
To investigate the putative physiological function of the nuclear localization of FADS and thus detect its enzymatic activity, we performed an alternative subcellular fractionation method starting from the abundant source represented by the rat liver, a tissue expected to require high levels of FAD because of its central role in metabolism. Thus, functionally active nuclei were isolated using a procedure based on differential centrifugation of liver homogenates in sucrose gradient (see “Experimental Procedures” for further details). The purity and functionality of the nuclear fractions were checked by following (i) the increase in the enzymatic activity of the nuclear enzyme nicotinamide-mononucleotide adenylyltransferase, a central player in the biosynthesis of NAD+ (46), and (ii) the decrease of the enzymatic activity of LDH, a marker enzyme of the cytosolic compartment (Fig. 5A). Immunoblotting experiments performed in parallel confirmed the purity of the nuclear fractions as indicated by the presence of lamin A/C (a nuclear marker), the lack of tubulin (a cytosolic marker), and the loss of dimethylglycine dehydrogenase (a mitochondrial marker) (Fig. 5B).
FIGURE 5.

Nuclear localization of FADS in rat liver. Pure nuclei (pNcl) were isolated from rat liver homogenate (Homo) as described under “Experimental Procedures.” A, distribution of the enzymatic activities of the cytosolic marker LDH and the nuclear marker nicotinamide-mononucleotide adenylyltransferase (NMNAT) between homogenate and nuclei. B, immunoblotting of rat liver homogenate and pure nuclei (0.03 mg protein each). The same PVDF membrane tested with the anti-FADS antiserum was tested with anti-tubulin, anti-lamin A/C, and anti-dimethylglycine dehydrogenase (DMGDH) antibodies.
Loading equal amounts (30 μg) of each fraction, different anti-FADS cross-reactive bands were found (Fig. 5B), with a clear enrichment of the 65-kDa migrating band in the nuclear fraction. An increase in the intensity of lower migrating immunoreactive bands (range 65–56 kDa) was also appreciable. Another faint band was visualized at ∼38 kDa, both in the homogenate and in the nuclear fractions. The specificity of all these bands was assessed by immunoinhibition assay (data not shown) carried out using the recombinant His6-hFADS2 as a competitor, essentially as in Ref. 23. Remarkably, the 38-kDa band was not detectable when the overall purification procedure was performed in the absence of CaCl2, suggesting that it could be an hydrolysis product induced by calcium activated proteases.
In the aim to further estimate the functional role of FADS in intact nuclei, we next explored nuclear FAD synthesis by both HPLC analyses and enzymatic assays. Thus, as first step, we revealed nuclear acid-extractable flavins by HPLC (Fig. 6A). In experiments performed with different preparations, the mean content levels of FAD, FMN, and Rf (that linearly depended on the protein amount; not shown) were 276 ± 25, 128 ± 31, and 71 ± 9 pmol·mg−1 protein, respectively. To understand whether the endogenous FAD pools found in the nuclear extracts were free or protein-bound, nuclei were ultrafiltered prior to perchloric acid extraction and HPLC analysis (data not shown). This procedure clearly revealed that the main acid-extractable nuclear flavins, i.e., FAD (∼65% of total flavins), is almost completely nonultrafilterable and thus presumably bound to apo-proteins. The less abundant pool of FAD precursors/hydrolysis products, i.e., FMN and Rf (26 and 9% of total flavins, respectively), were easy filterable (data not shown).
Next, we performed an enzymatic assay by incubating isolated pure nuclei with the FADS substrate pair (2 μm FMN and 5 mm ATP) at 37 °C for up to 10 min. A clear time-dependent increase of FAD concentration was observed (Fig. 6A), directly confirming the existence of a nuclear FADS activity. The initial rate for FAD synthesis, as measured in these experimental conditions, was of 12.4 pmol·min−1·mg−1 protein. Importantly, a strong inhibitory effect of HgCl2 (50 μm) on natural FADS activity was demonstrated, as previously observed in experiments performed with the recombinant protein (47) and in agreement with the high number of cysteines present in the putative isoforms of rat enzyme (see below).
To further confirm FAD synthesis by isolated nuclei, a coupled enzymatic assay was performed. Thus, the ability of newly synthesized FAD to continuously reactivate the apo-enzyme of d-amino acid oxidase and hence to induce d-alanine oxidation was measured spectrophotometrically at 340 nm as the rate of NADH oxidation in the LDH-coupled reaction (Fig. 6B). No FAD synthesis was measurable in the cuvette containing the ultrafiltered nuclear fraction in the absence of substrates (dotted line). The rate of absorbance decrease (0.008 ΔA·min−1) measured under these experimental conditions was, in fact, equal to the intercept on the y axis of the calibration curve (data not shown), i.e., to the residual holo-d-amino acid oxidase present in the apo-protein preparation. Also, no FAD was detectable when the substrates were added in the absence of MgCl2 (−MgCl2), in line with the existence of no free (but only protein-bound) FAD in the ultrafiltered nuclear fraction. In the presence of MgCl2 (+MgCl2), an essential cofactor of FADS (13, 24), FAD synthesis was found to occur at a rate of 0.014 ΔA·min−1, corresponding to 15 pmol of newly synthesized FAD·min−1·mg−1 protein. A value of 18.1 ± 4.8 pmol·min−1·mg−1 protein was measured in two different nuclear preparations, in good agreement with the HPLC analysis.
Evidence for a FAD Hydrolyzing Capacity of Rat Liver Nuclei
Thus, both the HPLC and the enzymatic assays performed demonstrated a robust nuclear FAD synthesis, suggesting, as previously described for NAD+ (46), a role for nuclear FADS in the biogenesis of the nuclear flavoproteins, among which there are lysine-specific demethylases (27).
In this perspective, we reasoned that one of the mechanisms potentially involved in the regulation of the nuclear flavin cofactor pool could have been the existence of a dynamic equilibrium between synthesis and hydrolysis of FAD. Thus, a last series of experiments aimed at assessing whether or not isolated nuclei can hydrolyze exogenously added FAD was performed.
Upon FAD (5 μm) addition to purified nuclei, flavin fluorescence emission spectra (excitation wavelength, 450 nm) were recorded for up to 250 min at different time intervals in the 500–600 nm wavelength range. Fluorescence emission at 520 nm (Fig. 7A), the wavelength at which all the flavins show their emission maximum, linearly increased with time for up to 100 min (Fig. 7A, inset). Because FMN and Rf fluorescence constants are ∼10-fold higher than that of FAD (see “Experimental Procedures”), the results reported in Fig. 7A strongly suggested that FAD to FMN/Rf conversion could have taken place. By measuring the rate of fluorescence increase at 520 nm in different nuclear preparations, a mean rate of 1.15 ± 0.14 nmol of FAD hydrolyzed·min−1·mg−1 protein was found. No increase in fluorescence intensity at 520 nm was measured when 5 mm EDTA was added (Fig. 7A, inset).
FIGURE 7.

FAD hydrolysis by isolated rat liver nuclei. A, rat liver nuclei (0.1 mg of protein) were incubated for 1 min as described under “Experimental Procedures,” and then 5 μm FAD was added. Flavin fluorescence emission spectra were monitored at different incubation times. In the inset, the fluorescence measured at 520 nm, as obtained either in the absence (●) or presence (○) of 5 mm EDTA, was plotted against time. B, chromatographic evidence of FAD hydrolysis catalyzed by pure nuclei. FAD, FMN, and Rf were revealed by HPLC in nuclear extracts obtained after 0 min (chromatograms a and a′), 4 min (chromatograms b and b′), and 120 min (chromatograms c and c′) of incubation with FAD, either in the absence (chromatograms a–c) or in the presence (chromatograms a′–c′) of 5 mm EDTA. C, some features of FAD hydrolysis are reported. The rate of FAD hydrolysis by pure nuclei (0.4 mg of proteins) was measured fluorimetrically. In chromatogram a, the pH profile of the rate of FAD (5 μm) hydrolysis is shown. 100 mm acetate/acetic acid and 50 mm Tris-HCl (with 5 mm MgCl2) at different pH values were used as buffering mixtures. A specific calibration curve was obtained at each pH value as described under “Experimental Procedures.” The values are reported as percentages of the maximum rate (6.3 nmol·min−1·mg−1 of protein) arbitrarily set equal to 100%. The inhibition by externally added GTP, ATP, and AMP (chromatogram b) or NADH and NAD+ (chromatogram c) on the rate of FAD (0.6 μm) hydrolysis is shown. The data shown in chromatograms b and c were fitted according to a single exponential decay equation using the Grafit software (version 3.00; Erithacus Software).
To further confirm FAD hydrolyzing activity in rat nuclei, FMN/Rf formation from FAD was assayed by HPLC with a similar experimental approach. Thus, exogenously added FAD was incubated with isolated nuclei either in the absence or in the presence of EDTA, and neutralized perchloric extracts of the reaction mixtures at different time intervals were analyzed by HPLC. As clearly shown by the typical chromatogram reported in the upper panel of Fig. 7B, the addition of FAD caused the appearance of newly formed FMN and Rf, whose amounts were found to increase over time, with a simultaneous decrease in FAD levels (chromatograms a–c). The rate of FAD to FMN hydrolysis was comparable with that measured by the fluorimetric assay (1.4 nmol·min−1·mg−1 of protein), whereas the rate of Rf formation was slower (0.15 nmol·min−1·mg−1 of protein). No FMN or Rf formation was found to occur in the suspension previously added with EDTA (chromatograms a′–c′).
These results show that nuclei can metabolize externally added FAD to give FMN, via a putative FAD pyrophosphatase, and more slowly Rf, via monoester hydrolases that are still not characterized. The kinetic behavior of the reaction and the correspondence between the two methods used to detect FAD hydrolysis demonstrate that the fluorimetric approach used here (more rapid and less expensive than the classical HPLC approaches) is a suitable model for the characterization of nuclear FAD pyrophosphatase.
FAD hydrolysis was then characterized with respect to the following features: dependence on substrate concentration (data not shown), sensitivity toward inhibitors, and pH profile (Fig. 7C). Hyperbolic characteristics were found with saturation kinetics as analyzed by the Michaelis and Menten equation. Thus, a Km equal to 0.44 ± 0.03 μm and a Vmax equal to 1.3 nmol·min−1·mg−1 of protein for FAD were calculated.
As far as the inhibition of FAD hydrolysis is concerned, a significant result of this part of the study is the inhibition found by purine nucleotides. In particular, ATP-induced inhibition of the process was found to be stronger than that obtained with GTP (being IC50 equal to 1.7 and 5.5 μm, respectively; see Fig. 7C). Interestingly, AMP (IC50 equal to 0.7 μm) and adenylate containing pyridine nucleotides, such as NADH and NAD+, powerfully inhibited FAD hydrolysis, with IC50 values of 0.6 and 3 μm, respectively. Among inorganic compounds, NaF was found to reduce by 50% the FAD hydrolysis reaction rate at 1 mm; at higher concentrations, NaF strongly inhibited the reaction. No inhibition was found by tartrate (used at concentrations up to 1 mm), a powerful inhibitor of the lysosomal acid phosphatase (48).
Concerning the pH profile, the rate of FAD hydrolysis was found to be quite slow in the pH range 4–7 and very fast at alkaline pH, with a maximum value at pH 9 (Fig. 7C). The marked inhibition exerted by the reduced nicotinamide nucleotide, by other dinucleotides (i.e., CoA and P1,P5-di-adenosine-5′-pentaphosphate), and by the inorganic compound NaF, together with the optimal activity registered at alkaline pH, strongly suggests that the hydrolyzing activity measured in pure nuclei could be due to one of the already characterized NUDIX hydrolases, prominent among the enzymes responsible for metabolizing nucleotides (49).
DISCUSSION
This paper deals with the role played by flavin cofactor metabolism and its compartmentalization in different subcellular domains. In opposition to pyridine nucleotide cofactors, whose metabolism has been extensively investigated in the last decades (50–52), the crucial function of flavin cofactors in cell biology has been quite neglected.
A piece of work in this direction has been made in our laboratory that has focused the research activity on the study of FAD biosynthesis and its subcellular localization in different organisms, including mammals (see Ref. 8 and references therein). Overcoming the assumption of an exclusive cytosolic localization for the enzymes responsible for FAD synthesis sustained by many laboratories (13, 14, 16, 17), we first proposed a mitochondrial localization for FADS both in rat liver and in mammalian cell lines (BHK-21 and Caco-2) transiently expressing hFADS1 (18, 19, 23, 53).
In this paper, we demonstrate for the first time a novel subcellular localization for this enzyme in different experimental rat models. In particular, we prove here the presence of FADS protein in the nuclei of primary cells and cell lines by classical immuno-based techniques and its functionality in intact nuclei from rat liver by biochemical assays.
The first direct demonstration of different FADS localization was obtained by confocal immunofluorescence experiments. The images reported in Fig. 2 clearly indicate that in cells in which the oxidative metabolism is expected to be crucial, i.e., neonatal rat ventricular cardiomyocytes, FADS localizes not only in the cytosol but also in mitochondria and nuclei, as already suspected when observing the corresponding immunofluorescence results obtained on BHK-21 fibroblasts (see nontransfected cells in Fig. 6 of Ref. 23 and Fig. 1 of this paper). The nuclear localization of FADS was further confirmed in different rat primary cultures (cardiac fibroblasts and astrocytes) and cell lines (insulin secreting β cells, INS1-E) subjected to the same immunofluorescence analysis (Figs. 3 and 4).
The presence of FADS and the enrichment of its activity in nuclei were further confirmed at the functional level in pure nuclei isolated by rat liver homogenate using both HPLC and spectrophotometric assays (Fig. 6). Pure rat liver nuclei, free from any trace of both cytosolic and mitochondrial marker enzymes (Fig. 5A), were shown to synthesize FAD with a specific activity equal to ∼18.1 pmol·min−1·mg−1 protein, corresponding to a total FADS activity of ∼0.2 nmol FAD·min−1. Because nuclear proteins represent ∼4% of the total proteins present in the homogenate, these results indicate that this enzyme contributes to the total activity of FAD synthesis less than cytosol and mitochondria (18).
As regards the flavin nuclear content, pure rat liver nuclei were shown to contain 3-fold less total flavin cofactors in respect to mitochondria (18). This observation tallies with the presence of a large number of flavoenzymes in mitochondria and with the subsequent high FAD autofluorescence measured in this subcellular compartment in living cells (54). Together with the evaluation of a significant presence of FAD tightly bound to proteins, the biochemical assay performed here allowed us to characterize in some detail the nuclear FAD forming enzyme, revealing its strict Mg2+ dependence and sensitivity to mercurial reagents, in agreement with what has already been observed for hFADS2 (38, 47).
Regarding the anti-FADS immunoreactive bands revealed in immunoblotting experiments on pure nuclei, although their specificity has been definitely demonstrated by immunoinhibition experiments, their classification as alternative splicing variants, post-translationally modified isoforms or degradation products, is still under investigation. In humans, at least four alternative splicing products of the FLAD1 gene have been demonstrated to be concurrently expressed in different cell lines/tissues (8). The corresponding products differ from each other at either the N- or C-terminal region, with the longest one being isoform 1, localized in mitochondria (8, 23). A FADS migrating on SDS-PAGE with a molecular mass of 65 kDa was also identified in human neurons (26). Different from humans, only a single transcript corresponding to a predicted product of 490 amino acids (54.6 kDa) is at the moment reported for rats in Ensembl (accession number ENSRNOP00000028030) and NCBI Entrez gene (gene identifier 751787.) databases. UniprotKB (Uniprot no. B1WBY2) reported an additional transcript corresponding to a predicted product of 474 amino acids (52.6 kDa). However, a wide BLAST search in nonredundant protein sequence database using as a query the human FADS isoforms 1 and 2 gave two additional products of 552 amino acids (61.5 kDa) and 371 amino acids (40.7 kDa), annotated in the GenBankTM database (accession numbers EDM00631.1 and EDM00632.1), both performing a 90% identity with the human proteins. The four putative rat isoforms differ from each other at the C-terminal region, and the 474-amino acid protein lacks 17 residues (corresponding to the 324–340 amino acid region). Unfortunately, a clear subcellular localization prediction using common bioinformatics analyses was possible for none of these proteins (23, 24). Thus, we can postulate that in rats, as in humans, a dynamic control by alternative splicing could regulate the expression/localization of specific FADS isoforms.
Also, it can be hypothesized that one of these isoforms may migrate to the nucleus in response to chemicophysical changes because of post-translational modifications induced, for example, by different metabolic states of the cell. Using the different post-translational modification prediction programs, available on the ExPASy tool, for all the rat isoforms a phosphorylation site at position 70 by protein kinase C was predicted with a high probability (score equal to 0.72). Also the CSS-Palm 3.0 software (55) set with the default threshold predicts two palmitoylation sites at positions 39 and 312 (with scores of 0.33 and 1.74, respectively) and an additional site at position 547 (with a score of 3.20) for the 552-amino acid isoform. Interestingly, this palmitoylation site is adjacent to a transmembrane helices predicted with high probability by the TMpred software. Other post-translational modifications were not predicted (i.e., O-glycosylations) or predicted with a low probability (i.e., sumoylation, using both the SUMOplotTM analysis program and Sumosp 2.0 software (56)). Tissue distribution and subcellular localization of these isoforms are still uncharacterized (8, 26) and represent one of the ongoing research efforts in our laboratory. Whatever the isoform details, to our knowledge this is the first evidence that FAD synthesis occurs in the nucleus, and we postulate that, as hypothesized by others (27), this event could be linked to the biogenesis and flavinylation of nuclear flavoproteome (Fig. 8).
FIGURE 8.

FAD homeostasis in mammalian cells. FAD cofactor is represented in this drawing by a star. Flavin transporters are indicated with circles (circled I, plasma membrane Rf transporter, i.e., RFVT1–3 described in Ref. 12; circled II, mitochondrial Rf transporter, still uncharacterized; circled III, mitochondrial FAD exporter). The question mark indicates the unknown origin of Rf in the nucleus. FAD-synthesizing enzymes are indicated with gray boxes (box 1, riboflavin kinase; box 2, FADS). FAD degrading enzymes are indicated with white boxes (box 3, FAD pyrophosphatase; box 4, FMN phosphohydrolase). Apo-flavoprotein (apo-Fp) into holoflavoprotein (holo-Fp) transition and FAD recycling are indicated with dotted lines. Some nuclear flavoenzymes are indicated with white boxes with a star. AIF, apoptosis inducing factor1; LSD1/2, lysine-specific demethylase 1/2; TXNRD1, thioredoxin reductase. The scheme summarizes the functional studies described in this and other previous papers (18, 61, 62).
In addition, our results indicate for the first time the existence of FAD hydrolyzing enzymes in the nucleus. The kinetic features of the FAD hydrolytic activity as characterized in pure rat liver nuclei suggest that it could be due to one of the already characterized NUDIX hydrolases. In humans, at least 24 genes and 5 pseudogenes encoding NUDIX hydrolases exist, and most of their protein products have been characterized to some extent. In rats, the corresponding orthologues exist (see the UniprotKB database), all of them inferred by homology. Although there is evidence at the transcript level for some of them, data at the protein level have been collected, and specific studies have been performed on just three of them: NUDT1 (57), NUDT6 (58), and NUDT3 (59). The members of this superfamily show diverse substrate specificity as well as different subcellular localizations (cytoplasm, nucleus, mitochondria, peroxisomes, plasma membrane) (60). A NUDIX hydrolase that efficiently hydrolyzes FAD has been characterized in mammals, namely NUDT12 (49), but its localization appears to be peroxisomal. Whether and how the same enzyme could be responsible for FAD hydrolysis in the nucleus remain to be established. Quite interesting is the ability of adenylate-containing nucleotides to inhibit nuclear FAD hydrolysis. In particular, the ability of the nuclear FAD hydrolyzing enzyme to discriminate between the redox states of pyridine nucleotides might suggest a novel role for nuclear NAD(H) redox status in regulating nuclear FAD homeostasis.
The results described in this paper, together with the intriguing, albeit still speculative, hypothesis that riboflavin kinase exists in rat liver nuclei, allow us to propose the occurrence of a Rf/FAD cycle in the nucleus, as previously suggested for mitochondria (18) (Fig. 8). Nuclear FADS could thus concur, together with cytosolic and mitochondrial FADS, to the creation of a “flavin network,” a scenario that is strictly in line with the recent literature demonstrating a fundamental role for cellular FAD biosynthesis in regulating cellular energy balance (27).
Comprehension of how and when the cell regulates flavin homeostasis, as well as the precise understanding of the physiological role exerted by FAD biosynthetic pathways in the three different subcellular compartments, requires further investigation. Answering such questions appears to be of special interest in light of the recent notion that impairment in flavoenzymes activity and flavin supply/metabolism could have a role in the pathogenesis of Rf-responsive myopathies (see review in Ref. 8 and references therein) and of the emerging role of FAD and other vitamin-derived cofactors as regulators of epigenetic events.
Acknowledgments
We thank G. Sirago who participated as a student in the early stages of this work and V. Giannoccaro (Università degli Studi di Bari “Aldo Moro”) for the technical assistance.
This work was supported by PON-Ricerca e Competitività 2007–2013 Project 01_00937 Modelli sperimentali Biotecnologici integrati per lo Sviluppo e la selezione di molecole di interesse per la salute dell'uomo (to M. B.).
- Rf
- riboflavin
- FADS
- FAD synthase
- hFADS
- human FAD synthase
- hFADS1
- human FAD synthase isoform 1
- hFADS2
- human FAD synthase isoform 2
- LDH
- lactate dehydrogenase.
REFERENCES
- 1. Massey V. (1995) Introduction. Flavoprotein structure and mechanism. FASEB J. 9, 473–475 [DOI] [PubMed] [Google Scholar]
- 2. Massey V. (2000) The chemical and biological versatility of riboflavin. Biochem. Soc. Trans. 28, 283–296 [PubMed] [Google Scholar]
- 3. McCormick D. B. (1989) Two interconnected B vitamins. Riboflavin and pyridoxine. Physiol. Rev. 69, 1170–1198 [DOI] [PubMed] [Google Scholar]
- 4. Macheroux P., Kappes B., Ealick S. E. (2011) Flavogenomics. A genomic and structural view of flavin-dependent proteins. FEBS J. 278, 2625–2634 [DOI] [PubMed] [Google Scholar]
- 5. Joosten V., van Berkel W. J. (2007) Flavoenzymes. Curr. Opin. Chem. Biol. 11, 195–202 [DOI] [PubMed] [Google Scholar]
- 6. De Colibus L., Mattevi A. (2006) New frontiers in structural flavoenzymology. Curr. Opin. Struct. Biol. 16, 722–728 [DOI] [PubMed] [Google Scholar]
- 7. Mansoorabadi S. O., Thibodeaux C. J., Liu H. W. (2007) The diverse roles of flavin coenzymes. Nature's most versatile thespians. J. Org. Chem. 72, 6329–6342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barile M., Giancaspero T. A., Brizio C., Panebianco C., Indiveri C., Galluccio M., Vergani L., Eberini I., Gianazza E. (2013) Biosynthesis of flavin cofactors in man. Implications in health and disease. Curr. Pharm. Des. 19, 2649–2675 [DOI] [PubMed] [Google Scholar]
- 9. Yao Y., Yonezawa A., Yoshimatsu H., Masuda S., Katsura T., Inui K. (2010) Identification and comparative functional characterization of a new human riboflavin transporter hRFT3 expressed in the brain. J. Nutr. 140, 1220–1226 [DOI] [PubMed] [Google Scholar]
- 10. Yamamoto S., Inoue K., Ohta K. Y., Fukatsu R., Maeda J. Y., Yoshida Y., Yuasa H. (2009) Identification and functional characterization of rat riboflavin transporter 2. J. Biochem. 145, 437–443 [DOI] [PubMed] [Google Scholar]
- 11. Yonezawa A., Masuda S., Katsura T., Inui K. (2008) Identification and functional characterization of a novel human and rat riboflavin transporter, RFT1. Am. J. Physiol. Cell Physiol. 295, C632–C641 [DOI] [PubMed] [Google Scholar]
- 12. Yonezawa A., Inui K. (2013) Novel riboflavin transporter family RFVT/SLC52. Identification, nomenclature, functional characterization and genetic diseases of RFVT/SLC52. Mol. Aspects. Med. 34, 693–701 [DOI] [PubMed] [Google Scholar]
- 13. McCormick D. B., Oka M., Bowers-Komro D. M., Yamada Y., Hartman H. A. (1997) Purification and properties of FAD synthetase from liver. Methods Enzymol. 280, 407–413 [DOI] [PubMed] [Google Scholar]
- 14. Oka M., McCormick D. B. (1987) Complete purification and general characterization of FAD synthetase from rat liver. J. Biol. Chem. 262, 7418–7422 [PubMed] [Google Scholar]
- 15. Yamada Y., Merrill A. H., Jr., McCormick D. B. (1990) Probable reaction mechanisms of flavokinase and FAD synthetase from rat liver. Arch. Biochem. Biophys. 278, 125–130 [DOI] [PubMed] [Google Scholar]
- 16. Wu M., Repetto B., Glerum D. M., Tzagoloff A. (1995) Cloning and characterization of FAD1, the structural gene for flavin adenine dinucleotide synthetase of Saccharomyces cerevisiae. Mol. Cell Biol. 15, 264–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deluca C., Kaplan N. O. (1958) Flavin adenine dinucleotide synthesis in animal tissues. Biochim. Biophys. Acta 30, 6–11 [DOI] [PubMed] [Google Scholar]
- 18. Barile M., Brizio C., Valenti D., De Virgilio C., Passarella S. (2000) The riboflavin/FAD cycle in rat liver mitochondria. Eur. J. Biochem. 267, 4888–4900 [DOI] [PubMed] [Google Scholar]
- 19. Barile M., Passarella S., Bertoldi A., Quagliariello E. (1993) Flavin adenine dinucleotide synthesis in isolated rat liver mitochondria caused by imported flavin mononucleotide. Arch. Biochem. Biophys. 305, 442–447 [DOI] [PubMed] [Google Scholar]
- 20. Bafunno V., Giancaspero T. A., Brizio C., Bufano D., Passarella S., Boles E., Barile M. (2004) Riboflavin uptake and FAD synthesis in Saccharomyces cerevisiae mitochondria. Involvement of the Flx1p carrier in FAD export. J. Biol. Chem. 279, 95–102 [DOI] [PubMed] [Google Scholar]
- 21. Pallotta M. L., Brizio C., Fratianni A., De Virgilio C., Barile M., Passarella S. (1998) Saccharomyces cerevisiae mitochondria can synthesise FMN and FAD from externally added riboflavin and export them to the extramitochondrial phase. FEBS Lett. 428, 245–249 [DOI] [PubMed] [Google Scholar]
- 22. Giancaspero T. A., Locato V., de Pinto M. C., De Gara L., Barile M. (2009) The occurrence of riboflavin kinase and FAD synthetase ensures FAD synthesis in tobacco mitochondria and maintenance of cellular redox status. FEBS J. 276, 219–231 [DOI] [PubMed] [Google Scholar]
- 23. Torchetti E. M., Brizio C., Colella M., Galluccio M., Giancaspero T. A., Indiveri C., Roberti M., Barile M. (2010) Mitochondrial localization of human FAD synthetase isoform 1. Mitochondrion 10, 263–273 [DOI] [PubMed] [Google Scholar]
- 24. Brizio C., Galluccio M., Wait R., Torchetti E. M., Bafunno V., Accardi R., Gianazza E., Indiveri C., Barile M. (2006) Over-expression in Escherichia coli and characterization of two recombinant isoforms of human FAD synthetase. Biochem. Biophys. Res. Commun. 344, 1008–1016 [DOI] [PubMed] [Google Scholar]
- 25. Galluccio M., Brizio C., Torchetti E. M., Ferranti P., Gianazza E., Indiveri C., Barile M. (2007) Over-expression in Escherichia coli. Purification and characterization of isoform 2 of human FAD synthetase. Protein Expr. Purif. 52, 175–181 [DOI] [PubMed] [Google Scholar]
- 26. Lin J., Diamanduros A., Chowdhury S. A., Scelsa S., Latov N., Sadiq S. A. (2009) Specific electron transport chain abnormalities in amyotrophic lateral sclerosis. J. Neurol. 256, 774–782 [DOI] [PubMed] [Google Scholar]
- 27. Hino S., Sakamoto A., Nagaoka K., Anan K., Wang Y., Mimasu S., Umehara T., Yokoyama S., Kosai K., Nakao M. (2012) FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat. Commun. 3, 758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Merglen A., Theander S., Rubi B., Chaffard G., Wollheim C. B., Maechler P. (2004) Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 145, 667–678 [DOI] [PubMed] [Google Scholar]
- 29. Lau B. W., Colella M., Ruder W. C., Ranieri M., Curci S., Hofer A. M. (2005) Deoxycholic acid activates protein kinase C and phospholipase C via increased Ca2+ entry at plasma membrane. Gastroenterology 128, 695–707 [DOI] [PubMed] [Google Scholar]
- 30. Dostal D. E., Rothblum K. N., Conrad K. M., Cooper G. R., Baker K. M. (1992) Detection of angiotensin I and II in cultured rat cardiac myocytes and fibroblasts. Am. J. Physiol. 263, C851–C863 [DOI] [PubMed] [Google Scholar]
- 31. Colella M., Pozzan T. (2008) Cardiac cell hypertrophy in vitro. Role of calcineurin/NFAT as Ca2+ signal integrators. Ann. N.Y. Acad. Sci. 1123, 64–68 [DOI] [PubMed] [Google Scholar]
- 32. Latronico T., Branà M. T., Gramegna P., Fasano A., Di Bari G., Liuzzi G. M. (2013) Inhibition of myelin-cleaving poteolytic activities by interferon-β in rat astrocyte cultures. Comparative analysis between gelatinases and calpain-II. PLoS One 8, e49656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cardone R. A., Bellizzi A., Busco G., Weinman E. J., Dell'Aquila M. E., Casavola V., Azzariti A., Mangia A., Paradiso A., Reshkin S. J. (2007) The NHERF1 PDZ2 domain regulates PKA-RhoA-p38-mediated NHE1 activation and invasion in breast tumor cells. Mol. Biol. Cell 18, 1768–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chauveau J., Moule Y., Rouiller C. (1957) [Technic for the isolation of cell nuclei based upon their density]. Bull. Soc. Chim. Biol. (Paris) 39, 1521–1533 [PubMed] [Google Scholar]
- 35. Cardone A., Lopez F., Affortunato F., Busco G., Hofer A. M., Mallamaci R., Martinelli C., Colella M., Farinola G. M. (2012) An aryleneethynylene fluorophore for cell membrane staining. Biochim. Biophys. Acta 1818, 2808–2817 [DOI] [PubMed] [Google Scholar]
- 36. Gerbino A., Maiellaro I., Carmone C., Caroppo R., Debellis L., Barile M., Busco G., Colella M. (2012) Glucose increases extracellular [Ca2+] in rat insulinoma (INS-1E) pseudoislets as measured with Ca2+-sensitive microelectrodes. Cell Calcium 51, 393–401 [DOI] [PubMed] [Google Scholar]
- 37. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ. 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Torchetti E. M., Bonomi F., Galluccio M., Gianazza E., Giancaspero T. A., Iametti S., Indiveri C., Barile M. (2011) Human FAD synthase (isoform 2). A component of the machinery that delivers FAD to apo-flavoproteins. FEBS J. 278, 4434–4449 [DOI] [PubMed] [Google Scholar]
- 39. Barile M., Brizio C., De Virgilio C., Delfine S., Quagliariello E., Passarella S. (1997) Flavin adenine dinucleotide and flavin mononucleotide metabolism in rat liver. The occurrence of FAD pyrophosphatase and FMN phosphohydrolase in isolated mitochondria. Eur. J. Biochem. 249, 777–785 [DOI] [PubMed] [Google Scholar]
- 40. Kunz W. S. (1988) Evaluation of electron-transfer flavoprotein and α-lipoamide dehydrogenase redox states by two-channel fluorimetry and its application to the investigation of β-oxidation. Biochim. Biophys. Acta 932, 8–16 [DOI] [PubMed] [Google Scholar]
- 41. King T. E., Howard R. L., Wilson D. F., Li J. C. (1962) The partition of flavins in the heart muscle preparation and heart mitochondria. J. Biol. Chem. 237, 2941–2946 [PubMed] [Google Scholar]
- 42. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 43. Bergmeyer H. U., Bernt E., Hess B. (1963) Lactate dehydrogenase, in Methods of Enzymatic Analysis (Bergmeyer H. U., ed.) pp. 736–743, Academic Press, New York [Google Scholar]
- 44. Barile M., Passarella S., Danese G., Quagliariello E. (1996) Rat liver mitochondria can synthesize nicotinamide adenine dinucleotide from nicotinamide mononucleotide and ATP via a putative matrix nicotinamide mononucleotide adenylyltransferase. Biochem. Mol. Biol. Int. 38, 297–306 [PubMed] [Google Scholar]
- 45. Ghosal A., Said H. M. (2012) Mechanism and regulation of vitamin B2 (riboflavin) uptake by mouse and human pancreatic beta-cells/islets. Physiological and molecular aspects. Am. J. Physiol. Gastrointest. Liver. Physiol. 303, G1052–G1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Koch-Nolte F., Fischer S., Haag F., Ziegler M. (2011) Compartmentation of NAD+-dependent signalling. FEBS Lett. 585, 1651–1656 [DOI] [PubMed] [Google Scholar]
- 47. Galluccio M., Giancaspero T. A., Miccolis A., Panebianco C., Indiveri C., Barile M. (2013) Relevance of Cys residues of human FAD synthase (isoform 2) in the protein function/structure as assessed by homology modeling and site directed mutagenesis, in FLAVINS and FLAVOPROTEINS 2011: Proceedings of the 17th International symposium on Flavins and Flavoproteins, July 24–29, 2011, University of California Berkeley (Miller S., Hille R., Palfey B., eds) pp. 509–514, Lulu, Raleigh, NC [Google Scholar]
- 48. Shibko S., Tappel A. L. (1963) Acid phosphatase of the lysosomal and soluble fraction of rat liver. Biochim. Biophys. Acta 73, 76–86 [DOI] [PubMed] [Google Scholar]
- 49. Abdelraheim S. R., Spiller D. G., McLennan A. G. (2003) Mammalian NADH diphosphatases of the Nudix family. Cloning and characterization of the human peroxisomal NUDT12 protein. Biochem. J. 374, 329–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chiarugi A., Dölle C., Felici R., Ziegler M. (2012) The NAD metabolome. A key determinant of cancer cell biology. Nat. Rev. Cancer 12, 741–752 [DOI] [PubMed] [Google Scholar]
- 51. Oka S., Hsu C. P., Sadoshima J. (2012) Regulation of cell survival and death by pyridine nucleotides. Circ. Res. 111, 611–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang H., Yang T., Baur J. A., Perez E., Matsui T., Carmona J. J., Lamming D. W., Souza-Pinto N. C., Bohr V. A., Rosenzweig A., de Cabo R., Sauve A. A., Sinclair D. A. (2007) Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130, 1095–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Giancaspero T. A., Torchetti E. M., Latronico T., Brizio C., Colella M., Liuzzi G. M., Barile M. (2010) Unravelling the relevance of fad synthetase in human physiopathology. A biotechnological approach. Eur. J. Clin. Invest. 40, 65–65 [Google Scholar]
- 54. Heikal A. A. (2010) Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies. Biomark. Med. 4, 241–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ren J., Wen L., Gao X., Jin C., Xue Y., Yao X. (2008) CSS-Palm 2.0. An updated software for palmitoylation sites prediction. Protein Eng. Des. Sel. 21, 639–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ren J., Gao X., Jin C., Zhu M., Wang X., Shaw A., Wen L., Yao X., Xue Y. (2009) Systematic study of protein sumoylation. Development of a site-specific predictor of SUMOsp 2.0. Proteomics 9, 3409–3412 [DOI] [PubMed] [Google Scholar]
- 57. Kasprzak K. S., Nakabeppu Y., Kakuma T., Sakai Y., Tsuruya K., Sekiguchi M., Ward J. M., Diwan B. A., Nagashima K., Kasprzak B. H. (2001) Intracellular distribution of the antimutagenic enzyme MTH1 in the liver, kidney and testis of F344 rats and its modulation by cadmium. Exp. Toxicol. Pathol. 53, 325–335 [DOI] [PubMed] [Google Scholar]
- 58. Sun L., Yu S., Wang H., Fan B., Liu B. (2012) NUDT6, the FGF-2's antisense gene, showed associations with fat deposition related traits in pigs. Mol. Biol. Rep. 39, 4119–4126 [DOI] [PubMed] [Google Scholar]
- 59. Safrany S. T., Caffrey J. J., Yang X., Bembenek M. E., Moyer M. B., Burkhart W. A., Shears S. B. (1998) A novel context for the ‘MutT’ module, a guardian of cell integrity, in a diphosphoinositol polyphosphate phosphohydrolase. EMBO J. 17, 6599–6607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McLennan A. G. (2006) The Nudix hydrolase superfamily. Cell Mol. Life. Sci. 63, 123–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brizio C., Brandsch R., Douka M., Wait R., Barile M. (2008) The purified recombinant precursor of rat mitochondrial dimethylglycine dehydrogenase binds FAD via an autocatalytic reaction. Int. J. Biol. Macromol. 42, 455–462 [DOI] [PubMed] [Google Scholar]
- 62. Brizio C., Otto A., Brandsch R., Passarella S., Barile M. (2000) A protein factor of rat liver mitochondrial matrix involved in flavinylation of dimethylglycine dehydrogenase. Eur. J. Biochem. 267, 4346–4354 [DOI] [PubMed] [Google Scholar]