

Abstract
Propionic acidemia (PA) is a rare autosomal recessive disorder of metabolism caused by deficient activity of the mitochondrial enzyme propionyl-CoA carboxylase. The clinical manifestations are metabolic acidosis, poor feeding, lethargy, vomiting, osteoporosis, neurological dysfunction, pancytopenia, developmental retardation and cardiomyopathy. Liver transplantation has recently been considered as one of the treatment options for patients with PA. This case report describes several anesthetic considerations for patients with PA undergoing liver transplantation. Understanding the patient's status and avoiding events that may precipitate metabolic acidosis are important for anesthetic management of patients with PA. In conclusion, anesthesia should be focused on minimizing the severity of metabolic acidosis with following considerations: (1) maintaining optimal tissue perfusion by avoiding hypotension, (2) preventing hypoglycemia, and (3) providing bicarbonate to compensate for the acidosis.
Keywords: Liver transplantation, Propionic acidemia
Propionic acidemia (PA) is a rare autosomal recessive disorder of metabolism with a prevalence of 1 in 100,000 [1]. The condition is caused by deficiency of the mitochondrial enzyme, propionyl-CoA carboxylase (PCC) which plays a key role in (1) the catabolism of branched chain amino acids such as leucine, isoleucine, valine, threonine, and methionine, (2) the beta-oxidation of odd chain fatty acids and (3) the metabolism of the cholesterol side chains. Derangements in these pathways result in the accumulation of potentially toxic metabolites such as propionic acid and methylmalonic acid. The onset of clinical manifestation is usually early from several hours to weeks after birth, and the affected infants present with a wide spectrum of features from poor feeding, vomiting, lethargy, metabolic acidosis, hyperammonemia to coma and death, depending on the severity of the enzyme impairment [2]. The liver is the major site of branched chain amino acid transamination and subsequent propionic acid production. Therefore, liver transplantation (LT) has recently been considered as one of the treatment options for patients with PA to lower the risk of acute metabolic decompensation and to improve the quality of life [3-5]. This case report describes several anesthetic considerations for patients with PA undergoing LT.
Case Report
A 22-month old male (body weight: 12.6 kg, height: 90 cm) with PA was scheduled for living donor LT. He was a full term infant born to a family with a history of genetic disease in which his elder sister died of a metabolic disorder at an age of 45 days. He was diagnosed with PA at 7 days of age when he was present with poor feeding and lethargy. Urine amino acid analysis and plasma amino acid analysis were positive for glycine. Urine organic acid analysis was positive for methylcitric acid, propionylglycine, 3-hydroxypropionic acid and triglycine. Plasma organic acid analysis was positive for 3-hydroxypropionic acid and propionylgycine. Specific mutations were identified in the PCC beta polypeptide gene. After the diagnosis, his medical management included low protein diet to reduce the load on amino acid catabolism and administration of sodium benzoate, L-carnitine, L-arginine thiazolidine carboxylate and biotin. He had been hospitalized in many occasions due to pneumonia, metabolic acidosis, and poor oral intake. One month prior to LDLT, he was admitted several times for decreased activity, altered mental status, hyperammonemia and ketoacidosis.
Preoperative vital signs showed blood pressure of 100/47 mmHg, a heart rate of 104 beats/min, and a respiratory rate of 24 breaths/min. Preoperative laboratory results were as follows: white blood cell 4.8 × 103/µl, hemoglobin 11.1 g/dl, platelet 202 × 103/µl, prothrombin time (international normalized ratio) 0.96, activated partial thromboplastin time 36.0 sec, ammonia 103.6 µmol/L, albumin 1.8 g/dl, protein 4.9 g/dl, total bilirubin 0.3 mg/dl, aspartate aminotransferase 24 U/L, alanine aminotransferase 16 U/L, and creatinine 0.25 mg/dl. The results of arterial blood gas analysis (ABGA) were pH 7.415, PaCO2 36.8 mmHg, PaO2 64.2 mmHg, HCO3- 23.1 mEq/L, BE -1.1 mEq and glucose 102 mg/dl. Chest X-ray and doppler echocardiogram were within normal limits. The patient received 5% DNK3 at 40 ml/hr, preoperatively.
Anesthesia was induced with midazolam 1 mg, thiopental sodium 75 mg, and rocuronium 10 mg and was maintained with desflurance in air and oxygen (inspiratory oxygen fraction of 0.5) and vecuronium (0.8 µg/kg/min). Ventilation was controlled with a tidal volume of 7-10 ml/kg and ventilatory rate was adjusted to maintain an end tidal CO2 of 35-40 mmHg. Anesthesia monitoring included electrocardiogram, noninvasive blood pressure, pulse oximetry, capnography, and esophageal core temperature. Invasive monitoring included continuous arterial pressures via radial and femoral arteries, femoral venous pressure, and central venous pressure via 5.5 Fr 3-lumen venous catheter placed in the right internal jugular vein. After the induction, the initial ABGA showed a glucose level of 58 mg/dl. We infused 5% dextrose 50 cc/hr, and 1 hour later, the glucose level increased to 122 mg/dl.
The operation started 40 minutes after the induction of anesthesia. During the preanhepatic period, the hemodynamic profile remained stable. 1 hour after the start of theoperation, ABGA showed pH 7.211 and BE -15.1 mEq/L, thus continuous infusion of bicarbonate 10 mEq/hr was started and titrated according to the hourly ABGA results. The anhepatic period began at 2 hour 15 minutes after the induction and continuous infusion of dopamine 8 µg/kg/min was initiated to maintain the mean BP above 55 mmHg. 1 hour after the start of the anhepatic period, the glucose level dropped to 45 mg/dl and we infused 5% dextrose 70 cc for 30 minutes. Reperfusion began at one hour and 10 minutes after portal vein clamping. Immediately after reperfusion, the mean BP dropped to 46 mmHg and then normalized within 3 minutes with bolus injections of atropine 0.3 mg and ephedrine 2 mg. During the postreperfusion period, metabolic acidosis still persisted thus requiring continued infusion of barcarbonate 10 mEq/hr. During the operation, the patient received 1,400 ml of crystalloid (normal saline), 450 ml of colloid (voluven®, Fresenius Kabi, Bad Homburg, Germany), and 1 unit of packed red blood cells. In this case, albumin was not administered in order to limit the exogenous source of protein intake. The total duration of operation was 8 hours and 51 minutes. Three hours and 30 minutes after the reperfusion, the patient was transferred to the intensive care unit. Intraoperative laboratory test results are summarized in Table 1.
Table 1.
Intraoperative Hemodynamic Profiles and ABGA Data
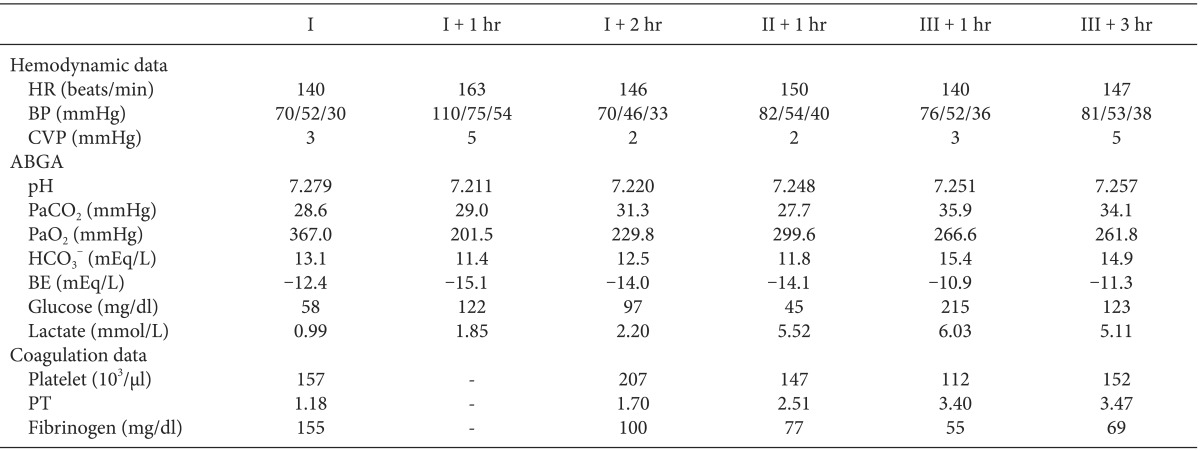
I: preanhepatic period, II: anhepatic period, III: postreperfusion period, HR: heat rate, BP: blood pressure (systolic/mean/diastolic), CVP: central venous pressure, ABGA: arterial blood gas analysis, BE: base excess, PT (international normalized ratio): prothrombin time.
The patient's postoperative course was complicated by persistent metabolic acidosis and hepatic failure due to hepatic vein obstruction (Table 2). On postoperative day (POD) 0, dopamine 20 µg/kg/min was continuously infused due to low BP and aliquots of bicarbonate 20 mEq were administered to correct metabolic acidosis. On POD 1, abdominal sonography revealed hepatic vein obstruction prompting the patient to undergo emergency re-operation for hepatic vein obstruction. Anesthesia was induced and maintained with isoflurane in air and oxygen (inspiratory oxygen fraction of 0.5) and vecuronium. Ventilation was controlled with a tidal volume of 7-10 ml/kg and ventilatory rate was adjusted to maintain an end tidal CO2 of 35-40 mmHg. After the induction, initial ABGA showed pH 7.135, BE -14.8mEq/L and a glucose level of 55 mg/dl. The patient received 5% dextrose 30 cc/hr and 0.45% normal saline 60 cc/hr. Dopamine 30 µg/kg/min was administered to maintain the mean BP above (55 mmHg) and continuous infusion of bicarbonate 8 mEq/hr was given for progressive metabolic acidosis. The surgery lasted for 2 hours, and the patient's ABGA at the end of surgery showed pH 7.179, BE -13.4 mEq/L and the glucose level of 55 mg/dl. In spite of re-operation, patient's liver function deteriorated further and metabolic acidosis worsened. Moreover, the patient developed hemodynamic compromise requiring continuous infusion of dopamine 30 µg/kg/min, epinephrine 1 µg/kg/min, and norepinephrine 5 µg/kg/min. On POD 4, the patient expired as a result of hepatic failure and subsequent severe metabolic acidosis.
Table 2.
Postoperative Hemodynamic Profiles, Liver Function Tests, and ABGA Data
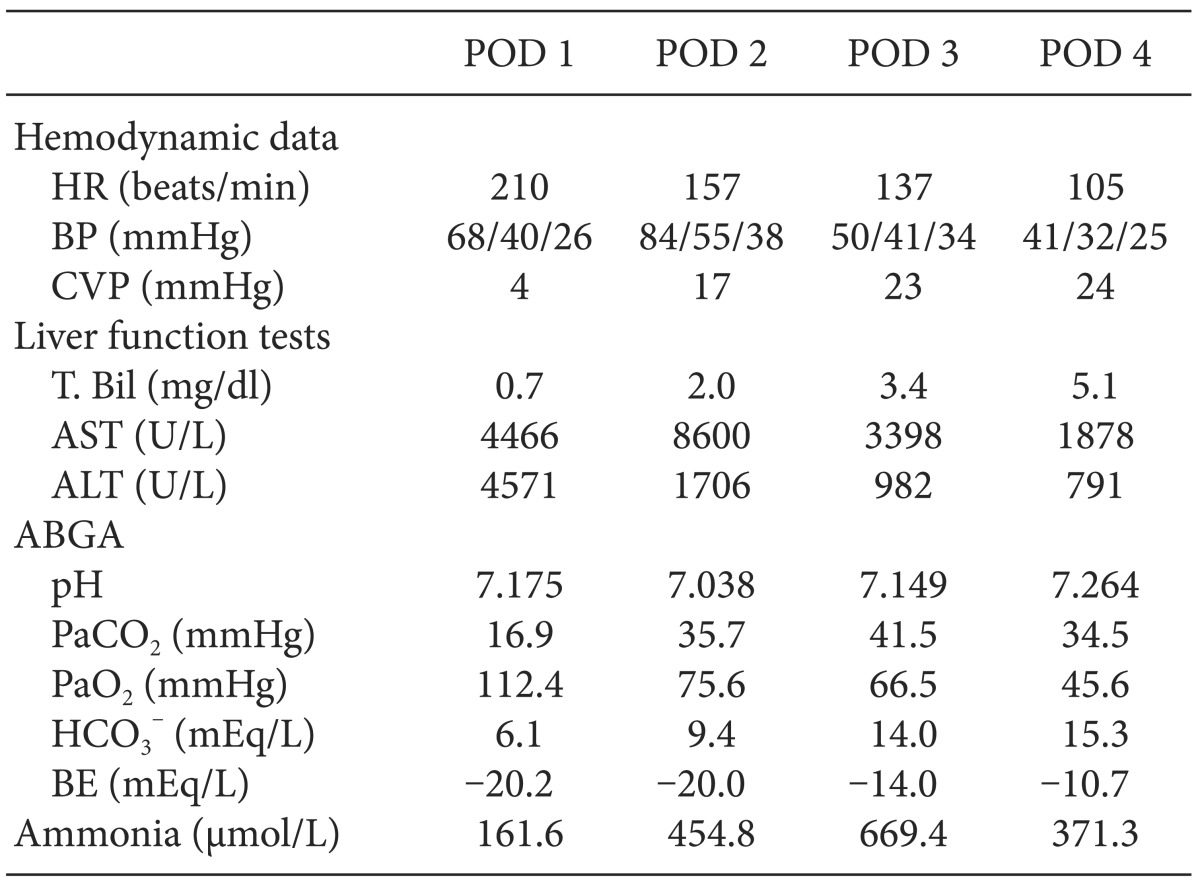
POD: postoperative day, HR: heat rate, BP: blood pressure (systolic/mean/diastolic), CVP: central venous pressure, T. Bil: total bilirubin, AST: aspartate aminotransferase, ALT: alanine aminotransferase, ABGA: arterial blood gas analysis, BE: base excess.
Discussion
PA is an autosomal recessive inborn defect of metabolism caused by deficient activity of the mitochondrial enzyme PCC. Two distinct genotypic forms exist in mutations in either alpha or beta genes encoding for corresponding alpha- and beta-subunits of PCC; however, there is no correlation between the genotypic forms and clinical severity. The prevalence of PA has been estimated in the Caucasian population to be 1 in 100,000 live births [1], and in the Korean population, it has been estimated to be 1 in 81,128 live births by tandem mass screening in 2007 [6]. The clinical manifestations are initiated as early as several hours after birth with the start of dietary protein intake. The hallmark of this disorder is recurrent metabolic acidosis with ketosis and hyperlactatemia usual precipitation of by excessive protein ingestion or infection. Ketoacidosis results from inhibition of the citric acid cycle enzymes by propionic acid. In addition, other clinical signs and symptoms are poor feeding, lethargy, vomiting, osteoporosis, neurological dysfunction (seizure, hypotonia, and coma), pancytopenia, developmental retardation and cardiomyopathy [2].
The management of PA includes primarily, dietary modifications with a low protein diet, along with metronidazole to reduce propionic acid-producing gut species and L-carnitine to promote propionic acid renal clearance. For acute management of acidosis and hyperammonemia, hydration with intravenous fluid containing dextrose and bicarbonate should be initiated promptly with concomitant treatment of precipitating events such as infection [7,8]. Despite the improvement in conservative management, patients with PA are prone to relapsing episodes of metabolic acidosis mandating frequent hospital admissions, and thus, the overall outcome of PA remains disappointing [9].
LT has been viewed as one of the potential treatment modalities for PA because liver is the major site of branched chain amino acid metabolism and subsequent propionic acid production. However, the role of LT in PA treatment is still controversial mostly due to the presence of extrahepatic sources (i.e., the gastrointestinal tract and the central nervous system) of propionic acid, and therefore, liver replacement offers only partial corrections of the metabolic defect. Early reports of LT for patients with PA showed unsatisfactory outcomes marked by significant mortality and transplantation-related complications [3,10]. However, more recent studies suggested that LT has an important role in the management of PA in reducing the risk of metabolic decompensation and improving cardiomyopathy, neurocognitive dysfunction, and quality of life [4,5,11]. Therefore, LT may have a key role in the course of PA treatment, as outcome of LT improve. In our case, the patient had a history of recurrent hospital admissions for poor oral intake, hyperammonemia and altered mental status, and thus, LT was planned in order to improve the quality of life.
There are several anesthetic considerations for patients with PA. Preoperative evaluation of the patient should include acid balance, nutritional state, muscle tone, mental status and gastrointestinal function and cardiac functions. In particular, events such as hypoxia, dehydration, and hypotension, and the use of inappropriate anesthetics may precipitate and/or aggravate metabolic acidosis and must be avoided. During the preoperative fasting period, patients require glucose in their intravenous (IV) fluids to suppress protein catabolism and subsequent acidosis. In addition, lactic acid containing fluids should be avoided because it contributes to the patient's acid load and is also poorly metabolized by dysfunctional mitochondria. In our case, patient's IV fluids consisted of 5% DNK3 at 40 ml/hr, preoperatively. For general anesthesia, rapid sequence induction should be considered in patients experiencing reflux or vomiting. Moreover, anesthesiologists should be aware of anesthetic medications that are metabolized to propionic acid, odd chain organic acids, odd chain alcohol, acrylic acid, and odd chain fatty acids. These drugs are as follows: muscle relaxants including succinylcholine, atracurium, cistatracurium, and mivacurium [12], propofol containing soybean oil rich in polyunsaturated fats [13]; and analgesics including ibuprofen, naproxen, and ketoprofen [14]. These drugs are likely to initiate problem in patient with PA and should be avoided. Therefore, in our case, thiopental, rocuronium and vecuronium were used as anesthetic agents.
For prolonged surgery such as LT, serial monitoring of arterial pH and blood glucose is strongly advised in order to make rapid adjustments in metabolic derangements. Accordingly, in our patient, ABGA was performed hourly during the operation. Based on the ABGA findings, dextrose and bicarbonate were administered as needed. During LT, albumin is commonly administered because LT recipients usually have preexisting low serum albumin levels. However, albumin should be avoided in PA patients since protein restriction is an important part of the treatment protocol [5,8]. Accordingly, in our patient, albumin was not used intraoperatively. During the postoperative period, patients with PA are at a high risk to develop respiratory distress secondary to fatigue or hypotonia and are prone to develop upper airway obstruction. Therefore, tracheal extubation should be delayed until patients regain adequate muscle strength. Additionally, humidified oxygen should be given to these patients [15]. Despite these anesthetic considerations, our patient expired on POD 4 mainly due to hepatic failure caused by hepatic vein obstruction.
In conclusion, understanding the patient's status and avoiding events that precipitate metabolic acidosis are important for anesthetic management for patients with PA undergoing LT. The mainstay of anesthesia should be focused on minimizing the severity of metabolic acidosis with following consierations: (1) maintaining optimal tissue perfusion by avoiding hypotension, (2) preventing hypoglycemia, and (3) providing bicarbonate to compensate for the acidosis.
References
- 1.Leonard JV, Vijayaraghavan S, Walter JH. The impact of screening for propionic and methylmalonic acidaemia. Eur J Pediatr. 2003;162(Suppl 1):S21–S24. doi: 10.1007/s00431-003-1345-1. [DOI] [PubMed] [Google Scholar]
- 2.Fernandes J, Saudubray JM, Berghe GVD, Walter JH. Inborn Metabolic Diseases. 4th ed. Berlin: Springer; 2006. pp. 247–256. [Google Scholar]
- 3.Leonard JV, Walter JH, McKiernan PJ. The management of organic acidaemias: the role of transplantation. J Inherit Metab Dis. 2001;24:309–311. doi: 10.1023/a:1010395724012. [DOI] [PubMed] [Google Scholar]
- 4.Barshes NR, Vanatta JM, Patel AJ, Carter BA, O'Mahony CA, Karpen SJ, et al. Evaluation and management of patients with propionic acidemia undergoing liver transplantation: a comprehensive review. Pediatr Transplant. 2006;10:773–781. doi: 10.1111/j.1399-3046.2006.00569.x. [DOI] [PubMed] [Google Scholar]
- 5.Vara R, Turner C, Mundy H, Heaton ND, Rela M, Mieli-Vergani G, et al. Liver transplantation for propionic acidemia in children. Liver Transpl. 2011;17:661–667. doi: 10.1002/lt.22279. [DOI] [PubMed] [Google Scholar]
- 6.Lee DH. The prevalence of pediatric endocrine and metabolic diseases in Korea. Korean J Pediatr. 2008;51:559–563. [Google Scholar]
- 7.Chapman KA, Gropman A, MacLeod E, Stagni K, Summar ML, Ueda K, et al. Acute management of propionic acidemia. Mol Genet Metab. 2012;105:16–25. doi: 10.1016/j.ymgme.2011.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sutton VR, Chapman KA, Gropman AL, MacLeod E, Stagni K, Summar ML, et al. Chronic management and health supervision of individuals with propionic acidemia. Mol Genet Metab. 2012;105:26–33. doi: 10.1016/j.ymgme.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 9.Lehnert W, Sperl W, Suormala T, Baumgartner ER. Propionic acidaemia: clinical, biochemical and therapeutic aspects. Experience in 30 patients. Eur J Pediatr. 1994;153(7 Suppl 1):S68–S80. doi: 10.1007/BF02138781. [DOI] [PubMed] [Google Scholar]
- 10.Schlenzig JS, Poggi-Travert F, Laurent J, Rabier D, Jan D, Wendel U, et al. Liver transplantation in two cases of propionic acidaemia. J Inherit Metab Dis. 1995;18:448–461. doi: 10.1007/BF00710056. [DOI] [PubMed] [Google Scholar]
- 11.Kayler LK, Merion RM, Lee S, Sung RS, Punch JD, Rudich SM, et al. Long-term survival after liver transplantation in children with metabolic disorders. Pediatr Transplant. 2002;6:295–300. doi: 10.1034/j.1399-3046.2002.02009.x. [DOI] [PubMed] [Google Scholar]
- 12.Evers AS, Maze M. Anesthetic pharmacology: physiologic principles and clinical practice: a companion to Miller's Anesthesia. New York: Churchill Livingstone; 2004. pp. 582–595. [Google Scholar]
- 13.Bame MA. A guide for the family of the child with methylmalonic acidemia. Columbus: Ross Products Division of Abbott Laboratories; 2011. p. 33. [Google Scholar]
- 14.Hardman J, Limbird LE, Molinoff P, Ruddon R, Gilman A. The pharmacological basis of therapeutics. 9th ed. New York: McGraw-Hill; 1996. pp. 617–657. [Google Scholar]
- 15.Harker HE, Emhardt JD, Hainline BE. Propionic acidemia in a four-month-old male: a case study and anesthetic implications. Anesth Analg. 2000;91:309–311. doi: 10.1097/00000539-200008000-00014. [DOI] [PubMed] [Google Scholar]