

Background: Multiple steps in the degradation of ubiquitinated proteins by the 26 S proteasome require ATP.
Results: The six ATPase subunits of the proteasome function in a cyclic manner. Rates of degradation of ubiquitinated proteins are directly proportional to rates of ATP hydrolysis.
Conclusion: A specific number of ATPs are consumed in degrading a ubiquitinated substrate.
Significance: Polypeptide structure determines the time required and ATP consumed in degrading ubiquitin conjugates.
Keywords: ATP, ATPases, Proteasome, Protein Degradation, Ubiquitin, 26 S Proteasome, AAA-ATPases, ATP Hydrolysis, Ubiquitin Conjugates
Abstract
The degradation of ubiquitinated proteins by 26 S proteasomes requires ATP hydrolysis. To investigate if the six proteasomal ATPases function independently or in a cyclic manner, as proposed recently, we used yeast mutants that prevent ATP binding to Rpt3, Rpt5, or Rpt6. Although proteasomes contain six ATPase subunits, each of these single mutations caused a 66% reduction in basal ATP hydrolysis, and each blocked completely the 2–3-fold stimulation of ATPase activity induced by ubiquitinated substrates. Therefore, the ATPase subunits must function in a ordered manner, in which each is required for the stimulation of ATPase activity by substrates. Although ATP is essential for multiple steps in proteasome function, when the rate of ATP hydrolysis was reduced incrementally, the degradation of Ub5-DHFR (where Ub is ubiquitin and DHFR is dihydrofolate reductase) decreased exactly in parallel. This direct proportionality implies that a specific number of ATPs is consumed in degrading a ubiquitinated protein. When the ubiquitinated DHFR was more tightly folded (upon addition of the ligand folate), the rate of ATP hydrolysis was unchanged, but the time to degrade a Ub5-DHFR molecule (∼13 s) and the energy expenditure (50–80 ATPs/Ub5-DHFR) both increased by 2-fold. With a mutation in the ATPase C terminus that reduced gate opening into the 20 S proteasome, the energy costs and time required for conjugate degradation also increased. Thus, different ubiquitin conjugates activate similarly the ATPase subunit cycle that drives proteolysis, but polypeptide structure determines the time required for degradation and thus the energy cost.
Introduction
Protein breakdown in eukaryotic cells requires ATP consumption at multiple steps. Proteins are targeted for degradation through the ATP-dependent addition of a ubiquitin chain, which leads to their ATP-dependent binding and proteolysis by the 26 S proteasome (1). This large proteolytic complex consists of one or two 19 S regulatory particles and the core 20 S proteasome, which sequesters the proteolytic sites inside its central chamber (2). ATP binding to the 19 S particle promotes the association with the 20 S particle and opens a gated channel into the 20 S particle that allows substrate entry and access to the peptidase sites (3, 4). This gate-opening mechanism involves the six 19 S ATPase subunits (Rpt1–6), which form a hexameric ring. Their C-terminal HbYX (hydrophobic-Tyr-X) motifs bind directly to pockets in the 20 S proteasome to trigger gate opening (5, 6). Translocation of proteins through this narrow gated channel requires substrates to be unfolded and linearized in an ATP-dependent process. Therefore, ATP binding and hydrolysis by the six ATPase subunits (Rpt1–6) play essential roles at multiple steps in the degradation of ubiquitinated substrates (7).
Nevertheless, it is currently unclear how these six ATPases are coordinated, how their rate of ATP consumption affects the rates of substrate degradation, and which step is rate-limiting during degradation of ubiquitinated proteins. Furthermore, it is unknown how much ATP is consumed in the degradation of a single ubiquitinated protein or if the ATP costs or the time required for degradation varies between ubiquitinated substrates with different structures. Therefore, we have systematically investigated how altering the rate of ATP hydrolysis and the capacity of the proteasomal ATPases for gate opening affect protein degradation rates.
In catalyzing proteolysis, the six ATPase subunits coordinate various steps, including substrate binding, deubiquitination, unfolding, and translocation. The binding of ubiquitin conjugates to the 26 S proteasome transiently activates the particle by enhancing gate opening and ATP hydrolysis, which presumably ensures maximum efficiency during degradation (8–10) and prevents nonspecific degradation of proteins. This stimulation of proteasome activity by ubiquitin conjugates requires their interaction with the associated deubiquitinating enzymes, and thus, substrate deubiquitination is coupled to conjugate degradation (10). Interestingly, this activation of gate opening is blocked by single mutations in the nucleotide-binding pocket of each of the six ATPase subunits (10). This observation suggests that the individual ATPase subunits function cooperatively in gate opening and probably also in protein and ATP hydrolysis. However, it is unclear how these six subunits contribute to the overall ATPase and proteolytic activities of the proteasome and how their function is regulated.
In the many AAA-ATPase complexes, which catalyze other cellular processes, the six subunits are identical, but the 19 S ATPases (Rpt1–6), although homologous, differ structurally and phenotypically (11). It is therefore important to understand the precise roles of the six 19 S ATPases and to determine whether they function stochastically as proposed for the homologous bacterial AAA-ATPases, ClpX (12), or in a specific ordered manner, as we recently proposed for the proteasome regulatory ATPases (13). In a stochastic mode of ATP hydrolysis, the loss of single subunit would only marginally reduce ATPase activity (perhaps by one-sixth). However, in an ordered cyclic mechanism (13), the failure of a single ATPase subunit would affect the behavior of other subunits, resulting in a more dramatic effect on the ATPase activity of the particle. Our recent findings on nucleotide binding to the proteasomal ATPases led us to propose a highly cooperative, cyclical mechanism for ATP-ADP exchange, in which ATP binding and hydrolysis by one subunit affect ATP binding and ADP release from the neighboring subunits throughout the hexameric ring (13). In contrast, based on studies on the highly mutated ClpX complex, it had been concluded that its six ATPase subunits hydrolyze ATP independently of one another (12). Because the six 26 S proteasomal ATPases are distinct gene products, the 26 S complex offers distinct advantages over the other hexameric AAA-ATPases for analyzing the actual mode of operation of this enzyme family. By analysis of the single mutants of the 26 S ATPase subunits, we have been able to test if there is cooperativity between the subunits both basally and upon stimulation of 26 S activity by ubiquitin conjugates.
An important factor determining the rate of hydrolysis of a substrate by the proteasome is its tightness of folding. Prior reports suggested that 26 S proteasomes require, in addition to ubiquitination, an unfolded region in the substrate to initiate degradation (14) and that tightly folded proteins seem to require some partial unfolding before they become tightly associated with the 26 S proteasome and committed to degradation (15). Therefore, it seemed important to determine how the degree of folding of a ubiquitinated substrate actually affects the time required and ATP cost for its degradation by the proteasome.
EXPERIMENTAL PROCEDURES
Purification of 26 S Proteasomes and Synthesis of Ubiquitin Conjugates
The yeast strains sub61 (WT), SP459 (rpt5YA), DY62 (rpt2RF), DY93 (rpt3R), DY65 (rpt5S), and DY100 (rpt6R) for 26 S proteasome purification were kindly provided by Dan Finley (Harvard Medical School). Mouse 26 S proteasomes were affinity-purified from mouse embryonic fibroblast cells as described previously (16) in the presence of 150 mm NaCl, whereas yeast 26 S particles were purified using the same method but without the addition of NaCl after harvesting the cells at A600 = 4 (10, 16). The E3 ligase E6AP was used for the generation of polyubiquitin conjugates as described previously (10, 17). Polyubiquitinated Sic1 was generated as reported (18, 19). Ub5-DHFR2 was a kind gift from Millennium Pharmaceuticals (Cambridge, MA).
Measurement of Proteasome Activity
ATP hydrolysis by 26 S proteasomes was measured using the malachite green assay, which detects the release of free phosphate (20, 21). Hydrolysis of the small peptide succinyl-GGL-7-amino-4-methyl coumarin (Bachem) by proteasomes was measured in the presence of 25 mm HEPES/KOH (pH 8), 2.5 mm MgCl2, 125 mm potassium acetate, 0.025% Triton X-100, 1 mm ATP, 1 mm DTT, and 0.1 mg/ml BSA (Sigma) as described previously (10). Stimulation of ATPase and gate opening was assayed under identical conditions using 26 S proteasomes at 10 nm together with a 100-fold excess of polyubiquitin conjugates or unmodified E3 ligases over 26 S particles unless stated otherwise. Degradation rates of Ub5-DHFR were measured after radiolabeling it with protein kinase A and [32P]ATP (22) and then following the conversion of the 32P-radiolabeled substrate to acid-soluble 32P-labeled peptides after TCA precipitation (22). Sic1 was radiolabeled before ubiquitination with [32P]ATP using CK2 (New England Biolabs), and its degradation was measured under the same conditions as described for Ub5-DHFR.
Each figure includes data from at least three independent experiments with at least three replicates for each condition. Every measurement of ATP hydrolysis included control reactions containing all reagents with and without the addition of the 26 S particles. Then, any background phosphate or minor contamination by other ATPases was subtracted. All values are means ± S.E. of three or more experiments after background subtraction.
RESULTS
Mutation of a Single ATPase Subunit Severely Impairs Proteasome Function
Initial experiments used 26 S proteasomes gently isolated from yeast (16) to determine whether they hydrolyze ATP in a stochastic or a highly cooperative manner, such as the cyclical ordered mechanism proposed recently in which nucleotides bind and exchange in pairs (13). We tested how mutations in the Walker A motifs, which prevent ATP binding to the different ATPase subunits (11), affect ATP hydrolysis by the 26 S complex and degradation of ubiquitinated proteins. Therefore, we affinity-purified 26 S particles from yeast strains carrying single mutations in individual RPT genes in which the conserved lysine residue in the Walker A box that interacts with the phosphate groups of ATP was replaced to block ATP binding (11). These mutant proteasomes all have a significantly reduced capacity for proteolysis, which is still sufficient to allow slow growth of cells under favorable conditions, but not during stress (11). Using proteasomes from these mutant yeast strains, we showed previously that the stimulation of gate opening (peptide entry) by ubiquitin conjugates is blocked after the loss of ATP binding to Rpt2, Rpt3, Rpt5, or Rpt6 (10), and generally similar results have been obtained recently with mammalian proteasomes (23). In addition, these mutant proteasomes showed a much smaller stimulation of gate opening upon nucleotide binding (ATPγS) compared with the wild-type particles (Fig. 1A).
FIGURE 1.
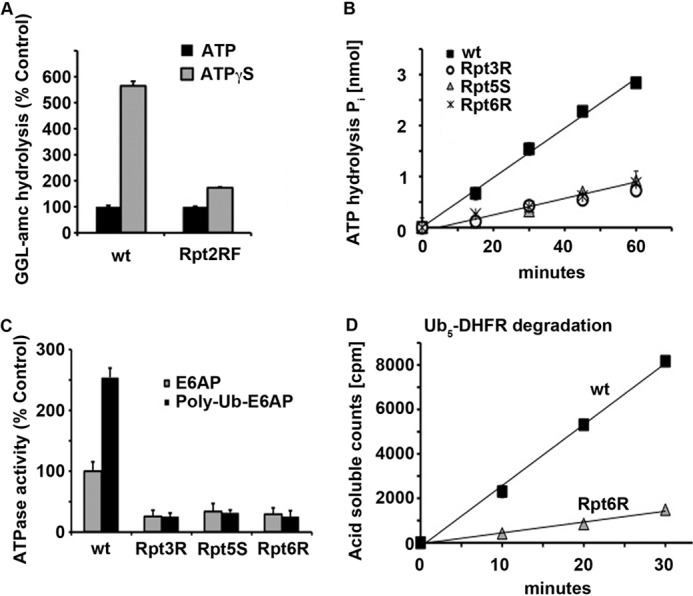
Binding to all 26 S ATPase subunits is required for stimulation of ATPase activity and maximum degradation. A, hydrolysis of the fluorescent tripeptide succinyl-GGL-7-amino-4-methyl coumarin (GGL-amc) was measured by WT or Rpt2RF yeast 26 S particles in the presence of ATP or ATPγS. The peptidase activity of 26 S particles in the presence of ATP was taken as 100%. B, basal ATP hydrolysis by WT, Rpt3R, Rpt5S, and Rpt6R yeast 26 S particles was measured. C, ATP hydrolysis by WT, Rpt3R, Rpt5S, and Rpt6R yeast 26 S particles was measured in the presence of E6AP or ubiquitinated E6AP. ATP hydrolysis of WT proteasomes in the presence of E6AP was taken as 100%. D, degradation of 32P-labeled Ub5-DHFR by WT or Rpt6R yeast 26 S proteasomes was assayed by measuring the increase in TCA-soluble radioactivity.
The mutations in the ATP-binding pockets of Rpt3, Rpt5, or Rpt6 all reduced basal ATP hydrolysis by ∼66% (Fig. 1B). This loss of most 26 S basal ATPase activity upon loss of function of a single subunit was much greater than the 16% reduction that would be expected if the six ATPase subunits functioned similarly and independently. More importantly, each of the single mutations completely blocked the 2–3-fold stimulation of ATP hydrolysis by ubiquitin conjugates (9, 21, 23) seen in WT particles (Fig. 1C). Thus, in the presence of a ubiquitinated substrate, the rate of ATP consumption in each of the three mutants tested was ∼84% lower than that in WT proteasomes. In other words, the basal activity of ATPases, as well as their activation, required the ATPase subunits to function similarly and cooperatively. Therefore, ATP hydrolysis by the 26 S proteasome is not a simple stochastic process in which the subunits function independently.
The effect of this decrease in ATPase activity on breakdown of ubiquitinated proteins was measured by assaying the conversion of 32P-labeled Ub5-DHFR to TCA-soluble peptides. Surprisingly, in the mutants, Ub5-DHFR degradation still occurred and at a linear rate. However, this rate was reduced by ∼84% below the WT levels (Fig. 1D), and thus, surprisingly, the net reduction in proteolysis corresponded exactly to the ∼84% decrease in ATPase rate. Very recently, DeMartino and co-workers (23) reported that ATP-binding mutants in Rpt5 and Rpt6 in mammalian proteasomes also fail to stimulate ATP hydrolysis upon binding ubiquitin conjugates and show a similar decreased degradation of ubiquitinated substrates. Thus, despite the many ATP-dependent steps in the degradation of ubiquitinated substrates by the 26 S proteasome, ATP hydrolysis and protein degradation appear to be tightly coupled, and both seem to require the concerted activity of all Rpt subunits.
The initial studies of the mutant proteasomes were technically challenging because these mutant strains grow slowly, and these 26 S proteasomes are less stable than the WT and after isolation rapidly lose their regulatory properties (11, 24) and their activity. (In fact, we could not study reliably the related Walker A mutation in Rpt1 because the proteasomes were particularly labile or in Rpt4 because of the appearance of secondary mutations in this ATPase subunit.) Nevertheless, because the mutations in Rpt3, Rpt5, or Rpt6 resulted in nearly identical losses of basal activity (by 66%), of all the conjugate-stimulated ATPase activity, and of conjugate degradation (by 84%), it is very likely that all six ATPases contribute very similarly to the ATP-ADP cycle, even though they have distinct structures and phenotypic consequences.
ATP Hydrolysis and Substrate Degradation Are Tightly Linked
To further analyze the coupling of ATP hydrolysis and conjugate degradation, we used WT mouse proteasomes, and by adding increasing amounts of the non-hydrolyzable ATP analog ATPγS in the presence of ATP to competitively inhibit ATP hydrolysis, we reduced ATPase activity in a graded controlled manner. Although this approach had been used to gradually reduce ATP hydrolysis by the 26 S proteasome by increasing the ATPγS/ATP ratios (9), it was unclear how the rates of ubiquitin conjugate degradation would be affected under these conditions. Using this method, we could determine for the first time precisely how rates of ATP hydrolysis influenced the rates of degradation of the model substrates Ub5-DHFR (Fig. 2A) (22, 25) and polyubiquitinated Sic1 (Fig. 2C) (19). Breakdown of both substrates was inhibited completely by proteasome inhibitors (e.g. bortezomib) and required ATP hydrolysis, as ATPγS alone prevented their degradation (19, 25) (data not shown).
FIGURE 2.

Rate of ubiquitinated substrate degradation is directly proportional to the rate of ATP hydrolysis. A, rates of Ub5-DHFR degradation by WT mouse 26 S proteasomes in the presence of 2 mm ATP were taken as 100%. ATP hydrolysis was measured under the same conditions and also taken as 100%. Increasing concentrations of the non-hydrolyzable ATP analog ATPγS (20, 200, or 500 μm) were added, and relative rates of substrate degradation or ATPase activity were calculated. B, values from A on the rates of Ub5-DHFR degradation plotted against the rate of ATP hydrolysis. C, rates of Ubn-Sic1 degradation by WT mouse 26 S proteasomes in the presence of 2 mm ATP were taken as 100%. ATP hydrolysis was measured under the same conditions and also taken as 100%. Increasing concentrations of the non-hydrolyzable ATP analog ATPγS (20, 200, or 500 μm) were added, and the relative rates of substrate degradation or ATPase activity were calculated. D, values from C on the rates of Ubn-Sic1 degradation plotted against the rate of ATP hydrolysis.
Surprisingly, when ATPase activity was decreased by up to 70%, degradation of both Ub5-DHFR and Ubn-Sic1 decreased exactly in parallel with the decrease in ATP hydrolysis. Thus, the rate of ATP hydrolysis determines the rate of substrate degradation. In fact, the extent of degradation of both ubiquitinated DHFR (Fig. 2B) and Sic1 (Fig. 2D) was directly proportional to the amount of ATP hydrolyzed. This linear relationship implies that a definite number of ATP molecules are consumed during degradation of each ubiquitinated substrate (i.e. an apparent stoichiometry exists for this process).
The two ubiquitinated proteins studied here differ markedly in length, type of the ubiquitin chain attached (Lys-48 on DHFR and Lys-63 on Sic1), and tightness of folding. Interestingly, with these gently isolated (16) 26 S preparations, the maximum rates of degradation of both Ub5-DHFR (∼4.7 molecules/min × 26 S) (Fig. 3A) and Ubn-Sic1 (∼2.3 molecules/min × 26 S) (Fig. 3B) by the mouse proteasomes, which were studied under substrate saturating conditions (Vmax), are ∼20-fold faster than those reported by previous investigators for Ub5-DHFR degradation (25), probably due to our use of more rapid gentle methods of proteasome isolation. As a consequence, we were able to calculate the ATP costs for the degradation of these two model substrates (see below; Table 1).
FIGURE 3.
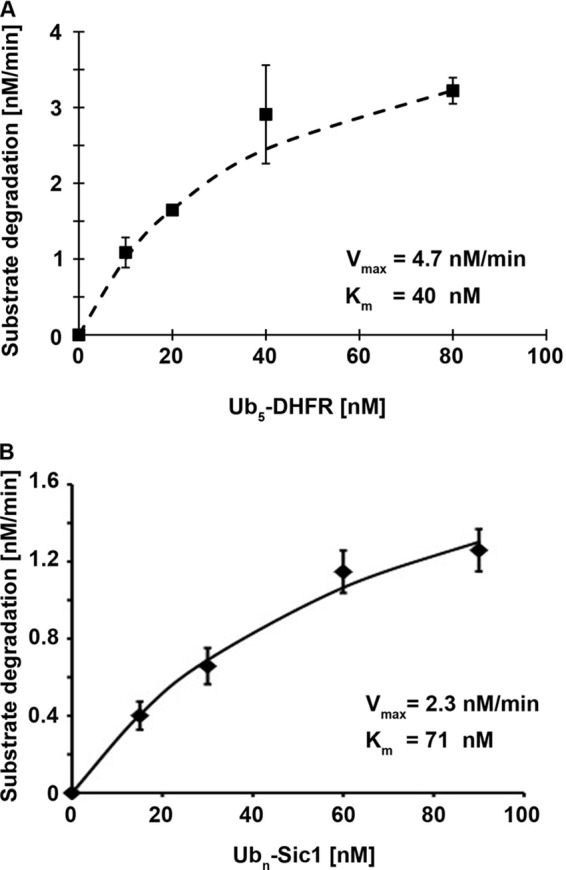
Measurement of degradation rates for Ub5-DHFR and Ubn-Sic1 to obtain Vmax values for proteasomal degradation. A, degradation of 32P-labeled Ub5-DHFR by WT mouse 26 S proteasomes (1 nm) was assayed by measuring the increase in TCA-soluble radioactivity using different substrate concentrations, and based on the rates of substrate degradation under these conditions, the Vmax and Km values were calculated. B, degradation of 32P-labeled Ubn-Sic1 by WT mouse 26 S proteasomes (1 nm) was assayed by measuring the increase in TCA-soluble radioactivity using different substrate concentrations, and based on the specific radioactivity of the substrate, the Vmax and Km values were calculated.
TABLE 1.
Calculated energy costs and time required for the degradation of ubiquitinated proteins by mammalian 26 S proteasomes
| Substrate |
|||
|---|---|---|---|
| Ub5-DHFR |
Ubn-Sic1 | ||
| −Folic acid | +Folic acid | ||
| Molecular weight (without ubiquitin) | 21,500 | 21,500 | 38,000 |
| Vmax (molecules/min × 26 S)a | 4.7 | 2.7 | 2.3 |
| Time to degrade (s) | 13 | 23 | 26 |
| Energy cost (ATP/molecule) | 50–80 | 90–140 | 100–160 |
Tighter Folding Reduces the Rate and Increases the ATP Cost for Degradation
An essential role of the 26 S ATPases is to catalyze substrate unfolding. Therefore, the folded structure of the polypeptide should affect the rate and energy costs for degradation by the 26 S proteasome. To learn how exactly different conformations of the same protein affect the amount of ATP consumed during proteolysis, we studied DHFR, whose conformation can be stabilized by the addition of the ligands folic acid and methotrexate (25). It has been shown previously that the very potent inhibitor methotrexate induces a tightly folded DHFR conformation and prevents degradation of ubiquitinated DHFR by the 26 S proteasome (24, 26). Methotrexate binding allows the association of Ub5-DHFR with the ubiquitin receptors on the 19 S ATPases but prevents the subsequent ATP-dependent step that commits to degradation and requires a loosely folded domain in the protein (24). In contrast, folic acid, which associates more weakly with DHFR and only partially inhibits its activity, only slowed the rate of DHFR hydrolysis by ∼2-fold (Fig. 4A) (25). No prior studies have investigated the ATP costs for the proteasome to degrade a more stably folded protein. Using folic acid, we thus could investigate simultaneously how increasing the stability of ubiquitinated DHFR affects both its rate of hydrolysis and the concomitant ATP consumption in WT mouse 26 S proteasomes. Like other ubiquitinated substrates (9, 21, 23), Ub5-DHFR stimulated 26 S ATP hydrolysis ∼2.5-fold (Fig. 4B), and increasing the stability of DHFR by the addition of folic acid did slow proteolysis 2-fold without affecting the stimulation of ATPase activity. In contrast, methotrexate completely blocked both DHFR degradation and stimulation of ATPase activity (Fig. 4B). Because stabilizing DHFR with folic acid reduced its degradation without affecting the rate of ATP hydrolysis, it increased the ATP cost for DHFR degradation by ∼2-fold.
FIGURE 4.
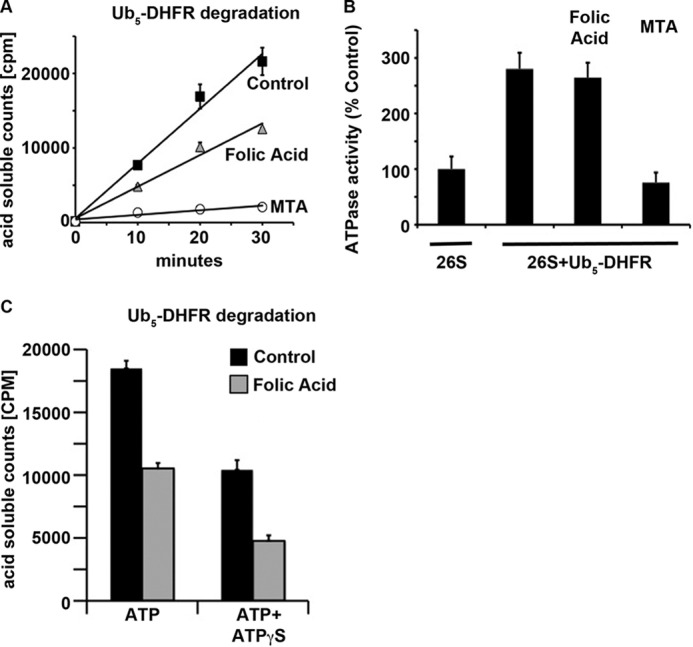
Tighter folding of the substrate decreases the rate of degradation but not of ATP consumption. A, Ub5-DHFR (∼40 nm) degradation by WT mouse 26 S proteasomes (1 nm) was measured in the presence of 25 μm folic acid or methotrexate (MTA). B, ATP hydrolysis by the 26 S proteasome was measured under the same conditions as described for A. ATPase activity without the addition of substrate was taken as 100%. C, Ub5-DHFR degradation (∼40 nm) by WT mouse 26 S proteasomes (1 nm) was measured in the presence of 2 mm ATP with and without the addition of 37.5 μm folic acid or 500 μm ATPγS. TCA-soluble radioactivity was measured after a 30-min incubation.
These findings indicate that the rates of conjugate degradation by the proteasome are determined by both the capacity of the particle for ATP hydrolysis and the tightness of folding of the substrate. However, it is unclear how these two factors may interact to determine the final rate of degradation. We therefore measured Ub5-DHFR degradation in the presence of folic acid to stabilize the substrate and at the same time used a ratio of ATPγS to ATP that by itself slows ATP hydrolysis by 50%. At the concentrations used, folic acid alone also slowed conjugate degradation by ∼50% (Fig. 4C). Upon addition of both together, degradation of Ub5-DHFR still occurred in a linear fashion, but its rate was reduced by 4-fold (by 75%). Thus, both the ATPase activity and the substrate folded structure determine the overall rate of proteolysis. Consequently, even under conditions of slow ATP hydrolysis, the 26 S proteasome is capable of degrading proteins that are difficult to unfold, although the process takes significantly longer.
Defects in Gate Opening in 20 S Proteasomes Influence the Rate and ATP Cost of Proteolysis
Binding of ubiquitin conjugates and ATP stimulates opening of the narrow gate for substrate entry into the 20 S proteasome (8–10). This gating process and its mechanism have thus far been studied only using small fluorogenic peptides, and consequently, the importance of gate opening in determining the rates and efficiency of degradation of ubiquitinated proteins has been unclear. To learn how a defect in gate opening might affect the duration and energy costs of proteolysis, we isolated 26 S proteasomes from a yeast strain harboring a mutation (rpt5YA) in the terminal HbYX domain of one of the 19 S ATPases (6). As expected, these proteasomes showed greatly reduced basal rates of hydrolysis of small peptides compared with WT particles due to their decreased capacity for gate opening (6, 8, 10) and a much smaller stimulation of peptide hydrolysis (i.e. gate opening) upon addition of ATPγS (Fig. 5A). In contrast, the basal rate of ATP hydrolysis and its stimulation by ubiquitin conjugates in these 26 S particles were not affected (Fig. 5B). However, this failure of Rpt5YA proteasomes to activate gate opening slowed the degradation of the ubiquitinated substrate Ub5-DHFR by ∼2-fold below WT rates (which is significantly less than the decrease observed in tripeptide hydrolysis by these proteasomes) (Fig. 5C). Thus, the ATP cost for the degradation of DHFR is increased if the gate is not fully open, presumably because more time is needed to translocate the unfolded protein into the 20 S proteasome. It is also noteworthy that the rates of ATP hydrolysis by proteasomes during breakdown of different ubiquitinated substrates were similar and that various perturbations (e.g. defects in gate opening) simply prolonged the time required to digest the substrates and thus the energy consumed in their degradation.
FIGURE 5.
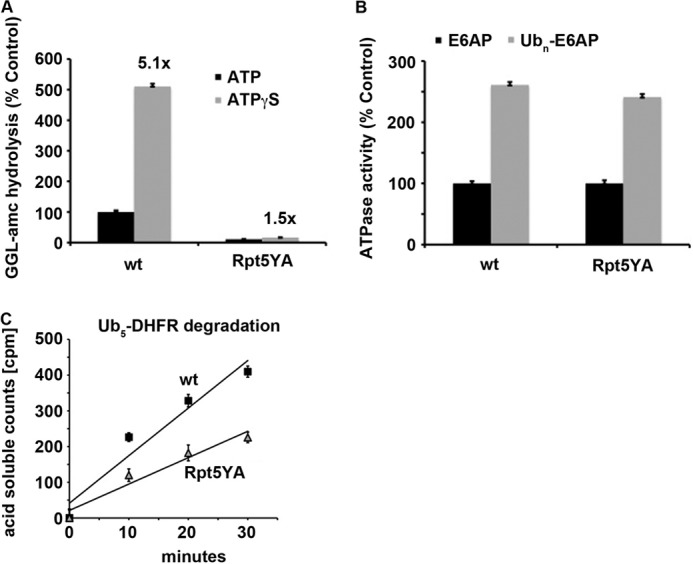
Decreased gate opening reduces the rate of degradation of ubiquitinated proteins. A, hydrolysis of the tripeptide succinyl-GGL-7-amino-4-methyl coumarin (GGL-amc) was measured by WT or Rpt5YA yeast 26 S particles in the presence of ATP or ATPγS. The peptidase activity of WT 26 S particles in the presence of ATP was taken as 100%. B, ATP hydrolysis by WT or Rpt5YA yeast 26 S particles was measured in the presence of E6AP or ubiquitinated E6AP. ATP hydrolysis of WT proteasomes in the presence of E6AP was taken as 100%. C, degradation of Ub5-DHFR by WT or Rpt5YA yeast 26 S proteasomes was assayed by measuring TCA-soluble radioactivity.
DISCUSSION
One of the most surprising and fundamental findings in this study is that even though proteolysis involves multiple ATP-activated steps that can be dissociated experimentally, the overall rates of degradation of ubiquitinated proteins and the rates of ATP hydrolysis are tightly coupled and directly proportional to each other. These processes were reduced to the same extent when ATP hydrolysis was inhibited by the addition of increasing amounts of ATPγS or by mutations affecting nucleotide binding to the individual ATPase subunits. For example, in the presence of ubiquitin conjugates, the WT yeast 26 S particles hydrolyzed ATP ∼6-fold faster, and their rates of Ub5-DHFR degradation were also ∼6-fold faster than those observed for Rpt6 mutant proteasomes. This tight linkage between ATP hydrolysis and substrate degradation was unexpected because the several ATP-activated steps contributing to conjugate degradation can be dissociated readily, some (e.g. unfolding) require nucleotide hydrolysis, and others (e.g. gate opening or conjugate binding) require only nucleotide binding (10, 23).
These findings confirm that ATP hydrolysis can be the rate-limiting step in substrate degradation, and therefore, the capacity of ubiquitinated proteins to stimulate the ATPases (9, 21, 23) appears important physiologically. Therefore, with the more stable conformations of DHFR resulting from the binding of folic acid or methotrexate, when degradation is slowed or blocked, substrate unfolding must become the rate-limiting step in the degradative process. Also with the proteasomal ATPase mutation in the HbYX motif that inhibits ATP-dependent gate opening, translocation becomes the rate-limiting step.
Another surprising and noteworthy finding was that the inability of a single ATPase subunit to bind ATP has such a large effect in decreasing the overall rate of ATP hydrolysis. In a random unordered reaction cycle, as the one proposed by Martin et al. (12), the loss of a single subunit contribution would result in a reduction of ATP hydrolysis by one-sixth (provided the six subunits function similarly). However, we found that the loss of Rpt3, Rpt5, or Rpt6 caused a much larger (66%) decrease in ATPase activity. Because the loss of nucleotide binding to any of the three different subunits caused a very similar loss of most of the ATPase activity of the particle, each of the six subunits in the ring is critical, and the hydrolytic cycle requires the coordinated function of all Rpt subunits, as we had proposed previously (13).
Even more dramatic was the effect of individual subunit mutations in blocking completely the stimulation of ATPase activity and gate opening by ubiquitin conjugates (10), resulting in an 88% decrease in proteolysis. After these studies were completed, Kim et al. (23) reported that the loss of ATP binding to Rpt5 and Rpt6 in mutant mammalian proteasomes also dramatically decreased proteolysis and prevented the activation of ATP hydrolysis by ubiquitin conjugates in accord with our findings. These workers did not observe a large reduction in basal ATP hydrolysis, but the low amount of basal ATPase activity and the variability in such assays between preparations (which depend on transfection and the purity of the preparations) could have masked such an effect on basal activity. In either case, the increased rate of ATP hydrolysis by the substrate-activated proteasome (9, 21, 23) and efficient proteolysis clearly required an ordered cooperative ATPase mechanism, rather than a stochastic process.
Substrate-mediated activation of ATP hydrolysis is a characteristic feature of the several homologous bacterial ATP-dependent proteases (LON, ClpAP, ClpXP, and HslU) (27, 28) and the proteasome regulatory ATPase complex PAN from archaea (the evolutionary predecessor of the 19 S ATPases) (29). These hexameric ATPases are all members of the AAA family, and presumably, these complexes all function by a common cyclical mechanism resembling that of the 26 S ATPases (13). Although the structural basis of this activation is unclear, its elimination by each of the single subunit mutants suggests an ordered ATP-ADP exchange cycle that requires cooperative cyclic functioning of all six Rpt subunits, as we had proposed (13). We had shown that nucleotides bind in pairs to the proteasomal ATPases, most likely to opposite sides of the hexameric ring; that two molecules of ATP and two of ADP are bound to the enzyme; and that two subunits are empty at any time (13). This cooperative binding of nucleotides strongly suggested an ordered reaction cycle for ATP binding and hydrolysis involving the six subunits (13).
The nucleotide-binding pockets of all the AAA-ATPase subunits contain an arginine finger extension from the neighboring subunits (30) that should enable ATP binding and hydrolysis by one subunit to influence the conformations and properties of the neighboring ones (13). Therefore, a mutation inactivating a single Walker A domain should not only block nucleotide binding to the mutant subunit but should also alter the behaviors of the adjacent subunits. This structural feature can help explain the cooperativity demonstrated here, and all these findings together clearly indicate that ATP hydrolysis must occur in a cyclical manner and that mutating any one of the subunits in the AAA ring disrupts the cycling. Moreover, the present measurements of ATP costs for conjugate degradation (see below) indicate that each Rpt subunit hydrolyzes an ATP molecule multiple times during the degradation of a ubiquitinated protein. In fact, according to the model proposed by Smith et al. (13), two ATPs are cleaved in each complete cycle around the Rpt ring, which must occur four to seven times during complete degradation of Ub5-DHFR and approximately twice as often with ubiquitinated Sic1.
The energy-dependent steps in degradation of proteins presumably have evolved to ensure efficient elimination of ubiquitinated proteins and also to minimize wasteful consumption of ATP. There has been no prior determination of the energy costs of degrading specific substrates by the 26 S proteasome or by the ubiquitin-proteasome pathway as a whole. In vivo, the energy costs for degradation of specific proteins must be significantly larger than the ATP consumption by the proteasome due to the ATP consumed in substrate ubiquitination and must depend on the number of ubiquitins added, the frequency of deubiquitination, and possible additional energy-requiring steps mediated by chaperones and the p97/valosin-containing protein complex (31). Nevertheless, our findings strongly suggest that for most substrates, the number of ATPs consumed by the proteasome, which most often must exceed 100 molecules/protein degraded (see below), will far exceed the ATPs consumed in protein ubiquitination.
To calculate the ATP consumption necessary to degrade a substrate molecule, we determined the Vmax for the hydrolysis of Ub5-DHFR and ubiquitinated Sic1 by the mammalian 26 S proteasome and obtained degradation rates of ∼4.7 molecules/min × 26 S for DHFR (Fig. 3A) and ∼2.3 molecules/min × 26 S for Sic1 (Fig. 3B). These rates of substrate degradation are much faster than those reported previously (25), probably because the rapid affinity purification used here (16) is more likely to yield proteasomes with intact catalytic and regulatory features than the traditional multistep chromatographic methods (16). The basal rate of ATP consumption in our assays was ∼60 ATPs/min × 19 S, which is very similar to the basal rate of ATP hydrolysis reported for the yeast 26 S proteasome by Coffino and co-workers (32), although these authors used non-ubiquitinated substrates and failed to observe a stimulation of ATP hydrolysis as we and others found with ubiquitinated proteins (21, 23). All ubiquitinated proteins used in this study that can be degraded by the 26 S proteasome stimulate ATP hydrolysis by 2–3-fold, which implies that ∼240–360 molecules are consumed per min during degradation of DHFR or Sic1 (Table 1). Thus, at Vmax, the cost of ATP per molecule of Ub5-DHFR degraded appears to be ∼50–80 ATPs, whereas breakdown of Ubn-Sic1 requires ∼100–160 ATPs (Table 1). Interestingly, Sic1 is almost twice the length of DHFR. Therefore, it is tempting to conclude that longer polypeptides require more time and more ATP to be degraded by the 26 S proteasome.
In addition to length, the tertiary structure of the protein also clearly affects the amount of ATP consumed in its destruction. The ATP costs for degradation of ubiquitinated DHFR increased by ∼2-fold after the addition of the stabilizing ligand folic acid. Efficient degradation requires not only ATPase activation and substrate unfolding, but also the HbYX-induced gate-opening mechanism. The mutations affecting this process probably slowed degradation by impeding the initial entrance of the polypeptide into the 20 S proteasome or by reducing the rate of polypeptide translocation. It is noteworthy that in these mutant particles with a gate-opening defect, the hydrolysis of small peptides was affected much more than the breakdown of ubiquitin conjugates, presumably because entry of the small peptides occurs by simple diffusion and depends only on the latency of the channel. In contrast, polypeptide substrates are actively translocated by the ATPase ring, probably in a specific orientation. Thus, their entry may be less dependent on the size of the channel, and once the polypeptide begins to enter, it may prevent gate closing and maintain it in an open conformation. Consequently, the lack of ATP-dependent gate opening would have lesser effects on the degradation of proteins than of small peptides. This coupling of ATP-activated gate opening to the ATP-dependent unfolding and translocation clearly enhances the efficiency of proteolysis and is further evidence that degradation of ubiquitinated proteins by the proteasome is a highly ordered, multistep process in which considerable ATP is consumed to ensure efficient destruction of different types of polypeptides.
It is noteworthy that the several ubiquitinated substrates examined here all stimulated ATPase activity in the WT 26 S proteasome and the gate-opening mutant particles similarly (2–3-fold). Because changes in rates of ATP hydrolysis over this range are directly proportional to rates of proteolysis, this activation of the ATPases (9, 21, 23) and gate opening (8–10) upon conjugate binding must be causing an acceleration of conjugate degradation. However, as discussed above, the actual rate of substrate degradation depends upon the tightness of protein folding and probably length. Thus, the hexameric ATPase ring seems to function in two modes: at a basal level and 2–3-fold faster after conjugate binding and commitment to degradation. However, the translocation of the hard-to-unfold proteins (and probably of longer polypeptides) requires the ATPases to be activated longer. Consequently, more ATP is consumed during conjugate digestion (24). Thus, the tightness of folding and probably polypeptide length determine the duration of the activated state (i.e. the time until translocation is completed) and thus the actual ATP consumed in the process.
Because this activation of the ATPases requires occupancy of the proteasome-associated deubiquitinating enzymes Usp14 and Uch37 by the ubiquitin chain, as well as interaction of the polypeptide with the ATPases (21), our observations suggest that the complete deubiquitination of the substrate is a late step during the degradation of the polypeptide so as to ensure that the proteasome remains as long as possible in the activated state that favors proteolysis. Accordingly, Rpn11 is located directly above the ATPase ring (33) and thus can remove the ubiquitin chain just before the polypeptide domain bearing the chain is translocated into the 20 S proteasome. Earlier removal of the chain would lead to premature termination of the ATPase activity enhancement and consequently could slow the degradative process. Alternatively, after translocation begins, ATPase activation (and gate opening) may become independent of the regulatory deubiquitinating enzymes and be triggered by the polypeptide itself, as occurs with the homologous AAA-ATPases that catalyze ubiquitin-independent proteolysis in prokaryotes. Thus, the active state would be maintained until substrate translocation into the 20 S proteasome is complete.
Acknowledgments
We are grateful to Mary Dethavong and Lisa Bacis for valuable assistance in preparing this manuscript; Galen Collins and Nikolay Kukushkin for critical input; and Dan Finley, Soyeon Park, and Suzanne Elsasser for providing the yeast strains.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM051923-17 from NIGMS.
- Ub
- ubiquitin
- DHFR
- dihydrofolate reductase
- ATPγS
- adenosine 5′-O-(thiotriphosphate).
REFERENCES
- 1. Finley D. (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Groll M., Bajorek M., Köhler A., Moroder L., Rubin D. M., Huber R., Glickman M. H., Finley D. (2000) A gated channel into the proteasome core particle. Nat. Struct. Biol 7, 1062–1067 [DOI] [PubMed] [Google Scholar]
- 3. Liu C. W., Li X., Thompson D., Wooding K., Chang T. L., Tang Z., Yu H., Thomas P. J., DeMartino G. N. (2006) ATP binding and ATP hydrolysis play distinct roles in the function of 26S proteasome. Mol. Cell 24, 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smith D. M., Kafri G., Cheng Y., Ng D., Walz T., Goldberg A. L. (2005) ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins. Mol. Cell 20, 687–698 [DOI] [PubMed] [Google Scholar]
- 5. Rabl J., Smith D. M., Yu Y., Chang S. C., Goldberg A. L., Cheng Y. (2008) Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol. Cell 30, 360–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smith D. M., Chang S. C., Park S., Finley D., Cheng Y., Goldberg A. L. (2007) Docking of the proteasomal ATPases' carboxyl termini in the 20S proteasome's α ring opens the gate for substrate entry. Mol. Cell 27, 731–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bar-Nun S., Glickman M. H. (2012) Proteasomal AAA-ATPases: structure and function. Biochim. Biophys. Acta 1823, 67–82 [DOI] [PubMed] [Google Scholar]
- 8. Bech-Otschir D., Helfrich A., Enenkel C., Consiglieri G., Seeger M., Holzhütter H. G., Dahlmann B., Kloetzel P. M. (2009) Polyubiquitin substrates allosterically activate their own degradation by the 26S proteasome. Nat. Struct. Mol. Biol. 16, 219–225 [DOI] [PubMed] [Google Scholar]
- 9. Li X., Demartino G. N. (2009) Variably modulated gating of the 26S proteasome by ATP and polyubiquitin. Biochem. J. 421, 397–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peth A., Besche H. C., Goldberg A. L. (2009) Ubiquitinated proteins activate the proteasome by binding to Usp14/Ubp6, which causes 20S gate opening. Mol. Cell 36, 794–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rubin D. M., Glickman M. H., Larsen C. N., Dhruvakumar S., Finley D. (1998) Active site mutants in the six regulatory particle ATPases reveal multiple roles for ATP in the proteasome. EMBO J. 17, 4909–4919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Martin A., Baker T. A., Sauer R. T. (2005) Rebuilt AAA+ motors reveal operating principles for ATP-fuelled machines. Nature 437, 1115–1120 [DOI] [PubMed] [Google Scholar]
- 13. Smith D. M., Fraga H., Reis C., Kafri G., Goldberg A. L. (2011) ATP binds to proteasomal ATPases in pairs with distinct functional effects, implying an ordered reaction cycle. Cell 144, 526–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Prakash S., Tian L., Ratliff K. S., Lehotzky R. E., Matouschek A. (2004) An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat. Struct. Mol. Biol. 11, 830–837 [DOI] [PubMed] [Google Scholar]
- 15. Beskow A., Grimberg K. B., Bott L. C., Salomons F. A., Dantuma N. P., Young P. (2009) A conserved unfoldase activity for the p97 AAA-ATPase in proteasomal degradation. J. Mol. Biol. 394, 732–746 [DOI] [PubMed] [Google Scholar]
- 16. Besche H. C., Haas W., Gygi S. P., Goldberg A. L. (2009) Isolation of mammalian 26S proteasomes and p97/VCP complexes using the ubiquitin-like domain from HHR23B reveals novel proteasome-associated proteins. Biochemistry 48, 2538–2549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim H. T., Kim K. P., Lledias F., Kisselev A. F., Scaglione K. M., Skowyra D., Gygi S. P., Goldberg A. L. (2007) Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J. Biol. Chem. 282, 17375–17386 [DOI] [PubMed] [Google Scholar]
- 18. Lee B. H., Lee M. J., Park S., Oh D. C., Elsasser S., Chen P. C., Gartner C., Dimova N., Hanna J., Gygi S. P., Wilson S. M., King R. W., Finley D. (2010) Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467, 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saeki Y., Isono E., Toh-e A. (2005) Preparation of ubiquitinated substrates by the PY motif-insertion method for monitoring 26S proteasome activity. Methods Enzymol. 399, 215–227 [DOI] [PubMed] [Google Scholar]
- 20. Lanzetta P. A., Alvarez L. J., Reinach P. S., Candia O. A. (1979) An improved assay for nanomole amounts of inorganic phosphate. Anal. Biochem. 100, 95–97 [DOI] [PubMed] [Google Scholar]
- 21. Peth A., Kukushkin N., Bosse M., Goldberg A. L. (2013) Ubiquitinated proteins activate the proteasomal ATPases by binding to Usp14 or Uch37 homologs. J. Biol. Chem. 288, 7781–7790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lam Y. A., Huang J. W., Showole O. (2005) The synthesis and proteasomal degradation of a model substrate Ub5DHFR. Methods Enzymol. 398, 379–390 [DOI] [PubMed] [Google Scholar]
- 23. Kim Y. C., Li X., Thompson D., DeMartino G. N. (2013) ATP binding by proteasomal ATPases regulates cellular assembly and substrate-induced functions of the 26 S proteasome. J. Biol. Chem. 288, 3334–3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Peth A., Uchiki T., Goldberg A. L. (2010) ATP-dependent steps in the binding of ubiquitin conjugates to the 26S proteasome that commit to degradation. Mol. Cell 40, 671–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thrower J. S., Hoffman L., Rechsteiner M., Pickart C. M. (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J. 19, 94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Johnston J. A., Johnson E. S., Waller P. R., Varshavsky A. (1995) Methotrexate inhibits proteolysis of dihydrofolate reductase by the N-end rule pathway. J. Biol. Chem. 270, 8172–8178 [DOI] [PubMed] [Google Scholar]
- 27. Hwang B. J., Woo K. M., Goldberg A. L., Chung C. H. (1988) Protease Ti, a new ATP-dependent protease in Escherichia coli, contains protein-activated ATPase and proteolytic functions in distinct subunits. J. Biol. Chem. 263, 8727–8734 [PubMed] [Google Scholar]
- 28. Waxman L., Goldberg A. L. (1986) Selectivity of intracellular proteolysis: protein substrates activate the ATP-dependent protease (La). Science 232, 500–503 [DOI] [PubMed] [Google Scholar]
- 29. Benaroudj N., Zwickl P., Seemüller E., Baumeister W., Goldberg A. L. (2003) ATP hydrolysis by the proteasome regulatory complex PAN serves multiple functions in protein degradation. Mol. Cell 11, 69–78 [DOI] [PubMed] [Google Scholar]
- 30. Zhang F., Hu M., Tian G., Zhang P., Finley D., Jeffrey P. D., Shi Y. (2009) Structural insights into the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol. Cell 34, 473–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meyer H., Bug M., Bremer S. (2012) Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 14, 117–123 [DOI] [PubMed] [Google Scholar]
- 32. Henderson A., Erales J., Hoyt M. A., Coffino P. (2011) Dependence of proteasome processing rate on substrate unfolding. J. Biol. Chem. 286, 17495–17502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beck F., Unverdorben P., Bohn S., Schweitzer A., Pfeifer G., Sakata E., Nickell S., Plitzko J. M., Villa E., Baumeister W., Förster F. (2012) Near-atomic resolution structural model of the yeast 26S proteasome. Proc. Natl. Acad. Sci. U.S.A. 109, 14870–14875 [DOI] [PMC free article] [PubMed] [Google Scholar]