

Background: PKC regulating Syk activity has been demonstrated in other cells but is unknown in platelets.
Results: PKCs regulate tyrosine phosphorylation and activity of Syk.
Conclusion: PKCβ-dependent differential regulation of Syk activity is seen in human but not in murine platelets.
Significance: Understanding this human pathway of platelet regulation might aid in development of anti-platelet therapy.
Keywords: Phosphotyrosine Signaling, Platelets, Protein Kinase C (PKC), Protein Phosphorylation, Protein-tyrosine Kinase (Tyrosine Kinase), GPVI, Syk
Abstract
Protein kinase C (PKC) isoforms differentially regulate platelet functional responses downstream of glycoprotein VI (GPVI) signaling, but the role of PKCs regulating upstream effectors such as Syk is not known. We investigated the role of PKC on Syk tyrosine phosphorylation using the pan-PKC inhibitor GF109203X (GFX). GPVI-mediated phosphorylation on Syk Tyr-323, Tyr-352, and Tyr-525/526 was rapidly dephosphorylated, but GFX treatment inhibited this dephosphorylation on Tyr-525/526 in human platelets but not in wild type murine platelets. GFX treatment did not affect tyrosine phosphorylation on FcRγ chain or Src family kinases. Phosphorylation of Lat Tyr-191 and PLCγ2 Tyr-759 was also increased upon treatment with GFX. We evaluated whether secreted ADP is required for such dephosphorylation. Exogenous addition of ADP to GFX-treated platelets did not affect tyrosine phosphorylation on Syk. FcγRIIA- or CLEC-2-mediated Syk tyrosine phosphorylation was also potentiated with GFX in human platelets. Because potentiation of Syk phosphorylation is not observed in murine platelets, PKC-deficient mice cannot be used to identify the PKC isoform regulating Syk phosphorylation. We therefore used selective inhibitors of PKC isoforms. Only PKCβ inhibition resulted in Syk hyperphosphorylation similar to that in platelets treated with GFX. This result indicates that PKCβ is the isoform responsible for Syk negative regulation in human platelets. In conclusion, we have elucidated a novel pathway of Syk regulation by PKCβ in human platelets.
Introduction
After vascular injury, platelet activation by exposed subendothelial collagen is a key step that initiates signaling cascades that lead to hemostatic plug formation and bleeding arrest (1). The platelet collagen receptor is a complex between glycoprotein VI (GPVI)3 and Fc receptor γ-chain (GPVI-FcRγ) (2). As the extracellular Ig domains of GPVI binds collagen, the immunoreceptor tyrosine activation motif (ITAM) within the cytoplasmic portion of FcRγ becomes phosphorylated by the GPVI-associated Src family kinases (SFKs) Fyn and Lyn (3, 4). The dual phosphorylated ITAM acts as a docking site for the spleen tyrosine kinase (Syk), which in turn undergoes phosphorylation at different tyrosine residues, including Tyr-323, Tyr-352, and Tyr-525/526. Although Syk−/− animals are embryonically lethal, a conditional platelet knock-out has been generated in which aggregation, secretion, and arachidonic acid release in response to GPVI agonists are defective (5). Thus, the role of Syk in GPVI signaling is undisputed. Upon Syk activation, a signaling cascade through PLCγ2 is initiated, resulting in Ca2+ mobilization, diacylglycerol production, and protein kinase C (PKC) activation, events that mediate platelet granule secretion and thromboxane A2 generation. Thromboxane A2 and secreted ADP will in turn activate nearby circulating platelets and act in an autocrine fashion to further enhance platelet activation through TP, P2Y1, and P2Y12 receptor activation.
PKC is a family of serine/threonine kinases that consist of 10 distinct isoforms categorized into three classes based on their lipid and cofactor requirements (6): the classical isoforms (α, β, and γ) that require diacylglycerol and Ca2+ for their activation, the novel isoforms (δ, θ, ϵ, and η) that require diacylglycerol but are Ca2+ insensitive, and the atypical isoforms (ζ and λ) that are activated in a diacylglycerol- and Ca2+-independent manner but require phosphatidylinositol trisphosphate (7). The role of PKC in platelets has been extensively studied. Seven isoforms are expressed in platelets (α, β, δ, θ, ϵ, η, and ζ), each one regulating different functional responses. In the context of GPVI signaling, our group has established that PKCδ negatively regulates dense granule secretion by means of a molecular complex that involves Lyn and SHIP-1 (8). PKCθ was also shown to regulate granule secretion and thromboxane generation downstream of GPVI receptors (9, 10). Furthermore, using GPVI-stimulated PKCθ−/− platelets, it was proposed that PKCθ down-regulates store-independent Ca2+ entry and thus modulates the Ca2+ signal (11). Recently, Gilio et al. (12) studied the differential role of PKC isoforms in collagen-induced thrombus formation. The authors demonstrated a differential role for conventional isoforms, PKC α and β in promoting thrombus formation by positively regulating granule secretion, whereas the novel isoform θ inhibited thrombus formation by regulating the Ca2+ signal and phosphatidylserine exposure.
The notion that PKC regulates Syk activity has been described in endothelial cells as in these cells, a PKCδ-dependent serine phosphorylation on Syk regulates thrombin-induced ICAM expression (13). In contrast, Pula et al. (14) postulated that active Syk is required for PKCα activation in platelets, locating Syk upstream of PKCα. A growing body of evidence suggests a close correlation between PKC-dependent serine/threonine phosphorylation and tyrosine phosphorylation events (15–17). In recent years our laboratory has been devoted to elucidating the signaling events downstream of GPVI and has described the role of signaling molecules that negatively regulate Syk activity (18, 19). In line with our previous research, in the present study we investigated the negative regulation of Syk by PKC in GPVI signaling. We show that, in human platelets, inhibition of PKC leads to Syk hyperphosphorylation on residues Tyr-525/526 whereas Tyr-323 and Tyr-352 phosphorylations are unaffected. PKC negatively regulates Syk activity, as evidenced by hyperphosphorylation of downstream targets, LAT and PLCγ2 upon PKC inhibition. The negative regulation of Syk is also observed downstream of other ITAM-containing receptors such as CLEC-2 and FcγRIIA receptors in human platelets. Importantly, we demonstrate that PKC-mediated negative regulation of Syk activity is seen in human but not in murine platelets. Finally, we identified PKCβ as the isoform responsible for Syk negative regulation in human platelets.
EXPERIMENTAL PROCEDURES
Materials
All reagents were from Sigma unless stated otherwise. Anti-Syk (4D10 and N19), anti-PLCγ2 (Q-20), and anti-LAT (LAT-01) were from Santa Cruz Biotechnology. Anti-phosphospecific Syk (Tyr-525/526, Tyr-323, and Tyr-352), anti-phosphospecific LAT (Tyr-191), and anti-phosphospecific PLCγ2 (Tyr-759) were from Cell Signaling Technology. Convulxin was purified according to the method of Polgár et al. (20). Collagen (type I, equine tendon) and CHRONO-LUME were from Chronolog (Havertown, PA). Collagen-related peptide was purchased from Dr. Richard Farndale. Fucoidan (80% pure) was obtained from Sigma. Fura-2/AM was obtained from Molecular Probes. Pan-PKC inhibitor GF109203X was from Biomol (now ENZO Life Sciences Inc., Plymouth Meeting, PA). PKCθ/δ and β inhibitors were from EMD Millipore. LY333531 was purchased from AG Scientific. All other reagents were reagent grade, and deionized water was used throughout.
Animals
Mice were maintained on a C57BL background, and experiments were performed in compliance with the Institutional Animal Care and Use Committee at Temple University and Thomas Jefferson University.
Preparation of Human and Murine Platelets
Human blood was obtained from healthy volunteers with informed consent in a one-sixth volume of acid-citrate-dextrose (2.5 g of sodium citrate, 1.5 g of citric acid, and 2.0 g of glucose in 100 ml of H2O). Blood was collected from anesthetized mice into syringes containing 1/10 blood volume of 3.8% sodium citrate as anticoagulant. Washed human and murine platelets were prepared by resuspending them in Tyrode's buffer containing 0.1 unit/ml apyrase as described previously (21). Aspirin-treated platelets (1 mm) were used throughout the studies.
Platelet Aggregations and Secretion
Platelet aggregation and secretion were measured using a lumiaggregometer (Chrono-Log, Havertown, PA) at 37 °C under stirring conditions as described previously (22).
Intracellular Ca2+ Mobilization
Platelet Ca2+ mobilization was also measured. Platelet-rich plasma was incubated with 5 μm Fura-2/AM and 1 mm aspirin. Fluorescence was measured, and the Ca2+ concentration was calculated as described previously (23).
Western Blotting
Platelets were stimulated with agonists for the indicated time, and the reaction was stopped by the addition of 3 × SDS sample buffer. Total cell lysate was electrophoresed on 8% SDS-polyacrylamide gels. Proteins were transferred to a nitrocellulose membrane (Millipore), subjected to Western blotting, blocked for 1 h with Odyssey blocking buffer, and incubated with the appropriate antibody 1:500 (v/v) in Odyssey blocking buffer with 0.1% (v/v) Tween 20 (16 h, 4 °C). After washing, membranes were incubated with the appropriate LICOR secondary antibody (1 h, 4 °C). Phosphorylation of proteins was visualized and quantified using the Odyssey system.
Statistical Analysis
Each experiment was repeated at least three times. Results are expressed as means ± S.E. Data were analyzed using GraphPad Prism 5 software. Significant differences were determined using two-way analysis of variance and Student's t test. Differences were considered significant at p ≤ 0.05.
RESULTS
Syk Tyrosine Phosphorylation Is Differentially Regulated by PKC
It has been established that different PKC isoforms positively and negatively regulate platelet functional responses downstream of GPVI signaling (8–10), but the role of PKC regulating upstream effectors such as Syk is not known. We investigated the role of PKC on Syk tyrosine phosphorylation by using the pan-PKC inhibitor GF109203X (GFX). As shown in Fig. 1A, convulxin (Cvx) stimulation causes platelet aggregation and secretion. GFX inhibited Cvx-induced aggregation and secretion even when higher concentrations of Cvx were used. Western blot analysis (Fig. 1Bi) has shown that Cvx induces phosphorylation of Syk at residues Tyr-525/526 which peaks at 30 s, followed by a TULA-2-dependent dephosphorylation of tyrosine residues (19). As shown in Fig. 1Bi and its quantitation (Fig. 1Bii), PKC inhibition causes Cvx-induced phosphorylation on Syk residues Tyr-525/526 to be significantly higher and more persistent than platelets stimulated in the absence of inhibitor. This potentiation of phosphorylation is also observed when human platelets were preincubated with GFX and stimulated with a physiological agonist, collagen (Fig. 1C) or collagen-related peptide (Fig. 1D). However, as shown in Fig. 1E, Syk Tyr-525/526 phosphorylation in murine platelets downstream of GPVI receptor is inhibited with pan-PKC inhibitor GFX, suggesting a pathway of regulation of Syk phosphorylation by PKCs only in human platelets.
FIGURE 1.

PKC inhibition reduces platelet aggregation and secretion as well as selectively modulates Syk tyrosine phosphorylation on Tyr-525/526. A, human platelets were incubated with 5 μm GFX for 5 min prior to stimulation with the GPVI agonist Cvx (200, 400, or 800 ng/ml). Platelet aggregation and secretion were measured for 2 min. Bi, platelets were treated as described in A, stimulated with 200 ng/ml Cvx for 0, 30, 60, and 180 s, and phosphotyrosine 525/526 Syk and total Syk were analyzed. Bii, quantitation of the blots with mean ± S.E. (error bars) of relative Syk phosphorylation for Cvx in untreated (●) and GFX treated (▴) platelets was plotted against time, and Student's t test was performed (p < 0.05, untreated versus GFX treated platelets). C and D, results are the same as B except that platelets were stimulated using GPVI agonist 10 μg/ml collagen (C) and 5 μg/ml collagen-related peptide (CRP) (D). E, washed murine platelets were treated same as in Bi but stimulated with 1 μg/ml Cvx and probed for pSyk Tyr-525/526 and total Syk. Blots are representative of at least three independent experiments.
In contrast to Syk Tyr-525/526 phosphorylation, Syk Tyr-323 (Fig. 2, Ai and Aii) and Tyr-352 (Fig, 2, Bi and Bii) phosphorylations were not affected by GFX in human platelets downstream of GPVI receptor. Similar results were obtained when collagen or collagen-related peptide was used as agonist (data not shown). Together, these data suggest that PKCs negatively regulate Syk Tyr-525/526 phosphorylation downstream of GPVI receptor in human platelets.
FIGURE 2.

PKC inhibition does not affect Syk Tyr-323 and Tyr-352 phosphorylation. Ai, human platelets were treated as described in Fig. 1Bi and analyzed for phospho-Syk Tyr-323 and total Syk. Bi, human platelets were treated as described in Fig. 1Bi and analyzed for phospho-Syk Tyr-352 and total Syk. Aii and Bii, quantitation of the blots with mean ± S.E. (error bars) of relative Syk phosphorylation for Cvx in untreated (●) and GFX-treated (▴) platelets was plotted against time, and Student's t test was performed (no significant difference was observed, untreated versus GFX-treated platelets). Blots are representative of at least three independent experiments.
Events Upstream of Syk Activation Are Unaffected by PKC Inhibition
In GPVI signaling, Syk is activated in a SFK-dependent manner. Fyn and Lyn phosphorylate tyrosine residues on the ITAM domain of FcRγ chain, creating docking sites for Syk recruitment and activation (24). In turn, Syk undergoes autophosphorylation on at least 10 different tyrosine residues including Tyr-323, Tyr-352, and Tyr-525/526 (25). The observation that Tyr-323, Tyr-352, and Tyr-525/526 are differentially phosphorylated upon PKC inhibition argues in favor of a regulation at the level of Syk and not at upstream events. In fact, SFK activation and subsequent FcRγ phosphorylation are not regulated by PKC, as tyrosine phosphorylation on SFK Tyr-416 (activation marker) (Fig. 3A) and phosphorylation on FcRγ (Fig. 3B) are not affected in platelets treated with GFX compared with control. This suggests that, in GPVI signaling, events upstream of Syk activation are not regulated by PKCs.
FIGURE 3.
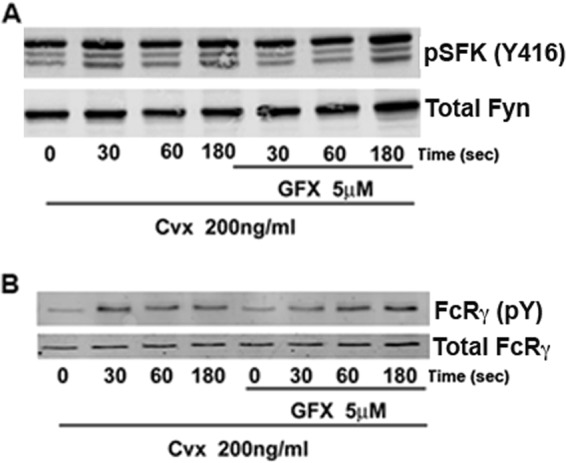
PKC inhibition does not affect Syk upstream molecules, SFKs, and FcRγ. A, human platelets were treated as described in Fig. 1Bi and analyzed for phospho-SFK Tyr-416 (activation marker) and total Fyn. B, analysis of phosphorylation of FcRγ using anti-phosphotyrosine antibody 4G10 was performed. Total FcRγ was used as loading control. Blots are representative of at least three independent experiments.
PKCs Regulate Syk Activity and Downstream Events
It has been well established that the GPVI signaling cascade is initiated by tyrosine kinases Fyn and Lyn. Further downstream, GPVI signaling requires Syk activation to initiate Ca2+ mobilization through the formation of the LAT signalosome, which is a complex of several proteins including LAT, SLP-76, Vav, and PLCγ2 (24). Although Syk Tyr-525/526 phosphorylation is suggested as a marker of its kinase activity, we investigated whether the observed negative regulation of its tyrosine phosphorylation by PKC is reflected in its kinase activity by analyzing the phosphorylation of downstream targets LAT and PLCγ2. LAT Tyr-191 (Fig. 4, A and B) and PLCγ2 Tyr-759 (Fig. 4, C and D) exhibit an increase in phosphorylations in human platelets pre-treated with GFX compared with those seen with convulxin alone, indicating that Syk activity toward LAT and PLCγ2 is up-regulated. An increase in PLCγ2 phosphorylation is in agreement with previous findings (12) that pan-PKC inhibitor potentiates GPVI-induced calcium responses in platelets.
FIGURE 4.

PKC inhibition negatively regulates Syk downstream targets, LAT and PLCγ2. Human platelets were treated as in Fig. 1Bi. A and C, analysis was performed of phosphorylation of LAT phosphotyrosine 191 and total LAT (A) or PLCγ2 phosphotyrosine 759 and total PLCγ2 (C). B and D, quantitation of the blots with mean ± S.E. (error bars) of relative Syk phosphorylation for Cvx in untreated (●) and GFX-treated (▴) platelets was plotted against time, and Student's t test was performed (p < 0.05, untreated versus GFX-treated platelets). Blots are representative of at least three independent experiments.
Secretion-mediated Feedback Does Not Regulate Syk Activity
As shown in Fig. 1A, our experimental design causes a complete inhibition of granule secretion with a pan-PKC inhibitor. During collagen-induced platelet activation, a cross-talk between GPVI and Gi protein signaling exists (26). Concomitant Gi-dependent signaling initiated by secreted ADP regulates many of the functional responses of GPVI signaling including PI3K activation (27). Furthermore, ADP signaling is required to obtain full platelet aggregation as evidenced by the effect of ADP receptor antagonists on platelet aggregation induced by GPVI agonists (23). To address whether Syk phosphorylation and activity were directly affected by PKC or by secretion-mediated feedback mechanism, we treated platelets with GFX and added ADP back into the experimental system to mimic granule secretion. As shown in Fig. 5A, addition of ADP to GFX-treated platelets augmented the aggregation response although not to the extent of nontreated platelets. When analyzing whether an ADP-induced feedback mechanism is responsible for Syk negative regulation, we observed that Syk phosphorylation on residues Tyr-525/526 remains significantly higher in platelets treated with GFX than untreated platelets even in the presence of ADP (Fig. 5, B and C). This result indicates that the negative regulation of Syk is directly mediated by PKCs.
FIGURE 5.

ADP partially restores aggregation response but not Syk Tyr-525/526 hyperphosphorylation in the presence of PKC inhibition. A, representative aggregation tracings of human platelets stimulated with 200 ng/ml Cvx in the absence and presence of the pan-PKC inhibitor GFX (5 μm). In some cases, samples were concomitantly stimulated with 200 ng/ml Cvx and 10 μm ADP. B, analyzed for Syk phosphotyrosine 526/526 and total Syk. C, quantitation of the blots with mean ± S.E. (error bars) of relative Syk phosphorylation for Cvx in untreated (●), GFX treated (■) and GFX with exogenous ADP addition (▴) platelets plotted against time. Student's t test was performed (p < 0.05, untreated versus GFX-treated platelets, and no significant difference was observed between GFX-treated platelets versus GFX+ADP added platelets). Blots are representative of at least three independent experiments.
Syk Phosphorylation Downstream of CLEC-2 and FcγRIIA Receptors Is Negatively Regulated by PKCs
Syk is a tyrosine kinase that regulates signaling downstream of several receptors (28, 29). Besides GPVI/FcRγ, human platelets express other ITAM-bearing receptors that act through a signaling cascade similar to GPVI resulting in Syk activation. One of these receptors is FcγRIIA, which mediates inside-out signaling in response to IgG immune complexes and has been recently demonstrated to be an ITAM-bearing receptor mediating Syk activation in platelet outside-in signaling to fibrinogen (30). Syk is also known to be involved in regulating hemi-ITAM-containing receptor CLEC-2-mediated signaling (31, 32). Thus, an important question is whether the PKC-mediated negative regulation of Syk activity is exclusive to GPVI or is shared by all pathways that utilize the kinase. We analyzed Syk phosphorylation following activation of platelet receptor CLEC-2. Recent studies from our laboratory have identified fucoidan as a selective agonist for CLEC-2 (33). Using this agonist, we evaluated the effect of Syk phosphorylation downstream of CLEC-2 with and without GFX. Fucoidan induces platelet aggregation that is inhibited with GFX (Fig. 6A). Interestingly, as shown in Fig. 6B, CLEC-2-mediated Syk Tyr-525/526 phosphorylation is potentiated with GFX. Similarly, CLEC-2-induced calcium mobilization is potentiated with GFX in human platelets (Fig. 6C) as shown with GPVI signaling (12).
FIGURE 6.

CLEC-2 mediated Syk tyrosine phosphorylation is negatively regulated by PKCs. A, 50 μg/ml fucoidan induced aggregation of human platelets, which is inhibited when platelets were preincubated with 5 μm GFX. B, the samples were probed for phospho-Syk Tyr-525/526 and total Syk. C, Fura-2/AM-loaded human platelets were treated with GFX or vehicle dimethyl sulfoxide (DMSO) and stimulated with 50 μg/ml fucoidan. Ca2+ mobilization was measured using a spectrofluorometer. The change in intracellular calcium was then measured, and mean ± S.E. (error bars) was plotted. Student's t test was performed (p < 0.05, untreated versus GFX-treated platelets). Blots are representative of at least three independent experiments.
Because Syk is activated downstream of FcγRIIA, we analyzed Syk tyrosine phosphorylation downstream of FcγRIIA. Cross-linking FcγRIIA antibodies with anti-mouse IgGs resulted in platelet aggregation that is inhibited with GFX (Fig. 7A). As shown in Fig. 7, B and C, Syk Tyr-525/526 phosphorylation downstream of FcγRIIA is potentiated with GFX, suggesting PKC regulation of Syk similar to GPVI and CLEC-2 receptors in human platelets. We conclude that PKCs negatively regulate Syk tyrosine phosphorylation downstream of GPVI-, CLEC-2, and FcγRIIA receptors in human platelets.
FIGURE 7.

FcγΡIIA-mediated Syk tyrosine phosphorylation is negatively regulated by PKCs. A, human platelets were stimulated with 5 μg/ml mAb IV.3 and 25 μg/ml anti-mouse IgG to cross-link and directly stimulate FcγRIIA signaling. FcγRIIA induced aggregation of human platelets, which was inhibited when platelets were preincubated with 5 μm GFX. B, the samples were probed for phospho-Syk Tyr-525/526 and total Syk. C, quantitation of the blots with mean ± S.E. (error bars) of relative Syk phosphorylation for Cvx in untreated (●) and GFX-treated (▴) platelets was plotted against time, and Student's t test was performed (p < 0.05, untreated versus GFX-treated platelets). Blots are representative of at least three independent experiments.
PKCβ Negatively Regulates Syk Tyrosine Phosphorylation
Negative regulation of Syk by PKCs is seen in human platelets but not in murine platelets, as we did not observe any hyperphosphorylation of Syk in GFX-treated wild type murine platelets (Fig. 1E). As PKC isoform-specific KO models cannot be used to address the isoform responsible for Syk negative regulation, we relied on a pharmacological approach. We inhibited the activity of PKCδ, θ, or β by using commercially available inhibitors. As shown in Fig. 8A, PKCβ inhibition either with PKCβ inhibitor (12) or LY333531 (another PKCβ inhibitor) (34) resulted in Syk hyperphosphorylation similar to that observed in platelets treated with GFX. All other isoform inhibitors tested did not exhibit Syk hyperphosphorylation. This result indicates that PKCβ is the isoform responsible for Syk negative regulation in human platelets. However, specificity can be a problem when using pharmacological inhibitors in platelets, as these inhibitors could exhibit off-target effects. To partially address this question, we performed a dose-response study with the PKCβ inhibitor with Cvx as an agonist. As shown in Fig. 8B, Cvx-induced aggregation and secretion were not completely inhibited with PKCβ inhibitor (3 μm) as opposed to the effect of pan-PKC inhibitor GFX, which abolished secretion and inhibited aggregation to a greater extent. Our findings are consistent with the idea that PKCβ inhibitor at the indicated concentration does not inhibit other PKC isoforms in platelets. As shown in Fig. 8C, at 3 μm concentration, PKCβ inhibitor potentiated GPVI-induced Syk tyrosine phosphorylation 525/526, consistent with the role of PKCβ in regulating tyrosine phosphorylation on Syk in human platelets.
FIGURE 8.
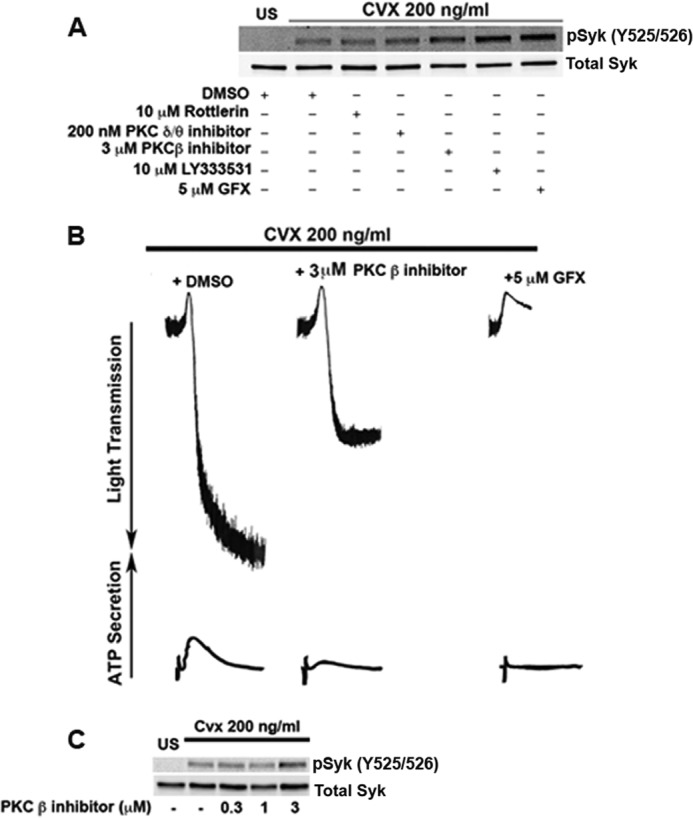
PKCβ is the isoform responsible for Syk negative regulation in human platelets. A, human platelets were stimulated with 200 ng/ml Cvx after preincubation with or without 10 μm Rottlerin, 200 nm PKCδ/θ inhibitor, 3 μm PKCβ inhibitor, 10 μm LY333531, or 5 μm GFX for 5 min and probed for Syk phosphotyrosine 525/526 and total Syk. B, human platelets preincubated with PKC isoform β inhibitor or GFX and were stimulated with 200 ng/ml Cvx for 180 s. Aggregation and secretion were measured by using a lumiaggregometer. C, human platelets were stimulated with 200 ng/ml Cvx after preincubation with or without 0.3 μm, 1 μm, or 3 μm PKCβ inhibitor and probed for Syk phosphotyrosine 525/526 and total Syk. Blots are representative of at least three independent experiments.
DISCUSSION
After vascular injury, subendothelial collagen is exposed, initiating important events leading to platelet activation and hemostasis through direct contact with the platelet collagen receptor, GPVI. Therefore, negative regulation of GPVI is of critical importance to prevent excessive platelet activation and uncontrolled thrombus formation. In this study, we have elucidated a novel role for PKC as a negative regulator of Syk tyrosine phosphorylation downstream of GPVI, CLEC-2, and FcγRIIA. Furthermore, we demonstrate that PKCβ is the isoform that regulates Syk tyrosine phosphorylation, which occurs in human platelets but not in murine platelets.
It has been demonstrated that tyrosine phosphorylation events are critical for platelet activation downstream of GPVI signaling. In this context, Syk plays a pivotal role as exemplified by the marked defects seen in platelet functional responses induced by GPVI agonist in Syk−/− platelets (5). However, in addition to tyrosine kinases, the PKC family of serine/threonine kinases is required to orchestrate important GPVI-mediated events such as granule secretion and thromboxane generation (8).
Studies in endothelial cells have demonstrated that PKCδ regulates Syk activity (13). In this study we have analyzed the role of PKC as a negative regulator of Syk activity in platelets. Initially we used the pan-PKC inhibitor GFX to address this question and used the Syk phosphorylation state on different tyrosine residues as our read-out. These initial analyses led us to identify a PKC-dependent negative regulation of tyrosine residues 525/526 on Syk. In sharp contrast, the phosphorylation on Tyr-323 and Tyr-352 was unaffected by PKC inhibition. As Syk residues Tyr-525/526 are located in the kinase domain, its phosphorylation is recognized as an activation marker. Our study also shows that in platelets, tyrosine phosphorylations on Syk downstream targets LAT and PLCγ2 are also hyperphosphorylated with GFX.
The realization that PKCβ differentially regulates Syk phosphorylation and function argues against a regulation on upstream events. As Syk tyrosine phosphorylation is a result of an autophosphorylation process (25), we checked whether the regulation was at the level of SFKs. We analyzed the phosphorylation state of SFK Tyr-416 and of its direct target, the FcRγ chain. As expected, no effect was seen on these two markers upon PKC inhibition (Fig. 3, A and B). This result is different from the one reported by Pula et al. (14) as they show a potentiation of Src activity when alboaggregin A-stimulated platelets were treated with the pan- PKC inhibitor GFX or the classical PKC inhibitor, Go6976. The reason for this difference is unknown, but it is important to note that our group has recently identified that Go6976 has nonspecific effects as it inhibits Syk activity (35). Although the effect on SFK was not tested, it is reasonable to think that the nonspecific effects of the inhibitor might be affecting other tyrosine kinases. With respect to GFX, Pula et al. (14) used a much higher concentration than ours (20 μm versus 5 μm), which might explain the observed difference.
In our experimental design, the lack of animal models and the use of pharmacological inhibitors of PKC, which affects several platelet functional responses (e.g. granule secretion), preclude us from further analyzing the physiological consequences of Syk hyperphosphorylation. Nonetheless, our laboratory has identified two molecules that negatively regulate Syk function in GPVI signaling: the E3 ubiquitin ligase c-Cbl (18) and the histidine tyrosine phosphatase TULA-2 (19). Both animal KO models exhibit Syk hyperphosphorylation, enhanced kinase activity, and importantly, more robust platelet functional responses. It is then logical to infer that abnormalities in Syk activity, as a result of PKCβ regulation, may lead to impaired functional responses. As per the mechanism involved in this negative regulation, it is noteworthy that the phenotypes observed in c-Cbl KO platelets and TULA-2 KO platelets seem similar to pharmacological inhibition of PKCβ in human platelets. Ongoing studies in our laboratory have identified both TULA-2 and c-Cbl as PKC substrates (data not shown). Further, it has been shown that TULA-2 can associate with Syk in platelets upon GPVI stimulation (19). Thus, a probable PKCβ-mediated phosphorylation event might regulate the association of these molecules leading to Syk ubiquitination and TULA-2-dependent Syk dephosphorylation. It is important to note that in murine platelets, TULA-2 is the phosphatase responsible for dephosphorylating Syk. All tyrosine residues tested in the TULA KO model, especially Syk Tyr-352, were found to be hyperphosphorylated. The reason that Syk Tyr-352 and Tyr-323 in human platelets do not undergo the same PKCβ-dependent regulation as Tyr-525/Tyr-526 is unknown, but the possibility exists for another phosphatase regulating these residues in human platelets.
The difference between human and murine platelet regulation of Syk phosphorylation is also not known at this time. We observed potentiation of Syk tyrosine phosphorylation in human platelets (Fig. 1Bi) with GFX, whereas we observed inhibition of Syk tyrosine phosphorylation in murine platelets (Fig. 1E) in agreement with the previous findings (17). However, the differential regulation can be attributed to the different phosphatases that are regulated by PKCβ in platelets; the phosphatase that differentially regulates Syk in human and murine platelets has yet to be identified. This study further highlights the difference in the signaling events between human and murine platelets, and hence some conclusions drawn from mouse platelet studies have to be considered cautiously.
Pasquet et al. (36) demonstrated a role for the phosphatase Shp1 in GPVI signaling. In their studies, Syk and Shp1 co-immunoprecipitated following GPVI stimulation, and Syk hyperphosphorylation was observed in Shp1-deficient platelets. More importantly, Shp1 regulation by GPVI signaling occurred only after Ca2+ mobilization. Therefore, a possible divergence in the phosphatases that regulate Syk function toward Ca2+-dependent and -independent functions might exist. Further studies are necessary to understand the mechanisms by which PKCβ regulates Syk and GPVI signaling in human platelets. In conclusion, we have elucidated a novel pathway of Syk regulation by PKCβ that is observed in human platelets but not in murine platelets.
This work was supported, in whole or in part, by National Institutes of Health Research Grant HL 93231 (to S. P. K.).
- GPVI
- glycoprotein VI
- CLEC-2
- C-type lectin-like receptor-2
- Cvx
- convulxin
- FcRγ
- Fc receptor γ-chain
- GFX
- GF109203X
- ITAM
- immunoreceptor tyrosine activation motif
- LAT
- linker of activated T cells
- PLCγ2
- phospholipase Cγ2
- SFK
- Src family kinase
- Syk
- spleen tyrosine kinase.
REFERENCES
- 1. Blockmans D., Deckmyn H., Vermylen J. (1995) Platelet activation. Blood Rev. 9, 143–156 [DOI] [PubMed] [Google Scholar]
- 2. Nieswandt B., Watson S. P. (2003) Platelet-collagen interaction: is GPVI the central receptor? Blood 102, 449–461 [DOI] [PubMed] [Google Scholar]
- 3. Ezumi Y., Shindoh K., Tsuji M., Takayama H. (1998) Physical and functional association of the Src family kinases Fyn and Lyn with the collagen receptor glycoprotein VI-Fc receptor γ-chain complex on human platelets. J. Exp. Med. 188, 267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Suzuki-Inoue K., Tulasne D., Shen Y., Bori-Sanz T., Inoue O., Jung S. M., Moroi M., Andrews R. K., Berndt M. C., Watson S. P. (2002) Association of Fyn and Lyn with the proline-rich domain of glycoprotein VI regulates intracellular signaling. J. Biol. Chem. 277, 21561–21566 [DOI] [PubMed] [Google Scholar]
- 5. Poole A., Gibbins J. M., Turner M., van Vugt M. J., van de Winkel J. G., Saito T., Tybulewicz V. L., Watson S. P. (1997) The Fc receptor γ-chain and the tyrosine kinase Syk are essential for activation of mouse platelets by collagen. EMBO J. 16, 2333–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Violin J. D., Newton A. C. (2003) Pathway illuminated: visualizing protein kinase C signaling. IUBMB Life 55, 653–660 [DOI] [PubMed] [Google Scholar]
- 7. Nakanishi H., Brewer K. A., Exton J. H. (1993) Activation of the ζ isozyme of protein kinase C by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 268, 13–16 [PubMed] [Google Scholar]
- 8. Chari R., Kim S., Murugappan S., Sanjay A., Daniel J. L., Kunapuli S. P. (2009) Lyn, PKC-δ, SHIP-1 interactions regulate GPVI-mediated platelet-dense granule secretion. Blood 114, 3056–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Harper M. T., Poole A. W. (2009) PKCθ in platelet activation. Blood 114, 489–491; author reply 491–492 [DOI] [PubMed] [Google Scholar]
- 10. Nagy B., Jr., Bhavaraju K., Getz T., Bynagari Y. S., Kim S., Kunapuli S. P. (2009) Impaired activation of platelets lacking protein kinase C-θ isoform. Blood 113, 2557–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harper M. T., Poole A. W. (2010) Protein kinase Cθ negatively regulates store-independent Ca2+ entry and phosphatidylserine exposure downstream of glycoprotein VI in platelets. J. Biol. Chem. 285, 19865–19873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gilio K., Harper M. T., Cosemans J. M., Konopatskaya O., Munnix I. C., Prinzen L., Leitges M., Liu Q., Molkentin J. D., Heemskerk J. W., Poole A. W. (2010) Functional divergence of platelet protein kinase C (PKC) isoforms in thrombus formation on collagen. J. Biol. Chem. 285, 23410–23419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bijli K. M., Fazal F., Minhajuddin M., Rahman A. (2008) Activation of Syk by protein kinase C-δ regulates thrombin-induced intercellular adhesion molecule-1 expression in endothelial cells via tyrosine phosphorylation of RelA/p65. J. Biol. Chem. 283, 14674–14684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pula G., Crosby D., Baker J., Poole A. W. (2005) Functional interaction of protein kinase Cα with the tyrosine kinases Syk and Src in human platelets. J. Biol. Chem. 280, 7194–7205 [DOI] [PubMed] [Google Scholar]
- 15. Konishi H., Yamauchi E., Taniguchi H., Yamamoto T., Matsuzaki H., Takemura Y., Ohmae K., Kikkawa U., Nishizuka Y. (2001) Phosphorylation sites of protein kinase Cδ in H2O2-treated cells and its activation by tyrosine kinase in vitro. Proc. Natl. Acad. Sci. U.S.A. 98, 6587–6592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kronfeld I., Kazimirsky G., Lorenzo P. S., Garfield S. H., Blumberg P. M., Brodie C. (2000) Phosphorylation of protein kinase Cδ on distinct tyrosine residues regulates specific cellular functions. J. Biol. Chem. 275, 35491–35498 [DOI] [PubMed] [Google Scholar]
- 17. Pears C. J., Thornber K., Auger J. M., Hughes C. E., Grygielska B., Protty M. B., Pearce A. C., Watson S. P. (2008) Differential roles of the PKC novel isoforms, PKCδ and PKCϵ, in mouse and human platelets. PLoS One 3, e3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dangelmaier C. A., Quinter P. G., Jin J., Tsygankov A. Y., Kunapuli S. P., Daniel J. L. (2005) Rapid ubiquitination of Syk following GPVI activation in platelets. Blood 105, 3918–3924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thomas D. H., Getz T. M., Newman T. N., Dangelmaier C. A., Carpino N., Kunapuli S. P., Tsygankov A. Y., Daniel J. L. (2010) A novel histidine tyrosine phosphatase, TULA-2, associates with Syk and negatively regulates GPVI signaling in platelets. Blood 116, 2570–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Polgár J., Clemetson J. M., Kehrel B. E., Wiedemann M., Magnenat E. M., Wells T. N., Clemetson K. J. (1997) Platelet activation and signal transduction by convulxin, a C-type lectin from Crotalus durissus terrificus (tropical rattlesnake) venom via the p62/GPVI collagen receptor. J. Biol. Chem. 272, 13576–13583 [DOI] [PubMed] [Google Scholar]
- 21. Buitrago L., Langdon W. Y., Sanjay A., Kunapuli S. P. (2011) Tyrosine-phosphorylated c-Cbl regulates platelet functional responses mediated by outside-in signaling. Blood 118, 5631–5640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bhavanasi D., Kim S., Goldfinger L. E., Kunapuli S. P. (2011) Protein kinase Cδ mediates the activation of protein kinase D2 in platelets. Biochem. Pharmacol. 82, 720–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Quinter P. G., Dangelmaier C. A., Quinton T. M., Kunapuli S. P., Daniel J. L. (2007) Glycoprotein VI agonists have distinct dependences on the lipid raft environment. J. Thromb. Haemost. 5, 362–368 [DOI] [PubMed] [Google Scholar]
- 24. Watson S. P., Auger J. M., McCarty O. J., Pearce A. C. (2005) GPVI and integrin αIIbβ3 signaling in platelets. J. Thromb. Haemost. 3, 1752–1762 [DOI] [PubMed] [Google Scholar]
- 25. Sada K., Takano T., Yanagi S., Yamamura H. (2001) Structure and function of Syk protein-tyrosine kinase. J. Biochem. 130, 177–186 [DOI] [PubMed] [Google Scholar]
- 26. Nieswandt B., Bergmeier W., Eckly A., Schulte V., Ohlmann P., Cazenave J. P., Zirngibl H., Offermanns S., Gachet C. (2001) Evidence for cross-talk between glycoprotein VI and Gi-coupled receptors during collagen-induced platelet aggregation. Blood 97, 3829–3835 [DOI] [PubMed] [Google Scholar]
- 27. Kim S., Mangin P., Dangelmaier C., Lillian R., Jackson S. P., Daniel J. L., Kunapuli S. P. (2009) Role of phosphoinositide 3-kinase β in glycoprotein VI-mediated Akt activation in platelets. J. Biol. Chem. 284, 33763–33772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kasirer-Friede A., Kahn M. L., Shattil S. J. (2007) Platelet integrins and immunoreceptors. Immunol. Rev. 218, 247–264 [DOI] [PubMed] [Google Scholar]
- 29. Mocsai A., Ruland J., Tybulewicz V. L. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat. Rev. Immunol. 10, 387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boylan B., Gao C., Rathore V., Gill J. C., Newman D. K., Newman P. J. (2008) Identification of FcγRIIa as the ITAM-bearing receptor mediating αIIbβ3 outside-in integrin signaling in human platelets. Blood 112, 2780–2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Séverin S., Pollitt A. Y., Navarro-Nuñez L., Nash C. A., Mourão-Sá D., Eble J. A., Senis Y. A., Watson S. P. (2011) Syk-dependent phosphorylation of CLEC-2: a novel mechanism of hem-immunoreceptor tyrosine-based activation motif signaling. J. Biol. Chem. 286, 4107–4116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Watson S. P., Herbert J. M., Pollitt A. Y. (2010) GPVI and CLEC-2 in hemostasis and vascular integrity. J. Thromb. Haemost. 8, 1456–1467 [DOI] [PubMed] [Google Scholar]
- 33. Manne B. K., Getz T. M., Hughes C. E., Alshehri O., Dangelmaier C., Naik U. P., Watson S. P., Kunapuli S. P. (2013) Fucoidan is a novel platelet agonist for the C-type lectin-like receptor 2 (CLEC-2). J. Biol. Chem. 288, 7717–7726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Konopatskaya O., Gilio K., Harper M. T., Zhao Y., Cosemans J. M., Karim Z. A., Whiteheart S. W., Molkentin J. D., Verkade P., Watson S. P., Heemskerk J. W., Poole A. W. (2009) PKCα regulates platelet granule secretion and thrombus formation in mice. J. Clin. Invest. 119, 399–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chari R., Getz T., Nagy B., Jr., Bhavaraju K., Mao Y., Bynagari Y. S., Murugappan S., Nakayama K., Kunapuli S. P. (2009) Protein kinase Cδ differentially regulates platelet functional responses. Arterioscler. Thromb. Vasc. Biol. 29, 699–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pasquet J. M., Quek L., Pasquet S., Poole A., Matthews J. R., Lowell C., Watson S. P. (2000) Evidence of a role for SHP-1 in platelet activation by the collagen receptor glycoprotein VI. J. Biol. Chem. 275, 28526–28531 [DOI] [PubMed] [Google Scholar]