

Abstract
To develop potent and selective nNOS inhibitors, new double-headed molecules with chiral linkers that derive from natural amino acids or their derivatives have been designed. The new structures contain two ether bonds, which greatly simplifies the synthesis and accelerates structure optimization. Inhibitor (R)-6b exhibits a potency of 32 nM against nNOS and is 475 and 244 more selective for nNOS over eNOS and iNOS, respectively. Crystal structures show that the additional binding between the aminomethyl moiety of 6b and the two heme propionates in nNOS, but not eNOS, is the structural basis for its high selectivity. This work demonstrates the importance of stereochemistry in this class of molecules, which significantly influences the potency and selectivity of the inhibitors. The structure-activity information gathered here provides a guide for future structure optimization.
Keywords: neuronal nitric oxide synthase, inhibition, chiral molecules, double-headed inhibitors, crystallography
Nitric oxide (NO) is produced from L-arginine by the nitric oxide synthase (NOS) family of enzymes, including neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS). NO plays important roles as a second-messenger molecule in neural and cardiovascular systems, but acts as a cytotoxic agent in the immune system. Extensive clinical research has shown that overexpression of nNOS is implicated in various neurodegenerative diseases, including Parkinson’s,1 Alzheimer’s,2 and Huntington’s diseases,3 also in the neuronal damage that results from stroke.4
A possible approach to prevent these neurodegenerative diseases is to inhibit nNOS, thereby blocking excess NO generation.5,6,7 However, inhibitors must be selective for nNOS over the other two isoforms to prevent side effects, as eNOS controls blood flow and blood pressure, and iNOS is involved with normal immunological functions. The structural similarity in the heme active site for the three isozymes of NOS has posed a challenge in the development of selective NOS inhibitors. Therefore, it is still necessary to develop nNOS inhibitors with high potency and selectivity.
We have developed two lead compounds, 1 and 2, which show very good potency against nNOS. Compound 1 provides an excellent dual-selectivity of nNOS over the other two isoforms,8 but, unfortunately, the tedious synthesis limits its structure/activity optimization to improve bioavailability and limits the quantities available for in vivo studies. The synthesis of double-headed inhibitor 2 is much easier than 1, but its selectivity needs to be improved.9 The previous results with the pyrrolidine-containing inhibitors for nNOS9 inspired us to explore the potential of incorporating stereogenic centers into the double-headed inhibitor design. In this work, we report results of utilizing some natural chiral building blocks for the composition of our double-headed inhibitors to improve selectivity. Because the inhibitors derive from chiral natural scaffolds, there is no chiral synthesis or resolution, which makes these new compounds easily accessible and ready for further optimization.
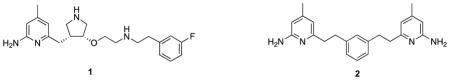
The synthesis of inhibitors 6 began with (S) or (R)-3-amino-1,2-propanediol, a derivative of isoserine (Scheme 1). Protection of the amino group with 2,5-hexanedione followed by a onestep double condensation with compound 4, connected the two aminopyridine heads simultaneously. After the removal of the protecting groups in the presence of NH2OH-HCl, inhibitors 6a-b were obtained. Achiral inhibitor 8 was synthesized via a similar pathway starting with ethylene glycol (Scheme 2).
Scheme 1.
Synthesis of 6a-b. Reagents and conditions: (a) p-TsOH, toluene, reflux, 77–79%; (b) NaH, NaI, DMF, 0 °C, 72–82%; (c) NH2OH-HCl, EtOH/H2O (2/1), 100 °C, 61–63%.
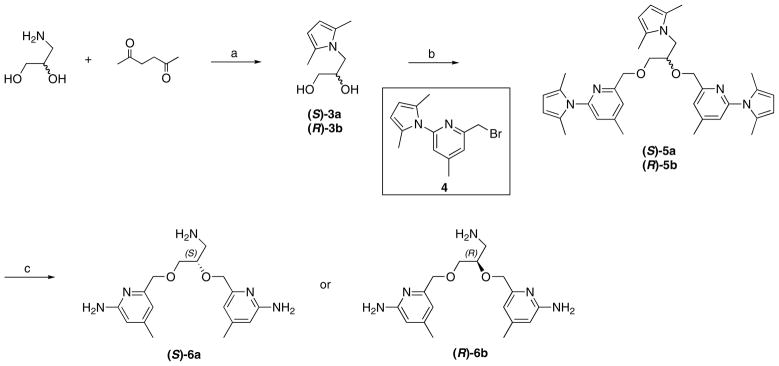
Scheme 2.
Synthesis of 8. Reagents and conditions: (a) 4, NaH, NaI, DMF, 0 °C, 84%; (b) NH2OH-HCl, EtOH/H2O (2/1), 100 °C, 71%.
(S) or (R)-4-Amino-2-hydroxybutyric acid was used as the starting material for the synthesis of 11. The carboxylic acid was reduced with LiBH4 in the presence of TMSCl in THF, providing diol compound 10. The two aminopyridine heads were connected to the linker at the same time via a one-step ether synthesis. Final compound 11 was obtained after deprotection of the three amino groups under the same conditions. The synthesis of compound 12 followed the same synthetic method starting with (S)-4-amino-2-hydroxybutyric acid.
To synthesize inhibitor 16, threonine-HCl was first neutralized with NaOH before protection of the amino group in toluene. The synthesis followed the same methodology as we developed for inhibitors 6, 11, and 12 (Scheme 4). Three isomers were obtained from the corresponding starting compounds; no chiral synthesis was used and no racemic mixtures were obtained. The (R,S) isomer was not available because of the absence of the starting material.
Scheme 4.
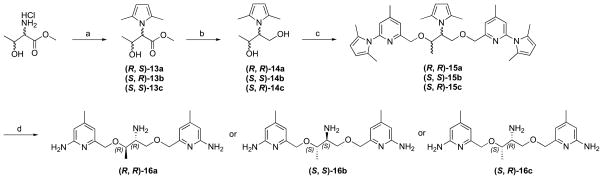
Synthesis of 16. Reagents and conditions: (a) (i) NaOH, CH3OH; (ii) 2,5-hexanedione, p-TsOH, toluene, reflux, 73%; (b) LiBH4, 96%; (c) 4, NaH, NaI, DMF, 0 °C, 73%; (d) NH2OH-HCl, EtOH/H2O (2/1), 100 °C, 63%. Note that the configuration change from 13 to 14 results from the R,S priority rules; there is no stereochemistry change.
All of the inhibitors were assayed against the three different isoforms of NOS including rat nNOS, bovine eNOS, and murine macrophage iNOS using L-arginine as a substrate. Ki values are shown in Table 1. Using compound 8 as a reference, which has a 6-atom linker bearing two ether oxygens between the two aminopyridine head groups, we have introduced either an aminoethyl or aminomethyl tail through a chiral center to generate inhibitors 11 or 6. The inhibitory assays indicate that neither of the two 11 isomers is potent and selective, but the two 6 isomers show either better potency or better selectivity compared to 8. More importantly, the stereochemistry of the chiral center dramatically influences the outcomes: inhibitor (R)-6b provides much better characteristics than (S)-6a in terms of both potency (32 nM) and selectivity (475 of e/n and 244 of i/n).
Table 1.
K ia values of inhibitors for rat nNOS, bovine eNOS and murine iNOS
| |||||
|---|---|---|---|---|---|
| Inhibitors |
Kia
(nM) |
Selectivity
|
|||
| nNOS | eNOS | iNOS | e/n | i/n | |
| (S)-6a | 382 | 26446 | 40829 | 69 | 107 |
| (R)-6b | 32 | 15213 | 7821 | 475 | 244 |
| 8 | 316 | 5264 | 2929 | 17 | 9 |
| (S)-11a | 1599 | 92756 | 53222 | 58 | 33 |
| (R)-11b | 881 | 66959 | 31410 | 76 | 35 |
| (S)-12 | 232 | 20196 | 10725 | 87 | 46 |
| (R,R)-16a | 47 | 2995 | 1857 | 64 | 39 |
| (S,S)-16b | 37 | 2542 | 2262 | 69 | 61 |
| (S,R)-16c | 384 | 17673 | 10996 | 46 | 28 |
The IC50 values were measured for three different isoforms of NOS including rat nNOS, bovine eNOS, and murine macrophage iNOS using L-arginine as a substrate with a standard deviation ±10%. The corresponding Ki values were calculated from the IC50 values using the equation Ki = IC50/(1 + [S]/Km) with known Km values (rat nNOS, 1.3 μM; iNOS, 8.3 μM; eNOS, 1.7 μM).
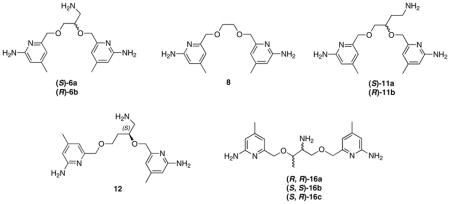
To understand the structure-activity relationship of these inhibitors, we have determined the crystal structures of both nNOS and eNOS in complex with either (S)-6a or (R)-6b. As shown in Figure 1, (S)-6a binds to the active site of nNOS and eNOS in totally different orientations. In nNOS, it is the aminopyridine with a 3-atom linker from the chiral center that hydrogen bonds to the active site Glu592, while in eNOS the inhibitor flips 180°, thereby placing the other aminopyridine with the 2-atom linker in position to H-bond with the active site Glu (Figure 1B). In nNOS the ether oxygen in the linker forms a weak H-bond (3.2 – 3.3 Å) with a water molecule that is in turn H-bonded with Glu592 (Figure 1A). The aminomethyl group off of the chiral center does not make any strong interactions with the protein, as evidenced by its weaker electron density. In nNOS the second aminopyridine is in position to H-bond with the heme propionate of the pyrrole ring D (propionate D). In eNOS the aminomethyl nitrogen is in position to form H-bonds with both heme propionates. However, this favorable amino group forces the second aminopyridine ring into position where it cannot make favorable protein interactions and is, therefore, partially disordered with poorly defined density. In summary, these structures show that (S)-6a does not gain much binding affinity from its aminomethyl group in nNOS while the inhibitor loses potential binding contributions from its second aminopyridine in eNOS.
Figure 1.
View of the active site of nNOS (A) (PDB code: 4K5D) and eNOS (B) (PDB code: 4K5H) in complex with inhibitor (S)-6a. Shown also are the omit 2Fo – Fc density maps for inhibitor contoured at the 2.5 σ level. Relevant hydrogen bonds are depicted as dash lines. All structural figures were prepared with PyMol (www.pymol.org).
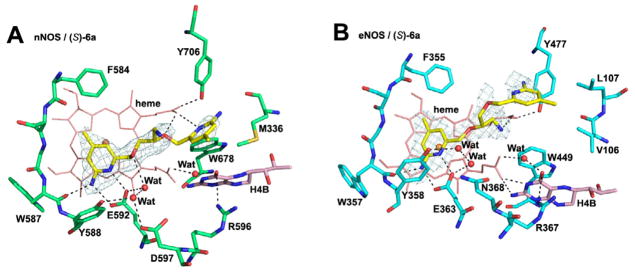
Despite having a different chirality, (R)-6b maintains the major anchoring H-bonds between one of the aminopyridines and the active site Glu in both eNOS (Glu363) and nNOS (Glu592) (Figure 2). In nNOS the aminomethyl nitrogen is situated between both heme propionates, where it can form H-bonding interactions with both (Figure 2A). As with (S)-6a the ether oxygen in the 3-atom linker H-bonds with the water that also H-bonds with Glu592 (Figure 2A). However, the second aminopyridine of (R)-6b extending out of the active site has weak electron density so its exact positioning is ill-defined. The binding mode of (R)-6b in eNOS is somewhat ambiguous. The electron density is well defined only up to the chiral center, so it is not possible to define the exact orientation of the aminomethyl group. As a result, it is not clear if (R)-6b binds in the same orientation as in nNOS or flips 180°. However, the model that best fits the density is to have the aminopyridine that bears the 3-atom linker to the chiral center H-bonded with Glu363, as shown in Figure 2B, suggesting that (R)-6b binds to eNOS in the same orientation as in nNOS. Although the same aminopyridine interacts with the active site Glu seen in nNOS (Figure 2A), the conformation of the 3-atom linker is different in nNOS and eNOS. In eNOS the ether oxygen bends away from Glu363; there is no H-bond from this ether oxygen to the water molecule next to Glu363. By having a more extended conformation for the 3-atom linker in eNOS, (R)-6b can no longer bring the aminomethyl nitrogen into a position that makes good interactions with both heme propionates. Neither the aminomethyl group nor the second aminopyridine ring has good electron density, indicative of high flexibility. In both eNOS and nNOS the second aminopyridine extending out of the active site exhibits weak electron density, suggesting that this ring does not contribute much to the binding affinity or selectivity. The main difference between nNOS and eNOS is the much stronger interaction of the aminomethyl group with the heme propionates in nNOS, which, very likely, is the structural basis for the excellent selectivity of nNOS over eNOS (Table 1). The structures of (S)-6a and (R)-6b complexed to eNOS and nNOS also provide an explanation for why (S)-11a and (R)-11b are poor inhibitors (Table 1). The bulkier aminoethyl group would result in steric clashes and the loss of important interactions with the heme propionates (Table 1).
Figure 2.
View of the active site of nNOS (A) (PDB code: 4K5E) and eNOS (B) (PDB code: 4K5I) in complex with inhibitor (R)-6b. Shown also the omit 2Fo – Fc density maps for the inhibitor contoured at 2.5 σ level. Relevant hydrogen bonds are depicted as dash lines.
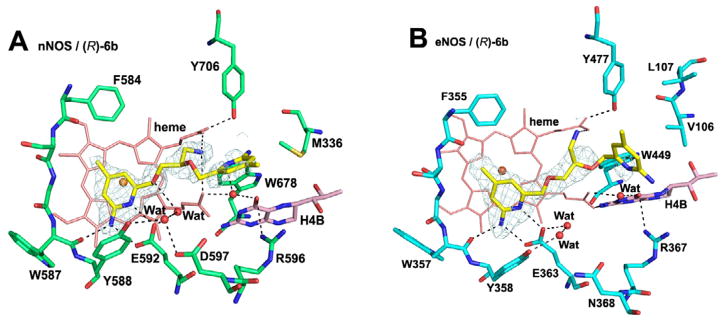
We next designed and synthesized inhibitor (S)-12, a modification of (R)-6b, by extending the 3-atom linker to 4-atoms. Although its stereochemical configuration is (S), compound (S)-12 has the same stereochemistry as (R)-6b, but the chain length modification improves neither potency nor selectivity (Table 1). The crystal structure of (S)-12 complexed to nNOS reveals that a different aminopyridine head, the one with the 2-atom linker, binds to Glu592 (Figure 3A), while the aminopyridine with the 3-atom linker in (R)-6b interacts with propionate D (Figure 2A). This binding mode enables the aminomethyl nitrogen to H-bond with Glu592 and is also about 5.3Å from Asp597. The position of the aminomethyl is very similar to how dipeptide amide inhibitors bind10 with the amino group situated between Glu592 and Asp597 for maximum electrostatic stabilization. The other aminopyridine group of (S)-12 H-bonds to heme propionate D (Figure 3A). In eNOS, (S)-12 flips relative to the nNOS binding mode (Figure 3B) so the aminopyridine head with a 4-atom linker H-bonds to Glu363 in eNOS. The main consequence of this different binding mode is that the second aminopyridine extends further out of the active site and is disordered. Also, the aminomethyl group is closer to heme propionate D, but weak electron density indicates, at most, a weak interaction. Although the binding mode of (S)-12 is totally different with nNOS and eNOS, the selectivity for nNOS over eNOS is only 87. (S)-12 may gain some affinity for eNOS from the H-bond between its ether oxygen and the water molecule next to Glu363 (Figure 3B) and possibly some favorable electrostatic interactions between the aminomethyl and heme propionate D.
Figure 3.
View of the active site of nNOS (A) (PDB code: 4K5F) and eNOS (B) (PDB code: 4K5J) in complex with inhibitor (S)-12. Shown also are the omit 2Fo – Fc density maps for the inhibitor contoured at the 2.5 σ level. Relevant hydrogen bonds are depicted as dash lines.
We have further explored the effects of having two chiral centers in the linker between the two aminopyridine heads, with one center bearing a primary amine and the other a methyl group. Three of four possible isomers were synthesized via the method described in Scheme 4. Inhibitors (R,R)-16a and (S,S)-16b show potencies of 47 and 37 nM, respectively, which is comparable to that of (R)-6b, but (S,R)-16c has a 10-fold decreased affinity (Table 1). The nNOS-(S,S)-16b crystal structure (Figure 4A) shows that the aminopyridine farthest from the methyl group in the linker H-bonds to Glu592 and that the amino nitrogen of one of the chiral centers H-bonds with both heme propionates. The second aminopyridine group H-bonds to heme propionate D, which requires that Tyr706 swings out into an alternate rotamer (Figure 4A). (S,S)-16b binds similarly to eNOS with the important exception that Tyr477 (Tyr706 in nNOS) remains in place, H-bonded to heme propionate D (Figure 4B). Although the density for the second aminopyridine group is weak, the 2-amino group can potentially H-bond with Tyr706 (Figure 4B). We have observed previously11,12 that other double-headed aminopyridine inhibitors with a 7- or 8-atom linker result in Tyr706 adopting the new rotamer conformation, thereby enabling one aminopyridine to interact with heme propionate D and that the movement of Tyr706 occurs most often in nNOS. Although we expected some isoform selectivity because the second aminopyridine group directly H-bonds with heme propionate D, (S,S)-16b, it is only 69-fold more selective for nNOS. The poor selectivity here is mainly the result of better affinity of (S,S)-16b to eNOS in comparison with (R)-6b (Table 1). This clearly indicates that having strong salt bridges from an amino group to both heme propionates significantly contributes to inhibitor binding affinity. In contrast, the interaction from the second aminopyridine group of (S,S)-16b to heme propionate D may only have limited impact on the overall potency. Another line of evidence to support this assumption is that no difference in potency is observed in nNOS when the additional H-bonds between the second aminopyridine group of (S,S)-16b and heme propionate D (Figure 4A) is compared to (R)-6b, where those H-bonds are missing (Figure 2A). The high flexibility of (S,S)-16b may be why there is partial disordering with weaker density for the second aminopyridine group in both nNOS and eNOS (Figure 4).
Figure 4.
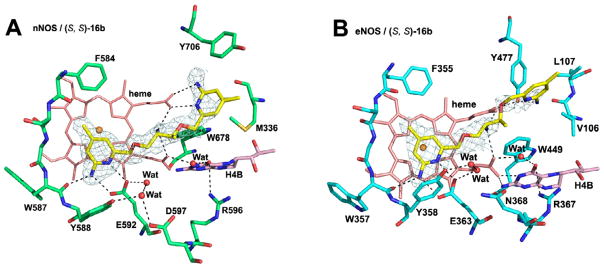
View of the active site of nNOS (A) (PDB code: 4K5G) and eNOS (B) (PDB code: 4K5K) in complex with inhibitor (S, S)-16b. Shown also are the omit 2Fo – Fc density maps for the inhibitor contoured at the 2.5 σ level. Relevant hydrogen bonds are depicted as dash lines.
In conclusion, we have designed and synthesized a series of double-headed inhibitors with a chiral linker derived from amino acids, which has substantially increased the ease of synthesis of the chiral inhibitors. Inhibitor (R)-6b affords a potency of 32 nM as well as a dual selectivity of 475 and 244 for nNOS over eNOS and iNOS, respectively. An important conclusion from this study is that to achieve low nanomolar affinity strong interactions between the inhibitor amine with both heme propionates is required, such as in the cases of (R)-6b and (S,S)-16b. Combined with our earlier work on double-headed inhibitors, a second important feature of potent and selective inhibitors relates to how well the second aminopyridine group extends out of the active site and interacts with heme propionate D. For optimal interactions, Tyr706 must swing out of the way in nNOS, and this occurs much more often in nNOS than eNOS, suggesting greater flexibility of Tyr706 in nNOS than the corresponding Tyr in eNOS. The flexible inhibitors developed in this study exhibit fairly weak electron density for this aminopyridine group, indicating that the interactions with heme proprionate D are weaker. The next generation of compounds must take this into account, but given the relative ease of synthesis, it should be possible to more readily develop compounds that incorporate these features.
Supplementary Material
Scheme 3.

Synthesis of 11 and 12. Reagents and conditions: (a) (i) LiBH4, TMSCl, THF; (ii) 2,5-hexanedione, p-TsOH, toluene, reflux, 61–65%; (b) 4, NaH, NaI, DMF, 0 °C, 78–92%; (c) NH2OH-HCl, EtOH/H2O (2/1), 100 °C, 58–67%.
Acknowledgments
The authors are grateful for financial support from the National Institutes of Health (GM049725 to RBS and GM057353 to TLP). We thank Dr. Bettie Sue Siler Masters (NIH grant GM52419, with whose laboratory P.M. and L.J.R. are affiliated). B.S.S.M. also acknowledges the Welch Foundation for a Robert A. Welch Distinguished Professorship in Chemistry (AQ0012). P.M. is supported by grants 0021620806 and 1M0520 from MSMT of the Czech Republic. We also thank the beamline staff at SSRL and ALS for their assistance during the remote X-ray diffraction data collections.
Footnotes
Supplementary data (including detailed synthetic procedures, full characterization (1H-NMR, 13C NMR) of compounds 6, 8, 11, 12, and 16, and detailed crystallographic data of 6, 12, 16b with nNOS and eNOS) associated with this article can be found, in the online version, at doi:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhang L, Dawson VL, Dawson TM. Pharmacol Ther. 2006;109:33. doi: 10.1016/j.pharmthera.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 2.Dorheim MA, Tracey WR, Pollock JS, Grammas P. Biochem Biophys Res Commun. 1994;205:659. doi: 10.1006/bbrc.1994.2716. [DOI] [PubMed] [Google Scholar]
- 3.Norris PJ, Waldvogel HJ, Faull RLM, Love DR, Emson PC. Neuronsicence. 1996;72:1037. doi: 10.1016/0306-4522(95)00596-x. [DOI] [PubMed] [Google Scholar]
- 4.Sims NR, Anderson MF. Neurochem Int. 2002;40:511. doi: 10.1016/s0197-0186(01)00122-x. [DOI] [PubMed] [Google Scholar]
- 5.Alderton WK, Cooper CE, Knowles RG. Biochem J. 2001;357:593. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Southan GJ, Szabo C. Biochem Pharmacol. 1996;51:383. doi: 10.1016/0006-2952(95)02099-3. [DOI] [PubMed] [Google Scholar]
- 7.Babu BR, Griffith OW. Curr Opin Chem Biol. 1998;2:491. doi: 10.1016/s1367-5931(98)80125-7. [DOI] [PubMed] [Google Scholar]
- 8.a) Ji H, Li H, Martásek P, Roman LJ, Poulos Ts L, Silverman RB. J Med Chem. 2009;52:779. doi: 10.1021/jm801220a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ji H, Delker SL, Li H, Martásek P, Roman LJ, Poulos TL, Silverman RB. J Med Chem. 2010;53:7804. doi: 10.1021/jm100947x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xue F, Delker SL, Li H, Fang J, Martásek P, Roman LJ, Poulos TP, Silverman RB. J Med Chem. 2011;54:2039. doi: 10.1021/jm101071n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flinspach ML, Li H, Jamal J, Yang W, Huang H, Hah JM, Gomez-Vidal JA, Litzinger EA, Silverman RB, Poulos TL. Nat Struct Mol Biol. 2004;11:54. doi: 10.1038/nsmb704. [DOI] [PubMed] [Google Scholar]
- 11.Delker SL, Ji H, Li H, Jamal J, Fang J, Xue F, Silverman RB, Poulos TL. J Am Chem Soc. 2010;132:5437. doi: 10.1021/ja910228a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang H, Li H, Martarsek P, Roman LJ, Poulos TL, Silverman RB. J Med Chem. 2013;56:3024. doi: 10.1021/jm4000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.