

Background: Sin resolvase is a site-specific DNA recombinase that catalyzes phosphotransfer without the use of divalent cations.
Results: Mutation of Arg-69 in the active site is partially rescued by a 3′ phosphorothiolate substrate.
Conclusion: Arg-69 is linked to protonation of the leaving group and most likely acts as a general acid catalyst.
Significance: Serine recombinases employ a different catalytic strategy than most other phosphoryl transfer enzymes.
Keywords: DNA Recombination, Enzyme Catalysis, Enzyme Mechanisms, Nucleic Acid Enzymology, Protein-DNA Interaction, Arginine, General Acid, Phosphotransfer, Serine Recombinase, Site-specific Recombination
Abstract
Members of the serine family of site-specific DNA recombinases use an unusual constellation of amino acids to catalyze the formation and resolution of a covalent protein-DNA intermediate. A recent high resolution structure of the catalytic domain of Sin, a particularly well characterized family member, provided a detailed view of the catalytic site. To determine how the enzyme might protonate and stabilize the 3′O leaving group in the strand cleavage reaction, we examined how replacing this oxygen with a sulfur affected the cleavage rate by WT and mutant enzymes. To facilitate direct comparison of the cleavage rates, key experiments used suicide substrates that prevented religation after cleavage. The catalytic defect associated with mutation of one of six highly conserved arginine residues, Arg-69 in Sin, was partially rescued by a 3′ phosphorothiolate substrate. We conclude that Arg-69 has an important role in stabilizing the 3′O leaving group and is the prime candidate for the general acid that protonates the 3′O, in good agreement with the position it occupies in the high resolution structure of the active site of Sin.
Introduction
Site-specific recombinases function by recognizing specific DNA sequences, cleaving the phosphodiester backbones, and religating the broken ends with new partners (1). Members of the serine recombinase family utilize a conserved serine residue for the initial nucleophilic attack on the scissile phosphate (2). The serine oxygen displaces the DNA 3′ oxygen, forming a 5′ phosphoserine linkage. After strand exchange, the chemical reaction is reversed, with a new 3′ oxygen attacking the phosphoserine linkage to reseal the DNA. The catalytic site of serine recombinases is unusual among phosphotransferases in that it requires no divalent metal ions and contains a cluster of arginine side chains (Fig. 1). Here we present evidence that a conserved arginine, Arg-69, acts as a general acid to protonate the 3′ oxygen during the initial cleavage reaction.
FIGURE 1.

Recombinase mechanism and active site. A, simplified serine recombinase reaction scheme. The recombinase generally binds crossover site DNA as a dimer (yellow and green lines). Two DNA-bound dimers then assemble into a catalytically competent tetramer. Cleavage occurs, forming a transient 5′ phosphoserine linkage (red dots) followed by a putative 180° subunit rotation, which aligns the cleaved DNA ends for religation, yielding a recombinant product. Although WT resolvases usually require accessory factors to trigger tetramerization, the pseudo-WT Sin used here (Q115R/R54E) does not. B, structure of the tetrameric catalytic domain of Sin Q115R/R54E (Protein Data Bank ID code 3PKZ) (16). The nucleophilic serine is shown as red spheres, and bound sulfate ions as sticks. Side chains are also shown for the activating mutation (Q115R, in blue) and the solubilizing mutation (R54E, in yellow). C, active site of Sin. Stereo view shows the Sin active site from the structure in B, in which all of the conserved residues implicated in catalysis form a hydrogen-bonded network surrounding a sulfate ion that lies in the scissile phosphate group-binding pocket (16). The path of a modeled DNA backbone is sketched as a yellow line.
We addressed this question using Sin resolvase, a member of the serine recombinase family for which extensive structural, biochemical, and genetic data are available. It was first identified on the pI9789 plasmid in Staphylococcus aureus, which contains a variety of resistance genes (3, 4). In vitro, and presumably in vivo, Sin resolves plasmid dimers in which two copies of the plasmid sequence are connected in a head-to-tail orientation (5, 6).
To simplify kinetic studies of Sin, we mutationally circumvented a key regulatory step that might otherwise be rate-limiting. Sin-mediated recombination is normally regulated by a multicomponent synaptic complex that forms on supercoiled substrates and controls formation of a catalytically active species. The DNA cleavage and rejoining reactions occur within the context of a tetramer, but Sin and other serine recombinases bind their cognate crossover sites as dimers (7). The dimer-to-tetramer conformational change normally requires the full synaptic complex. However, that requirement can be bypassed by activating mutations that tip the conformational equilibrium from the inactive dimer to the activated tetramer in the absence of regulatory sites or proteins (8–13). The most effective such mutation, Q115R, allows Sin to recombine isolated linear crossover sites efficiently (Fig. 1A) (13). Once the catalytic tetramer is formed, the nucleophilic serine of each protomer attacks the scissile phosphate, forming 5′ phosphoserine linkages and two-nucleotide 3′ overhangs. Two protomers of the protein are then thought to rotate 180° to align the broken DNA ends (14–17). Religation to yield recombinant duplexes occurs via the reverse of the cleavage reaction.
The key catalytic residues of serine recombinases have been identified, but their specific roles have not been clearly defined. Alignment of a large set of serine recombinase sequences identified a number of highly conserved charged and/or polar residues, and experiments on Tn3 resolvase determined which ones, when mutated, most drastically impair recombination activity both in vivo and in vitro (18). These key residues cluster near the catalytic serine in several crystal structures of serine recombinases (15, 19, 20). The structure that appears closest to a catalytically competent state is that of the tetrameric Sin Q115R catalytic domain, in which all of the key residues form a hydrogen-bonded network surrounding a sulfate ion that lies in the scissile phosphate-binding pocket (16) (Fig. 1, B and C). Directly interacting with the sulfate ion are the nucleophilic Ser-9, a glutamine (Gln-13), and three arginines (Arg-7, Arg-66, and Arg-69).
Enzymes can use several strategies to catalyze phosphotransfer reactions: activating the nucleophile, stabilizing the leaving group, reducing electrostatic repulsion, stabilizing the transition state, and reducing entropy by optimally orienting the reactants (21). For phosphodiesters such as DNA, the transition state is expected to be pentavalent, with partial bond formation to both the nucleophile and the leaving group. Many, but not all, phosphotransferases utilize divalent metal ion(s) in catalysis (22–25). Although there are many variations, in general, one metal ion coordinates to the nucleophile, orienting it and lowering its pKa to help generate the more reactive alkoxide anion, whereas the other metal ion stabilizes the developing negative charge on the leaving group, and both ions can stabilize the charge and geometry of the pentacovalent transition state. In contrast to the metal ion containing active sites, the tyrosine recombinases such as Flp and Cre, and the related topoisomerase Ib use a different mechanism, which involves a lysine general acid to protonate the leaving 5′ oxygen and either a histidine or a water to deprotonate the nucleophilic tyrosine. They also contain conserved arginines that probably orient the scissile phosphate and stabilize the transition state (26–28).
For the serine recombinases, which also do not require metal ions, the catalytic mechanism is less well studied. As with the tyrosine recombinases, structures suggest that the transition state is probably stabilized by one or more conserved arginines. However, the mechanisms for activation of the Ser-9 nucleophile and stabilization of the 3′-oxygen leaving group of the DNA (which has a pKa ∼15) have been unclear. Residues commonly used for acid/base catalysis such as histidine are not present at the active site; instead, the active site contains a cluster of conserved arginines. In addition to the three mentioned above (Arg-7, Arg-66, and Arg-69) that the Sin tetramer structure shows hydrogen-bonded to the sulfate in the active site pocket, three more (Arg-43, Arg-125, and Arg-131) sit within ∼14 Å of the catalytic site. Arg-125 and Arg-131 are not visible in the Sin catalytic domain structure, but their equivalents in γδ resolvase lie near the scissile phosphate in a postcleavage DNA-bound complex structure (15). Arginine is not usually considered a likely general acid/base catalyst due to its high pKa (∼12 in water). However, the pKa could be lowered by a positively charged environment such as that of the serine recombinase active site, and there is precedent for arginine playing such a role, for example in IMP dehydrogenase (29).
To identify the general acid in the Sin active site, we compared the DNA cleavage rates of a series of mutant proteins on WT and modified DNA substrates. We replaced the 3′- bridging oxygen with sulfur at the scissile position, which is predicted to alleviate the need for a general acid to protonate the leaving group during cleavage. Similar logic was used to investigate the catalytic mechanism of type Ib topoisomerases, where the leaving group during cleavage is the 5′O (27). More recently, a 3′ phosphorothiolate was used to study the roles of metal ions in type II topoiomerases (30–32), and both 3′ and 5′ bridging phosphorothiolates have been critical in studying general acid/base catalysis by ribozymes (32, 33). In this study, we find that mutations of Arg-69, but not of other active site residues, were partially rescued by the modified substrate. We conclude that this highly conserved arginine residue likely contributes to catalysis of the cleavage reaction by protonating the 3′ oxygen leaving group.
EXPERIMENTAL PROCEDURES
Sin Protein Purification
All mutants used in this paper were derived from the plasmid pSA1122 (kindly provided by Sally J. Rowland), which encodes full-length WT Sin of pI9789 and a C-terminal His6 tag. pSA1122 was first mutagenized to incorporate the R54E and Q115R mutations, yielding pSA1147. All subsequent point mutations were created via the QuikChange Lightning site-directed mutagenesis kit (Stratagene). The pSA1147 derivatives were then transformed into Escherichia coli Rosetta (DE3)/pLysS cells (Novagen) and grown at 225 rpm and 37 °C in Luria-Bertani medium containing 50 μg/ml kanamycin and 33 μg/ml chloramphenicol. The cells were grown to an A600 of ∼0.6 and induced with 0.5 mm isopropyl-β-d-thiogalactopyranoside. Following induction, the cells were grown for an additional 3 h and harvested by centrifugation for 10 min at 8000 rpm. The cell pellets were stored at −20 °C for later use. The pellet from 2 liters of culture (∼10 g) was resuspended in 50 ml of lysis buffer (25 mm Tris, pH 7.5, 100 mm NaCl, 2.5 mm EDTA, 10 mm β-mercaptoethanol, and protease inhibitor mixture (Complete; Roche Applied Science), sonicated, and centrifuged at 18,000 rpm in an SS34 rotor for 60 min. Sin was insoluble and remained in the pellet and was thus resuspended in buffer 2 (lysis buffer + 400 mm NaCl) and centrifuged again. The pellet was resuspended in buffer 3 (lysis buffer + 0.5% Triton X-100) and centrifuged again. The pellet was then resuspended in 50 ml of buffer A (20 mm NaPO4, pH 7.5, 6 m urea, 0.5 m NaCl) and spun again, yielding solubilized, denatured Sin. The supernatant was filtered (0.22-μm pore size) and loaded onto a HisTrap nickel column (Amersham Biosciences) and eluted with buffer B (buffer A + 0.5 m imidazole). Pooled fractions were dialyzed against 3 × 1 liter of buffer C (25 mm Tris, pH 7.5, 0.5 mm EDTA, 1 mm β-mercaptoethanol, and 6 m urea) and loaded onto a Mono S column and eluted with buffer D (buffer C + 1 m NaCl). Pooled fractions containing purified Sin (∼95%) were then refolded via dialysis against 2 × 1 liter of buffer E (25 mm Tris, pH 7.5, 0.5 mm EDTA, 2 m NaCl, and 20% glycerol) and then 2 × 1 liter of buffer F (25 mm Tris, pH 7.5, 0.5 mm EDTA, 400 mm (NH4)2SO4, and 20% glycerol). The protein was concentrated to ∼10 mg/ml in centrifugal filters (molecular weight cutoff 10,000; Amicon) and stored in 10-μl aliquots at −80 °C. The final yield was ∼10 mg of protein/liter of culture. Concentrated proteins were diluted to 10 μm stocks in buffer containing 10 mm Tris, pH 8.0, 1 m NaCl, 5% glycerol, and 100 μg/ml BSA for use in cleavage assays.
Synthesis of the 3′S Substrate
Oligodeoxynucleotides were synthesized (1 μmol scale) on a Millipore Expedite Nucleic Acid Synthesis system. 5′-O-Monomethoxytrityl-2′-deoxy-3′-thiouridine 3′S-phosphoramidite was prepared according to the method described for the synthesis of the corresponding thymidine analog (34). The phosphoramidite was used as described previously (35, 36) to synthesize the 32-mer DNA substrate containing 3′S-dU: 5′-TTGTGAAATTTGGGTA(dU)sACCCTAATCATACAA via solid phase synthesis and manual coupling at the modified position. The 3′S modified 32-mer was then purified via gel electrophoresis and dialyzed against TE buffer (10 mm Tris, pH 8.0, 1 mm EDTA). The pooled DNA was concentrated to ∼1 mg/ml in centrifugal filters (molecular weight cutoff 3000 Amicon) and stored in 30-μl aliquots at −20 °C.
Site I Duplexes
Unmodified gel-purified oligonucleotides were purchased from IDT (Coralville, IA). The 32-nucleotide top strand contained either a 3′S or a 3′O at the scissile position. It was annealed to two oligonucleotides to create a duplex with a 45-nucleotide bottom strand that contains a nick at the scissile position. One bottom strand was 5′-phosphorylated so that only the covalent bond is missing at the nick: 32-mer, 5′-TTGTGAAATTTGGGTAXACCCTAATCATACAA-3′; where X is 3′S dU or unmodified dU 45-mer_left (25-mer), 5′-TTGTTGCATTGTATGATTAGGGTAT-3′; 45-mer_right (20-mer), 5′-/Phos/ACCCAAATTTCACAACGCGT-3′ (contains 5′ phosphate).
All oligonucleotides were annealed in 100 mm NaCl by slow cooling from 85 °C to room temperature in a water bath overnight. Duplexes are cleavable by Sin only on the intact 32-mer strand whereas the nicked 45-mer strand is bound but not cleaved by the protein. Cleavage of the 32-mer strand presumably leads to dissociation of the two half-sites of the 45-mer strand.
Substrate Design Considerations
An initial substrate design featuring a fully intact duplex was discarded because cleavage of the unmodified strand is fully reversible (7). Determining the rate of cleavage of such a substrate would require a more complicated analysis based on the overall equilibrium, similar to that used by Krogh and Shuman to study topoisomerase 1B (27). Unfortunately, such analysis is more difficult to carry out in our case because it is difficult to know whether the very slow mutants ever achieve equilibrium and because the stoichiometry is more complicated for Sin. The active species is a tetramer than can synapse two DNA segments, and our previous studies showed that the equilibrium between cleavage and ligation can be affected by the protein:DNA ratio (7).
To observe kcl directly for both modified and unmodified substrates, we used a system where we can measure cleavage rates directly on both substrates without the interference of religation rates. Experiments on Tn3 showed that putting a nick at the scissile position on the opposite strand renders cleavage of the intact strand irreversible (18). The top strand contains either a 3′O or 3′S linkage, both of which are irreversibly cleaved, whereas the “45-mer” contains a nick at the scissile position.
Cleavage Assays
Cleavage assays were done as described previously with a few modifications (7). Gel-purified 32-mer oligonucleotides (either 3′O or 3′S) were 5′ 32P-labeled by T4 polynucleotide kinase and annealed in trace amounts with known amounts of unlabeled 32-mer and unlabeled, nicked 45-mer. A master reaction mix containing 500 nm site I duplex and 10 μm Sin protein was incubated with reaction buffer containing 50 mm Tris, pH 8.0, 7% glycerol, 10 mm DTT, 2 mm EDTA, 10 μg/ml poly(dI-dC), and an additional 100 mm NaCl from the added protein. Reactions were incubated at room temperature while 4.5-μl aliquots at appropriate time points were removed to tubes containing 0.5 μl of 10% SDS (1% final concentration w/v) and boiled for 1 min at 95 °C. For the pseudo-WT protein, cleavage occurred on the order of seconds, so time points were taken within the first 80 s. For the Arg-69 mutants, time points were extended out to 1 h whereas for the dual labeled substrate used in Fig. 3, the time points extended to 3 h (to obtain the equilibrium product distribution). Only time points where there was less than ∼10% cleavage were used for calculation of initial cleavage rates except for the pseudo-WT protein where ∼25% of the substrate was cleaved after 60 s. Proteinase K was added to a final concentration of 1 mg/ml and incubated at 37 °C for 30 min. An equal amount of formamide was added to the samples, which were treated for 1 min at 95 °C. All samples were loaded onto a denaturing (7 m urea) 20% (w/v) polyacrylamide (19:1 w/w acrylamide/bisacrylamide) gels and electrophoresed for 2 h at 20 watts and 2000 volts. Gels were visualized by overnight exposure on a Fuji PhosphorScreen and autoradiographed on a Typhoon PhosphorImager scanner. For clarity in the gels, neither the 20-mer nor 25-mer was labeled, so only the 32-mer is visible, which can be cleaved into a labeled 17-mer and an unlabeled 15-mer. Band intensities were quantified with the Quantity One software suite (Bio-Rad), and cleavage fractions were calculated by dividing the amount of substrate in the cleaved form (17-mer) by the amount in the cleaved and intact (32-mer) forms. Cleavage rates were calculated by plotting fraction cleaved versus time and fitted by regression to a straight line through the origin using the OriginLab 6.0 software suite. Error bars represent S.D. from experiments performed in duplicate.
FIGURE 3.

A phosphorylated nick prevents religation of the paired strand. A, schematic of the recombination assay used to test the suicide substrate. The substrate used was the same as that shown in Fig. 2, except that both strands were 5′ 32P-labeled. A tetramer of Sin synapses two duplexes in either parallel or antiparallel fashion. Cleavage of the 32-nucleotide intact strand results in an unlabeled 15-mer that is covalently linked to Sin and a labeled 17-mer with a free 3′OH. The other strand has a phosphorylated nick at the scissile position (gap and small circle), which prevents formation of a covalent Sin-DNA intermediate. A putative 180° subunit rotation aligns the broken ends for religation. Subsequent nucleophilic attack by the free 3′ hydroxyls on the phosphoserine linkages yields religated recombinant products, the length of which depends on the orientation of the synapse (parallel versus antiparallel). If the phosphorylated nick prevents religation of the strand that is paired to it, 17-mer will accumulate because it cannot be ligated to the partner duplex in either synapse. In contrast, the labeled 25-mer cannot be religated in the parallel synapse (left pathway) but can in the antiparallel synapse (right pathway). Thus ∼50% of the 25-mer should be converted to 40-mer at equilibrium. This agrees with the distribution of products seen. B, fate of the dual-labeled 3′O substrate. Upper, denaturing gel analyzing a 3-h time course of the assay diagrammed in A. >90% of 32-mer is cleaved after 3 h. Based on this, we assume that cleavage of the 32-mer strand is either irreversible or that the religation rate is much slower than the cleavage rate. However, ligation of the unlabeled half-site (15-mer) to the labeled 25-mer half-site does occur as evidenced by the appearance of a 40-mer product. As the reaction approaches equilibrium, the labeled 25-mer is distributed nearly evenly between a recombinant 40-mer and the original 25-mer half-site. No products involving the initial 5′ phosphorylated 20-mer are seen because the nick prevents Sin from forming the initial phosphoserine linkage to that strand. Lower, graph quantifying the fate of the labeled half-sites over the course of 3 h.
pH Screen
Cleavage assays for the pH screen were done as above except with different buffers. The buffers (50 mm) used were MES, pH 5.5; bis-Tris,4 pH 6.0; bis-Tris, pH 6.5; HEPES, pH 7.0; Tris, pH 7.5; Tris, pH 8.0; Tris, pH 8.5; and CHES, pH 9.0.
RESULTS
Design of the DNA Cleavage Assay
The DNA substrate for all cleavage assays was similar to that used in previous Sin studies, but modified to render cleavage essentially irreversible regardless of the leaving group (7). Briefly, the 5′ 32P-labeled top strand (as shown in Fig. 2) contains either a 3′O or 3′S at the scissile position, and the unlabeled bottom strand has a nick at the scissile position. The nick prevents religation of the top strand after the normally reversible cleavage reaction, presumably by hindering realignment of the broken ends (18). Because sulfur more easily stabilizes a negative charge than oxygen, a 3′ thiolate is a better leaving group than a 3′ alkoxide; it may therefore not require protonation during catalysis and is predicted to be a better leaving group in the absence of a general acid.
FIGURE 2.
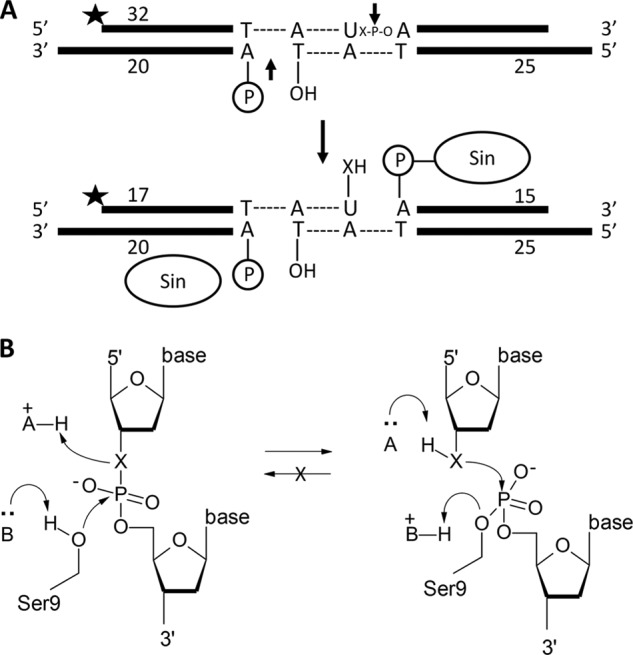
Cleavage assay. A, substrate used in all DNA cleavage assays. Two Sin protomers can bind this crossover site. The top strand (32 nucleotides) is 5′ 32P-labeled, and the scissile position (arrow) contains either a 3′ oxygen or 3′ sulfur (X). The unlabeled bottom strand is nicked at the position of cleavage. Cleavage of the top strand forms a 5′ phosphoserine linkage and a free 3′OH or 3′SH. B, reaction diagram. This represents a hypothetical catalytic mechanism for either the 3′O or 3′S substrate (schematized as X). A general base deprotonates the nucleophilic Ser-9 whereas a general acid protonates the 3′ leaving group. The nick on the opposite strand prevents the reverse (religation) reaction, rendering it a suicide substrate regardless of the identity of X.
All proteins used in these assays contained the Q115R mutation, which bypasses the need for regulatory sites and/or proteins, the R54E mutation, which enhances solubility for in vitro experiments, and a C-terminal His6 tag. The C terminus, Gln-115, and Arg-54 are all distant from the active site, and the R54E mutation has no effect on reactions in the absence of regulatory sites. Henceforth, the His6-tagged R54E/Q115R construct will be referred to as “pseudo-WT,” and all additional mutations were made in this activated background. All six conserved arginines were individually mutated to Ala and Lys.
Cleavage reactions were set up under conditions where only a single turnover is expected: with 50 nm DNA duplex and a 10-fold excess (1 μm) of Sin monomers (note that each duplex contains binding sites for two Sin subunits). As Sin binds this site with a Kd,app of ∼2.5–5 nm, the DNA should be fully bound. The protein concentration was chosen to ensure that it would be in the active tetrameric form. Previous work showed that the cleavage rate plateaus at protein concentrations above ∼500 nm (7). Time points were chosen for each variant such that cleavage rates could be fit to the initial, approximately linear portion of the product versus time curve. The results are summarized in Table 1.
TABLE 1.
Cleavage rates
The rates are described as kcl values, fitted from the slope of the reaction velocities from two independent experiments. The -fold reduction is the reduction of the rate relative to that of the pseudo-WT with the same substrate. NA is data not available.
| Mutant | k3′O (×10−3) | k3′S (×10−3) | k3′S/k3′O | k3′O | k3′S |
|---|---|---|---|---|---|
| % cleaved/s | % cleaved/s | % cleaved/s | -fold reduction | -fold reduction | |
| Pseudo-WT | 420 ± 22 | 281 ± 2.8 | 0.67 | ≡1 | ≡1 |
| R7A | ∼0 | ∼0 | NA | NA | NA |
| R7K | ∼0 | ∼0 | NA | NA | NA |
| R43A | 18.8 ± 0.84 | 1.86 ± 0.16 | 0.099 | 22.4 | 151 |
| R43K | 53.1 ± 2.7 | 1.02 ± 0.65 | 0.019 | 7.9 | 275 |
| R66A | ∼0 | ∼0 | NA | NA | NA |
| R66K | ∼0 | ∼0 | NA | NA | NA |
| R69A | 0.0518 ± 0.0033 | 0.490 ± 0.025 | 9.5 | 8110 | 573 |
| R69K | 1.52 ± 0.072 | 2.22 ± 0.044 | 1.5 | 276 | 127 |
| R125A | 187 ± 5.3 | 103 ± 2.8 | 0.56 | 2.25 | 2.73 |
| R125K | 170 ± 4.8 | 39.0 ± 1.7 | 0.23 | 2.47 | 7.21 |
| R131A | 85.5 ± 2.7 | 55.2 ± 1.2 | 0.65 | 4.91 | 5.09 |
| R131K | 206 ± 7.8 | 87.3 ± 2.6 | 0.42 | 2.04 | 3.22 |
The suicide substrate was tested first with the pseudo-WT enzyme. Throughout the time course, disappearance of the 32-mer substrate was concurrent with the appearance of a 17-mer cleavage product. Quantitative analysis (of duplicate experiments) showed that the protein cleaves the 3′S substrate ∼1.5 times more slowly than the 3′O substrate. The pseudo-WT enzyme was expected to cleave the 3′S substrate more slowly because it has evolved to act on a substrate containing a 3′ oxygen rather than a 3′ sulfur, and thus the active site may accommodate the larger sulfur atom less efficiently. To verify that the nicked bottom strand prevents religation of the top strand without otherwise affecting recombination, we examined the products of reactions where both strands were 5′ 32P-labeled (Fig. 3). Cleavage of the 34-nucleotide top strand neared completion: 80–90% was rapidly converted to a 17-nucleotide cleavage product, indicating that even if any religation did occur, the religation rate was very slow compared with the cleavage rate. In contrast, the 20-nucleotide precleaved labeled bottom strand equilibrated to near 50% with a 40-nucleotide recombination product. This was as expected: recombination of DNAs synapsed in a parallel orientation would yield products identical to the substrates (and thus not ligatable), whereas recombination of antiparallel DNAs would allow ligation of the labeled half of the bottom strand (Fig. 3).
R69A and R69K are Partially Rescued by the 3′S Substrate
All mutants were assayed in the same way as the pseudo-WT protein, except that for some, longer time points were needed. All mutants displayed a defect in cleavage relative to the pseudo-WT enzyme (Table 1). In general, the alanine substitutions were more impaired than the corresponding lysine substitutions. This pattern was not particularly surprising, considering that a mutation from arginine to lysine is a more conservative change in terms of both charge and shape.
All of the mutants tested except those at position 69 cleaved the 3′O substrate faster than the 3′S substrate (Table 1 and Fig. 4). The most deleterious mutants were those at positions 7 and 66, which exhibited almost no detectable cleavage activity even after 6 h. Mutants of Arg-43 showed measurable activity but with rates reduced by 7.9–280-fold. Changes at positions 125 and 131 were less deleterious: cleavage rates for these variants were no worse than 5-fold lower than that of the pseudo-WT enzyme. The trend of Arg-7, Arg-66, and Arg-69 being the most important of the conserved Arg residues is consistent with experiments on an activated mutant of Tn3 resolvase (18).
FIGURE 4.
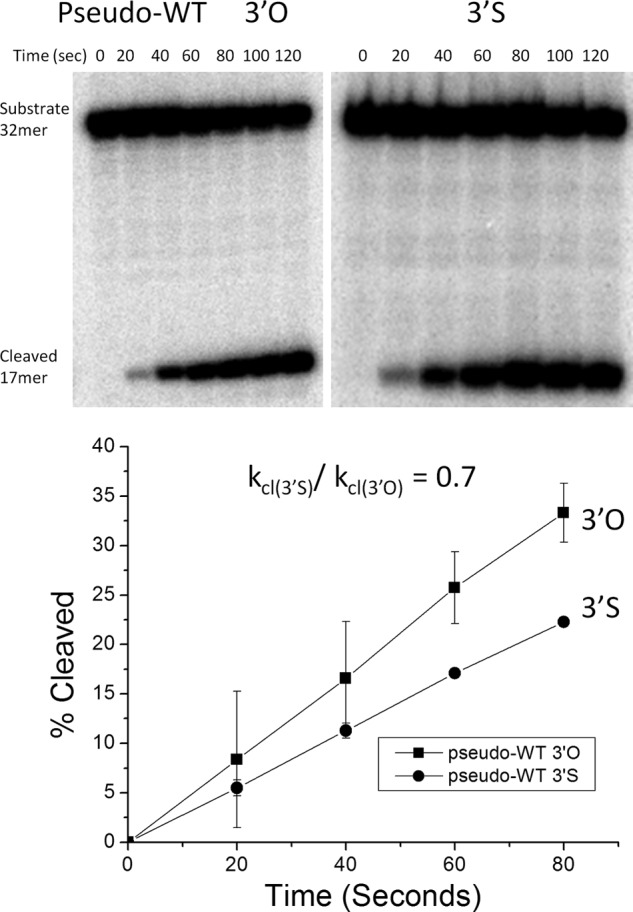
Cleavage assay with pseudo-WT Sin. DNA cleavage by Sin Q115R/R54E (pseudo-WT) was followed over 2 min on the 3′O unmodified (left) or the 3′ phosphorothiolate modified substrate (right). Time points were taken every 20 s and the 17-nucleotide product separated from the 32-nucleotide substrate on denaturing gels. The graph below is a quantification of each gel where the percentage cleavage is calculated by taking the amount of product in the cleaved form and dividing by the total amount of product and substrate. The averages and S.D. (error bars) from two replicates are shown. Only the first few data points that were used for rate calculation are shown. The ratio of the slopes was 0.7.
Variants at position 69 also cleaved DNA slowly but differed from all the other Arg mutants studied in that they cleaved the 3′S-containing substrate more rapidly than the 3′O one (Fig. 5 and Table 1). Whereas the other mutants cleaved the 3′S-containing substrate up to ∼50-fold more slowly than the 3′O-containing one, R69K and R69A cleaved it 1.5 and 9.5 times faster, respectively. As changing the leaving group from an oxyanion to a thioanion was expected to partially rescue a defect in general acid catalysis, we conclude that Arg-69 is a good candidate for the general acid that protonates the leaving group during the cleavage reaction.
FIGURE 5.

Cleavage assay with Sin R69A and R69K. Upper, representative denaturing gels showing the time courses of DNA cleavage by Arg-69 mutants. Lower, plots showing the percentage DNA cleaved versus time by R69A (left) and R69K (right). The averages ± S.D. (error bars) from two replicates are shown. Lines shown on the plots simply connect the points to aid the eye; see Table 1 for the slopes and errors resulting from fitting these points to a straight line.
pH Dependence of DNA Cleavage Rates
As described above, the pseudo-WT enzyme catalyzed cleavage of the 3′O and 3′S substrates at similar rates at pH 8.0. Our conclusion that Arg-69 mediates general acid catalysis in the Sin active site hinges upon the faster reaction of 3′S versus 3′O in the R69A mutant (k3′S/k3′O = 9.5) relative to pseudo-WT enzyme (k3′S/k3′O = 0.7). To control for the possibility that different rate-limiting steps for the 3′S and 3′O substrates could mask the true k3′S/k3′O effect in the pseudo-WT enzyme, we attempted to expose potential changes in the rate-limiting step by monitoring the reactions of each substrate at different pH values. Furthermore, it remained possible that the phosphorothiolate altered the response of the enzyme to changes in pH. Assays were performed as before, except that the pH was changed from 8.0 to values between 5.5 and 9.0. Assays were performed on the pseudo-WT protein and both Arg-69 variants with both the 3′O and 3′S substrates (Fig. 6). Cleavage rates were not calculated for R69A with the 3′O substrate because insufficient cleavage product was produced over the duration of the experiment at most pH values.
FIGURE 6.
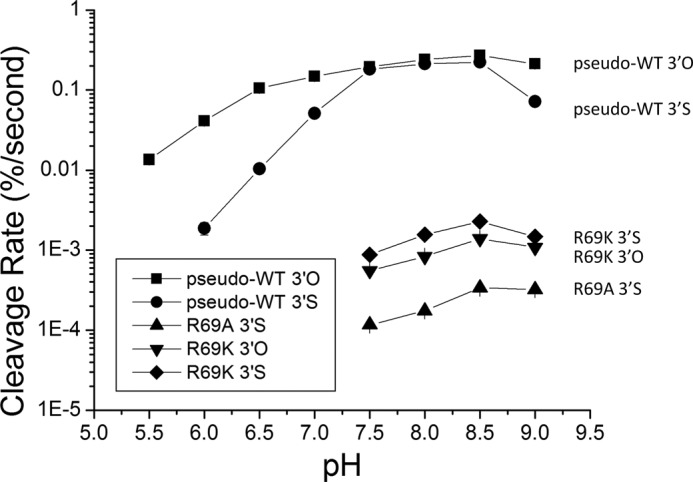
pH profile of Sin mutants. This graph is a summary of a set of experiments conducted at various pH screens for three different mutants and two different substrates which are listed next to each curve on the graph. The x axis is the pH value, and the y axis is the cleavage rate in percentage substrate cleaved per second. The y axis is on a log scale to visualize all experiments over a wide range of rates, and the error bars indicate errors of linear rate fits. Missing data points indicate a rate equal to ∼0 due to the sensitivity limitations of these experiments. The points are connected to aid the eye but the quantifiable data were not complete enough to be meaningfully fit to an ionization model.
The pH rate profiles imply that when the chemical rate is the fastest, as for cleavage of the 3′O substrate by the pseudo-WT enzyme at optimal pH values, some nonchemical step does limit the observed rate. Comparison of the 3′O versus 3′S curves for this enzyme shows that at most pH values cleavage of the 3′S substrate is roughly an order of magnitude slower. However, for 3′O substrate there is a broad plateau in the middle of the curve, such that cleavage rates on the two substrates are nearly equal between pH 7.5 and 8.5. From pH 5.5 to pH 7.0, the log linear increase in rate with pH is a hallmark for rate-limiting chemistry. The shift in the pH rate profile from the log linear region to the plateau could represent either a change in rate-limiting step (for example, from chemistry to a conformational change) or a change in the group undergoing titration (either complete deprotonation of the group mediating the rate-stimulating effect or the onset of a second titration that inhibits the reaction rate). Because the 3′O and 3′S substrates react at very different rates at low pH but plateau at nearly the same rate, we favor the model in which a conformational change governs the rate in the plateau region of these profiles.
The shapes of the curves for the R69A and R69K mutants were similar and were consistent with the trends reported in Table 1 for reactions at pH 8.0: replacement of R69K is less deleterious than replacement with Ala, and both variants always cleave the 3′S substrate faster than the 3′O substrate. Unfortunately, the pH rate data could not be collected in enough detail to derive the apparent pKa of the presumed general acid and base: reactions below pH 7.5 were too slow for accurate rate measurement, as were most reactions with R69A on the 3′O substrate. Also, the decrease in all rates at pH 9.0 may be due to instability of the protein at high pH. However, these observations do strengthen the conclusion that Arg-69 is functionally linked to the leaving group and likely serves as a general acid: first by showing that, for the pseudo-WT enzyme, the difference between intrinsic cleavage rates of the 3′S and 3′O substrates is probably larger than that reported in Table 1 due to the masking effect of an alternative rate-limiting step, and second, by showing that the rescue effect of the 3′S substrate for both Arg-69 mutants occurs at multiple pH values.
DISCUSSION
The pKa of arginine in water is ∼12, which is quite high for a general acid catalyst, but the environment of an enzyme active site can dramatically shift the pKa values of critical residues. The pKa of a lysine or arginine could be lowered by surrounding it with a cluster of positive charges that would alter the equilibrium between the neutral and protonated forms of that residue. For example, acetoacetate decarboxylase appears to lower the pKa of an active site lysine to ∼6 with the presence of an adjacent positively charged lysine contributing to this effect (37). However, newer structural information on this enzyme shows that the adjacent lysine is actually positioned away from the active site lysine, but aids in lowering its pKa by orienting it within a hydrophobic protein core which leads to a desolvation effect (38). Also, arginines have been implicated in general base catalysis in enzymes such as IMP dehydrogenase, likely indicative of their ability to undergo a large pKa shift to increase the fraction of the neutral (basic) form (29, 39).
Our data suggest that the six arginines in the vicinity of the active site of Sin (Fig. 1C) exemplify this effect. Whenever measurable, the k3′S/k3′O ratio was larger for the alanine substitution than the corresponding lysine one. Considering the long range nature of electrostatic interactions, every positive charge in the active site could aid the general acid by helping to lower its pKa. Thus, substitution of any conserved arginine to alanine would be expected to slow the cleavage rate for a 3′O leaving group more than that for a 3′S leaving group. Note that for substitutions at Arg-69, the difference in k3′S/k3′O for lysine (1.5) versus alanine (9.5) probably reflects the lysine acting directly as a general acid even though its positively charged primary amine group may not be optimally oriented.
Our data reveal a functional link between Arg-69 and the 3′ oxygen leaving group, implying that Sin and other serine recombinases use this highly conserved arginine to directly protonate the 3′ bridging oxygen during the cleavage event. It remains possible that Arg-69 helps lower the pKa of either or both Arg-7 and Arg-66 and that one of those arginines act as the general acid itself; but based on the crystal structure, this seems less likely. The identity of the general base that deprotonates Ser-9 is more difficult to address experimentally; but as noted previously, Arg-7 is the best candidate due to its location and the nearly undetectable activity of mutants at that position (18).
The other conserved arginines probably help lower the pKa values of the general acid and base but may play other roles as well. Within this group, mutations of Arg-66 have the most dramatic effect. Arg-66 may stabilize the transition state, by coordinating the nonbridging oxygens and stabilizing the buildup of negative charge, but we have not addressed this experimentally. Its position at the bottom of the catalytic pocket may also provide considerable catalytic power simply by pulling the scissile phosphate into the active site and orientating it properly. Other enzymes likely use an arginine residue that directly contacts the scissile phosphate in this capacity, which has been reviewed extensively (21, 40). Arg-43 participates in a second shell of hydrogen bonding around the active site and may serve a primarily structural and electrostatic role. Finally, Arg-125 and Arg-131 probably bind the phosphate backbone of neighboring nucleotides and help to dock the DNA properly into the active site (15, 19, 20).
Although divalent metal ions are often used to catalyze phosphotransfer, serine recombinases and many other enzymes have evolved different strategies that do not require cofactors (21–23). Such enzymes often catalyze reactions where the nucleophile is macromolecular, e.g. a protein side chain or an RNA 2′OH. Macromolecular nucleophiles are presumably less difficult to localize, which may obviate one function of metal ions. Nevertheless, general acid and base catalysts are still important in these nucleotidyl transferases, and a multitude of functional groups, including, water, lysine, histidine, and even nucleotides themselves, have been shown to play those roles. For example, in some catalytic RNAs nucleobases themselves play a role in acid/base catalysis. The HDV (hepatitis delta virus) ribozyme contains a catalytic cytosine with a protonated N3-imino nitrogen, which functions as a general acid to protonate the leaving 5′ oxygen (41). Also, the hairpin and VS (Varkud satellite) ribozymes have been proposed to use a guanosine and an adenosine as the general base and general acid, respectively (42). The type Ib topoisomerases and tyrosine recombinases (which share a conserved catalytic domain) use a lysine as a general acid to protonate the 5′ oxygen and water or histidine to deprotonate the nucleophilic tyrosine (26–28). Additional data support a potential proton relay through a conserved arginine in topoisomerase Ib, but a direct proton transfer from the guanidinium group to the oxygen leaving group was not proposed (43). Despite this variety, serine recombinases appear to be quite unusual in using arginine as a general acid. The only other enzyme, to our knowledge, for which arginine has been proposed to act as a general acid in nucleotidyl transfer is staphylococcal nuclease, but that idea has not been rigorously tested (44). This work thus adds to the repertoire of strategies that enzymes can use in catalyzing such reactions.
Acknowledgments
We thank Ying Zhang Pigli for protein purification advice, Adrianna Zhang for training in radioactive assays, Sherwin Montaño for helpful discussion and advice, and W. Marshall Stark for comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM086826 (to P. A. R.) and R01GM088656 (to J. A. P.).
- bis-Tris
- bis(2-hydroxyethyl)iminotris(hydroxymethyl)methane
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid.
REFERENCES
- 1. Grindley N. D., Whiteson K. L., Rice P. A. (2006) Mechanisms of site-specific recombination. Annu. Rev. Biochem. 75, 567–605 [DOI] [PubMed] [Google Scholar]
- 2. Hatfull G. F., Grindley N. D. (1986) Analysis of γδ resolvase mutants in vitro: evidence for an interaction between serine 10 of resolvase and site I of res. Proc. Natl. Acad. Sci. U.S.A. 83, 5429–5433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Derbise A., Dyke K. G., el Solh N. (1995) Rearrangements in the staphylococcal β-lactamase-encoding plasmid, pIP1066, including a DNA inversion that generates two alternative transposons. Mol. Microbiol. 17, 769–779 [DOI] [PubMed] [Google Scholar]
- 4. Highlander S. K., Hultén K. G., Qin X., Jiang H., Yerrapragada S., Mason E. O., Jr., Shang Y., Williams T. M., Fortunov R. M., Liu Y., Igboeli O., Petrosino J., Tirumalai M., Uzman A., Fox G. E., Cardenas A. M., Muzny D. M., Hemphill L., Ding Y., Dugan S., Blyth P. R., Buhay C. J., Dinh H. H., Hawes A. C., Holder M., Kovar C. L., Lee S. L., Liu W., Nazareth L. V., Wang Q., Zhou J., Kaplan S. L., Weinstock G. M. (2007) Subtle genetic changes enhance virulence of methicillin resistant and sensitive Staphylococcus aureus. BMC Microbiol. 7, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Paulsen I. T., Gillespie M. T., Littlejohn T. G., Hanvivatvong O., Rowland S. J., Dyke K. G., Skurray R. A. (1994) Characterisation of sin, a potential recombinase-encoding gene from Staphylococcus aureus. Gene 141, 109–114 [DOI] [PubMed] [Google Scholar]
- 6. Rowland S. J., Dyke K. G. (1989) Characterization of the staphylococcal β-lactamase transposon Tn552. EMBO J. 8, 2761–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mouw K. W., Steiner A. M., Ghirlando R., Li N. S., Rowland S. J., Boocock M. R., Stark W. M., Piccirilli J. A., Rice P. A. (2010) Sin resolvase catalytic activity and oligomerization state are tightly coupled. J. Mol. Biol. 404, 16–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arnold P. H., Blake D. G., Grindley N. D., Boocock M. R., Stark W. M. (1999) Mutants of Tn3 resolvase which do not require accessory binding sites for recombination activity. EMBO J. 18, 1407–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haffter P., Bickle T. A. (1988) Enhancer-independent mutants of the Cin recombinase have a relaxed topological specificity. EMBO J. 7, 3991–3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klippel A., Cloppenborg K., Kahmann R. (1988) Isolation and characterization of unusual gin mutants. EMBO J. 7, 3983–3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olorunniji F. J., He J., Wenwieser S. V., Boocock M. R., Stark W. M. (2008) Synapsis and catalysis by activated Tn3 resolvase mutants. Nucleic Acids Res. 36, 7181–7191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haykinson M. J., Johnson L. M., Soong J., Johnson R. C. (1996) The Hin dimer interface is critical for Fis-mediated activation of the catalytic steps of site-specific DNA inversion. Curr. Biol. 6, 163–177 [DOI] [PubMed] [Google Scholar]
- 13. Rowland S. J., Boocock M. R., McPherson A. L., Mouw K. W., Rice P. A., Stark W. M. (2009) Regulatory mutations in Sin recombinase support a structure-based model of the synaptosome. Mol. Microbiol. 74, 282–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dhar G., McLean M. M., Heiss J. K., Johnson R. C. (2009) The Hin recombinase assembles a tetrameric protein swivel that exchanges DNA strands. Nucleic Acids Res. 37, 4743–4756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li W., Kamtekar S., Xiong Y., Sarkis G. J., Grindley N. D., Steitz T. A. (2005) Structure of a synaptic γδ resolvase tetramer covalently linked to two cleaved DNAs. Science 309, 1210–1215 [DOI] [PubMed] [Google Scholar]
- 16. Keenholtz R. A., Rowland S. J., Boocock M. R., Stark W. M., Rice P. A. (2011) Structural basis for catalytic activation of a serine recombinase. Structure 19, 799–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stark W. M., Sherratt D. J., Boocock M. R. (1989) Site-specific recombination by Tn3 resolvase: topological changes in the forward and reverse reactions. Cell 58, 779–790 [DOI] [PubMed] [Google Scholar]
- 18. Olorunniji F. J., Stark W. M. (2009) The catalytic residues of Tn3 resolvase. Nucleic Acids Res. 37, 7590–7602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang W., Steitz T. A. (1995) Crystal structure of the site-specific recombinase γδ resolvase complexed with a 34 bp cleavage site. Cell 82, 193–207 [DOI] [PubMed] [Google Scholar]
- 20. Kamtekar S., Ho R. S., Cocco M. J., Li W., Wenwieser S. V., Boocock M. R., Grindley N. D., Steitz T. A. (2006) Implications of structures of synaptic tetramers of γδ resolvase for the mechanism of recombination. Proc. Natl. Acad. Sci. U.S.A. 103, 10642–10647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lassila J. K., Zalatan J. G., Herschlag D. (2011) Biological phosphoryl-transfer reactions: understanding mechanism and catalysis. Annu. Rev. Biochem. 80, 669–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim E. E., Wyckoff H. W. (1991) Reaction mechanism of alkaline phosphatase based on crystal structures: two-metal ion catalysis. J. Mol. Biol. 218, 449–464 [DOI] [PubMed] [Google Scholar]
- 23. Steitz T. A., Steitz J. A. (1993) A general two-metal ion mechanism for catalytic RNA. Proc. Natl. Acad. Sci. U.S.A. 90, 6498–6502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. O'Brien P. J., Herschlag D. (2002) Alkaline phosphatase revisited: hydrolysis of alkyl phosphates. Biochemistry 41, 3207–3225 [DOI] [PubMed] [Google Scholar]
- 25. Beese L. S., Steitz T. A. (1991) Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two-metal ion mechanism. EMBO J. 10, 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Whiteson K. L., Chen Y., Chopra N., Raymond A. C., Rice P. A. (2007) Identification of a potential general acid/base in the reversible phosphoryl transfer reactions catalyzed by tyrosine recombinases: Flp H305. Chem. Biol. 14, 121–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krogh B. O., Shuman S. (2000) Catalytic mechanism of DNA topoisomerase Ib. Mol. Cell 5, 1035–1041 [DOI] [PubMed] [Google Scholar]
- 28. Gibb B., Gupta K., Ghosh K., Sharp R., Chen J., Van Duyne G. D. (2010) Requirements for catalysis in the Cre recombinase active site. Nucleic Acids Res. 38, 5817–5832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guillén Schlippe Y. V., Hedstrom L. (2005) Is Arg-418 the catalytic base required for the hydrolysis step of the IMP dehydrogenase reaction? Biochemistry 44, 11700–11707 [DOI] [PubMed] [Google Scholar]
- 30. Deweese J. E., Burgin A. B., Osheroff N. (2008) Using 3′-bridging phosphorothiolates to isolate the forward DNA cleavage reaction of human topoisomerase IIα. Biochemistry 47, 4129–4140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deweese J. E., Burch A. M., Burgin A. B., Osheroff N. (2009) Use of divalent metal ions in the DNA cleavage reaction of human type II topoisomerases. Biochemistry 48, 1862–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Deweese J. E., Burgin A. B., Osheroff N. (2008) Human topoisomerase IIα uses a two-metal ion mechanism for DNA cleavage. Nucleic Acids Res. 36, 4883–4893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li N. S., Frederiksen J. K., Piccirilli J. A. (2011) Synthesis, properties, and applications of oligonucleotides containing an RNA dinucleotide phosphorothiolate linkage. Acc. Chem. Res. 44, 1257–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cosstick R., Vyle J. S. (1990) Synthesis and properties of dithymidine phosphate analogues containing 3′-thiothymidine. Nucleic Acids Res. 18, 829–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vyle J. S., Connolly B. A., Kemp D., Cosstick R. (1992) Sequence- and strand-specific cleavage in oligodeoxyribonucleotides and DNA containing 3′-thiothymidine. Biochemistry 31, 3012–3018 [DOI] [PubMed] [Google Scholar]
- 36. Sun S., Yoshida A., Piccirilli J. A. (1997) Synthesis of 3′-thioribonucleosides and their incorporation into oligoribonucleotides via phosphoramidite chemistry. RNA 3, 1352–1363 [PMC free article] [PubMed] [Google Scholar]
- 37. Highbarger L. A., Gerlt J. A., Kenyon G. L. (1996) Mechanism of the reaction catalyzed by acetoacetate decarboxylase: importance of lysine 116 in determining the pKa of active-site lysine 115. Biochemistry 35, 41–46 [DOI] [PubMed] [Google Scholar]
- 38. Ho M. C., Ménétret J. F., Tsuruta H., Allen K. N. (2009) The origin of the electrostatic perturbation in acetoacetate decarboxylase. Nature 459, 393–397 [DOI] [PubMed] [Google Scholar]
- 39. Guillén Schlippe Y. V., Hedstrom L. (2005) A twisted base? The role of arginine in enzyme-catalyzed proton abstractions. Arch. Biochem. Biophys. 433, 266–278 [DOI] [PubMed] [Google Scholar]
- 40. O'Brien P. J., Lassila J. K., Fenn T. D., Zalatan J. G., Herschlag D. (2008) Arginine coordination in enzymatic phosphoryl transfer: evaluation of the effect of Arg-166 mutations in Escherichia coli alkaline phosphatase. Biochemistry 47, 7663–7672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Das S. R., Piccirilli J. A. (2005) General acid catalysis by the hepatitis δ virus ribozyme. Nat. Chem. Biol. 1, 45–52 [DOI] [PubMed] [Google Scholar]
- 42. Pinard R., Hampel K. J., Heckman J. E., Lambert D., Chan P. A., Major F., Burke J. M. (2001) Functional involvement of G8 in the hairpin ribozyme cleavage mechanism. EMBO J. 20, 6434–6442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Krogh B. O., Shuman S. (2002) Proton relay mechanism of general acid catalysis by DNA topoisomerase IB. J. Biol. Chem. 277, 5711–5714 [DOI] [PubMed] [Google Scholar]
- 44. Weber D. J., Gittis A. G., Mullen G. P., Abeygunawardana C., Lattman E. E., Mildvan A. S. (1992) NMR docking of a substrate into the x-ray structure of staphylococcal nuclease. Proteins 13, 275–287 [DOI] [PubMed] [Google Scholar]