

Background: Cross-talk between GPCRs is an important but undercharacterized mechanism regulating receptor responsiveness.
Results: Co-activation of β and α2ARs accelerates α2AAR endocytosis in a PKA- and spinophilin-dependent fashion.
Conclusion: βAR-mediated signaling modulates α2AAR endocytosis via PKA-dependent disruption of α2AAR/spinophilin interaction.
Significance: Cross-talk from β to α2ARs may have important implications in basal adrenergic tone and the pharmacology of commonly used adrenergic therapeutics.
Keywords: Adrenergic Receptor, Endocytosis, G Protein-coupled receptors (GPCR), Neurons, Protein Kinase A (PKA), Cross-talk, Spinophilin
Abstract
Inter-regulation of adrenergic receptors (ARs) via cross-talk is a long appreciated but mechanistically unclear physiological phenomenon. Evidence from the AR literature and our own extensive studies on regulation of α2AARs by the scaffolding protein spinophilin have illuminated a potential novel mechanism for cross-talk from β to α2ARs. In the present study, we have characterized a mode of endogenous AR cross-talk in native adrenergic neurons whereby canonical βAR-mediated signaling modulates spinophilin-regulated α2AAR endocytosis through PKA. Our findings demonstrate that co-activation of β and α2AARs, either by application of endogenous agonist or by simultaneous stimulation with distinct selective agonists, results in acceleration of endogenous α2AAR endocytosis in native neurons. We show that receptor-independent PKA activation by forskolin is sufficient to accelerate α2AAR endocytosis and that α2AAR stimulation alone drives accelerated endocytosis in spinophilin-null neurons. Endocytic response acceleration by β/α2AAR co-activation is blocked by PKA inhibition and lost in spinophilin-null neurons, consistent with our previous finding that spinophilin is a substrate for phosphorylation by PKA that disrupts its interaction with α2AARs. Importantly, we show that α2AR agonist-mediated α2AAR/spinophilin interaction is blocked by βAR co-activation in a PKA-dependent fashion. We therefore propose a novel mechanism for cross-talk from β to α2ARs, whereby canonical βAR-mediated signaling coupled to PKA activation results in phosphorylation of spinophilin, disrupting its interaction with α2AARs and accelerating α2AAR endocytic responses. This mechanism of cross-talk has significant implications for endogenous adrenergic physiology and for therapeutic targeting of β and α2AARs.
Introduction
The physiological phenomenon of cross-talk between β- and α2-adrenergic receptor (AR)2 subtypes has long been indicated by reports in the G protein-coupled receptor (GPCR) literature. Inter-regulation of β and α2ARs has been described in in vitro cell models (1–5), in vivo central (6–12), and peripheral (13) nervous systems and rodent development (14). Despite this accumulation of evidence, a clear picture of the mechanisms underlying AR cross-talk has yet to emerge, particularly as regards the unidirectional influence of βAR activity on α2AR function. Such information is vital given that the ARs are an important GPCR family responsible for mediating responses to the endogenous agonists epinephrine (Epi) and norepinephrine. These receptors exhibit wide distribution in the body and have myriad well appreciated functions, with most cell types expressing some combination of AR subtypes. Perhaps unsurprisingly, there is significant overlap in both the physiology and pharmacology of the ARs (15–17). Given that overlap, any new insights into AR inter-relationships and the mechanisms underlying AR cross-talk will contribute to a better understanding of adrenergic physiology and pharmacology.
We have previously carried out extensive studies on the function of the α2AAR subtype and its regulation by non-G protein-interacting partners. Our work has identified the scaffolding protein spinophilin (18–20) as an α2AAR interacting partner (21–23), and we have characterized a novel regulatory mechanism whereby spinophilin serves as a functional antagonist at the α2AAR to the traditional GPCR-interacting partners GPCR kinase and arrestin (24). We have demonstrated the importance of this regulatory mechanism both in vitro and in vivo, finding that a number of agonist-dependent α2AAR-mediated responses are enhanced and/or accelerated in the absence of spinophilin (24–26). Intriguingly, our work has also pinpointed spinophilin as a potential link between α2A and βARs. Spinophilin is known to be a substrate for phosphorylation by PKA (27), and we have shown that this modification disrupts spinophilin/α2AAR interaction and accelerates agonist-driven receptor endocytosis (28). Meanwhile, canonical βAR signal transduction results in activation of PKA downstream of Gαs-containing heterotrimeric G proteins (29). Therefore, in the present study, we hypothesize that co-activation of βARs will accelerate agonist-driven α2AAR endocytosis via PKA-dependent phosphorylation of spinophilin, disrupting its interaction with α2AARs.
In the present study, we have elected to utilize endocytosis of endogenous receptors as a functional readout that can be examined cleanly and specifically through the use of our previously reported novel epitope-tagged α2AAR knock-in mouse model (30). By culturing from the superior cervical ganglia (SCG), we can obtain a 98% pure population of adrenergic sympathetic neurons (31), allowing us to investigate endocytic responses in a native cell type with endogenous expression of α2AARs, βARs, and interacting protein partners. Furthermore, endocytosis is itself an important GPCR response, under tight and complex control, which is intimately involved in determining acute and long term neuronal responsiveness to both endogenous neuromodulators and exogenous therapeutics (32, 33). Indeed, our past findings have underscored the importance of spinophilin/arrestin-regulated endocytosis for α2AAR signal transduction (24, 34).
Our results indicate that co-activation of α2A and βARs, either by application of endogenous agonist or by simultaneous stimulation with distinct selective agonists, results in an acceleration of α2AAR endocytosis in native adrenergic neurons. This acceleration occurs in a PKA- and spinophilin-dependent fashion, whereas a similar acceleration of agonist-driven α2AAR endocytosis is observed either with receptor-independent activation of PKA by forskolin or in spinophilin-null neurons. We further show that βAR co-activation disrupts agonist-dependent α2AAR/spinophilin interaction in a PKA-dependent fashion. In sum, our data establish a novel mechanism for unidirectional cross-talk from β to α2ARs affecting α2AAR responsiveness in a setting with significant physiological and pharmacological importance.
EXPERIMENTAL PROCEDURES
Animals
Mice were housed in an Association for Assessment and Accreditation of Laboratory Animal Care-accredited animal resources program facility at the University of Alabama at Birmingham in accordance with the Animal Welfare Act and the 1989 amendments to that act. All studies followed protocols approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee. The generation of HA-tagged α2AAR knock-in (30), spinophilin-null (35), and arrestin3-null (36) mice has been previously described. These transgenic lines were backcrossed over 10 generations to a pure C57BL/6 genetic background. HA-α2AAR mice were crossed with spinophilin-null mice to generate a HA+/+/spinophilin−/− line (which will be referred to as Sp−/− in the interest of simplicity), as well as with arrestin3-null mice to generate a HA+/+/arrestin3−/− line (which will be referred to as Arr3−/− in the interest of simplicity).
Primary Culture of SCG Neurons
SCG neurons were cultured from mouse pups at postnatal day 4–6 as described previously (30, 37) with slight modifications. Briefly, SCG were dissected and placed into Hanks' balanced salt solution (Invitrogen) containing 25 mm glucose and 20 mm HEPES, pH 7.3, and subjected to enzymatic digestion with 3 mg/ml collagenase and 1 mg/ml trypsin (Sigma). Neurons were dissociated by trituration with a fire-polished siliconized Pasteur pipette and, after a preplating step to reduce non-neuronal cell types in the final culture, plated onto coverslips treated with poly-d-lysine and laminin (Sigma). Growth medium was L-15 base medium (Invitrogen) plus 10% Nu-Serum (Clontech), 30% glucose, 2% GlutaMAX (Invitrogen), 1% insulin/transferrin/selenium supplement (Invitrogen), 25 ng/ml nerve growth factor (Sigma), and 24 mm NaHCO3. Medium changes were on days in vitro 1, 4, and 6, with the addition of 10 μm 5-fluoro-2′-deoxyuridine (Sigma) on days 1 and 4 to control non-neuronal cell growth, and 1 μm yohimbine (α2AR antagonist; Sigma) on days 4 and 6 to protect surface α2AARs. For immunofluorescent staining, neurons were plated at a ganglion to coverslip ratio of 1:1. All experiments were performed on day in vitro 8, a time point at which α2AARs have robust somatodendritic and axonal surface expression in SCG neurons (37).
Immunofluorescent Staining
Internalization of HA-α2AARs was assessed by a prelabeling method that has been well described previously (28, 30). All staining experiments detected HA-tagged α2AARs. As an initial step prior to antibody prelabeling/drug treatments, neurons were washed thoroughly to remove yohimbine. HA-α2AARs were detected with HA.11 primary antibody (Covance, 1:100 dilution), which was used for a 20-min prelabeling of surface α2AAR population at room temperature prior to agonist stimulation. Cells were then permeabilized, blocked, and incubated with AlexaFluor 488-conjugated goat anti-mouse secondary antibody (Invitrogen, 1:1,000 dilution) for 1 h at room temperature. Images were obtained using a Zeiss LSM 710 confocal microscope (Carl Zeiss) at 63× magnification. For quantitative assessment of receptor internalization, images were analyzed with MetaMorph software (Molecular Devices) to determine total and intracellular fluorescent intensities as described previously (22). A “relative internalization unit” for stimulated cells was then calculated as a ratio of intracellular to total fluorescent intensity normalized to matched unstimulated controls (30). A minimum of 12–14 neurons collected over at least three independent samples were analyzed for each data group, with the exception of clonidine + SAL (n = 10).
For the HA-α2AAR double-labeling experiment, surface receptors were prelabeled as above. Nonpermeabilized neurons were then incubated with AlexaFluor 488-conjugated anti-mouse secondary antibody (1:250 dilution) for 1 h at room temperature to saturate prelabeled surface receptors. After permeabilization/blocking, neurons were incubated with AlexaFluor 594-conjugated secondary antibody (Invitrogen, 1:1,000 dilution) for 1 h at room temperature to detect prelabeled cytosolic (endocytosed) receptors.
Immunostaining of LAMP1 was performed together with the prelabeling method to detect both HA-α2AARs and LAMP1. After HA prelabeling and permeabilization/blocking, neurons were incubated with anti-LAMP1 primary antibody (University of Iowa Hybridoma Bank, 1:400 dilution) overnight at 4 °C. The cells were then subjected to secondary labeling with AlexaFluor 488-conjugated anti-mouse and AlexaFluor 594-conjugated anti-rat (Invitrogen) antibodies (1:1,000 dilution) for 1 h at room temperature.
For adenylyl cyclase activation, neurons were pretreated with forskolin or vehicle (Me2SO), and forskolin/vehicle was maintained during stimulation with Epi (Sigma, 100 μm final). Epi stimulation was done either alone (for simultaneous activation of β and α2AARs) or in combination with the non-subtype-selective βAR antagonist propranolol (Sigma, 1 μm final) for activation of α2AARs only. For βAR co-activation, neurons were pretreated for 10 min with the non-subtype-selective agonist isoproterenol (ISO, 100 μm final) or one of several agonists with varying selectivity for β1 versus β2ARs: dobutamine (DOB, 1 μm final), albuterol (ALB, 1 μm final), or salmeterol (SAL, 100 nm final). ISO/DOB/ALB/SAL was maintained during stimulation with the α2AR agonist clonidine (Sigma, 1 μm final). For PKA inhibition, neurons were subjected to a 10-min pretreatment with myristoylated protein kinase A inhibitor 14–22 amide (PKI, Calbiochem, 8.3 μm final), with PKI then maintained through ISO pretreatment and clonidine stimulation. Previous enzymological evidence indicates that this PKI concentration achieves effectively complete inhibition of PKA (38, 39). Prazosin (Sigma, 1 μm final) was included in all experiments to block potential activation of α1 and α2B/CAR subtypes.
Determination of Arrestin Dependence
For these experiments, SCG neurons were cultured from our HA+/+/arrestin3−/− line (referred to as Arr3−/− in the interest of simplicity) described above. Arrestin redistribution was examined through the use of a rabbit polyclonal antibody against endogenous arrestin2 (a generous gift of Dr. Jeffrey Benovic, Thomas Jefferson University) and AlexaFluor 594-conjugated anti-rabbit secondary antibody (Invitrogen, 1:1,000 dilution). Arrestin2 knockdown was achieved by lentiviral constructs encoding shRNA against mouse arrestin2, purchased from Open Biosystems and packaged using the ViraPower Lentiviral Packaging System (Invitrogen) according to the manufacturer's instructions. Arrestin3-null SCG neurons were transduced on day in vitro 3, and experiments were performed on day in vitro 8 as above.
FLIM-FRET
FLIM-based FRET experiments were utilized to directly observe α2AAR/spinophilin interaction in live cells according to a previously described method (34). The C-terminally CFP-tagged α2AAR construct has been reported (34), and the N-terminally YFP-tagged spinophilin construct was prepared by PCR amplification and cloning of cDNA encoding spinophilin into the pEYFP-C1 vector (Clontech), with the construct verified by sequencing prior to use. HEK293 cells were transiently transfected using Lipofectamine 2000 (Sigma) with the plasmids containing either CFP-α2AAR alone (2 μg/60-mm plate) or in combination with YFP-spinophilin (1 μg/60-mm plate). For each treatment group, five or six individual cells from two or three independent samples were imaged and analyzed. FLIM-FRET efficiency (E) was calculated as: E = 1 − (tFRET/tCFP), where tFRET and tCFP are CFP lifetime values obtained from cells expressing CFP and YFP together and CFP alone, respectively (34). Cells were stimulated with clonidine plus prazosin at 1 μm, with βAR co-activation achieved as described above.
Mouse Embryonic Fibroblasts (MEFs)
The isolation of MEFs from spinophilin-null mouse mice and generation of a spinophilin-null MEF line stably expressing HA-tagged α2AARs have been previously described (40). Spinophilin-null MEFs were transfected using Lipofectamine 2000 with plasmids encoding GFP-tagged wild-type spinophilin (referred to as WT-Sp) or GFP-tagged mutant spinophilin lacking PKA phosphorylation sites (Sp94A, 177A, referred to as mut-Sp), 3 μg/60-mm plate. Transfected MEFs were split to coverslips on a 24-well culture plate ∼24 h post-transfection, and immunostaining for α2AAR endocytosis was performed 48 h post-transfection as described above, with the substitution of AlexaFluor 594-conjugated anti-mouse (Invitrogen) secondary labeling. MEF cells were visualized via confocal microscopy using a Nikon A1 scope (Nikon) at 60× magnification.
cAMP Production Assay
cAMP production in MEFs was measured using the AlphaScreen cAMP assay kit (PerkinElmer Life Sciences) according to the manufacturer's instructions. A total of 2 × 104 cells/assay well were used, and cells were exposed to 100 μm ISO for 30 min at room temperature to determine cAMP response mediated by endogenous βARs. MEFs isolated from WT and spinophilin-null mice of matched genetic background were compared in parallel. AlphaScreen cAMP reporter readings were acquired using a Synergy 2 microplate reader (Biotek). Relative cAMP production was calculated by converting absolute value change in raw AlphaScreen signal to a fold change over control cells.
RESULTS
Epinephrine-mediated α2AAR Endocytosis in Native Neurons Is Arrestin-dependent and Accelerated in Spinophilin-null Neurons
SCG neurons exhibit extensive expression of both α2AAR and α2CAR subtypes (37) in addition to β and α1 AR subtypes. To focus on the α2A subtype, we have made use of our HA-tagged α2AAR knock-in mouse model for clean and specific detection of α2AARs, and we have included appropriate AR blockers (prazosin for α1 and α2B/C ARs, propranolol for βARs) to ensure stimulation of α2AARs alone. We have previously demonstrated that α2AAR expression level, distribution, localization, and functional properties are unaltered in the knock-in line (30). Additionally, we have made exclusive use of immunostaining and imaging methods to assay endocytosis, because SCG culture yields (less than 2,000 cells/ganglion) limit our ability to perform other biochemical or ELISA-based techniques.
As shown in Fig. 1A, stimulation with Epi (plus prazosin/propranolol) drives endocytosis of endogenously expressed α2AARs in the native SCG neurons. Our primary antibody prelabeling plus double secondary antibody labeling method (see “Experimental Procedures”) detects internalized receptors by the appearance of cytosolic staining in stimulated but not control cells.
FIGURE 1.
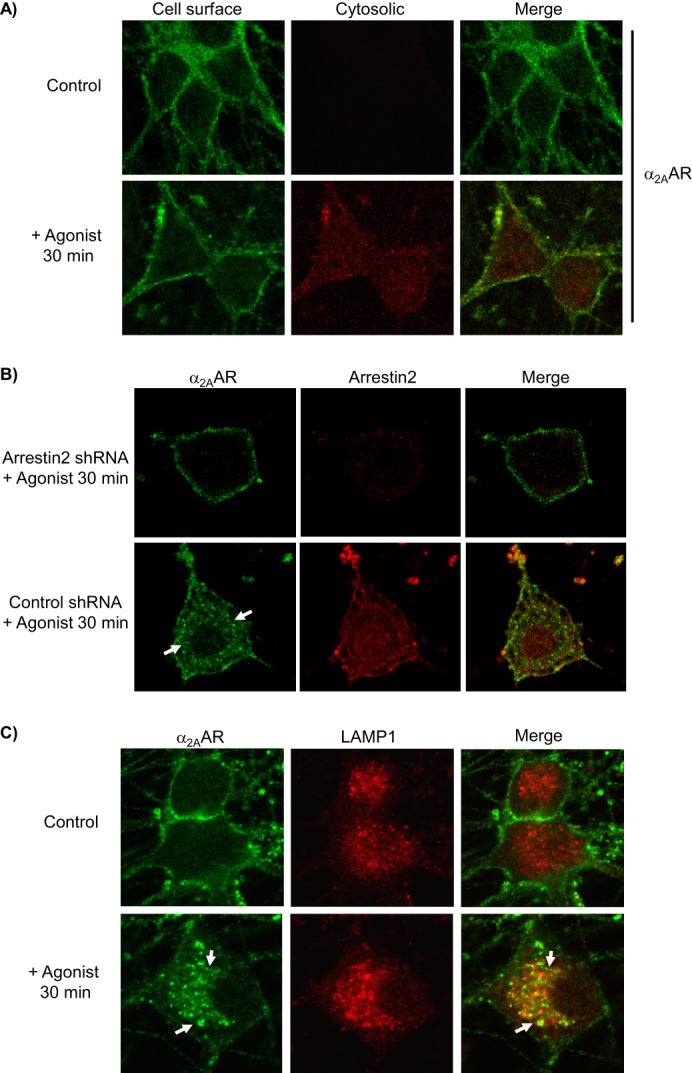
Evaluation of arrestin-dependent agonist-stimulated α2AAR endocytosis in native neurons. All immunostaining experiments detect endogenously expressed HA-α2AARs. A, neurons were subjected to primary antibody prelabeling of surface α2AARs prior to stimulation, followed by saturation of labeled surface receptors with AlexaFluor488-conjugated secondary labeling, and then permeabilization and cytosolic AlexaFluor594-conjugated secondary labeling. Cytosolic labeling was seen only in stimulated cells, indicating endocytosis of prelabeled surface α2AARs. B, endocytosis was assayed in arrestin3-null neurons, transduced with either arrestin2 shRNA (to achieve complete ablation of arrestins) or control shRNA. Analysis revealed that arrestin2 shRNA resulted in a 69 ± 5.8% reduction in arrestin2 immunoreactivity. α2AARs were detected by primary antibody prelabeling followed by permeabilization and AlexaFluor488-conjugated secondary labeling. In control but not arrestin2 shRNA cells, agonist stimulation resulted in significant α2AAR endocytosis, indicated by the appearance of intracellular punctae containing internalized receptors (arrows), as well as a partial co-localization of α2AARs with arrestin2. C, endocytosis was additionally observed by monitoring α2AAR co-localization with the late endolysosomal pathway marker LAMP1; prelabeling of α2AARs was done as in A, with the addition of co-immunostaining for LAMP1. In agonist-stimulated but not unstimulated control neurons, endocytosis was seen as indicated by the appearance of intracellular punctae which exhibited co-localization with LAMP1 (arrows). Confocal images are representative of at least three independent samples.
The nonvisual arrestins (arrestin2 and 3, also known as β-arrestin1 and 2) are key mediators of classical GPCR endocytosis via clathrin-coated pits (32, 41, 42). To determine whether this classical endocytosis was being driven for endogenous α2AARs in SCG neurons, we cultured neurons from arrestin3-null mice; we have found no difference in α2AAR density between arrestin3-null and WT mice (34). Those cells were then transduced with either arrestin2 shRNA (to achieve ablation of both arrestins) or control shRNA. We then utilized our single prelabeling method (see “Experimental Procedures”), which allows us to monitor the initial surface α2AAR population. Agonist stimulation drove significant α2AAR endocytosis, indicated by the appearance of characteristic intracellular punctae containing internalized receptors, in control shRNA neurons but not in arrestin2 shRNA neurons (Fig. 1B). These data demonstrate that arrestins are required for agonist-mediated endocytosis of endogenously expressed α2AARs.
To provide further confirmation of our ability to observe α2AAR endocytosis in SCG neurons via our prelabeling method, we performed co-immunostaining for lysosomal-associated membrane protein 1 (LAMP1), a late endolysosomal pathway marker. As shown in Fig. 1C, following agonist stimulation, prelabeled internalized α2AARs (intracellular punctae seen clearly in the lower left panel) exhibit partial co-localization with LAMP1 (lower right panel), strongly suggesting that they are internalized receptors that have entered the endolysosomal pathway. Collectively, these results establish agonist-mediated arrestin-dependent endocytosis of endogenously expressed α2AARs in our native SCG cell model.
We have previously established a clear regulatory mechanism for α2AAR endocytosis involving functional antagonism of arrestin functions by spinophilin (24). However, this regulatory mechanism has not been reported for endogenously expressed α2AARs in native SCG neurons. We therefore compared the kinetics of endocytosis induced by Epi (plus prazosin/propranolol) in neurons with and without spinophilin expression (SpWT and Sp−/−, respectively). As would be predicted by our regulatory model, we observed a clear acceleration of α2AAR endocytosis in spinophilin-null neurons, with significantly enhanced endocytosis at the 5-min time point in Sp−/− versus SpWT neurons (Fig. 2). Two-way ANOVA revealed significant effects of genotype (p = 0.0045) and time (p < 0.0001) and a significant genotype × time interaction (p < 0.0001). It should be noted that our past work indicates no difference in α2AAR density between spinophilin-null and WT mice (25). These results are consistent with our previous findings in heterologous cells and can likely be attributed to unimpeded agonist-dependent binding of arrestins to the α2AAR in the absence of opposition from spinophilin.
FIGURE 2.
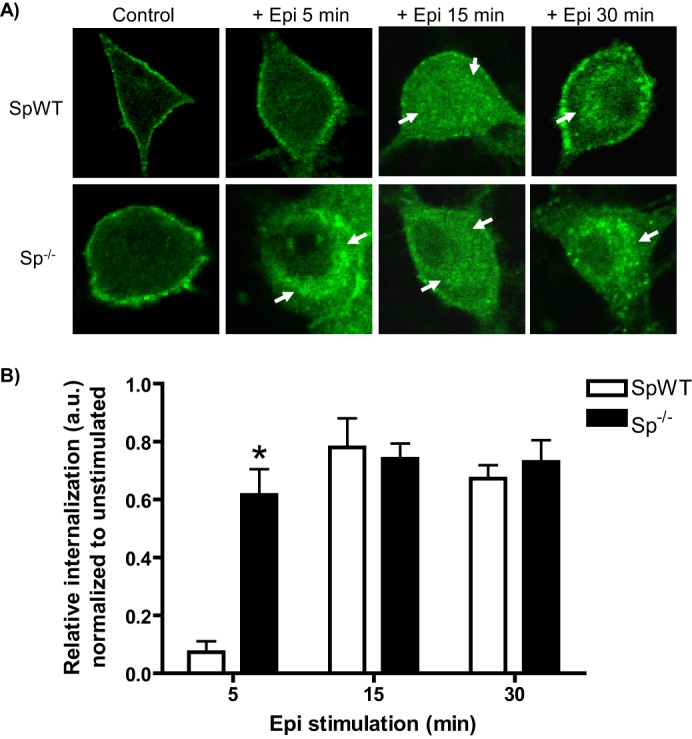
Epinephrine-mediated α2AAR endocytosis is regulated by spinophilin. A, neurons with (SpWT) and without (Sp−/−) spinophilin expression were stimulated by application of Epi (100 μm, plus prazosin and propranolol) as indicated. Neurons were subjected to α2AAR prelabeling method as in Fig. 1 (B and C). Endocytosis, indicated by the appearance of intracellular punctae containing internalized receptors (arrows), was observed beginning at 5 min in Sp−/− neurons and at 15 and 30 min in both SpWT and Sp−/− neurons, consistent with endocytic acceleration in the absence of spinophilin. B, quantitation of agonist-mediated α2AAR endocytosis in Epi-stimulated SpWT and Sp−/− neurons, with relative internalization determined as described under “Experimental Procedures.” Confocal images are representative of at least three independent samples, and quantitation was performed over at least 12–14 neurons. *, p < 0.01, SpWT versus Sp−/−.
Co-activation of β and α2AARs Accelerates α2AAR Endocytosis in a PKA-dependent Fashion
Given the preponderance of evidence suggesting β/α2AR cross-talk described at the outset, and endogenous expression of a full range of AR subtypes in the adrenergic SCG neurons, we decided to investigate whether simultaneous activation of both β and α2AARs would affect the α2AAR endocytic response.
We began by attempting to more closely model the physiological setting with Epi stimulation in the absence βAR blockade, although prazosin was maintained in these experiments. Under these conditions, both β and α2AARs will be co-activated by Epi. As shown in Fig. 3, Epi stimulation in the absence of propranolol (i.e., non-subtype-selective βAR blockade) drove significant α2AAR endocytosis at the early time points of 5 and 10 min. When propranolol was added, this endocytosis was effectively blocked at 5 min and significantly attenuated at 10 min (Fig. 3), indicating that the endocytic enhancement was dependent on βAR activation and suggesting the existence of modulatory cross-talk from β to α2AARs. Two-way ANOVA revealed significant effects of propranolol exposure and time (p < 0.0001) and a significant propranolol × time interaction (p = 0.0002).
FIGURE 3.
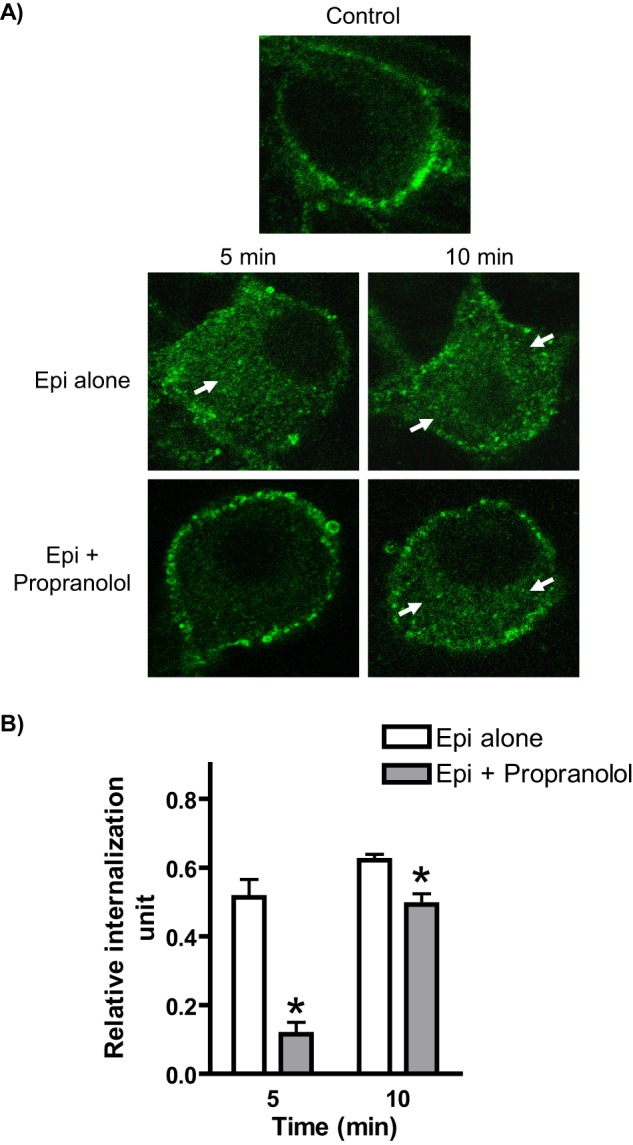
Simultaneous activation of α2A and βARs by application of Epi results in accelerated endocytosis of endogenously expressed α2AARs in native neurons. A, neurons (SpWT, subjected to α2AAR prelabeling as in Fig. 1, B and C) were stimulated by application of Epi (plus prazosin) in the absence or presence of the βAR antagonist propranolol, conditions that allow for activation of both α2A and β ARs or α2AARs alone, respectively. Significant endocytosis, as indicated by the appearance of intracellular punctae containing internalized receptors (arrows), was observed in response to Epi alone at both 5 and 10 min, whereas Epi together with propranolol drove observable endocytosis only at 10 min. B, quantitation of agonist-mediated α2AAR endocytosis in Epi-stimulated neurons the absence or presence of propranolol, with relative internalization determined as described under “Experimental Procedures.” Internalization by Epi alone was significantly attenuated by propranolol at both 5 and 10 min. Confocal images are representative of at least three independent samples, and quantitation was performed over at least 12–14 neurons. *, p < 0.01 versus Epi alone.
To further confirm the ability of βAR co-activation to regulate α2AAR endocytosis via cross-talk, we next investigated the ability of βAR stimulation by β-specific agonists to affect α2 agonist-induced α2AAR endocytosis. Based upon our previous experience with the therapeutic partial α2 agonist clonidine, we chose to focus on stimulation for 10 and 30 min, time points when clonidine drives little detectable α2AAR endocytosis and a maximal endocytic response, respectively.
We first utilized the non-subtype-selective βAR agonist ISO as a tool for co-activation. Importantly, we failed to detect any significant endocytosis of endogenous α2AARs with ISO treatment alone (Fig. 4A). In comparison with clonidine stimulation alone, we found a dramatic enhancement of α2AAR endocytosis at the 10-min time point with ISO co-treatment (Fig. 4, B and C). Intriguingly, this endocytic enhancement was prevented by the PKA inhibitor PKI, raising the possibility that canonical cAMP signaling by βARs is involved. We observed no further enhancement of clonidine-induced α2AAR endocytosis by ISO at the 30-min time point (Fig. 4, B and C), indicating that βAR co-activation does not increase the efficacy of agonist-mediated α2AAR endocytosis but rather accelerates the kinetics.
FIGURE 4.
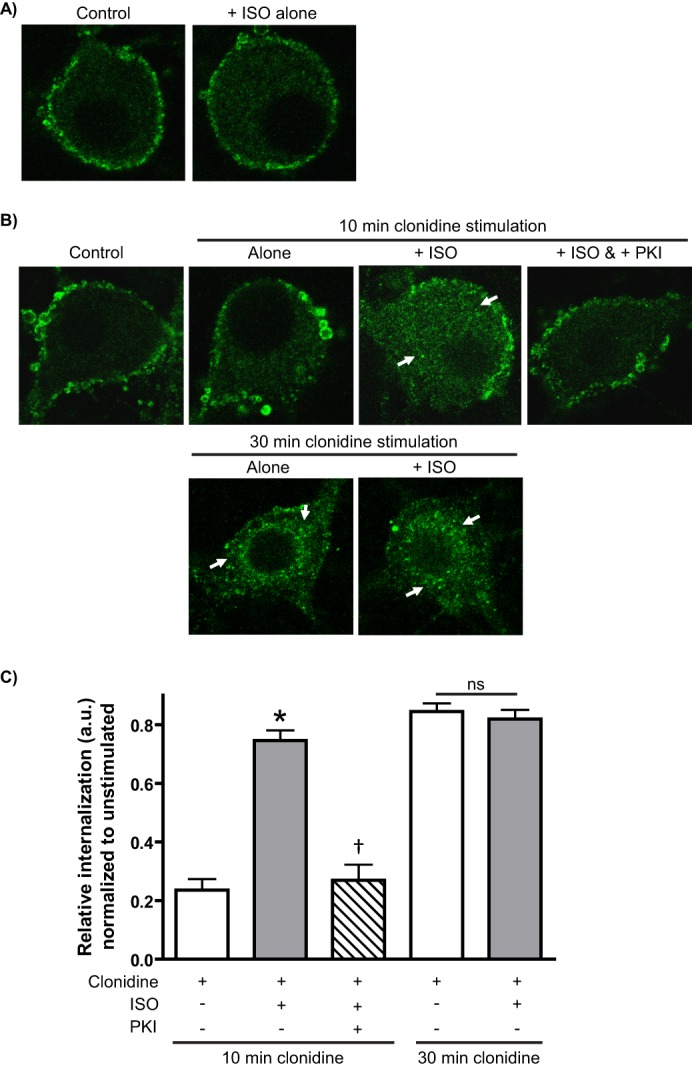
βAR co-activation by ISO accelerates α2AR agonist-mediated α2AAR endocytosis in native neurons in a PKA-dependent fashion. Neurons (SpWT) were subjected to α2AAR prelabeling method as in Fig. 1 (B and C) with endocytosis indicated by the appearance of intracellular punctae containing internalized receptors (arrows). A, ISO alone was not found to drive any significant α2AAR endocytosis. B and C, neurons were subjected to the indicated treatments; pretreatment with the non-subtype-selective βAR agonist ISO (100 μm) and/or the PKA inhibitor PKI (8.3 μm) was done prior to stimulation with the therapeutic partial α2AR agonist clonidine (1 μm). 10 min of clonidine stimulation alone was not sufficient to drive any significant α2AAR endocytosis, whereas pretreatment with ISO drove a significant response that was blocked when PKA was inhibited with PKI. No further enhancement of clonidine-mediated α2AAR endocytosis by ISO was observed at the 30-min time point, consistent with a kinetic acceleration of α2AAR endocytosis by βAR co-activation. Confocal images (A and B) are representative of at least three independent samples, and quantitation (C) was performed over at least 12–14 neurons. *, p < 0.001 versus clonidine alone; †, p < 0.001 versus clonidine plus ISO.
We additionally performed βAR co-activation using a panel of clinically relevant agonists with varying degrees of β1 versus β2AR subtype selectivity. Our results indicated that DOB, ALB, and SAL were all capable of enhancing clonidine-induced α2AAR endocytosis (Fig. 5). DOB and ALB were slightly less effective than the non-subtype-selective ISO at enhancing endocytosis, whereas SAL was similarly effective. Although there is some debate regarding the relative selectivity of DOB and ALB, SAL is widely accepted as a potent and highly β2AR-selective agonist. These results further support that activation of both βAR subtypes is capable of driving the acceleration of α2AAR endocytosis and that the effects observed in Fig. 4 are not unique to ISO only among the βAR agonists.
FIGURE 5.
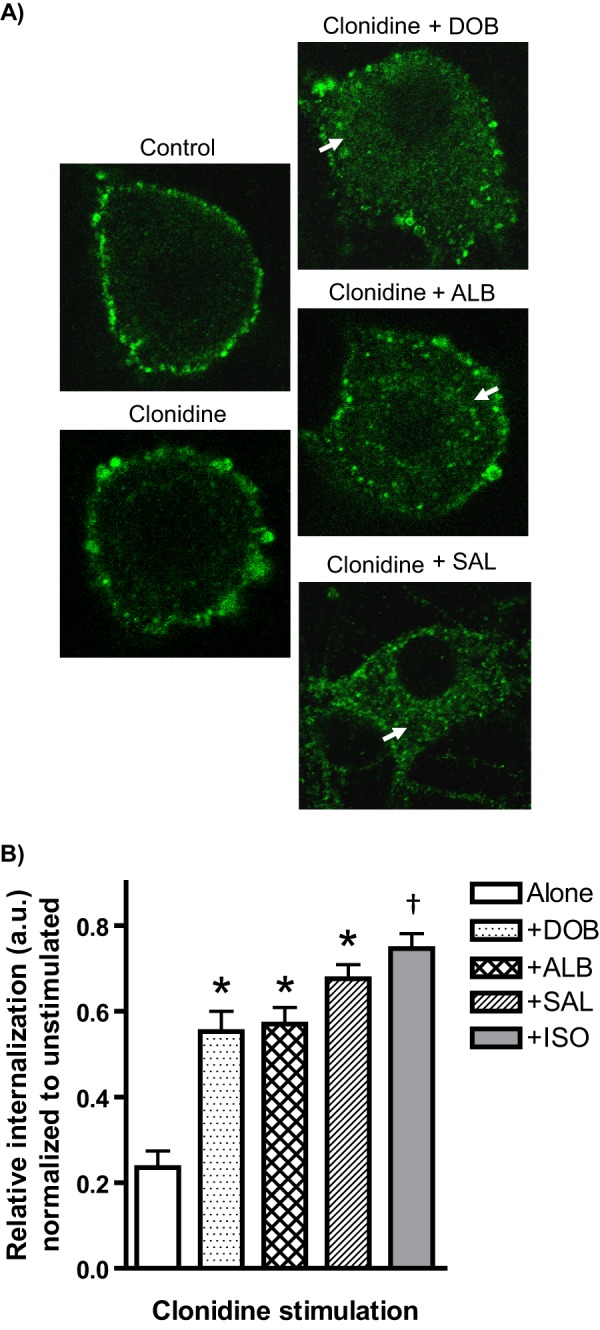
Co-activation with βAR subtype-selective agonists accelerates α2AR agonist-mediated α2AAR endocytosis in native neurons. A, neurons (SpWT) were subjected to treatment with a panel of clinically relevant agonists with varying degrees of β1 versus β2AR subtype selectivity (1 μm DOB, 1 μm ALB, or 100 nm SAL) and 10 min of stimulation with clonidine (1 μm). α2AAR endocytosis (detected by prelabeling method), as indicated by the appearance of intracellular punctae containing internalized receptors (arrows), was observed with co-activation by DOB, ALB, and SAL, but not with clonidine alone. B, quantitation of agonist-mediated α2AAR endocytosis, with relative internalization determined as described under “Experimental Procedures.” Internalization by clonidine alone was significantly enhanced with co-activation by DOB, ALB, and SAL. The data for clonidine plus ISO from Fig. 4 are included for comparison. Confocal images are representative of at least three independent samples, and quantitation was performed over at least 12–14 neurons (except for clonidine plus SAL, n = 10). *, p < 0.01 versus clonidine alone. †, p < 0.01 versus clonidine plus DOB or clonidine plus ALB.
Collectively, the above results demonstrate that co-activation of endogenous β and α2AARs results in an acceleration of agonist-mediated α2AAR endocytosis in native neurons. These data establish a phenomenon of cross-talk from β to α2AARs modulating α2AAR endocytic responses and suggest that such cross-talk may rely on canonical βAR/Gαs/cAMP signal transduction linking to PKA activation.
Receptor-independent PKA Activation Is Sufficient to Accelerate Agonist-mediated α2AAR Endocytosis
The present data indicate that PKA activation is critically involved in accelerating agonist-mediated α2AAR endocytosis in SCG neurons (Fig. 4). Indeed, we have previously demonstrated that receptor-independent activation of PKA by forskolin accelerates α2AAR endocytosis in heterologous cells (28). Such a finding in our native neurons would support our contention that βAR/Gαs/cAMP signal transduction linking to PKA activation underlies the α2AAR endocytic acceleration by βAR co-activation.
We therefore ascertained whether forskolin treatment would affect endocytosis of endogenous α2AARs in SCG neurons. Consistent with our previous findings, we observed an acceleration of endocytosis as stimulated by Epi (plus prazosin/propranolol) with significantly higher levels of internalization for forskolin-treated cells compared with vehicle controls (Fig. 6). The effect of forskolin was most dramatic at the 5- and 10-min time points, with a diminishing effect at 30 min of Epi stimulation. Two-way ANOVA revealed significant effects of forskolin pretreatment and time (p < 0.0001), but no significant forskolin × time interaction. These results demonstrate that PKA activation alone in the absence of βAR co-activation is sufficient to accelerate agonist-mediated endocytosis of endogenous α2AARs.
FIGURE 6.

Activation of PKA by forskolin treatment is sufficient to accelerate agonist-mediated α2AAR endocytosis in native neurons. A, neurons (SpWT) were pretreated with either vehicle or forskolin and then subjected to α2AAR prelabeling as in Fig. 1B&C followed by stimulation with Epi (100 μm, plus prazosin and propranolol) as indicated. Endocytosis, as indicated by the appearance of intracellular punctae containing internalized receptors (arrows), was observed in beginning at 5 min in forskolin-treated but not vehicle-treated neurons and continued to be significant, although less dramatic endocytic enhancement was observed at 15 and 30 min. B, quantitation of agonist-mediated α2AAR endocytosis in Epi-stimulated forskolin- and vehicle-treated neurons, with relative internalization determined as described under “Experimental Procedures.” Confocal images are representative of at least three independent samples, and quantitation was performed over at least 12–14 neurons. *, p < 0.05; **, p < 0.01 versus vehicle-treated.
βAR Co-activation Results in PKA-dependent Disruption of α2AAR/Spinophilin Interaction
Having established the clear importance of PKA activity in accelerating endogenous α2AAR endocytosis in SCG neurons, we sought to further investigate the mechanistic link underlying cross-talk from β to α2AARs. Spinophilin is a substrate for PKA phosphorylation at serine residues 94 and 177 (27), and our previous work has demonstrated that this PKA phosphorylation of spinophilin disrupts its ability to interact with and regulate α2AARs (28). Furthermore, we have shown that the interaction of α2AARs with spinophilin occurs in an agonist-dependent fashion both in vitro and in vivo (21, 23–25). We therefore postulated that our findings of endocytic acceleration (Figs. 3 and 4) could be explained by βAR co-activation leading to canonical Gαs/cAMP signal transduction, PKA activation, phosphorylation of spinophilin, and disrupted α2AAR/spinophilin interaction. To provide direct evidence for this mechanism, we utilized a FLIM-FRET technique to observe the α2AAR/spinophilin interaction under various conditions in live cells. As shown in Fig. 7, we observed a significant clonidine-dependent interaction between α2AARs and spinophilin, indicated by the large increase in FRET efficiency (Fig. 7A). Importantly, this clonidine-dependent interaction was abolished when βARs were co-activated by ISO treatment, and this effect was in turn reversed with inhibition of PKA activity by PKI, which rescued the clonidine-dependent interaction (Fig. 7A). Taken together, these data provide support for our postulated mechanism of β to α2AAR cross-talk and provide a plausible explanation for the observed endocytic acceleration in native neurons.
FIGURE 7.
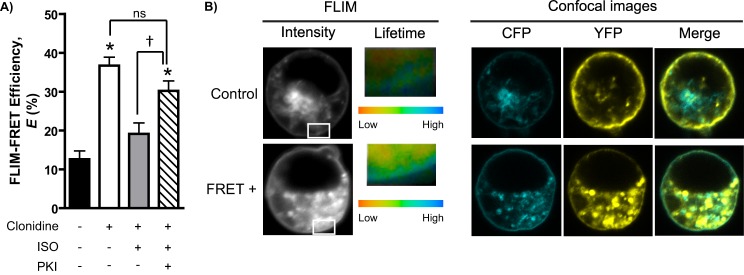
Agonist-dependent α2AAR/spinophilin interaction is blocked by βAR co-activation in a PKA-dependent fashion. A, FLIM-FRET efficiency (E) values calculated based on CFP lifetime values as described under “Experimental Procedures.” The values were obtained over five or six cells from two or three independent samples. Live HEK293 cells transiently transfected with CFP-α2AAR and YFP-spinophilin constructs were subjected to ISO (100 μm) and/or PKI (8.3 μm) pretreatment prior to stimulation with clonidine (1 μm) as in Fig. 2(A and B). B, representative images from nonstimulated control and stimulated FRET positive (FRET+) cells. FLIM images show CFP fluorescent intensity (gray scale) and lifetime values (represented in pseudocolors). Inset areas are cell surface regions selected for CFP lifetime analysis. A single measurement point corresponding to a median CFP lifetime value (green in the pseudocolor range) was the data point extracted from each cell. Confocal images show the cellular localization of CFP-α2AAR and YFP-spinophilin. *, p < 0.0001 versus clonidine alone; †, p < 0.0001 versus clonidine + ISO.
Expression of WT and Mutant Spinophilin in Spinophilin-null MEFs Supports Involvement of βAR-mediated Phosphorylation
Given the impracticality of biochemically assaying for spinophilin phosphorylation in SCG neurons, we elected to provide additional support for our proposed mechanism using MEFs with endogenous βAR expression as a substitute model system. Using transient transfection, we expressed GFP-tagged WT spinophilin (WT-Sp) or GFP-tagged mutant spinophilin with PKA phosphorylation sites at Ser-94 and Ser-177 mutated to Ala (Sp94A, 177A, referred to as mut-Sp) into spinophilin-null MEFs. Based on our extensive experience studying α2AAR endocytosis, we know that response kinetics are accelerated in MEFs versus SCG neurons, and so we used 5 min of stimulation as the early time point instead of 10 min. As shown in Fig. 8, when compared with nonexpressing control (spinophilin-null) cells, expression of WT-Sp appears to rescue the phenotype of these cells. Endocytosis can be detected in spinophilin-null but not WT-Sp cells after 5 min of clonidine stimulation (Fig. 8B, compare left and middle panels). Co-stimulation with ISO caused detectable endocytosis in WT-Sp cells but no additional effect in spinophilin-null cells (compare left and middle images; Fig. 8C). By contrast, cells expressing mut-Sp exhibited no α2AAR endocytosis following 5-min clonidine stimulation alone or in combination with ISO (Fig. 8, B and C, right panels). All cells exhibited α2AAR endocytosis following 30 min of clonidine stimulation (Fig. 8D). These data underline the importance of phosphorylation of spinophilin at the Ser-94/Ser-177 sites to the α2AAR endocytic acceleration observed with βAR co-activation. We additionally utilized an assay for cAMP production to compare the canonical Gαs/cAMP response for endogenous βARs in our spinophilin-null MEFS versus WT MEFs (matched genetic background). As shown in Fig. 8E, MEFs from the spinophilin-null line do not exhibit any deficit in ISO-stimulated cAMP response; in fact, the response to ISO is slightly enhanced in the spinophilin-null cells. This result indicates that loss of spinophilin does not affect βAR responsiveness at the ISO concentration used throughout our study.
FIGURE 8.

Effects of expressing wild-type (WT-Sp) or PKA phosphorylation mutant (mut-Sp) spinophilin in spinophilin-null MEF cells stably expressing HA-α2AARs. A–D, confocal images representative of numerous cells observed over n = 3 independent samples. Spinophilin-null MEFs were transiently transfected with GFP-tagged spinophilin (WT-Sp or mut-Sp) as described under “Experimental Procedures.” Nonexpressing cells were also recorded as controls. MEFs were treated as indicated: no stimulation control (A), 5 min of clonidine stimulation alone (B), 5 min of clonidine + ISO (C), and 30 min of clonidine stimulation alone (D). Cells were then subjected to the antibody prelabeling method for assaying α2AAR endocytosis as in previous figures, with the substitution of AlexaFluor594-conjugated secondary labeling. α2AAR endocytosis is indicated by the appearance of intracellular punctae containing internalized receptors (arrows). E, cAMP assay in spinophilin WT (SpWT) and spinophilin-null (Sp−/−) MEFs, matched for genetic background. cAMP accumulation was measured in response to 100 μm ISO to compare endogenous βAR responsiveness in SpWT versus Sp−/− cells. n = 3 replicates were obtained for each group. *, p < 0.001, +ISO versus control; †, p < 0.01, Sp−/− versus SpWT. Ctl, control.
Acceleration of α2AAR Endocytosis by βAR Co-activation Is Lost in Spinophilin-null Neurons
As a final step, we sought to provide validation of our proposed mechanism relying on disruption of the α2AAR/spinophilin interaction in native neurons. We first characterized the kinetics of clonidine-mediated α2AAR endocytosis in spinophilin-null neurons, finding that endocytosis of endogenous α2AARs by clonidine is accelerated in Sp−/− versus SpWT neurons (Fig. 9, A and B). Two-way ANOVA revealed significant effects of genotype and time (p < 0.0001) and a significant genotype × time interaction (p < 0.0001).
FIGURE 9.

Acceleration of endogenously expressed α2AAR endocytosis by βAR co-activation requires spinophilin. A, neurons with (SpWT) and without (Sp−/−) spinophilin expression were stimulated with clonidine (1 μm) as indicated. Neurons were subjected to α2AAR prelabeling method as in Fig. 1 (B and C), with endocytosis indicated by the appearance of intracellular punctae containing internalized receptors (arrows). ISO pretreatment to co-activate βARs was done as in Fig. 4. B, quantitation showing an acceleration of clonidine-mediated α2AAR endocytosis basally in Sp−/− neurons, with significantly enhanced internalization at both the 10- and 15-min time points. C, quantitation showing that no enhancement of clonidine-mediated α2AAR endocytosis by βAR co-activation with ISO is observed in Sp−/− neurons. Confocal images are representative of at least three independent samples, and quantitation was performed over at least 12–14 neurons. *, p < 0.01; **, p < 0.0001 versus SpWT.
We then repeated our clonidine plus ISO experiment in the Sp−/− neurons. Using the same conditions as in Fig. 4, we found no additional enhancement of clonidine-mediated α2AAR endocytosis by βAR co-activation in Sp−/− neurons (Fig. 9, A and C, SpWT included for comparison). This result indicates that spinophilin is required for βAR co-activation to modulate endogenous α2AAR endocytosis and supports our proposed mechanism for β to α2AAR cross-talk relying on βAR signaling to PKA and disruption of α2AAR/spinophilin interaction.
DISCUSSION
The present study provides a novel example of adrenergic receptor cross-talk wherein co-activation of endogenously expressed β and α2AARs results in accelerated α2AAR endocytic responses to agonist stimulation in native adrenergic neurons. Our findings support an acceleration of agonist-stimulated α2AAR endocytosis under βAR co-activating conditions, as well as in other scenarios in which α2AAR/spinophilin interaction is disrupted or abolished, as seen with forskolin-driven cAMP signaling or in spinophilin-null cells, respectively. Based upon the data presented here and our past findings, we have constructed a working model for PKA- and spinophilin-dependent cross-talk from β to α2ARs affecting α2AAR responsiveness, which is reliant upon βAR-Gαs signal transduction (Fig. 10). Importantly, our data support the existence of this cross-talk in a native adrenergic neuronal cell type with endogenous AR expression, and we have further shown that this cross-talk is associated with stimulation by a panel of physiological and clinical β and α2AR ligands.
FIGURE 10.
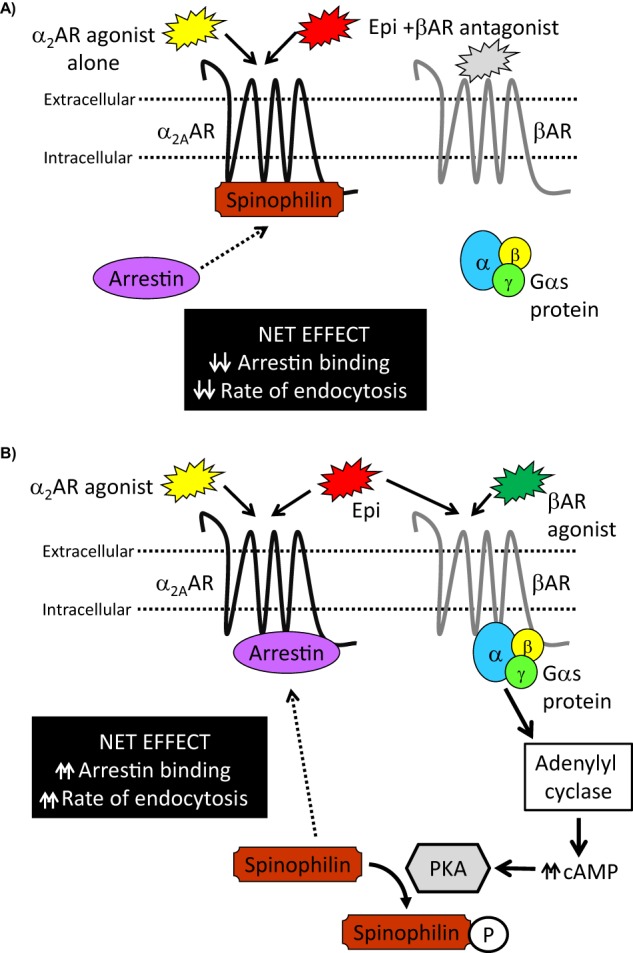
Working model for spinophilin- and PKA-dependent β to α2AAR cross-talk modulating endogenous α2AAR endocytosis. A, when α2AARs are activated alone, the spinophilin regulatory mechanism will be fully engaged, and α2AAR responsiveness will be determined by interplay between spinophilin and arrestin binding to the receptor. Relative to co-activating conditions, there would be a net effect of decreased arrestin binding and decreased rate of agonist-stimulated α2AAR endocytosis. B, under β and α2AAR co-activating conditions, canonical βAR/Gαs signal transduction coupled to PKA activation will result in PKA-dependent phosphorylation of spinophilin. This phosphorylation attenuates the ability of spinophilin to interact with α2AARs, which would lead to a predominance of arrestin binding and a net effect of increased rate of agonist-stimulated α2AAR endocytosis.
Simultaneous Activation of β and α2AARs Accelerates α2AAR Endocytosis
We have demonstrated that an acceleration of agonist-driven α2AAR endocytosis occurs under various conditions of β and α2AAR co-activation in native adrenergic neurons. First, we showed that stimulation of neurons with the endogenous nonselective full AR agonist Epi has differential effects in the presence or absence of the βAR antagonist propranolol. In the presence of propranolol, a condition that allows for only α2AAR activation, Epi drives little or no α2AAR endocytosis at early time points, particularly at 5 min (Figs. 2 and 3). However, in the absence of propranolol, a condition that allows for Epi to simultaneously activate β and α2AARs, endocytosis is significantly enhanced at these early time points (Fig. 3). Furthermore, the use of agonists that distinctly target β or α2AARs reveals similar findings of cross-talk. Co-stimulation of SCG neurons with the non-subtype-selective βAR agonist ISO (Fig. 4, B and C) or with agonists of varying subtype selectivity for either β1 or β2ARs (Fig. 5) together with the α2AR agonist clonidine results in an enhancement of early time point α2AAR endocytosis. Collectively, these results establish the phenomenon of accelerated endogenous α2AAR endocytic responses to agonist under conditions of βAR co-activation. Although work by the Hall laboratory (1) has established the occurrence of heterodimerization between β1 and α2AARs, we believe that dimerization does not provide an adequate mechanistic explanation for our data. In fact, their study as well as our own (Fig. 4A) found that stimulation with β-agonist only is not sufficient to drive endocytosis of α2AARs, and so our present findings of cross-talk are unlikely to be explained by simple physical co-internalization of β and α2AARs.
Modulation of α2AAR endocytosis would be expected to have consequences for cellular α2AAR responsiveness. First of all, the accelerated endocytosis means a corresponding acceleration of receptor desensitization, with receptors being removed from the cell surface at a faster rate. Furthermore, we have previously shown that removal of spinophilin results in accelerated kinetics of α2AAR-mediated MAPK signal transduction along with accelerated endocytosis (24). Collectively, these effects will translate into a dramatically altered functional profile for the α2AAR under conditions of βAR cross-talk.
βAR-stimulated PKA-dependent Phosphorylation of Spinophilin Provides a Mechanistic Basis for Cross-talk with α2AARs
Our previous work has established a regulatory mechanism for α2AARs involving interplay between the non-G protein receptor interacting partners spinophilin and arrestin (24, 25). We have further shown that this regulatory mechanism can be disrupted by PKA-dependent phosphorylation of spinophilin, which in turn prevents α2AAR/spinophilin interaction (28). The present results expand the application of this regulatory mechanism to endogenously expressed α2AARs in a native adrenergic neuronal cell type, whereas our previous work was done in heterologous cell systems.
First, we have demonstrated that ligand-dependent α2AAR endocytosis in SCG neurons is arrestin-dependent (Fig. 1B). Additionally, as predicted by our established mechanistic model, the kinetics of this endocytosis are altered in the absence of spinophilin, with an acceleration of the time course observed with application of both the endogenous agonist Epi (Fig. 3) and the therapeutic partial agonist clonidine (Fig. 9, A and B). Next, we have shown that under conditions of PKA activation, either in a receptor-independent fashion by forskolin stimulation of adenylyl cyclase (Fig. 6) or by stimulation of Gαs-coupled βARs (Figs. 4 and 5), the kinetics of agonist-driven α2AAR endocytosis are accelerated. In the case of βAR agonist-mediated acceleration of endocytosis, we have shown that the effects are blocked by inhibition of PKA (Fig. 4, B and C) and are not observed in spinophilin-null neurons (Fig. 9, A and C). In fact, in each case presented here, both PKA activation and genetic deletion of spinophilin have similar effects on α2AAR responses, with PKA activation by βARs having no additional effect in the spinophilin-null system. To further support our mechanistic contention, we have provided direct evidence that co-activation of βARs results in a disruption of ligand-dependent α2AAR/spinophilin interaction and that this disruption depends critically on PKA activity (Fig. 7). Finally, our experiments utilizing expression of WT and phospho-mutant spinophilin in spinophilin-null MEFs (Fig. 8) highlight the importance of the phosphorylation state of spinophilin to its α2AAR regulatory function under β/α2AAR co-activating conditions.
Taken together, our present data are strongly supportive of a clear mechanistic link underlying cross-talk from β to α2AARs. As described above, we propose a model whereby activation of βARs results in a disruption of ligand-dependent α2AAR/spinophilin interaction (Fig. 10). This disruption occurs as a consequence of canonical βAR/Gαs signal transduction linking to activation of PKA, which in turn catalyzes a phosphorylation of spinophilin, disrupting its interaction with α2AARs. The removal of spinophilin from the regulatory scheme would lead to a predominance of α2AAR/arrestin interaction and, subsequently, to the observed acceleration of agonist-dependent α2AAR endocytic responses and, potentially, resulting effects on signal transduction. Interestingly, previous reports have suggested the existence of PKA-dependent neuronal current modulation by combinatorial β/α AR stimulation in the CA1 region of hippocampus (9, 10), a brain region with particularly high spinophilin expression and function (18, 35).
Potential Importance of β/α2AAR Cross-talk to Adrenergic Physiology and Pharmacology
We believe that our present findings have a number of significant implications for clinically relevant adrenergic physiology and pharmacology. All of the present experiments have utilized clinically relevant therapeutic β and α2AAR ligands, the effects of which have been examined in a native cell model with endogenous expression of receptors and the players in the proposed mechanism of cross-talk. These drugs, including beta blockers, sympathomimetics, and other adrenergic agonists, have a wide array of clinical applications and are among the most frequently used GPCR-directed therapeutics (17, 43, 44).
Indeed, certain in vivo physiological responses to Epi administration are consistent with an enhancement of α2AAR endocytosis resulting from β/α2AAR cross-talk. As an example, ISO is known to drive a slight hypotensive response, which is not easily explained in terms of βAR activation (17). In light of our data, this response may be explained as a potentiation of α2AAR signaling drive resulting from basal adrenergic tone. Additionally, the clinical usage of β2AR-selective agonists to delay preterm labor raises the possibility of developmental relevance for β/α2AAR cross-talk, especially given evidence that administration of the β2AR-selective agonist terbutaline to pregnant rats affects α2AR expression levels in various tissues at different developmental stages (14). This finding could now potentially be explained as a modulation of α2AAR expression patterns through cross-talk with β2ARs affecting α2AAR trafficking and localization. In a long term sense, chronically reduced α2AAR/spinophilin interaction and corresponding enhancement of arrestin interaction would be expected to potentiate receptor down-regulation, a process that we have previously characterized as occurring in an arrestin-mediated fashion in vivo (34).
Another area in which our cross-talk may be relevant is ocular pharmacology. Here, the SCGs are directly involved in the neural circuitry, and α2AR agonists have therapeutic benefit in the management of intraocular pressure in glaucoma (45). The clinical application of α2AR agonists in this setting has been somewhat limited by side effects, especially sedation, resulting from activation of central α2AARs. Given our present findings, it may be advisable to consider combination treatment with low doses of β and α2AR agonists, thereby more effectively engaging local α2AARs while limiting spillover into more systemic effects. In a broader sense, our results suggest that modulation of α2AAR responsiveness should be considered in the mechanisms of both beta blockers, which may additionally attenuate basal α2AAR tone, and βAR agonists, which may additionally potentiate basal α2AAR tone.
Follow-up studies will be necessary to validate the existence of β/α2AAR cross-talk in in vivo physiological settings. Additionally, the relative importance of our proposed mechanism of cross-talk will vary depending on the relative expression of β and α2AAR subtypes. Also, more recently appreciated noncanonical βAR signaling mediated by non-Gαs-containing G proteins or arrestins (29) would not be expected to engage the PKA-dependent cross-talk mechanism. Furthermore, the expression of spinophilin is developmentally regulated and varies widely across different tissues (18, 19). At the cellular level, spinophilin localizes exclusively to somatodendritic neuronal compartments (18, 46, 47), precluding the involvement of our regulatory mechanism in axonal terminals. Finally, it should be noted that spinophilin is a multifunctional scaffolding protein with an ever-growing list of physical and regulatory interactions appropriately termed the “spinophilin interactome” (48), raising the possibility that our findings may involve a more complex mechanism than we have proposed here.
Nevertheless, our study has provided new and valuable insight into the physiological and pharmacological inter-relationship among the ARs. Furthermore, we have extended the application of our previous findings on regulation of α2AARs by PKA and spinophilin into native adrenergic neurons with endogenous expression of all of the players involved. Integrating our past and present data, we have constructed a novel mechanism for cross-talk between β and α2AAR subtypes, a mechanism that we believe has great physiological and therapeutic importance.
Acknowledgments
We thank Drs. Robert Lefkowitz (Duke University) and Paul Greengard (Rockefeller University) for generously providing the arrestin3-null and spinophilin-null mouse lines, respectively; Dr. Jeffrey Benovic (Thomas Jefferson University) for providing the antibody against endogenous arrestin2; Shawn Williams and the University of Alabama at Birmingham High Resolution Imaging Facility for assistance with FLIM-FRET and confocal imaging; Tana Birky for assistance with data entry and manuscript editing; and Dr. Yunjia Chen for assistance in preparing the YFP-spinophilin construct and performing the cAMP assay.
This work was supported, in whole or in part, by National Institutes of Health Grant MH081917 (to Q. W.).
- AR
- adrenergic receptor
- ALB
- albuterol
- CFP
- cyan fluorescent protein
- DOB
- dobutamine
- Epi
- epinephrine
- FLIM
- fluorescence lifetime imaging
- GPCR
- G protein-coupled receptor
- ISO
- isoproterenol
- PKI
- protein kinase A inhibitor
- SAL
- salmeterol
- SCG
- superior cervical ganglion
- YFP
- yellow fluorescent protein
- MEF
- mouse embryonic fibroblast
- ANOVA
- analysis of variance.
REFERENCES
- 1. Xu J., He J., Castleberry A. M., Balasubramanian S., Lau A. G., Hall R. A. (2003) Heterodimerization of α2A- and β1-adrenergic receptors. J. Biol. Chem. 278, 10770–10777 [DOI] [PubMed] [Google Scholar]
- 2. Prinster S. C., Holmqvist T. G., Hall R. A. (2006) α2C-Adrenergic receptors exhibit enhanced surface expression and signaling upon association with β2-adrenergic receptors. J. Pharmacol. Exp. Ther. 318, 974–981 [DOI] [PubMed] [Google Scholar]
- 3. Bawa T., Altememi G. F., Eikenburg D. C., Standifer K. M. (2003) Desensitization of α2A-adrenoceptor signalling by modest levels of adrenaline is facilitated by β2-adrenoceptor-dependent GRK3 up-regulation. Br. J. Pharmacol. 138, 921–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bawa-Khalfe T., Altememi G. F., Mandyam C. D., Schwarz L. A., Eikenburg D. C., Standifer K. M. (2007) The presence of β2-adrenoceptors sensitizes α2A-adrenoceptors to desensitization after chronic epinephrine treatment. BMC Pharmacol. 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Desai A. N., Standifer K. M., Eikenburg D. C. (2004) Simultaneous α2B- and β2-adrenoceptor activation sensitizes the α2B-adrenoceptor for agonist-induced down-regulation. J. Pharmacol. Exp. Ther. 311, 794–802 [DOI] [PubMed] [Google Scholar]
- 6. Maggi A., U'Prichard D. C., Enna S. J. (1980) β-Adrenergic regulation of α2-adrenergic receptors in the central nervous system. Science 207, 645–647 [DOI] [PubMed] [Google Scholar]
- 7. Nomura Y., Kawai M., Segawa T. (1984) The interaction between β- and α2-adrenoceptors in cerebral cortical membranes. Isoproterenol-induced increase in [3H]clonidine binding in rats. Brain Res. 302, 101–109 [DOI] [PubMed] [Google Scholar]
- 8. Nakamura T., Tsujimura R., Nomura J. (1991) Interaction between α2- and β-adrenergic receptors in rat cerebral cortical membranes. Clonidine-induced reduction in agonist and antagonist affinity for β-adrenergic receptors. Brain Res. 542, 181–186 [DOI] [PubMed] [Google Scholar]
- 9. Pedarzani P., Storm J. F. (1996) Interaction between α- and β-adrenergic receptor agonists modulating the slow Ca2+-activated K+ current IAHP in hippocampal neurons. Eur. J. Neurosci. 8, 2098–2110 [DOI] [PubMed] [Google Scholar]
- 10. Pedarzani P., Storm J. F. (1993) PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 11, 1023–1035 [DOI] [PubMed] [Google Scholar]
- 11. Northam W. J., Mobley P. (1985) Clonidine pretreatment enhances the sensitivity of the β-noradrenergic receptor coupled adenylate cyclase system in astrocytes. Eur. J. Pharmacol. 113, 153–154 [DOI] [PubMed] [Google Scholar]
- 12. Atkinson B. N., Minneman K. P. (1992) Preferential desensitization of β- versus α2-adrenergic receptors accelerates loss of response to norepinephrine in primary glial cultures. Mol. Pharmacol. 41, 688–694 [PubMed] [Google Scholar]
- 13. Apparsundaram S., Eikenburg D. C. (1996) Prejunctional α adrenoceptor desensitization in rat heart after chronic epinephrine treatment. J. Pharmacol. Exp. Ther. 278, 862–870 [PubMed] [Google Scholar]
- 14. Kreider M. L., Seidler F. J., Slotkin T. A. (2004) β-Adrenoceptor modulation of transiently overexpressed α2-adrenoceptors in brain and peripheral tissues. Cellular mechanisms underlying the developmental toxicity of terbutaline. Brain Res. Bull. 62, 305–314 [DOI] [PubMed] [Google Scholar]
- 15. Molinoff P. B. (2011) Neurotransmission and the central nervous system, in Goodman & Gilman's The Pharmacological Basis of Therapeutics, online edition (Brunton L. L., ed) 12th Ed., McGraw-Hill Inc., New York [Google Scholar]
- 16. Westfall T. C., Westfall D. P. (2011) Neurotransmission. The autonomic and somatic motor nervous systems, in Goodman & Gilman's The Pharmacological Basis of Therapeutics, online edition (Brunton L. L., ed) 12th Ed., McGraw-Hill Inc., New York [Google Scholar]
- 17. Westfall T. C., Westfall D. P. (2011) Adrenergic agonists and antagonists, in Goodman & Gilman's The Pharmacological Basis of Therapeutics, online edition (Brunton L. L., ed) 12th Ed., McGraw-Hill Inc., New York [Google Scholar]
- 18. Allen P. B., Ouimet C. C., Greengard P. (1997) Spinophilin, a novel protein phosphatase 1 binding protein localized to dendritic spines. Proc. Natl. Acad. Sci. U.S.A. 94, 9956–9961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Satoh A., Nakanishi H., Obaishi H., Wada M., Takahashi K., Satoh K., Hirao K., Nishioka H., Hata Y., Mizoguchi A., Takai Y. (1998) Neurabin-II/spinophilin. An actin filament-binding protein with one pdz domain localized at cadherin-based cell-cell adhesion sites. J. Biol. Chem. 273, 3470–3475 [DOI] [PubMed] [Google Scholar]
- 20. Sarrouilhe D., di Tommaso A., Métayé T., Ladeveze V. (2006) Spinophilin. From partners to functions. Biochimie 88, 1099–1113 [DOI] [PubMed] [Google Scholar]
- 21. Wang Q., Limbird L. E. (2002) Regulated interactions of the α2A adrenergic receptor with spinophilin, 14-3-3ζ, and arrestin 3. J. Biol. Chem. 277, 50589–50596 [DOI] [PubMed] [Google Scholar]
- 22. Brady A. E., Wang Q., Allen P. B., Rizzo M., Greengard P., Limbird L. E. (2005) α2-adrenergic agonist enrichment of spinophilin at the cell surface involves βγ subunits of Gi proteins and is preferentially induced by the α2A-subtype. Mol. Pharmacol. 67, 1690–1696 [DOI] [PubMed] [Google Scholar]
- 23. Richman J. G., Brady A. E., Wang Q., Hensel J. L., Colbran R. J., Limbird L. E. (2001) Agonist-regulated interaction between α2-adrenergic receptors and spinophilin. J. Biol. Chem. 276, 15003–15008 [DOI] [PubMed] [Google Scholar]
- 24. Wang Q., Zhao J., Brady A. E., Feng J., Allen P. B., Lefkowitz R. J., Greengard P., Limbird L. E. (2004) Spinophilin blocks arrestin actions in vitro and in vivo at G protein-coupled receptors. Science 304, 1940–1944 [DOI] [PubMed] [Google Scholar]
- 25. Lu R., Chen Y., Cottingham C., Peng N., Jiao K., Limbird L. E., Wyss J. M., Wang Q. (2010) Enhanced hypotensive, bradycardic, and hypnotic responses to α2-adrenergic agonists in spinophilin-null mice are accompanied by increased G protein coupling to the α2A-adrenergic receptor. Mol. Pharmacol. 78, 279–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cottingham C., Li X., Wang Q. (2012) Noradrenergic antidepressant responses to desipramine in vivo are reciprocally regulated by arrestin3 and spinophilin. Neuropharmacology 62, 2354–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hsieh-Wilson L. C., Benfenati F., Snyder G. L., Allen P. B., Nairn A. C., Greengard P. (2003) Phosphorylation of spinophilin modulates its interaction with actin filaments. J. Biol. Chem. 278, 1186–1194 [DOI] [PubMed] [Google Scholar]
- 28. Xu J., Chen Y., Lu R., Cottingham C., Jiao K., Wang Q. (2008) Protein kinase A phosphorylation of spinophilin modulates its interaction with the α2A-adrenergic receptor (AR) and alters temporal properties of α2AAR internalization. J. Biol. Chem. 283, 14516–14523 [DOI] [PubMed] [Google Scholar]
- 29. Kobilka B. K. (2011) Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol. Sci. 32, 213–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu R., Li Y., Zhang Y., Chen Y., Shields A. D., Winder D. G., Angelotti T., Jiao K., Limbird L. E., Zhou Y., Wang Q. (2009) Epitope-tagged receptor knock-in mice reveal that differential desensitization of α2-adrenergic responses is because of ligand-selective internalization. J. Biol. Chem. 284, 13233–13243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yamauchi A., Lever J. D., Kemp K. W. (1973) Catecholamine loading and depletion in the rat superior cervical ganglion. A formol fluorescence and enzyme histochemical study with numerical assessments. J. Anat. 114, 271–282 [PMC free article] [PubMed] [Google Scholar]
- 32. Hanyaloglu A. C., von Zastrow M. (2008) Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu. Rev. Pharmacol. Toxicol. 48, 537–568 [DOI] [PubMed] [Google Scholar]
- 33. von Zastrow M., Williams J. T. (2012) Modulating neuromodulation by receptor membrane traffic in the endocytic pathway. Neuron. 76, 22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cottingham C., Chen Y., Jiao K., Wang Q. (2011) The antidepressant desipramine is an arrestin-biased ligand at the α2A-adrenergic receptor driving receptor down-regulation in vitro and in vivo. J. Biol. Chem. 286, 36063–36075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Feng J., Yan Z., Ferreira A., Tomizawa K., Liauw J. A., Zhuo M., Allen P. B., Ouimet C. C., Greengard P. (2000) Spinophilin regulates the formation and function of dendritic spines. Proc. Natl. Acad. Sci. U.S.A. 97, 9287–9292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bohn L. M., Lefkowitz R. J., Gainetdinov R. R., Peppel K., Caron M. G., Lin F. T. (1999) Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 286, 2495–2498 [DOI] [PubMed] [Google Scholar]
- 37. Brum P. C., Hurt C. M., Shcherbakova O. G., Kobilka B., Angelotti T. (2006) Differential targeting and function of α2A and α2C adrenergic receptor subtypes in cultured sympathetic neurons. Neuropharmacology 51, 397–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Glass D. B., Cheng H. C., Mende-Mueller L., Reed J., Walsh D. A. (1989) Primary structural determinants essential for potent inhibition of cAMP-dependent protein kinase by inhibitory peptides corresponding to the active portion of the heat-stable inhibitor protein. J. Biol. Chem. 264, 8802–8810 [PubMed] [Google Scholar]
- 39. Harris T. E., Persaud S. J., Jones P. M. (1997) Pseudosubstrate inhibition of cyclic AMP-dependent protein kinase in intact pancreatic islets. Effects on cyclic AMP-dependent and glucose-dependent insulin secretion. Biochem. Biophys. Res. Commun. 232, 648–651 [DOI] [PubMed] [Google Scholar]
- 40. Brady A. E., Wang Q., Colbran R. J., Allen P. B., Greengard P., Limbird L. E. (2003) Spinophilin stabilizes cell surface expression of α2B-adrenergic receptors. J. Biol. Chem. 278, 32405–32412 [DOI] [PubMed] [Google Scholar]
- 41. Shenoy S. K., Lefkowitz R. J. (2003) Multifaceted roles of β-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem. J. 375, 503–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tan C. M., Brady A. E., Nickols H. H., Wang Q., Limbird L. E. (2004) Membrane trafficking of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 44, 559–609 [DOI] [PubMed] [Google Scholar]
- 43. Biaggioni I., Robertson D. (2012) Adrenoceptor agonists & sympathomimetic drugs, in Basic & Clinical Pharmacology (Katzung B. G., Masters S. B., Trevor A. J., eds) 12th Ed., McGraw-Hill Inc., New York [Google Scholar]
- 44. Robertson D., Biaggioni I. (2012) Adrenoceptor antagonist drugs, in Basic & Clinical Pharmacology (Katzung B. G., Masters S. B., Trevor A. J., eds) 12th Ed., McGraw-Hill Inc., New York [Google Scholar]
- 45. Henderer J. D., Rapuano C. J. (2011) Ocular pharmacology, in Goodman & Gilman's The Pharmacological Basis of Therapeutics, online edition (Brunton L. L., ed) 12th Ed., McGraw-Hill Inc., New York [Google Scholar]
- 46. Ouimet C. C., Katona I., Allen P., Freund T. F., Greengard P. (2004) Cellular and subcellular distribution of spinophilin, a PP1 regulatory protein that bundles F-actin in dendritic spines. J. Comp. Neurol. 479, 374–388 [DOI] [PubMed] [Google Scholar]
- 47. Muly E. C., Smith Y., Allen P., Greengard P. (2004) Subcellular distribution of spinophilin immunolabeling in primate prefrontal cortex. Localization to and within dendritic spines. J. Comp. Neurol. 469, 185–197 [DOI] [PubMed] [Google Scholar]
- 48. Baucum A. J., 2nd, Jalan-Sakrikar N., Jiao Y., Gustin R. M., Carmody L. C., Tabb D. L., Ham A. J., Colbran R. J. (2010) Identification and validation of novel spinophilin-associated proteins in rodent striatum using an enhanced ex vivo shotgun proteomics approach. Mol. Cell Proteomics 9, 1243–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]