

Abstract
Background:
The epidermal growth factor receptor (EGFR) is expressed in ovarian cancer, but agents targeting this pathway have shown little effect as single agents. This may be due to the presence of alternative pathways, particularly activation of the PI3K/Akt/MTOR pathway.
Methods:
We have therefore examined the effect of inhibitors of this pathway (ZSTK474 and sirolimus) in combination with the EGFR inhibitors erlotinib and gefitinib in ovarian cancer primary cell cultures.
Results:
The single-agent EGFR inhibitors showed little activity, although some activity was seen with the single-agent PI3K inhibitor, ZSTK474. Combinations of ZSTK474 with EGFR inhibitors showed enhanced activity with some evidence of synergy, whereas sirolimus combinations were less active. The results were not explicable on the basis of PIK3CA mutation or amplification, or PTEN loss, although one tumour with a KRAS mutation showed resistance to EGFR inhibitors. However, there was correlation of the EGFR expression with sensitivity to EGFR and resistance to PI3K active agents, and inverse correlation in the sensitivity of individual tumours to agents active against these pathways, suggesting a mechanism of action for the combination.
Conclusion:
Phase I/II clinical trials with these agents should include further pharmacodynamic endpoints and molecular characterisation to identify patients most likely to benefit from this strategy.
Keywords: MTOR, erlotinib, gefinitib, ZSTK474, sirolimus, ovarian cancer
Epidermal growth factor receptor (EGFR) is expressed in ovarian cancer (Niikura et al., 1997), but use of anti-EGFR antibodies and small molecule inhibitors in ovarian cancer patients has had little or no success in terms of clinical response (Blank et al., 2005; Murphy and Stordal, 2011). Consistent with this, we have previously examined a number of tyrosine kinase inhibitors (TKIs) for activity in ovarian cancer primary cell cultures, and have only seen occasional evidence of gefitinib activity, although not at a level likely to produce clinical response (Knight et al., 2004). Although data from non-small-cell lung cancer (NSCLC) suggest that this may be due to a lack of activating mutations, it has also been suggested that an anti-EGFR strategy may also fail due to the presence of alternate activated pathways (Niikura et al., 1997; Qiu et al., 2005). Inhibition of a solitary signal transduction pathway is often inefficient due to the activation of alternative signalling cascades or receptor switching (Bianco et al., 2008; Jain et al., 2010; Larsen et al., 2011; Yang et al., 2011). The PI3K/Akt/mTOR pathway is of importance in ovarian cancer (Sain et al., 2006) and is activated by a number of known molecular defects, particularly PIK3CA mutation or amplification in up to 30% and PTEN loss in up to 40% patients (Campbell et al., 2004; Kolasa et al., 2009). In addition, the IGF pathway as well as other human epidermal growth factor receptor (HER) pathways are present and thought to be active in ovarian cancer (Hamburger, 2001; Mayr et al., 2006).
Given the observation of limited activity of gefitinib and the potential for combination with inhibitors of the PI3K/Akt/mTOR pathway, we decided to examine the possibility that EGFR-targeted agents would be more effective against ovarian cancer in combination with other specific pathway inhibitors. These combinations were tested against primary cell cultures from human ovarian tumours to determine whether this strategy might have potential for clinical application.
Methods
Ovarian cancers
Tumour material (ascitic fluid) from nine ovarian carcinomas (five adenocarcinomas not otherwise specified, three serous carcinomas and one papillary carcinoma) with patient consent. The age of the patients ranged from 44 to 82, mean 66 years. Three patients had no prior treatment, whereas five patients with recurrence had been previously been treated with carboplatin+paclitaxel and one patient with recurrence had received carboplatin alone. All were staged as T3c tumours. Ethics approval was given for the research use of surplus tissue not required for diagnosis.
Samples from effusions were transported in 250 ml bottles containing 25 ml Dulbecco's modified Eagle's medium (DMEM; Sigma, Poole, UK) with antibiotics (100 IU ml−1 penicillin and 100 μg ml−1 streptomycin) and 1M HEPES, but as a precaution to prevent blood clots forming while in transit, 5000 IU heparin sodium (Monoparin, CP Pharmaceuticals Ltd, Wales, UK) was also added. The samples were split equally and aliquoted into 50 ml universals under sterile conditions in a BioQ Microfuge (PecLab, Southampton, UK) and spun at 300 g for 10 min to pellet the cells.
Following centrifugation, Ficoll-Hypaque density gradient separation (Histopaque; Sigma-Aldrich, Poole, UK) was used to separate dead cells, erythrocytes and debris from viable cells and was performed according to the manufacturer's instructions. Briefly, 10 ml of Ficoll-Hypaque was pipetted into sterile polystyrene 30 ml universal containers. Equal volumes of the ascites-derived cells were carefully layered on top of the Ficoll-Hypaque and centrifuged at 400 g for 30 min. The interface containing the purified tumour cell suspension was transferred into a separate sterile 30 ml universal using a sterile Pasteur pipette. The cells were washed twice in DMEM supplemented with 1 M HEPES and antibiotics. After the second wash the cells were resuspended in 10 ml complete assay medium (CAM; DCS Innovative Diagnostik Systeme, Hamburg, Germany) for a viable cell count, which was performed using the trypan blue exclusion method. Any cell that was not required immediately was cryopreserved in DMEM supplemented with antibiotics, 10% fetal calf serum (FCS; Labtech International, East Sussex, Uckfield, UK) and 10% DMSO (Sigma).
ATP-TCA
The ATP-based tumour chemosensitivity assay (ATP-TCA) was performed as previously described (Andreotti et al., 1995; Glaysher and Cree, 2011).
Preparation of chemotherapeutic agents
Gefitinib, erlotinib, ZSTK474 and sirolimus were purchased from LC Laboratories (Woburn, MA, USA). All drugs were diluted in CAM to concentrations thought to be clinically achievable (gefitinib 0.06–2 μM, erlotinib 0.2–6.5 μM, ZSTK474 0.07–2.2 μM, sirolimus 0.06–2 μM). Combinations were tested by simultaneous addition as previously published (Andreotti et al., 1995; Glaysher and Cree, 2011). All of the chemotherapeutic drugs or combinations were tested in triplicate at six dilutions in 96-well round-bottomed polypropylene microplate (Corning Life Sciences, Amsterdam, The Netherlands; 3790), allowing four drugs or drug combinations to be tested. Two controls were included in one row of each plate: a no drug control consisting of media only (MO) and a maximum inhibitor (MI) control, which kills all cells present giving a zero ATP count.
Cell culture
Cells obtained from the malignant effusions were adjusted to 100 000 cells ml−1 in CAM. Cell suspension (100 μl) was then added to each well of the 96-microwell plate giving final cell concentrations of 10 000 cells per well. Plates were incubated in a 95% humidified, 37 °C, 5% CO2 incubator for 6 days and checked periodically for overgrowth and infection.
ATP extraction and measurement
ATP was extracted from cells by the addition of 50 μl of ATP extraction reagent (DCS) to each well of the 96-well plate. Plates were incubated at room temperature for a minimum of 20 min and a maximum of 1 h before the ATP was read. The ATP in the wells was measured using a luciferin–luciferase counting reagent as previously described (Andreotti et al., 1995; Glaysher and Cree, 2011). Light output was measured using a Berthold Diagnostic Systems MPL1 luminometer (Berthold Diagnostic Systems, Pforzheim, Germany). All luminescence measurements were performed using the manufacturer's instructions and an ATP standard curve run before each read using Adenosine 5′-triphosphate standard disodium salt hydrate (Sigma).
Viability analysis
The data produced from each ATP-TCA plate was entered into an Excel (Microsoft, Redmond, WA, USA) spreadsheet that calculated the percentage tumour inhibition at each concentration from the ATP measurements, the IC50 and IC90 (concentration of drug required to cause 50% and 90% inhibition, respectively) for each drug. The percentage tumour inhibition at each drug concentration was used to plot curves for each drug or combination. As the variation between the wells that were averaged to calculate the percentage tumour inhibition is small (typical coefficient of variance of <10%), error bars have not been included on most graphs as they are usually smaller than the markers on the graphs. The percentage tumour growth inhibition was calculated as follows:
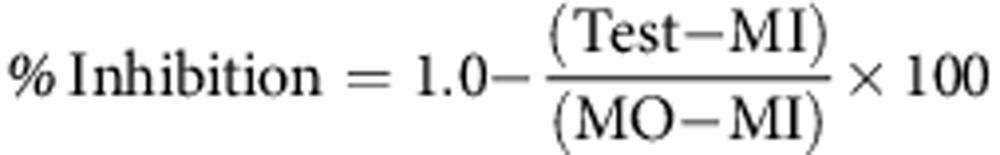 |
where, Test=mean counts for test drug wells; MI=mean counts for maximum inhibitor wells; MO=mean counts for medium only wells
To allow comparison between different tumours, a sensitivity index (IndexSUM) ranging from 0 to 600 for each individual drug or combination was calculated by summing the percentage tumour growth inhibition at each of the six drug concentrations tested and subtracting the result from 600 (IndexSUM=600−Sum [Inhibition 6.25…200]). In previous papers, we have used values <300 (i.e. a 50% reduction in IndexSUM) as an indication of sensitivity (Knight et al., 2004).
Combination indices (CI) calculated by the Chou and Talalay methods (Chou and Talalay, 1984) were determined at 50% and 90% cell death. These were defined as follows:
where CIA+B=CI for a fixed effect (F=50% or 90%) for the combination of cytotoxic A and cytotoxic B; DA/A+B=concentration of cytotoxic A in the combination A+B giving an effect F; DB/A+B=concentration of cytotoxic B in the combination A+B giving an effect F; DA=concentration of cytotoxic A alone giving an effect F; DB=concentration of cytotoxic B alone giving an effect F; α= parameter with value 0 when A and B are mutually exclusive and 1 when A and B are mutually non-exclusive. CI calculates synergism <0.8; additivity between >0.8 and <1.2; antagonism >1.2 (Greco et al., 1995).
Taqman array
Aliquots of cells used in primary cell culture experiments were stored in RA1 2ME for later RNA extraction using the Ambion RecoverALL nucleic acid isolation kit (Life Technologies, Paisley, UK) using their total nucleic acid isolation protocol as previously described (Glaysher et al., 2011). DNAse treatment was used to obtain total RNA for measurement of gene expression by Taqman array (Life Technologies). Samples were then quantified and purity checked using a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE, USA). Isolated RNA was immediately converted into cDNA using ABI High Capacity cDNA conversion kits (Life Technologies). Samples were diluted when necessary in nuclease-free water to give a working concentration of approximately 100 ng μl−1 cDNA. Data from NanoDrop readings were used to inform dilution calculations necessary to complete cDNA and RT-negative reaction mixes. These mixes were added and chilled in a cooling block until ready to load into the thermal cycler. Thermal cycler (PeqLab) conditions were set to the manufacturer's ‘manual method' settings. After the run was completed samples were pulse microfuged at 13 000 r.p.m. for 30 s before cDNA quantity was checked via the NanoDrop.
Gene expression was assessed using Taqman arrays as previously published (Glaysher et al., 2010; Glaysher et al., 2011), which were run in an Applied Biosystems 7900HT Fast Real-Time PCR System (Life Technologies) according to the manufacturer's instructions. Each sample was made up with Taqman × 2 Universal Master Mix (Life Technologies) and mixed with an equal volume of cDNA to give a final concentration of 300 ng μl−1, and samples were then pipetted into loading ports (100 μl per port) of the 384-well cards. The loaded Taqman array was run in the 7900HT thermal cycler according to the manufacturer's instructions. PCR was performed for 90 min with the following conditions.
AmpErase UNG activation for 2 min at 50 °C; AmpliTaq Gold DNA polymerase activation for 10 min at 94.5 °C; followed by 40 cycles each of melt anneal/extend for 30 s at 97 °C and 1 min at 59.7 °C. The ‘auto threshold cycle' function was performed at the end of the run and resulting Ct data from the array card was transferred to a Microsoft Excel spreadsheet. Controls were checked and the data transferred to a Microsoft Access database for further analysis.
Ct values were standardised by reference to PBGD, the least variable housekeeping gene of four present on the array. Standard methods of PCR analysis (delta Ct) were inappropriate for this array as evaluation of these methods often requires a good dilution curve for both standard and reference genes on every plate. The array format makes this type of analysis impractical. In these studies a logarithmic gene expression ratio (GER) was calculated as ln(2−Ct(test)/2−Ct(PBGD)) and used for comparison with ATP-TCA data, as previously published (Glaysher et al., 2010). Periodically during the course of the study, standard control material obtained from pooled cDNA samples were run alongside non-template controls (NTC) and RT-negative sample testing card and sample efficacy. Pooled cDNA was generated from FFPE blocks (>50 anonymous patients with a range of cancers) to provide control material for the entirety of the study.
Mutation analysis
For DNA extraction, the Ambion RecoverALL nucleic acid isolation kit using their total nucleic acid isolation protocol was used with RNAse according to the manufacturer's instructions. Samples were quantified and purity checked using 1.3 μl of each undiluted sample with the NanoDrop spectrophotometer. Rapid mutation screening for common mutations in EGFR, PI3K and BRAF was performed using Therascreen (Qiagen Ltd, Manchester, UK) kits, according to the manufacturer's instructions. PCR was performed in an Applied Biosystems 7500 Fast Dx PCR machine (Life Technologies) and results downloaded to an Excel spreadsheet for analysis.
Immunocytochemistry
Sections from FFPE cell pellets were cut at 4 μm and stained using at a dilution of 1:100 and a standard immunocytochemistry protocol on the Bond automated immunostainer (Leica, Newcastle, UK) using monoclonal mouse anti-human PTEN clone 6H2.1 (Dako, Ely, UK). The slides were viewed by a pathologist (IAC) and scored on a scale of 0−3+ for intensity of cytoplasmic staining.
Fluorescence in situ hybridisation (FISH)
Unless stated otherwise, procedures were performed at room temperature. Paraffin wax-embedded sections (4 μm thick) mounted on X-tra slides (Leica Biosystems, Newcastle-upon-Tyne, UK) were dewaxed for 10 min in 50 ml xylene at room temperature, dehydrated for 5 min in 50 ml ethanol, then washed for 23 min in 50 ml of 0.2 N HCl. The slides were then washed in 50 ml double distilled water (DDW) for 2 min and transferred to 50 ml Zymed (South San Francisco, CA, USA) heat pretreatment solution at 95 °C for 160 min. The slides were then washed in 50 ml of DDW for 2 min; 60 μl of Zymed digestion enzyme solution was then applied to a 22 mm × 22 mm coverslip and overlayed on to the slide. The slide was then incubated at 38 °C in a wet box for 2 × 35 min. Slides were then washed in 50 ml DDW for 3 × 2 min before being dehydrated through a 50-ml ethanol series: 70%, 95% and 100% for 2 min each. The slides were then left to dry before probe application.
FISH probes used were bought from Empire Genomics (Buffalo, NY, USA). A two-colour probe set was used, a probe for PIK3CA labelled with Gold 5(6)-carboxyrhodamine dUTP and a chromosome 3 centromere-specific probe labelled with green-fluorescein dUTP. Probes were made up before use according to the manufacture's recommendations.
The sample DNA and probe DNA were co-denatured at 72 °C for 5 min and then allowed to hybridise at 37 °C overnight on a PTC-200 thermal cycler (MJ Research, Waltham, MA, USA). The slides were then washed in 50 ml of 0.4 × saline sodium citrate/Tween 20 at 73 °C for 2 min and then transferred to 50 ml 2 × saline sodium citrate/Tween 20 at room temperature for 30 s. Ethanol series dehydration was performed as before and the slides were air dried in darkness. The slides were coverslipped with 20 μl of counterstain (20 μl mounting medium with DAPI in 1000 μl mounting medium for fluorescence; Vector Laboratories, Burlingame, CA, USA).
A dual analysis was performed by two independent clinical scientists using scoring and analysis guidelines currently recommended for HER2 ISH analysis (Bartlett et al., 2011).
Data analysis
The sensitivity of primary cell cultures to the single agents tested was compared using Pearson linear correlation, which was also used to compare the sensitivity to single agents with gene expression ratios.
Results
The ATP-TCA was used to determine the effect of EGFR inhibitors (gefitinib and erlotinib) alone and in combination with inhibitors of the Akt/PI3K/mTOR pathway (Figure 1). These data have been compared with RT–PCR gene expression profiles and relevant mutation analysis to determine mechanisms of sensitivity and resistance.
Figure 1.

Example ovarian tumour primary cell culture results, showing the effect on cell viability as assessed by an ATP-based chemosensitivity assay. There is relatively little activity of the single agents tested. However, the combinations are more active. (A) Tumour 9 shows synergy to erlotinib+ZSTK474 and (B) tumour 1 to gefitinib+sirolimus. In (A) the degree of inhibition in the ATP-TCA reaches 90% at clinically relevant concentrations, whereas in (B) the degree of inhibition is somewhat less impressive. The % inhibition (y-axis) is calculated from the ATP counts from medium only (MO) wells as described in the methods section, whereas the final concentration of each drug is given on the x-axis. Each point represents the mean ATP count derived from triplicate wells, normalised to the mean MO well ATP count as described.
Effects of single agents
Heterogeneity between ovarian tumours was observed for all four targeted agents. The majority of ovarian tumours tested were found to be resistant to EGFR inhibitors with only 2 out of 9 for gefitinib and 1 out of 9 for erlotinib showing strong single-agent activity with an IndexSUM <300 (Figure 2). Greater sensitivity was seen with the PI3K inhibitor ZSTK474 with 4 out of 9 tumours showing single-agent sensitivity (Figure 2). Inhibitors of mTOR (sirolimus) have been shown to elicit a predominately cytostatic response (Meric-Bernstam and Gonzalez-Angulo, 2009) and hence, not surprising only 1 out of 9 tumours showed any sensitivity in this type of assay where cell death is the end point (Figure 2).
Figure 2.

Waterfall plots of sensitivity index (IndexSUM) in ovarian cancer samples for (A) single agents and (B) combinations. A sensitivity index of 0=complete inhibition and 600=no inhibition. The tables below each plot show the gene expression ratio (GER) data for the tumours identified by number below each bar. The darker shading indicates a low gene expression, whereas the lighter shading shows higher GER. Higher expression of EGFR, HER2 or HER4 appears to be associated with increased sensitivity to EGFR inhibitors, whereas the reverse is true for PI3K pathway inhibitors. This relationship is not observed with combinations. Tumour 8 had a KRAS mutation and was resistant to EGFR inhibition, although it remained relatively sensitive to PI3K inhibition.
Effect of combinations
The combination of EGFR inhibitors with PI3K and sirolimus inhibitors showed considerably greater sensitivity compared with their corresponding single-agent activity (Figures 1 and 2).
Some of the activity observed with gefitinib and ZSTK474 was due to both synergism and additivity at lower concentrations (50% and 20%, respectively), whereas 50% of tumours also showed additivity at higher doses. This is reflected by the CI calculated by the Chou and Talalay method on the median dose–response curve (Table 1) (Chou and Talalay, 1984). Antagonism was seen more predominately for this combination at higher concentrations where 50% of samples had >1.2 CI for 90% inhibition.
Table 1. Effect of combining EGFR inhibitors with PI3K and mTOR inhibitors using Chou and Talalay combination index (CI) calculated for 50% cell death (CI50) and for 90% cell death (CI90), where synergy is <0.8, additivity is between 0.8 and 1.2, and antagonism is >1.2 showing number of tumours in each category.
| |
CI50 |
CI90 |
||||||
|---|---|---|---|---|---|---|---|---|
| Tumour number | Gefitinib+ZSTK474 | Erlotinib+ZSTK474 | Gefitinib+sirolimus | Erlotinib+sirolimus | Gefitinib+ZSTK474 | Erlotinib+ZSTK474 | Gefitinib+sirolimus | Erlotinib+sirolimus |
| 1 | 0.32 | 0.16 | 0.23 | 0.09 | 1.56 | 1.23 | 1.09 | 0.97 |
| 2 | 0.56 | 0.70 | 0.92 | 0.91 | 0.95 | 1.02 | 0.92 | 0.91 |
| 3 | 0.83 | 1.00 | 4.63 | — | 1.046 | 1.53 | 1.23 | 1.15 |
| 4 | 0.23 | 0.72 | — | 1.27 | 1.06 | 1.42 | 1.03 | 1.48 |
| 5 | 1.22 | 1.26 | 1.06 | 0.51 | 1.26 | 1.21 | 1.25 | 1.18 |
| 6 | 0.53 | 0.44 | 0.96 | 0.46 | 1.13 | 1.24 | 1.15 | 1.35 |
| 7 | 1.28 | 1.10 | 1.59 | 1.40 | 1.38 | 1.10 | 1.59 | 1.40 |
| 8a | 1.36 | 1.66 | 23.31 | 1.03 | 1.10 | 1.01 | 1.51 | 1.37 |
| 9 | 0.31 | 0.29 | 0.17 | 0.92 | 1.70 | 1.71 | 1.58 | 1.55 |
Tumour 8 had a KRAS mutation.
The combination of erlotinib and ZSTK474 showed similar effects to that of the gefitinib and ZSTK474 combination (Figure 1). A pronounced increase in sensitivity could be seen with co-administration of these agents (Figures 1 and 2). Results showed synergy at lower concentrations, while antagonism was seen at 90% inhibition where 70% of samples had >1.2 CI (Table 1).
The combination of sirolimus and EGFR inhibitors, although not as effective as the EGFR and PI3K combinations, showed some increase in sensitivity to its combination compared with individual single-agent activity (Figures 1 and 2). Although some synergy was observed, the sensitivity increase seen with this combination was predominantly due to an additive effect (Table 1). This was not observed with ZSTK474. An inverse relationship could be seen in the single-agent activity of both EGFR inhibitors and sirolimus, where sensitivity to one referred resistance to the other (Figure 3).
Figure 3.

Comparison of sensitivity by IndexSUM for all nine ovarian tumours for (A) erlotinib and sirolimus; (B) gefitinib and sirolimus; (C) gefitinib and ZSTK474; (D) erlotinib and ZSTK474. There is an inverse correlation for each comparison (r2=0.52, 0.66, 0.51 and 0.57, respectively; all P <0.05). Each symbol represents an individual tumour. IndexSUM calculated as 600−Sum[Inhibition 6.25…200] as previously described (Andreotti et al., 1995), such that complete inhibition=0 and complete resistance=600.
Mutation status
Tumours were tested for common mutations within EGFR, KRAS, BRAF and PI3K genes using Therascreen kits for each gene (Qiagen Ltd), details of which are available at http://www.qiagen.com/products/artus/genetictesting.aspx. All tumours tested negative for the common mutations of EGFR, PI3K and BRAF. A single serous adenocarcinoma (tumour 8) was shown to have a KRAS c.35G>T (G12V) mutation. The G12V mutation results in an amino-acid substitution at position 12 in KRAS, from a glycine (G) to a valine (V). This tumour showed decreased sensitivity to all EGFR, PI3K and mTOR inhibitors tested, with IndexSUM values indicative of resistance (504 gefitinib, 455 erlotinib, 347 ZSTK474 and 396 sirolimus). Interestingly, gefitinib showed greater resistance than erlotinib.
In addition, amplification of PIK3CA was checked by FISH. None of the tumours were shown to have amplification >2.0 in comparison with centromeric signals, although some variation in copy number was noted.
Gene alteration and sensitivity
Relative gene expression data for ovarian tumours showed varying expression levels of multiple growth factor receptors assessed by quantitative RT–PCR. EGFR and PI3K or mTOR inhibitors showed contrasting correlating gene expression data (Table 2). There was positive correlation of EGFR and HER2 expression with gefitinib sensitivity, whereas there was negative correlation of EGFR and HER2 expression with PI3K inhibition (Figure 4). The reverse relationship was observed for IGF and IGF2R (Figure 4). However, it should be noted that the correlation of individual genes in the EGFR and PI3K pathways with single-agent activity was not strong because of the small number of tumours studied and the effect of other genes.
Table 2. Linear correlation (Pearson) of gene expression ratio (GER) with sensitivity to single agents, shown as r 2.
| Gene | Gefitinib | Erlotinib | ZSTK474 | Rapamycin |
|---|---|---|---|---|
| Akt | 0.06 | 0.24 | 0.15 | 0.28 |
| EGFR | 0.46 | 0.36 | 0.48 | 0.37 |
| HER2 | 0.45 | 0.44 | 0.40 | 0.29 |
| HER3 | 0.44 | 0.54 | 0.82 | 0.66 |
| HER4 | 0.15 | 0.17 | 0.00 | 0.07 |
| IGF1 | 0.52 | 0.69 | 0.23 | 0.44 |
| IGF1R | 0.10 | 0.00 | 0.05 | 0.00 |
| IGF2 | 0.07 | 0.07 | 0.02 | 0.02 |
| IGF2R | 0.50 | 0.55 | 0.37 | 0.55 |
| mTOR | 0.00 | 0.08 | 0.11 | 0.16 |
| NFkB | 0.13 | 0.31 | 0.19 | 0.27 |
| PIK3CA | 0.00 | 0.04 | 0.00 | 0.08 |
| PTEN | 0.24 | 0.15 | 0.00 | 0.06 |
| STAT3 | 0.07 | 0.12 | 0.00 | 0.07 |
| VEGF | 0.31 | 0.26 | 0.34 | 0.16 |
Abbreviations: EGFR=epidermal growth factor receptor; HER=human epidermal growth factor receptor; IGF1=insulin-like growth factor 1; IGF1R=insulin-like growth factor 1 receptor; mTOR=mammalian target of rapamycin; NFkB=nuclear factor kappa B; VEGF=vascular endothelial growth factor.
None of these are regarded as significant following Bonferroni correction, although several genes show high r2 values with the EGFR inhibitors, notably EGFR, HER2, HER3, IGF1 and IGF2R.
Figure 4.

Comparison of single-agent sensitivity (IndexSUM) with gene expression ratio (GER) showing (A) correlation of increasing gefitinib activity with increased EGFR expression (Pearson's correlation, r2=0.46, P <0.05); (B) decreasing ZSTK474 activity with increased EGFR expression (r2=0.48, P <0.04); (C) increasing gefitinib activity with increased HER2 expression (r2=0.45, P <0.05); (D) decreasing ZSTK474 activity with increased HER2 expression (r2=0.43, NS); (E) decreasing gefinitib activity with increased IGF1 expression (r2=0.52, P <0.03); (F) increasing ZSTK474 activity with increased IGF1 expression (r2=0.23, NS); (G) decreasing gefitinib activity with increased IGF2R expression (r2=0.50, P <0.04); (H) increasing ZSTK474 activity with increased IGF2R expression (r2=0.36, NS). While the numbers are small and the results are of borderline statistical significance, there is clearly an inverse relationship between the sensitivity to EGFR and PI3K inhibitors with expression of key genes in these pathways.
There was no clear pattern in PTEN expression by qRT–PCR or immunohistochemistry in this small number of tumours and no relationship to sensitivity was noted (Figure 2).
Tumours with high PIK3CA copy numbers showed the highest sensitivity to EGFR inhibitors. The reverse seemed to be true for the PI3K or mTOR inhibitors (Figure 2). However, there appeared to be no relationship between PIK3CA copy number and activity of the combinations.
Discussion
Combinations of EGFR and PI3K inhibitors showed synergistic activity against ovarian cancer. Sensitivity was seen despite lack of EGFR mutations in this tumour type and with some heterogeneity of activity of the different drugs between tumours. Similar heterogeneity is seen in clinical trials of single agents in ovarian cancers, and it will be important to define the pathways to stratify patients for optimal treatment. Despite the small number of tumours studied, there was positive correlation of EGFR gene expression with sensitivity to EGFR inhibitors and inverse correlation of EGFR gene expression with sensitivity to PI3K or MTOR inhibitors. This is as predicted from the inverse correlation in activity of the single agents. However, it should be noted that the correlation of activity with the gene expression results is weak, probably due in part to the small number of tumours studied, but suggesting that determinants of resistance may not be limited to the target pathways, incorporating both the apoptotic potential of the cell and classical drug resistance mechanisms relating to drug efflux pumps, as has been seen with other anti-cancer drugs (Glaysher et al., 2010).
This study identified varied inhibition of ovarian tumours when exposed to EGFR, PI3K and mTOR inhibitors. Greatest single-agent activity was observed with the PI3K inhibitor ZSTK474 confirming the importance of PI3K signalling within these tumours. Evidence for the deregulation of PI3K/AKT signalling in ovarian cancer includes gain-of-function mutations, amplifications of PI3K and AKT genes as well as allelic imbalance and mutations in PTEN (Huang et al., 2011).
In ovarian tumours, all combinations showed greater activity than the single agents, with ZSTK474 and EGFR inhibitor combinations most active. Antagonism was seen at higher concentrations of EGFR and PI3K inhibitors and suggests that there may be a biologically optimal dose beyond which activity is lost. The strategy to hit multiple aspects of signalling pathways, thought to be key to ovarian cancer control of growth and survival, proved to be more effective than single-agent administration in most but not all of the tumours studied. The most effective combinations were of EGFR and PI3K inhibitors. However, with inhibition of EGFR alone, continued expression of HER2, HER3, IGFR and c-MET goes unchecked. Subsequent heterodimerisation of these receptors has been shown to increase downstream signalling via the PI3K-Akt pathway (Wheeler et al., 2008). Therefore, the addition of further agents or multi-targeted HER inhibitors (e.g., lapatinib) targeting such PI3K compensatory pathways might result in greater effects. Such a strategy would of course require phase I trials with careful monitoring of toxicity, but is feasible.
Expression of EGFR and PI3K pathway genes were seen to correlate with greater and lesser activity, respectively, of single-agent EGFR inhibitors. With these agents, increased levels of EGFR, HER2 or HER3 inferred sensitivity to these inhibitors, whereas increased IGF or IGF2R expression inferred greater resistance. The inverse relationship was seen with ZSTK474 and sirolimus sensitivity where expression of IGF or IGF2R led to greater sensitivity and expression of EGFR pathway genes to lesser sensitivity. The inverse relationship between the effects of gene expression for these receptors may give rise to some of the antagonism seen with the combination of these inhibitors and is explicable in terms of the pathway (Figure 5). For instance, tumour 9 has low EGFR expression and shows resistance to EGFR inhibitors, but sensitivity to PI3K pathway inhibition (Figure 2).
Figure 5.

Human epidermal growth factor receptor and PI3K pathways in ovarian cancer.
In the presence of gefitinib, PI3K signalling can be maintained by an activated IGF1R pathway (Jones et al., 2006; Guix et al., 2008). Therefore it could be suggested that for single-agent anti-EGFR therapy to be effective in EGFR wild-type ovarian cancers, it would require cells to have EGFR-dominant HER signalling in the absence of IGFR signalling pathways. In anti-EGFR-resistant tumours, this profile may indicate the employment of alternate signalling mechanisms such as continued signalling via Met by driving HER3 (ErbB3)-dependent activation of PI3K.
Cellular inhibition with the PI3K inhibitor ZSTK474 was shown to be more effective in tumours with low EGFR receptor expression. It has been shown that inhibition of PI3K can result in feedback upregulation of expression and phosphorylation of multiple receptor tyrosine kinases in breast cancer (Chakrabarty et al., 2012). The identification of these feedback mechanisms emphasises the need for multiple inhibitor approach to treatment, and in ovarian cancer, combination of HER and PI3K inhibitors may prove useful. Unfortunately, the number of tumours studied and the rarity of mutations in relevant genes in ovarian cancer meant that there were very few tumours with activating mutations in our series, and no conclusions can be drawn from the one tumour with an activating KRAS mutation, which showed resistance to both EGFR inhibitors but only moderate sensitivity to PI3K inhibitors.
In conclusion, this study shows encouraging activity of a combination of EGFR and PI3K inhibitors: combined HER inhibitors such as lapatinib may also be interesting although they were not tested here. Phase I/II clinical trials with these agents should include pharmacodynamic endpoints and molecular characterisation to identify patients most likely to benefit from this strategy.
Acknowledgments
We are grateful to CanTech Ltd for funding this work.
The authors declare no conflict of interest.
Footnotes
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
References
- Andreotti PE, Cree IA, Kurbacher CM, Hartmann DM, Linder D, Harel G, Gleiberman I, Caruso PA, Ricks SH, Untch M, et al. Chemosensitivity testing of human tumors using a microplate adenosine triphosphate luminescence assay: clinical correlation for cisplatin resistance of ovarian carcinoma. Cancer Res. 1995;55 (22:5276–5278. [PubMed] [Google Scholar]
- Bartlett JM, Starczynski J, Atkey N, Kay E, O'Grady A, Gandy M, Ibrahim M, Jasani B, Ellis IO, Pinder SE, Walker RA. HER2 testing in the UK: recommendations for breast and gastric in-situ hybridisation methods. J Clin Pathol. 2011;64 (8:649–653. doi: 10.1136/jcp.2011.089847. [DOI] [PubMed] [Google Scholar]
- Bianco R, Garofalo S, Rosa R, Damiano V, Gelardi T, Daniele G, Marciano R, Ciardiello F, Tortora G. Inhibition of mTOR pathway by everolimus cooperates with EGFR inhibitors in human tumours sensitive and resistant to anti-EGFR drugs. Br J Cancer. 2008;98 (5:923–930. doi: 10.1038/sj.bjc.6604269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank SV, Chang R, Muggia F. Epidermal growth factor receptor inhibitors for the treatment of epithelial ovarian cancer. Oncology (Williston Park) 2005;19 (4:553–559. [PubMed] [Google Scholar]
- Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, Cristiano BE, Pearson RB, Phillips WA. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;64 (21:7678–7681. doi: 10.1158/0008-5472.CAN-04-2933. [DOI] [PubMed] [Google Scholar]
- Chakrabarty A, Sanchez V, Kuba MG, Rinehart C, Arteaga CL. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad SciUSA. 2012;109:2718–2723. doi: 10.1073/pnas.1018001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Glaysher S, Cree IA. Cell sensitivity assays: the ATP-based tumor chemosensitivity assay. Methods Mol Biol. 2011;731:247–257. doi: 10.1007/978-1-61779-080-5_21. [DOI] [PubMed] [Google Scholar]
- Glaysher S, Gabriel FG, Cree IA. Measuring gene expression from cell cultures by quantitative reverse-transcriptase polymerase chain reaction. Methods Mol Biol. 2011;731:381–393. doi: 10.1007/978-1-61779-080-5_31. [DOI] [PubMed] [Google Scholar]
- Glaysher S, Gabriel FG, Johnson P, Polak M, Knight LA, Parker K, Poole M, Narayanan A, Cree IA. Molecular basis of chemosensitivity of platinum pre-treated ovarian cancer to chemotherapy. Br J Cancer. 2010;103 (5:656–662. doi: 10.1038/sj.bjc.6605817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco WR, Bravo G, Parsons JC. The search for synergy: a critical review from a response surface perspective. Pharmacol Rev. 1995;47 (2:331–385. [PubMed] [Google Scholar]
- Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, Rinehart C, Seidel B, Yee D, Arteaga CL, Engelman JA. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest. 2008;118 (7:2609–2619. doi: 10.1172/JCI34588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger AW. Detection of ErbB receptor family tyrosyl phosphorylation in ovarian carcinoma cells. Methods Mol Med. 2001;39:571–575. doi: 10.1385/1-59259-071-3:571. [DOI] [PubMed] [Google Scholar]
- Huang J, Zhang L, Greshock J, Colligon TA, Wang Y, Ward R, Katsaros D, Lassus H, Butzow R, Godwin AK, Testa JR, Nathanson KL, Gimotty PA, Coukos G, Weber BL, Degenhardt Y. Frequent genetic abnormalities of the PI3K/AKT pathway in primary ovarian cancer predict patient outcome. Genes Chromosomes Cancer. 2011;50 (8:606–618. doi: 10.1002/gcc.20883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Penuel E, Mink S, Schmidt J, Hodge A, Favero K, Tindell C, Agus DB. HER kinase axis receptor dimer partner switching occurs in response to EGFR tyrosine kinase inhibition despite failure to block cellular proliferation. Cancer Res. 2010;70 (5:1989–1999. doi: 10.1158/0008-5472.CAN-09-3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones HE, Gee JM, Hutcheson IR, Knowlden JM, Barrow D, Nicholson RI. Growth factor receptor interplay and resistance in cancer. Endocr Relat Cancer. 2006;13 (Suppl 1:S45–S51. doi: 10.1677/erc.1.01275. [DOI] [PubMed] [Google Scholar]
- Knight LA, Di Nicolantonio F, Whitehouse P, Mercer S, Sharma S, Glaysher S, Johnson P, Cree IA. The in vitro effect of gefitinib (‘Iressa') alone and in combination with cytotoxic chemotherapy on human solid tumours. BMC Cancer. 2004;4:83. doi: 10.1186/1471-2407-4-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolasa IK, Rembiszewska A, Felisiak A, Ziolkowska-Seta I, Murawska M, Moes J, Timorek A, Dansonka-Mieszkowska A, Kupryjanczyk J. PIK3CA amplification associates with resistance to chemotherapy in ovarian cancer patients. Cancer Biol Ther. 2009;8 (1:21–26. doi: 10.4161/cbt.8.1.7209. [DOI] [PubMed] [Google Scholar]
- Larsen AK, Ouaret D, El Ouadrani K, Petitprez A. Targeting EGFR and VEGF(R) pathway cross-talk in tumor survival and angiogenesis. Pharmacol Ther. 2011;131 (1:80–90. doi: 10.1016/j.pharmthera.2011.03.012. [DOI] [PubMed] [Google Scholar]
- Mayr D, Kanitz V, Amann G, Engel J, Burges A, Lohrs U, Diebold J. HER-2/neu gene amplification in ovarian tumours: a comprehensive immunohistochemical and FISH analysis on tissue microarrays. Histopathology. 2006;48 (2:149–156. doi: 10.1111/j.1365-2559.2005.02306.x. [DOI] [PubMed] [Google Scholar]
- Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol. 2009;27 (13:2278–2287. doi: 10.1200/JCO.2008.20.0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M, Stordal B. Erlotinib or gefitinib for the treatment of relapsed platinum pretreated non-small cell lung cancer and ovarian cancer: a systematic review. Drug Resist Updat. 2011;14 (3:177–190. doi: 10.1016/j.drup.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Niikura H, Sasano H, Sato S, Yajima A. Expression of epidermal growth factor-related proteins and epidermal growth factor receptor in common epithelial ovarian tumors. Int J Gynecol Pathol. 1997;16 (1:60–68. doi: 10.1097/00004347-199701000-00010. [DOI] [PubMed] [Google Scholar]
- Qiu L, Di W, Jiang Q, Scheffler E, Derby S, Yang J, Kouttab N, Wanebo H, Yan B, Wan Y. Targeted inhibition of transient activation of the EGFR-mediated cell survival pathway enhances paclitaxel-induced ovarian cancer cell death. Int J Oncol. 2005;27 (5:1441–1448. [PubMed] [Google Scholar]
- Sain N, Krishnan B, Ormerod MG, De Rienzo A, Liu WM, Kaye SB, Workman P, Jackman AL. Potentiation of paclitaxel activity by the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin in human ovarian carcinoma cell lines with high levels of activated AKT. Mol Cancer Ther. 2006;5 (5:1197–1208. doi: 10.1158/1535-7163.MCT-05-0445. [DOI] [PubMed] [Google Scholar]
- Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, Gondi V, Hsu KT, Harari PM. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27 (28:3944–3956. doi: 10.1038/onc.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Li J, Ran L, Pan F, Zhao X, Ding Z, Chen Y, Peng Q, Liang H. Phosphorylated insulin-like growth factor 1 receptor is implicated in resistance to the cytostatic effect of gefitinib in colorectal cancer cells. J Gastrointest Surg. 2011;15 (6:942–957. doi: 10.1007/s11605-011-1504-z. [DOI] [PubMed] [Google Scholar]