

Abstract
To improve future drug development and patient management for patients with castration-resistant prostate cancer (CRPC), surrogate biomarkers that are linked to relevant outcomes are urgently needed. A biomarker must be measurable, reproducible, linked to relevant clinical outcomes, and demonstrate utility. This is a rapidly evolving area, with recent trials in CRPC incorporating the detection of circulating tumour cells (CTCs), imaging, and patient-reported outcome biomarkers. We discuss the framework for the development of biomarkers for CRPC, including different categories and contexts of use. We also highlight the requirements of analytical validation, the sequence of trials needed for clinical validation and regulatory approval, and the future outlook for imaging and CTC biomarkers.
Introduction
To establish a new treatment standard of care requires demonstration of a clinical benefit or that the treatment alters an outcome measure known to be a substitute or surrogate for that benefit. The success of recent phase III trials for castration-resistant prostate cancer (CRPC) has led to the approval of several agents with diverse mechanisms of action1–6 and new treatment standards. However, there were also notable failures,7–11 which highlight the challenges in developing new treatments and improving outcomes for patients with CRPC. For example, sipuleucel-T showed an overall survival benefit, despite a modest effect on prostate-specific antigen (PSA) levels and no effect on disease progression.1 This example illustrates that clinical outcome was not correlated with the studied biomarker. Furthermore, a placebo-controlled trial demonstrated a survival benefit for radium-223 chloride and a delay in time to PSA progression,12 although there was no significant difference in PSA response rate (>50% decline from baseline) in the study-drug arm relative to placebo.13 Finally, androgen receptor (AR) signalling inhibitors can lower PSA without prolonging survival.14
Bone is the most-common site of metastatic spread in patients with CRPC. Assessment of bone metastases remains problematic because of the lack of standards for using and interpreting imaging modalities to detect and monitor disease in bone. The need for new biomarkers becomes all the more crucial as additional life-prolonging treatment options emerge, making overall survival trial results difficult to interpret because downstream therapies after trial participation may alter the survival equation.15 This crowded therapeutic landscape increases the difficulty of demonstrating a survival benefit for the next promising approach.
All of these factors highlight the need for clinically relevant intermediate end points that are surrogates for overall survival, and that can reliably inform phase III outcomes and/or lead to drug approvals in their own right. Validated intermediate end point biomarkers would shorten the time to complete a clinical trial and enable a greater number of therapies to be tested within a given time frame. Predictive biomarkers are also needed to enable trials to enroll and treat patients most likely to respond to a particular treatment based on the patient’s disease characteristics. Although the need to explore new biomarkers is apparent, there is too little appreciation and understanding of the rigorous structure that is required to develop a new biomarker for a specific context of use. We provide a detailed framework for biomarker testing in CRPC that is focused on determining prognosis and assessing treatment effects. In 2008, the Prostate Cancer Working Group (PCWG2) presented a new framework for clinical trial conduct in CRPC16 in response to a challenge by the FDA. The new paradigm more-directly aligned trial objectives with clinical practice and patient benefit by reframing early post-treatment response outcomes as the control, relief or elimination of disease manifestations present when treatment is initiated, and reframing time-to-event outcomes indicative of progression as preventing or delaying disease manifestations, including death from disease, from occurring in the future. The indications for drug approvals in CRPC are consistent with this paradigm (Table 1). PCWG2 stated that trials should be designed for patients in discrete clinical states which represent key milestones and decision points in the disease continuum which for CRPC, are focused primarily on prior chemotherapy exposure. This Review builds upon the PCWG2 framework and terminology by considering trial eligibility (the decision to treat a patient) and outcomes (endpoints) by their usefulness (utility). We focus on the analytical validity of the specific biomarker measurement, and the level of evidence needed to clinically validate its use in a specific context to inform a medical decision.
Table 1.
Biomarkers of clinical benefit for successful or approved agents in CRPC
| Biomarker end point* | Biomarker | Therapy |
|---|---|---|
| Control or relieve or eliminate existing disease manifestations‡ | Pain | Strontium-89 |
| Samarium-153 lexidronam | ||
| Mitoxantrone + prednisone | ||
| Elevated PSA | None | |
| Tumour regression | None | |
| CTCs | None | |
|
| ||
| Delay or prevent future disease manifestations§ | Death from disease | Docetaxel |
| Sipuleucel-T | ||
| Cabazitaxel | ||
| Abiraterone | ||
| Radium-223 chloride|| | ||
| Enzalutamide | ||
| Skeletal-related events | Zoledronic acid | |
| Denosumab | ||
| Time to PSA progression | None | |
| Time to radiographic progression | Abiraterone¶ | |
Control or relieve or eliminate, and delay or prevent end points were defined by the Prostate Cancer Working Group 2 guidelines.16
Response end points.
Time-to-event end points.
Treatment showing a survival benefit in a phase III trial, but not FDA-approved.
Co-primary end point with overall survival.
Abbreviations: CRPC, castration-resistant prostate cancer; CTC,circulating tumour cell; PSA, prostate-specific antigen.
Overview of biomarker development
Biomarkers are characteristics that can be objectively measured and evaluated as indicators of normal processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention.17 Biomarkers can be clinical parameters (such as age, performance status), laboratory measures (such as PSA), imaging-based measures, or genetic and molecular determinants. Biomarker development consists of two separate components: analytical validation and clinical validation. 18,19
For analytical validation of a biomarker, data are generated to describe the performance characteristics of the biomarker measurement itself; these include the device, imaging modality, or assay, and the range of conditions under which the measurement gives reproducible and accurate results. Analytical validation includes the pre-analytical assessment of specimen and/or image acquisition, processing and storage; determining the consistency and reproducibility of the analysis each time it is performed at the same or independent laboratories, and quality control measures; the post-analytical data reduction, including how the results will be recorded, reported, and the specific measures that will be associated with clinical outcome. An example of a measure of reproducibility is the concordance correlation coefficient, which determines the variation of replicate pairs plotted around a 45-degree line through the origin (Figure 1).20 After analytical validity has been established, clinical studies are initiated to establish clinical validity: the demonstration that the biomarker is fit for purpose for the specific context of use—that is, that the results will inform the medical decision.21 Establishing clinical validity requires trials designed to address the biomarker question.
Figure 1.

Correlation Log (1+CTC) values for 67 patients plotted using CellSearch® and Flow CTC assays.99 The concordance correlation coefficient is computed to assess the reproducibility of the two assays. With the assays plotted against each other, the concordance correlation coefficient measures the variation of the points around a 45 degree line through the origin. The concordance correlation coefficient for the two assays equals 0.82.
Clinical utility demonstrates how much additional information the biomarker provides relative to what is currently available;22 both cost and clinical utility affect reimbursement. Although the FDA does not formally request clinical utility in the biomarker development process, it is a vital consideration that will impact on how widely the marker is used and ultimately reimbursed by private and public payers. For example, clinical utility would be a deciding factor when comparing the value of a costly molecular analysis of a tumour compared to inexpensive clinical parameters routinely available in practice to assess prognosis.
Clinical qualification is an additional step in biomarker development that has regulatory implications. A clinically qualified biomarker is one for which sufficient evidence has been generated for FDA acceptance for use in regulatory submissions, without a re-review of the data supporting the use of the biomarker. A biomarker that qualifies for a clinical benefit end point (an improvement in survival time or patient function) could potentially lead to accelerated drug approval.23 The level of evidence needed for clinical qualification involves prospective testing of the biomarker in multiple phase III trials.24
The FDA has four specific categories for contexts of biomarker use: prognostic, predictive, response-indicator, and efficacy-response (Table 2). Considered in the PCWG2 framework,16 prognostic and predictive biomarkers include pretreatment characteristics of the patient and the tumour. Such biomarkers may be identified retrospectively after a trial has been completed and may form part of the eligibility criteria for a future trial. Response-indicator and efficacy-response biomarkers are those occurring after treatment and represent an effect of the treatment. Alternatively, biomarker use can be classified by the PCWG2 recommendations as pretreatment or post-treatment (Table 3).
Table 2.
FDA biomarker classification based on contexts of use
| Biomarker type | When biomarker is measured | What biomarker indicates |
|---|---|---|
| Prognostic | Prior to treatment | Indicates (estimates) the risk or likelihood that a patient who receives no further cancer-directed therapy will experience a specified clinical outcome, such as recurrence, progression, or death. |
| Predictive | Prior to treatment | Interpreted with defined criteria to identify patients who are likely to benefit from a specific treatment compared to patients who do not meet the specified criteria. |
| Response-indicator | During or after treatment | Demonstrates a pharmacological or physiological response to the treatment, but does not necessarily signify patient benefit. Examples are declines in prostate-specific antigen, measures of tumour shrinkage, or pharmacodynamic changes in a parameter to show the on-target effect of a drug as proof of mechanism or to optimize dosing. |
| Efficacy-response (surrogate) | After treatment | Provides an early and accurate prediction of both a clinical end point, and the effects of the treatment on that end point. |
Table 3.
Biomarkers correlating with timing of treatment and outcome measure
| Biomarker type | When biomarker is measured | What biomarker indicates |
|---|---|---|
| Pretreatment | Prior to treatment | Includes prognostic and predictive biomarkers. |
| Post-treatment: control, relieve, eliminate | After treatment | Indicates response to the treatment, that is, shows a change in a disease manifestation that was present prior to therapy. Such biomarkers include response-indicator pharmacodynamic measures, changes in tumour size or tumour markers, and efficacy-response types of biomarkers. |
| Post-treatment: delay, prevent | After treatment | Represents time-to-event biomarkers, that is, a delay in or the prevention of a manifestation that would have occurred had treatment not been given. For example, a treatment that prolongs life is one that delays or prevents death from disease. Such biomarkers include response-indicator and efficacy-response types of biomarkers. |
A recent change in the regulatory requirements for clinical trials evaluating predictive biomarkers of sensitivity to a drug is noteworthy.25 Under the revised guidance, if the results of a pretreatment biomarker assay are used to guide the choice of one treatment versus another in a clinical trial, the assay and the device used to measure it, are subject to regulatory review for an Investigational Device Exemption (IDE) and must be formally approved for investigational use by the FDA. Once clinical development starts, the assay itself cannot be changed. A biomarker that informs the choice of a specific therapy on an investigational protocol is called an integral biomarker and, if it is shown to be predictive, may require a companion diagnostic assay that meets regulatory requirements before the drug itself can be approved for clinical use.26
Biomarkers and trial design issues
Pretreatment biomarkers
Prognostic biomarkers
Prognostic biomarkers are often part of a study’s eligibility criteria; one example is a nomogram for estimating survival time.27 Several nomograms incorporate baseline clinical parameters that are categorical (such as performance status, Gleason score, prior treatment, or sites of disease), and biological determinants that are continuous (such as PSA and lactate dehydrogenase [LDH]).27–29 In a nomogram, each parameter is assigned a separate score, and the sum of these scores is used to estimate the probability of an event in a certain number of years. Although the use of nomograms has improved our ability to estimate prognosis, when the discriminatory accuracy of CRPC nomograms was examined, the known prognostic factors for overall survival and progression-free survival explained only a modest understanding of patient risk. 30,31 This lack of predictive accuracy suggests that the practice of using nomogram-based prognoses to identify a ‘similar’ historical control group, rather than randomizing a contemporary control group, might lead to inaccurate clinical decisions.
Predictive biomarkers
Predictive biomarkers are typically considered to be biological or molecular determinants; for example, mutations that are associated with sensitivity to tyrosine kinase inhibitors.32 They can also include clinical parameters such as prior exposure to a specific therapy, response to therapy, or laboratory measurements such as serum testosterone levels.4 The PCWG2 definition of castration—testosterone levels ≤50 ng/ml in the blood—was largely consensus-based, but also reflected the varied sensitivity of the testosterone assays available.16 In association with the recent approval of the CYP17 inhibitor abiraterone acetate, it was shown that much lower androgen levels (1–2 ng/ml) can be detected.33 Now that mass spectroscopy-based assays that can measure androgens in this lower range are available, the definition of castration will change. As a result, and with the demonstrated survival benefit of the androgen receptor signalling inhibitor enzalutamide,5 more studies are considering both prior hormonal exposure as well as measured testosterone levels as part of eligibility for trials.
Common molecular alterations in CRPC include changes in the AR and AR signalling axis, such as AR overexpression, increased androgen biosynthesis, splice variants and mutations, altered PTEN signalling, and translocations that allow the ETS transcription factor to be under the control of androgen.34 Mutations in specific receptor tyrosine kinases including the EGFR or BRAF, common in other tumour types, are infrequent in prostate cancer. 35–37 Reciprocal feedback inhibition between the AR and PTEN axes has also been described.38 These and other molecular changes are being studied as both prognostic and predictive biomarkers. Unfortunately, none of the assays for these determinants in prostate cancer has been analytically validated yet, limiting our ability to explore the association of these changes with outcomes.
Post-treatment biomarkers
Response-indicator biomarkers
A response-indicator biomarker shows that there has been a change following treatment, but does not necessarily indicate that a patient has benefitted from the treatment. Such biomarkers include pharmacodynamic measures to assess the on-target effects of a drug, a change in tumour size, and change in response criteria. For instance, outcomes for most solid tumours are reported using RECIST.39,40 In most patients with metastatic prostate cancer, RECIST is not particularly useful because it does not adequately address PSA changes or the assessment of disease in bone.39 It is for these reasons that PCWG2 recommended reporting the outcomes for each disease manifestation independently and specifically recommended against using the grouped categorizations of response in RECIST.16
Response-indicator biomarkers in CRPC include the control and/or relief of symptoms;41 post-therapy changes in PSA, which are now reported using waterfall plots; changes in bone scans (reported as improved, unchanged or stable, or progression); and changes in soft-tissue disease reported using RECIST 1.1, but recognizing its limitations. Reporting outcomes individually for each disease manifestation enables a better understanding of the association between a specific parameter and clinical outcomes.16
Efficacy-response biomarkers
The control and relief of pain in patients with CRPC is a clinical benefit and has led to drug approvals. 41–43 Pain relief has been reported with docetaxel,41 cabazitaxel,2 abiraterone,44 radium-223 chloride,12 and enzalutamide,5 although none of these drugs has a formal pain relief indication. Biomarkers to assess presence of pain at baseline and change in pain post-treatment typically begin as individual measures on quality-of-life instruments—such as the Brief Pain Inventory46 or QLC-30,46—which then undergo a detailed validation process similar to the process performed for laboratory assays. Once validated, individual measures cannot be modified or taken out of context. 45,47 Of the measures reported, maximal pain intensity in the previous 24 h has been used as an efficacy-response indicator of clinical benefit for drug approval.45 More recently, dramatic improvements in bone scan lesional number, area, and intensity, along with significant pain relief, were reported with cabozantinib.48 Based on these findings, the COMET-2 trial was developed using a primary end point of pain palliation.49 Docetaxel in combination with custirsen, an inhibitor of clusterin, is also under study for the same indication.50
Prevent or delay biomarkers
Owing to the difficulties assessing response and the frequent lack of association between a change in an individual disease parameter and clinical benefit, PCWG2 recommended focusing less on whether a treatment was working and more on when it had failed.16 This shift has more closely aligned the objectives of clinical research and clinical practice. To apply this approach requires a prospectively defined primary end point that can be measured with minimal bias and that is in itself a clinical benefit or can serve as a surrogate for that benefit. Prolonging life—defined as the delay or prevention of death from disease—is the gold standard for drug approval, as is a reduction or delay in the development of skeletal-related events. PSA progression, regardless of how it is defined, associates poorly with survival,31 and no radiographic progression biomarkers have been validated analytically.
To assess the adequacy of a biomarker-based time-to-progression end point, the association between the end point and survival time must be evaluated using a statistical measure that allows for censoring in both the time to death and the time to biomarker-derived progression; one example is the generalized version of Kendall rank correlation coeffcient.51 If a biomarker that closely associates with survival time is identified, its adequacy as a surrogate end point must be tested using multiple independent randomized clinical trials, the Prentice criteria being the most common conceptual framework for this.50 In addition to randomized trials demonstrating a treatment effect and the biomarker’s association with survival, the Prentice criteria require that the treatment effect is fully explained by the newly constructed biomarker. However, this requirement is thought to be too high a standard to achieve: alternative continuous metrics have been proposed that measure the amount of the treatment effect explained by the biomarker.23
The way forward—emerging biomarkers
Imaging biomarkers
Bone scintigraphy and PCWG2
To establish the performance characteristics of an imaging modality, each step of the process must be standardized, including tracer manufacturing, tracer administration, the time to image acquisition, the methodology for acquiring the images, the definition and selection of data elements, and data collection. Qualitative and subjective descriptors are not sufficient. To ensure correlations can be performed consistently, researchers at Memorial Sloan-Kettering Cancer Center (MSKCC) focus their initial studies on patients in a particular disease state, such as those with disease progression or who have metastatic disease visible on radionuclide bone scan.53
Intuitively, it follows that prognosis varies inversely with disease extent; apart from considering the distribution of lesions (axial versus appendicular),54 or groupings based on the number of lesions,55 there remains no standard method of recording osseous disease, despite decades of bone scan use.56 Therefore, PCWG2 first focused on post-treatment outcomes, and recommended that post-treatment effects on bone scintigraphy be characterized as improved, stable or worse (progression), a highly subjective classification. At the same time, PCWG2 proposed a definition of progression as the appearance of two or more new lesions, with the recognition that a first follow-up scan might show new lesions that reflect bone healing and favorable effects of treatment, rather than treatment failure. To account for this flare phenomenon, and to ensure that an effective treatment was not discontinued prematurely, PCWG2 advised that the appearance of two or more new lesions on the first post-treatment scans should be confirmed with a follow-up scan showing two additional new lesions before the patient’s disease can be considered as having progressed. After the follow-up (second) scan, the appearance of two new lesions on any scan is considered to show disease progression.16
To test the PCWG2 criteria clinically, a bone scan interpretation assay was developed to systematically collect and aggregate new lesions so that a definitive PCWG2-compliant date of progression could be designated. The instrument was developed, tested, refined and validated at prostate cancer imaging research centres in the Prostate Cancer Clinical Trials Consortium over a period of years. It allows radiologists to record the seminal data points of bone imaging and record disease progression uniformly across study sites and across studies.57
Following development of the bone-scan assay, the progression measure was embedded into three phase III registration trials of active AR-targeted therapies powered to detect a survival advantage.4 The first has been completed 4 and showed excellent concordance for interpretation between individual sites and central readers.58 The results of the first trial were a supporting factor for the approval of abiraterone plus prednisone for chemotherapy pretreated patients,57 and a revision of the indication for the drugs to treat CRPC. The results from the other two studies are pending.
Bone scan index
Recognizing that the distribution of bone metastases mirrors the distribution of the adult bone marrow, researchers at MSKCC developed the Bone Scan Index (BSI), a quantitative measure of tumour burden as percentage of skeletal mass. The first clinical study of the BSI focused on reproducibility and showed an interobserver variability of less than 10%, and intraobserver correlation between a first and second reading of the same scan after a 2-year period of 0·97 and 0·94.60 A study of 191 patients with progressive CRPC divided into equal tertiles showed inferior survival as BSI increased from <1·4%, 1·4–5·1%, and >5·1% (P=0·0079). The prognostic significance of BSI was retained in a multivariate analysis that also included age, haemoglobin, and LDH.60
Although simply counting new lesions is a reproducible metric, it does not capture increases in the size of lesions. To address this, we studied serial changes in BSI over time as an outcome. In an initial series of 88 patients, pretreatment and post-treatment BSI was highly associated with survival, and in a multivariate model in which post-treatment changes in BSI were compared with post-treatment PSA changes, BSI was the most significant marker correlated with outcome. A doubling of BSI conferred an almost twofold increase in the risk of death.61 Further study in ongoing phase III trials is planned.
Automation of quantitative bone imaging
Conventional bone scans in patients with prostate cancer result in subjective interpretation, which is dependent on the skill of individual readers. For this reason, quantitative methods that are inherently more objective, such as BSI, have a definite appeal for assessing treatment effects. Recently, two computer-aided detection systems have been described: EXINI bone62 (Exini Diagnostics, Lund, Sweden) and MedQIA63 (MedQIA, Los Angeles, CA, USA). Both methods reproducibly segment and quantify lesions within a single scan and allow for the comparison of lesion measurements between consecutive scans. The EXINI system provides a BSI value that represents percentage of total skeletal mass involved by the tumour, whereas the MedQIA system assesses the lesion count, the area (Bone Scan Lesion Area [BSLA]), and the intensity (Bone Scan Lesion Intensity [BSLI]). The parameters of lesion count, BSLA, and BSLI were used to quantitatively demonstrate the significant post-treatment changes seen with cabozantinib,48,63 which have in turn resulted in the opening of a randomized placebo-controlled trial with overall survival as the primary endpoint (COMET-1).64 It is likely that the introduction of these automated methods will make quantitative methods of bone scan evaluation more practical and readily standardized.63
PET imaging
Molecular imaging can assess both soft tissue and bone with a single modality, and inform on the biology of the tumour itself rather than its impact on the surrounding tissues. A variety of tracers are being explored that are at different stages of development. Each must follow the same process to justify regulatory approval.
Analytic validation
To use PET imaging as a biomarker, the tracer itself must undergo analytical validation that includes uniform tracer production and administration; image acquisition, data collection, and post-scanning data processing; defining its pharmacokinetic properties; establishing correlations with other imaging modalities and tissue; and data interpretation.53 This validation is accomplished by maintaining the image biomarker as the primary variable in a prospectively defined cohort of patients.
Performance characteristics
The importance of controlling for confounding factors cannot be underestimated. For example, using a heterogeneous population, it was originally postulated that 18F-fluorodeoxyglucose (FDG)–PET correlated poorly with known sites of disease.65 By contrast, when imaging was restricted to patients with progressive disease, and the scans performed at fixed intervals, the outcomes were very different. In an analysis of 43 patients with metastatic CRPC, 1,720 bones were examined by bone scintigraphy and compared with PET findings: 1,079 (63%) bones were negative on both modalities, and 400 (23%) were positive on both, a 90% concordance rate.66,67 Of the 121 mismatches where FDG–PET was positive and the bone scan was negative, 105 had follow-up bone scans and 84 (80%) were positive, suggesting that PET was superior in terms of detecting early metastatic disease.
Reproducibility can be further defined by performing two scans before any treatment is delivered. Such test and re-test studies are now underway in the Prostate Cancer Clinical Trials Consortium for sodium fluoride PET–,66 a bone-imaging PET tracer likely to replace technetium bone scintigraphy. The 18F-fluorodihydrotesterone (FDHT)–PET tracer is being studied in a similar manner.53 Such studies of reproducibility within a modality and between modalities must be performed with each imaging method studied (Figure 2).
Figure 2.
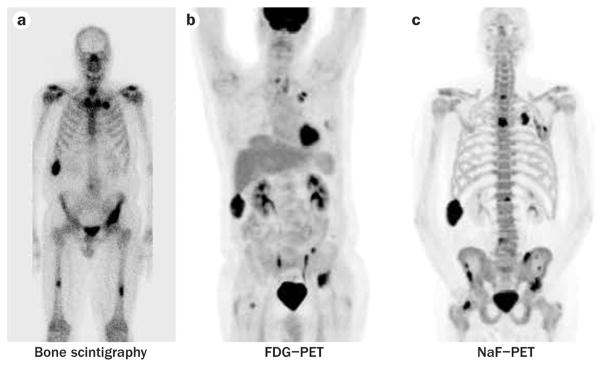
Radionuclide imaging of a patient with castration-resistant prostate cancera | Tc-99m bone scan, b | FDG–PET, c | NaF–PET. Since the three scans are measuring different aspects of osseous metastases, biomarkers unique to each modality must be developed, analytically validated, and tested independently and prospectively before they achieve clinical validity. Abbreviation: FDG, fluorodeoxyglucose.
Prognosis
PET imaging has the potential to serve as both a prognostic and response-indicator biomarker because it lends itself to quantitation. Previous studies examined the hottest slice of the hottest lesion on the scan (SUVmax), the average of five index lesions (SUVmaxavg), or the uptake in a region around the maximal pixel (SUVpeak), and other measures.69 In a multivariate analysis of a series of 96 patients with metastatic CRPC, only LDH and FDG–PET SUVmaxavg were significantly associated with overall survival.70
Response indicator
Explorations of PET imaging as a response-indicator are also ongoing. 69,71 The PET measure that will be followed across the treatment interval is the biomarker—be it a single hottest slice such as the SUVmax or the average of the hottest slices of a selected group of index lesions such as the SUVmaxavg. In one study of 22 patients with progressive CRPC undergoing treatment with antimicrotubule chemotherapy who were scanned at baseline, 4 weeks, and 12 weeks, SUVmaxavg PET imaging, as a single measure captured the post-treatment effects usually identified by an amalgam of PSA, bone imaging, and cross-sectional imaging.71
These studies are labour intensive as they require manual measurements of individual lesions followed across time. Now, with an increase in automation and computing power, it is feasible to examine all of the lesions as opposed to a handful of index lesions. Figure 3, depicts a Larson–Fox–Gonen plot72 that shows the individual lesions in a patient at baseline and after 1 month of treatment with a novel anti-androgen, enzalutamide. Two tracers were administered, 18FDHT to visualize the androgen receptor, and 18FDG to visualize glucose metabolism. The Larson-Fox-Gonen plot illustrates the heterogeneity of response in individual sites of metastases using the two tracers, as well as the heterogeneity of the lesions demonstrated by each single tracer.
Figure 3.

Larson–Fox–Gonen plot showing SUVmax for individual lesions in a patient at baseline and following 1 month of treatment with a novel anti-androgen, enzalutamide. The Larson–Fox–Gonen plot visually demonstrates changes in lesional uptake for the totality of the patient’s disease burden over time. By using different tracers, one can illuminate the heterogeneity of a patient’s response to treatment pathway by pathway, depending on the tracers used. Shown here are two tracers, 18FDHT to demonstrate the presence of the androgen receptor, and 18FDG to demonstrate glucose metabolism. Abbreviations: FDG, fluorodeoxyglucose; FDHT, fluorodihydrotestosterone; SUVmax, standardized uptake value (maximum).
Circulating tumour cells
Circulating tumour cells (CTCs) are estimated to represent less than one in a billion of the circulating mononuclear cells in the blood.73 Methods to detect and isolate these cells typically include an enrichment step followed by a variety of techniques to enable enumeration, profiling and and/or biological characterization.73,74 Enrichment methods include those based on: physical characteristics such as density gradient centrifugation, filtration, dielectric focusing, or that exploit differences in cell plasticity; direct capture methods with a single antibody or antibody cocktail to cell-surface markers that are conjugated to magnetic ferrofluids, magnetic beads, microposts on chips,75,76 or for separation by flow cytometric techniques; indirect methods that deplete CD45-expressing mononuclear cells that leave the CTC population behind; a red-cell lysis followed by the direct deposit of the buffy coat on a microscope slide for future imaging;77,78 or the ability of cells to grow in vitro.79
Detection and characterization can be achieved with tumour-specific or tissue-specific antibodies visualized with semi-automated microscopes, laser-scanning techniques, or other DNA-based and RNA-based methods.80,81 No two assays measure the same CTC biomarker. Even two antibodies to different epitopes on the same protein may not give equivalent results in the clinic. As such, there is no single definition for a CTC or CTC biomarker to represent the full spectrum of tumour-derived cells in the circulation. These cells range from those with stem or stem-cell-like properties to those that have undergone an epithelial–mesenchymal transition (EMT) to fully differentiated cells. The selection of an assay is dependent on the clinical context for which it will be used: no single assay can address all of the unmet needs that can be addressed with CTC biomarkers.
Analytical validation
The collection tubes for assays can differ: some have fixatives to preserve the cells, while those for ribonucleic acids, short-term culture, xenograft development, or functional characterization generally do not. Some methods require sample processing within hours, while others provide consistent results up to several days after the blood draw. Sensitivity and specificity are issues for all of the techniques owing to the heterogeneity of tumour cells with respect to size, density, and marker expression. EpCAM is the most widely used capture antibody, and cytokeratins (CK8 or CK18) are the most widely used marker for the detection of CTCs from epithelial prostate tumours.82 Either method might not capture or detect cells that have undergone EMT or that have stem cell or stem cell–like properties, and variations in expression levels83 can lead to false-negatives.
Currently, CellSearch® (Veridex LLC, Warren, NJ, USA) is the only assay that is analytically valid and FDA-approved for patient use.84 With this assay, CTCs are captured using an EpCAM conjugated ferrofluid, and are defined after immunofluorescent staining as morphologically intact cells with a 4′,6-diamidino-2-phenylindole (DAPI)-positive nucleus surrounded by cytoplasm that express the cytokeratins CK8, CK18 or CK19, but do not express CD-45.85 The assay reports the number of cells in 7·5 ml of blood that meet the defined criteria; notably, this repeated value represents a small proportion of the EpCAM-expressing cells captured (Figure 4).
Figure 4. Identifying CTCs using CellSearch®.
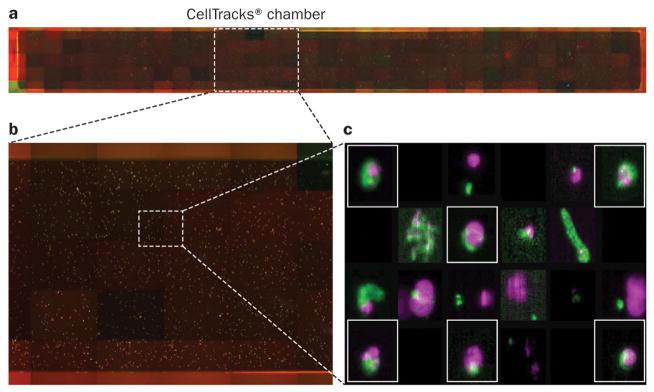
The CellSearch® system uses a glass-surfaced cartridge. Immunomagnetically labelled cells expressing EpCAM are fluorescently stained and pulled to the surface of the chamber using a targeted magnetic field. a | The semiautomated imaging system captures four fluorescent images at each of 175 locations in order to create a 4-composite photograph of the entire surface of the chamber. b | A close-up of approximately 25 of the 175 locations, showing a large number of fluorescently labelled events present within each location. c | The CellTracks® software analyzes individual events in the image frames, creating a gallery of images including only those events where DAPI and cytokeratin fluorescence are co-located. The user examines these events for the presence of DAPI (purple), cytokeratin (green), and the absence of CD-45 (red) fluorescence. Events that meet the criteria of a CTC assay based on morphology, an intact nucleus, and cytokeratin expression, and which are negative for CD-45 expression (events boxed in red), are only a proportion of cytokeratin-positive events in the chamber. Other events (not boxed) that do not meet these criteria are not scored as CTCs. Permission obtained from Robert McCormack, Veridex, LLC. Abbreviations: CTC, circulating tumour cell; DAPI, 4′,6-diamidino-2-phenylindole. (Image courtesy of Robert McCormack, Veridex, LLC.)
The assay was analytically validated using cell-line-spiking experiments, and with over 450 duplicate breast cancer patient samples showing consistent results over a wide range of cell numbers analysed in the same laboratory by different technicians, and in a reference laboratory compared to a local laboratory.84 We showed that the cells captured expressed PSA and AMACR racemase, and that prostate cancer samples had unique copy number alterations and AR amplification by FISH.85
Prognosis
The prognostic significance of CTCs detected by CellSearch® was demonstrated in sequential studies of similar design (the IMMC38 series) that enrolled patients with breast,86 colorectal87 and prostate88 cancer about to receive a new first-line or second-line chemotherapy regimen. In the prostate cancer study, CTC enumeration and PSA determinations were performed before treatment and at the start of a cycle,89; assessment while imaging was discretionary. The prostate cancer results established a baseline value of four or fewer cells per 7·5 ml of blood as a favourable prognosis, whereas five or more cells per 7·5 ml of blood was associated with an unfavourable prognosis.88 This finding was used to show the activity of hormonal agents,96 and in sequential phase I trials of different targeted therapies.91 The cut-off point of five cells was based on differences in the hazard ratio (HR) for survival at different cut-off points at the lower end of the measurement scale. A similar association with survival was shown for CTC numbers analysed as a continuous variable.89,92 The wide range of survival times for patients with low CTC counts demonstrates that a low count alone does not assure a long survival.92,93 The predictive accuracy of CTC with survival analysed with the concordance probability estimate showed the discriminatory power of CTCs equal to 0·71, which increased to 0·74 with the addition of baseline LDH levels.90
Response indicator
A similar association between favourable versus unfavourable CTC counts and survival was shown at different time points (2–5 weeks, 6–8 weeks, 9–12 weeks, and 16–20 weeks) post-treatment.88 The association between the presence of CTCs with decreased overall survival led to the FDA clearance of the test “as an aid in the monitoring of [these] patients in conjunction with other clinical methods.”84 A subsequent analysis of patients receiving their first cytotoxic therapy showed that CTC count was associated with overall survival as a continuous variable. In a multivariate analysis, only CTC count and LDH were prognostic, but PSA was no longer prognostic.92 Similar results were observed in an analysis of men enrolled on a series of phase I and phase II trials of molecular-targeted agents.94 Importantly, the FDA clearance does not establish CTC enumeration as an efficacy-response surrogate, and as such this biomarker cannot be used in regulatory submissions. However, it does establish the ability of the test result to inform prognosis pretreatment and post-treatment.
Efficacy-response qualification
Qualifying CTC enumeration as an efficacy-response surrogate marker for survival requires consistent results in multiple phase III clinical trials where the biomarker is embedded. Based on the IMMC38 results, CTC enumeration (CellSearch®) was studied independently in two phase II abiraterone trials that enrolled men who had received prior chemotherapy for CRPC. It was notable that in this closely defined patient population the different trials (conducted at different sites) showed a similar frequency of unfavourable CTC counts at baseline and conversion rates post-treatment.90,95 The trial led by the Royal Marsden Hospital showed 79% of patients with unfavourable CTC counts at baseline and 41% converting to favourable CTC counts post-treatment.90 The trial led by MSKCC showed 69% of patients with unfavourable CTC counts at baseline and 34% and converting to favourable CTC counts post-treatment,95 a consistency in outcome that strongly supports the reliability of the results. Based on these data, CTC enumeration was studied in the abiraterone phase III registration trial as an efficacy-response biomarker.96 The final analysis of the phase III trial showed a median overall survival difference of 4·6 months in favour of abiraterone compared with placebo (15·8 months versus 11·2 months; HR = 0·74; P< 0·0001). CTC conversion from unfavourable (CTC ≥ 5) to favourable (CTC < 5) counts was predictive of overall survival as early as 4 weeks after beginning treatment, and its inclusion significantly explained the treatment effect at all post-treatment time points (HR: from 0·74–0·97), a key component of the Prentice criteria.96 An analysis of whether the prognostic significance of CTC conversion is increased if it is combined with other biomarkers (such as LDH, haemaglobin, PSA levels) is planned. If it is, then the best combination of markers will be used as a CTC-based biomarker panel in subsequent trials.97,98 A goal of these studies is to determine whether this biomarker panel may be used as a surrogate end point in future CRPC clinical trials.
Prediction and precision medicine
Optimal use of a targeted approach requires a demonstration that the target is present in an individual patient’s tumour when therapy is considered. CTCs can originate from the primary tumour itself or from the metastatic sites, and potentially could be measured non-invasively, using a real-time liquid biopsy, for disease characterization to guide treatment selection. The specific CTC assay needed will depend on the determinant being studied. Presently, there are a wide range of assays, many of which are being used in patients without any analytical or clinical validity. Caution must be exercised by both investigators and practitioners in the routine clinical use of these assays.
CONCLUSIONS
The development of biomarkers to inform drug development requires a systematic and stepwise approach. It begins with an assessment of the performance characteristics and reproducibility of the measurement itself, followed by a series of prospectively designed studies with adequate statistical support to demonstrate a clinical effect within a well-defined context. Analytical and clinical validation for a particular context of use are required steps for FDA approval (clinical qualification). At present, CTCs and imaging are promising candidates for informative new biomarkers. Together, they present the opportunity to examine a patient’s entire disease burden, disease aggressiveness and disease biology. With each technology enhancement comes the burden of starting the biomarker validation and qualification process from the beginning. This requirement includes the analytical validation of devices, assays, and software, as well as the data elements and outputs. The statistical and computational methodologies to describe this wealth of data are also rapidly evolving. There are no shortcuts, but through focused efforts significant advances have been realized and promise to continue.
Key Points.
Improving current treatment for patients with castration-resistant prostate cancer requires new biomarkers and surrogate endpoints for clinical trials.
Of highest priority are biomarkers that reflect clinical benefit, and predictive biomarkers to guide the selection of treatment most likely to work in the individual patient.
The development and approval processes for biomarkers are rigorous and lengthy, requiring analytically valid assays and a sequence of trials that support the biomarker’s use in a specific context of use
The investment of resources and time will be recovered by achieving streamlined clinical trials and better selection of new therapies to develop.
Promising emerging biomarkers for castration-resistant prostate cancer include circulating tumour cells and new methods for imaging bone metastases.
Review Criteria.
A formal literature search was performed using the PubMed database and the following terms: prostate cancer biomarkers, clinical trials, trial design, response and endpoints. The authors used their own judgment about which papers to include from the literature search based on the relevance of the article to the clinical scenario. This Review also includes a summary of the authors’ work and knowledge based on reading the oncology literature. Knowledge gained from regular attendance at conferences, workshops, and other national and international meetings was also included.
Acknowledgments
The work in this article was supported by: The Sidney Kimmel Center for Prostate and Urologic Cancers. Supported in part by the MSKCC SPORE in Prostate Cancer (P50 CA92629), the Department of Defense Prostate Cancer Research Program (PC051382), The Research and Therapeutics Program for Prostate Cancer, The Prostate Cancer Foundation, Mr. William H. and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and the Experimental Therapeutics Center of Memorial Sloan-Kettering Cancer Center. In addition, Steven Larson was supported by P50 CA85438. We thank Amy Plofker, MSKCC editor, for manuscript editing.
Biographies
Howard I. Scher, MD, obtained his medical degree in 1976 from New York University School of Medicine. He is currently Chief of the Genitourinary Oncology Service at Memorial Sloan-Kettering Cancer Center and Professor of Medicine at Weill Cornell Medical College. An internationally recognized investigator, he has helped elucidate key molecular and genetic features of prostate cancer and translated these insights into the evaluation of novel targeted agents in the clinic. His work focuses on developing non-invasive methods to identify predictive biomarkers of drug sensitivity to guide treatment selection, and clinical trial endpoints that reflect clinical benefit to patients. Dr. Scher is widely published and the recipient of numerous awards.
Dr. Morris is a member of the Genitourinary Oncology Service at Memorial Sloan-Kettering Cancer Center. His research focus lies in the field of prostate cancer therapeutics and biomarker development, with a particular focus on bone imaging and bone-directed therapies. He earned his medical degree from the Mount Sinai School of Medicine in New York, performed his residency in internal medicine at Columbia Presbyterian Medical Center, fellowship at Memorial Sloan-Kettering Cancer Center. He holds leadership positions in the Department of Defense/Prostate Cancer Foundation Prostate Cancer Clinical Consortium and the Alliance for Clinical Trials in Oncology.
Dr. Larson obtained his medical degree in 1968 from the University of Washington, Seattle, and subsequently obtained board certification in both internal medicine and nuclear medicine. He has published more than 500 articles in various aspects of nuclear medicine, especially radiopharmaceutical development, novel quantitative nuclear imaging analysis methodologies clinical Positron Emission Tomography and radio labeled antibody development. Since 1988, after arriving at Memorial Sloan Kettering Cancer center, he has focused his research on translational aspects of molecular imaging and targeted radionuclide therapy applied to oncology.
Dr. Heller is a biostatistician at Memorial Sloan-Kettering Cancer Center. He advises clinical and laboratory investigators on study design and data analysis. His current research interests include the areas of prediction models for patient risk, analysis of microarray data, survival analysis, the design of small sample animal studies, and clinical trial design. Dr. Heller received his PhD degree in statistics at New York University in 1987 and joined Memorial Sloan-Kettering Cancer Center the same year. He was elected a fellow of the American Statistical Association in 2007.
Footnotes
Competing interests
H. I. Scher is a consultant for Bristol-Myers Squibb, Dendreon, Endo/Orion Pharmaceuticals, Genentech, Novartis, Ortho Biotech Oncology Research & Development, and Sanofi Aventis. He receives research funding from Aragon, Bristol-Myers Squibb, Exelixis, Janssen Research & Development, Janssen Services, and Medivation. M. J. Morris is a consultant for Millennium. He receives research funding from Agensys, Algeta, Bayer, Genta, Medivation, and Sanofi Aventis. S. Larson is a consultant for ImaginAb, Perceptive, and Progenics. He receives research funding from GE Medical Systems. G. Heller declares no competing interests.
References
- 1.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 2.de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376:1147–54. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 3.de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ryan CJ, Smith MR, De Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–48. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scher HI, Fizazi K, Saad F, et al. Enzalutamide prolongs survival in men with prostate cancer following chemotherapy. N Engl J Med. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 6.Fizazi K, Carducci M, Smith M, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet. 2011;377:813–22. doi: 10.1016/S0140-6736(10)62344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scher HI, Jia X, Chi K, et al. Randomized, open-label phase III trial of docetaxel plus high-dose calcitriol versus docetaxel plus prednisone for patients with castration-resistant prostate cancer. J Clin Oncol. 2011;29:2191–8. doi: 10.1200/JCO.2010.32.8815. [DOI] [PubMed] [Google Scholar]
- 8.Kelly WK, Halabi S, Carducci M, et al. Randomized, double-blind, placebo-controlled phase III trial comparing docetaxel and prednisone with or without bevacizumab in men with metastatic castration-resistant prostate cancer: CALGB 90401. J Clin Oncol. 2012;30:1534–40. doi: 10.1200/JCO.2011.39.4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michaelson MD, Oudard S, Ou Y, et al. Randomized, placebo-controlled, phase III trial of sunitinib in combination with prednisone (SU+P) versus prednisone (P) alone in men with progressive metastatic castration-resistant prostate cancer (mCRPC). 2011 ASCO Annual Meeting; June 3–7, 2011; Chicago, IL. p. Abstract 4515. [Google Scholar]
- 10.Nelson JB, Fizazi K, Miller K, et al. Phase 3, randomized, placebo-controlled study of zibotentan (ZD4054) in patients with castration-resistant prostate cancer metastatic to bone. Cancer. 2012;118:5709–18. doi: 10.1002/cncr.27674. [DOI] [PubMed] [Google Scholar]
- 11.Petrylak DP, Fizazi K, Sternberg CN, et al. A phase 3 study to evaluate the efficacy and safety of docetaxel and prednisone (DP) with or without lenalidomide (LEN) in patients with castrate-resistant prostate cancer (CRPC): the MAINSAIL trial. European Society for Medical Oncology Congress; September 28 – October 2, 2012; Vienna, Austria. p. Abstract LBA24. [Google Scholar]
- 12.Parker C, et al. Overall survival benefit of radium-223 chloride (alpharadin) in the treatment of patients with symptomatic bone metastases in castration-resistant prostate cancer (CRPC): a phase III randomized trial (ALSYMPCA). Program and abstracts of the 2011 European Multidisciplinary Cancer Congress; September 23–27, 2011; Stockholm, Sweden. p. Abstract LBA1. [Google Scholar]
- 13.Nilsson S, Franzén L, Parker C, et al. Bone-targeted radium-223 in symptomatic, hormone-refractory prostate cancer: a randomised, multicentre, placebo-controlled phase II study. Lancet Oncol. 2007;8:587–94. doi: 10.1016/S1470-2045(07)70147-X. [DOI] [PubMed] [Google Scholar]
- 14.Small EJ, Halabi S, Dawson NA, et al. Antiandrogen withdrawal alone or in combination with ketoconazole in androgen-independent prostate cancer patients: a phase III trial (CALGB 9583) J Clin Oncol. 2004;22:1025–33. doi: 10.1200/JCO.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 15.Saad ED, Buyse M. Overall survival: patient outcome, therapueitc objective, clinical trial end point, or public health measure? J Clin Oncol. 2012;30:1750–4. doi: 10.1200/JCO.2011.38.6359. [DOI] [PubMed] [Google Scholar]
- 16.Scher HI, Halabi S, Tannock I, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26:1148–59. doi: 10.1200/JCO.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- 18.Goodsaid FM, Mendrick DL. Translational medicine and the value of biomarker qualification. Sci Transl Med. 2010;2:47p, s44. doi: 10.1126/scitranslmed.3001040. [DOI] [PubMed] [Google Scholar]
- 19.Khleif SN, Doroshow JH, Hait WN. AACR-FDA-NCI Cancer Biomarkers Collaborative consensus report: advancing the use of biomarkers in cancer drug development. Clin Cancer Res. 2010;16:3299–318. doi: 10.1158/1078-0432.CCR-10-0880. [DOI] [PubMed] [Google Scholar]
- 20.Lin LI. A concordance correlation coefficient to evaluate reproducibility. Biometrics. 1989;45:255–68. [PubMed] [Google Scholar]
- 21.Cummings J, Raynaud F, Jones L, Sugar R, Dive C. Fit-for-purpose biomarker method validation for application in clinical trials of anticancer drugs. Br J Cancer. 2010;103:1313–7. doi: 10.1038/sj.bjc.6605910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evidence for clinical utility of molecular diagnostics in oncology: a workshop. Institute of Medicine; Washington, DC: May 24, 2012. [Google Scholar]
- 23.Buyse M, Sargent DJ, Grothey A, Matheson A, de Gramont A. Biomarkers and surrogate end points--the challenge of statistical validation. Nat Rev Clin Oncol. 2010;7:309–17. doi: 10.1038/nrclinonc.2010.43. [DOI] [PubMed] [Google Scholar]
- 24.U.S. Food and Drug Administration. [Accessed January 30, 2013];Draft guidance for industry: Qualification process for drug development tools. Issued October 2010. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM230597.pdf.
- 25.U.S. Food and Drug Administration. [Accessed January 30, 2013];Draft guidance for industry and FDA staff: In vitro companion diagnostic devices. Issued July 14, 2011. http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm262292.htm.
- 26.Schilsky RL, Doroshow JH, Leblanc M, Conley BA. Development and use of integral assays in clinical trials. Clin Cancer Res. 2012;18:1540–6. doi: 10.1158/1078-0432.CCR-11-2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smaletz O, Scher HI, Small EJ, et al. Nomogram for overall survival of patients with progressive metastatic prostate cancer after castration. J Clin Oncol. 2002;20:3972–82. doi: 10.1200/JCO.2002.11.021. [DOI] [PubMed] [Google Scholar]
- 28.Armstrong AJ, Garrett-Mayer ES, Yang YC, et al. A contemporary prognostic nomogram for men with hormone-refractory metastatic prostate cancer: a TAX327 study analysis. Clin Cancer Res. 2007;13:6396–403. doi: 10.1158/1078-0432.CCR-07-1036. [DOI] [PubMed] [Google Scholar]
- 29.Halabi S, Small EJ, Kantoff PW, et al. Prognostic model for predicting survival in men with hormone-refractory metastatic prostate cancer. J Clin Oncol. 2003;21:1232–7. doi: 10.1200/JCO.2003.06.100. [DOI] [PubMed] [Google Scholar]
- 30.Halabi S, Vogelzang NJ, Ou SS, et al. Progression-free survival as a predictor of overall survival in men with castrate-resistant prostate cancer. J Clin Oncol. 2009;27:2766–71. doi: 10.1200/JCO.2008.18.9159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scher HI, Warren M, Heller G. The association between measures of progression and survival in castrate-metastatic prostate cancer. Clin Cancer Res. 2007;13:1488–92. doi: 10.1158/1078-0432.CCR-06-1885. [DOI] [PubMed] [Google Scholar]
- 32.Ong FS, Das K, Wang J, et al. Personalized medicince and pharmacogenetic biomarkers: progress in molecular oncology testing. Expert Rev Mol Diagn. 2012;12:593–602. doi: 10.1586/erm.12.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Attard G, Reid AH, A’Hern R, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27:3742–8. doi: 10.1200/JCO.2008.20.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 35.Baca SC, Garraway LA. The genomic landscape of prostate cancer. Front Endocrinol (Lausanne) 2012;3:69. doi: 10.3389/fendo.2012.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prensner JR, Rubin MA, Wei JT, et al. Beyond PSA: the next generation of prostate cancer biomarkers. Sci Transl Med. 2012;4:127rv3. doi: 10.1126/scitranslmed.3003180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carver BS, Chapinski C, Wongvipat J, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–86. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scher HI, Morris MJ, Kelly WK, et al. Prostate cancer clinical trial end points: “RECIST”ing a step backwards. Clin Cancer Res. 2005;11:5223–32. doi: 10.1158/1078-0432.CCR-05-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gignac GA, Morris MJ, Heller G, et al. Assessing outcomes in prostate cancer clinical trials: a twenty-first century tower of Babel. Cancer. 2008;113:966–74. doi: 10.1002/cncr.23719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tannock IF, Osoba D, Stockler MR, et al. Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: a Canadian randomized trial with palliative end points. J Clin Oncol. 1996;14:1756–64. doi: 10.1200/JCO.1996.14.6.1756. [DOI] [PubMed] [Google Scholar]
- 42.Berthold DR, Pond GR, Roessner M, et al. Treatment of hormone-refractory prostate cancer with docetaxel or mitoxantrone: relationships between prostate-specific antigen, pain, and quality of life response and survival in the TAX-327 study. Clin Cancer Res. 2008;14:2763–7. doi: 10.1158/1078-0432.CCR-07-0944. [DOI] [PubMed] [Google Scholar]
- 43.Sartor O, Reid RH, Hoskin PJ, et al. Samarium-153-Lexidronam complex for treatment of painful bone metastases in hormone-refractory prostate cancer. Urology. 2004;63:940–5. doi: 10.1016/j.urology.2004.01.034. [DOI] [PubMed] [Google Scholar]
- 44.Logothetis CJ, Basch E, Molina A, et al. Effect of abiraterone acetate and prednisone compared with placebo and prednisone on pain control and skeletal-related events in patients with metastatic castration-resistant prostate cancer: exploratory analysis of data from the COU-AA-301 randomised trial. Lancet Oncol. 2012;13:1210–7. doi: 10.1016/S1470-2045(12)70473-4. [DOI] [PubMed] [Google Scholar]
- 45.Atkinson TM, Mendoza TR, Sit L, et al. The Brief Pain Inventory and its “pain at its worst in the last 24 hours” item: clinical trial endpoint considerations. Pain Med. 2010;11:337–46. doi: 10.1111/j.1526-4637.2009.00774.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Osoba D, Tannock IF, Ernst DS, Neville AJ. Health-related qualify of life in men with metastatic prostate cancer treated with prednisone alone or mitoxantrone and prednisone. J Clin Oncol. 1999;17:1654–63. doi: 10.1200/JCO.1999.17.6.1654. [DOI] [PubMed] [Google Scholar]
- 47.Sternberg CN, Petrylak DP, Sartor O, et al. Multinational, double-blind, phase III study of prednisone and either satraplatin or placebo in patients with castrate-refractory prostate cancer progressing after prior chemotherapy: the SPARC trial. J Clin Oncol. 2009;27:5431–8. doi: 10.1200/JCO.2008.20.1228. [DOI] [PubMed] [Google Scholar]
- 48.Smith DC, Smith MR, Sweeney C, et al. Cabozantinib in patients with advanced prostate cancer: results of a phase II randomized discontinuation trial. J Clin Oncol. 2013;31:412–9. doi: 10.1200/JCO.2012.45.0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.US National Library of Medicine. ClinicalTrials.gov [online] 2013 http://clinicaltrials.gov/show/NCT01522443.
- 50.US National Library of Medicine. ClinicalTrials.gov [online] 2012 http://clinicaltrials.gov/show/NCT01083615.
- 51.Lakhal L, Rivest L-P, Beaudoin D. IPCW estimator for Kendall’s Tau under bivariate censoring. Int J Biostat. 2009;5:1–20. [Google Scholar]
- 52.Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med. 1989;8:431–40. doi: 10.1002/sim.4780080407. [DOI] [PubMed] [Google Scholar]
- 53.Fox JJ, Morris MJ, Larson SM, Schöder H, Scher HI. Developing imaging strategies for castration resistant prostate cancer. Acta Oncol. 2011;50 (Suppl 1):39–48. doi: 10.3109/0284186X.2011.572914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Logothetis CJ, Samuels ML, von Eschenbach AC, et al. Doxorubicin, mitomycin-C, and 5-fluorouracil (DMF) in the treatment of metastatic hormonal refractory adenocarcinoma of the prostate, with a note on the staging of metastatic prostate cancer. J Clin Oncol. 1983;1:368–79. doi: 10.1200/JCO.1983.1.6.368. [DOI] [PubMed] [Google Scholar]
- 55.Soloway MS, Hardeman SW, Hickey D, et al. Stratification of patients with metastatic prostate cancer based on extent of disease on initial bone scan. Cancer. 1988;61:195–202. doi: 10.1002/1097-0142(19880101)61:1<195::aid-cncr2820610133>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 56.Rigaud J, Tiguert R, Le Normand L, et al. Prognostic value of bone scan in patients with metastatic prostate cancer treated initially with androgen deprivation therapy. J Urol. 2002;168:1423–6. doi: 10.1016/S0022-5347(05)64465-5. [DOI] [PubMed] [Google Scholar]
- 57.Morris MJ, Farrelly JS, Fox JJ, et al. The Prostate Cancer Clinical Trials Consortium (PCCTC) bone scan data capture tool for clinical trials using Prostate Cancer Working Group 2 (PCWG2) criteria: Effect on data accuracy and workload. ASCO Genitourinary Cancers Symposium; February 17–19, 2011; Orlando, FL. p. Abstract 121. [Google Scholar]
- 58.Ryan CJ, Morris M, Molina A, et al. Association of radiographic progression-free survival (rPFS) adapted from Prostate Cancer Working Group 2 (PCWG2) consensus criteria (APCWG2) with overall survival (OS) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): result. European Society for Medical Oncology Congress; September 28 – October 2, 2012; Vienna, Austria. p. Abstract 8940. [Google Scholar]
- 59. [Accessed January 8, 2013];FDA expands Zytiga’s use for late-stage prostate cancer: drug can now be used before treatment with chemotherapy. [press release December 10, 2012]. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm331492.htm.
- 60.Imbriaco M, Larson SM, Yeung HW, et al. A new parameter for measuring metastatic bone involvement by prostate cancer: the bone scan index. Clin Cancer Res. 1998;4:1765–72. [PubMed] [Google Scholar]
- 61.Dennis ER, Jia X, Mezheritskiy IS, et al. Bone scan index: a quantitative treatment response biomarker for castration-resistant metastatic prostate cancer. J Clin Oncol. 2012;30:519–24. doi: 10.1200/JCO.2011.36.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ulmert D, Kaboteh R, Fox JJ, et al. A novel automated platform for quantifying the extent of skeletal tumour involvement in prostate cancer patients using the Bone Scan Index. Eur Urol. 2012;62:78–84. doi: 10.1016/j.eururo.2012.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown MS, Chu GH, Kim HJ, et al. Computer-aided quantitative bone scan assessment of prostate cancer treatment response. Nucl Med Commun. 2012;33:384–94. doi: 10.1097/MNM.0b013e3283503ebf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.US National Library of Medicine. ClinicalTrials.gov [online] 2013 http://clinicaltrials.gov/show/NCT01605227.
- 65.Yeh SD, Imbriaco M, Larson SM, et al. Detection of bony metastases of androgen-independent prostate cancer by PET-FDG. Nucl Med Biol. 1996;23:693–7. doi: 10.1016/0969-8051(96)00044-3. [DOI] [PubMed] [Google Scholar]
- 66.Meirelles GS, Schoder H, Ravizzini GC, et al. Prognostic value of baseline [18F] fluorodeoxyglucose positron emission tomography and 99mTc-MDP bone scan in progressing metastatic prostate cancer. Clin Cancer Res. 2010;16:6093–9. doi: 10.1158/1078-0432.CCR-10-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morris MJ, Akhurst T, Osman I, et al. Fluorinated deoxyglucose positron emission tomography imaging in progressive metastatic prostate cancer. Urology. 2002;59:913–8. doi: 10.1016/s0090-4295(02)01509-1. [DOI] [PubMed] [Google Scholar]
- 68.US National Library of Medicine. ClinicalTrials.gov [online] 2012 http://clinicaltrials.gov/show/NCT01516866.
- 69.Wahl RL, Jacene H, Kasamon Y, et al. From RECIST to PERCIST: Evolving Considerations for PET response criteria in solid tumors. J Nucl Med. 2009;50 (Suppl 1):122S–50S. doi: 10.2967/jnumed.108.057307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morris MJ, Fox JJ, Dennis ER, et al. Pretreatment fluorodeoxyglucose (FDG) PET and survival for castration-resistant metastatic prostate cancer (CRMPC). ASCO Genitourinary Cancers Symposium; March 5–7, 2010; San Francisco, CA. p. Abstract 108. [Google Scholar]
- 71.Morris MJ, Akhurst T, Larson SM, et al. Fluorodeoxyglucose positron emission tomography as an outcome measure for castrate metastatic prostate cancer treated with antimicrotubule chemotherapy. Clin Cancer Res. 2005;11:3210–6. doi: 10.1158/1078-0432.CCR-04-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fox JJ, Autran-Blanc E, Morris MJ, et al. Practical approach for comparative analysis of multilesion molecular imaging using a semiautomated program for PET/CT. J Nucl Med. 2011;52:1727–32. doi: 10.2967/jnumed.111.089326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pantel K, Alix-Panabieres C. Circulating tumour cells in cancer patients: challenges and perspectives. Trends Mol Med. 2010;16:398–406. doi: 10.1016/j.molmed.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 74.Yu M, Stott S, Toner M, et al. Circulating tumor cells: approaches to isolation and characterization. J Cell Biol. 2011;192:373–82. doi: 10.1083/jcb.201010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Danila DC, Pantel K, Fleisher M, et al. Circulating tumors cells as biomarkers: progress toward biomarker qualification. Cancer J. 2011;17:438–50. doi: 10.1097/PPO.0b013e31823e69ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gleghorn JP, Pratt ED, Denning D, et al. Capture of circulating tumor cells from whole blood of prostate cancer patients using geometrically enhanced differential immunocapture (GEDI) and a prostate-specific antibody. Lab Chip. 2010;10:27–9. doi: 10.1039/b917959c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tan SJ, Lakshmi RL, Chen P, et al. Versatile label free biochip for the detection of circulating tumor cells from peripheral blood in cancer patients. Biosens Bioelectron. 2010;26:1701–5. doi: 10.1016/j.bios.2010.07.054. [DOI] [PubMed] [Google Scholar]
- 78.Tan SJ, Yobas L, Lee GYH, et al. Microdevice for isolating viable circulating tumor cells. 2008 International Conference on Biocomputation, Bioinformatics, and Biomedical Technologies; June 29 - July 5, 2008; Singapore. pp. 109–13. biotechno. [Google Scholar]
- 79.Alix-Panabieres C, Rebillard X, Brouillet JP, et al. Detection of circulating prostate-specific antigen-secreting cells in prostate cancer patients. Clin Chem. 2005;51:1538–41. doi: 10.1373/clinchem.2005.049445. [DOI] [PubMed] [Google Scholar]
- 80.Katz RL, He W, Khanna A, et al. Genetically abnormal circulating cells in lung cancer patients: an antigen-independent fluorescence in situ hybridization-based case-control study. Clin Cancer Res. 2010;16:3976–87. doi: 10.1158/1078-0432.CCR-09-3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marrinucci D, Bethel K, Luttgen M, et al. Circulating tumor cells from well-differentiated lung adenocarcinoma retain cytomorphologic features of primary tumor type. Arch Pathol Lab Med. 2009;133:1468–71. doi: 10.1043/1543-2165-133.9.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pantel K, Brakenhoff RH, Brandt B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat Rev Cancer. 2008;8:329–40. doi: 10.1038/nrc2375. [DOI] [PubMed] [Google Scholar]
- 83.Willipinski-Stapelfeldt B, Riethdorf S, Assmann V, et al. Changes in cytoskeletal protein composition indicative of an epithelial-mesenchymal transition in human micrometastatic and primary breast carcinoma cells. Clin Cancer Res. 2005;11:8006–14. doi: 10.1158/1078-0432.CCR-05-0632. [DOI] [PubMed] [Google Scholar]
- 84.FDA Clearance Document for Veridex LLC. CellSearch(TM) Circulating Tumor Cell Kit. [Accessed February 1, 2013];Premarket notification - expanded indications for use - metastatic prostate cancer. 2008 Feb 26; http://www.accessdata.fda.gov/cdrh_docs/pdf7/K073338.pdf.
- 85.Shaffer DR, Leversha MA, Danila DC, et al. Circulating tumor cell analysis in patients with progressive castration-resistant prostate cancer. Clin Cancer Res. 2007;13:2023–9. doi: 10.1158/1078-0432.CCR-06-2701. [DOI] [PubMed] [Google Scholar]
- 86.Cristofanilli M, Budd GT, Ellis MJ, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004;351:781–91. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- 87.Cohen SJ, Punt CJ, Iannotti N. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:3213–21. doi: 10.1200/JCO.2007.15.8923. [DOI] [PubMed] [Google Scholar]
- 88.de Bono JS, Scher HI, Montgomery RB, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res. 2008;14:6302–9. doi: 10.1158/1078-0432.CCR-08-0872. [DOI] [PubMed] [Google Scholar]
- 89.Danila DC, Heller G, Gignac GA, et al. Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer. Clin Cancer Res. 2007;13:7053–8. doi: 10.1158/1078-0432.CCR-07-1506. [DOI] [PubMed] [Google Scholar]
- 90.Reid AH, Attard G, Danila DC, et al. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol. 2010;28:1489–95. doi: 10.1200/JCO.2009.24.6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Olmos D, Baird RD, Yap TA, et al. Baseline circulating tumor cell counts significantly enhance a prognostic score for patients participating in phase I oncology trials. Clin Cancer Res. 2011;17:5188–96. doi: 10.1158/1078-0432.CCR-10-3019. [DOI] [PubMed] [Google Scholar]
- 92.Scher HI, Jia X, de Bono JS, et al. Circulating tumour cells as prognostic markers in progressive, castration-resistant prostate cancer: a reanalysis of IMMC38 trial data. Lancet Oncol. 2009;10:233–9. doi: 10.1016/S1470-2045(08)70340-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Goodman OB, Jr, Fink LM, Symanowski JT, et al. Circulating tumor cells in patients with castration-resistant prostate cancer baseline values and correlation with prognostic factors. Cancer Epidemiol Biomarkers Prev. 2009;18:1904–13. doi: 10.1158/1055-9965.EPI-08-1173. [DOI] [PubMed] [Google Scholar]
- 94.Olmos D, Arkenau HT, Ang JE, et al. Circulating tumour cell (CTC) counts as intermediate end points in castration-resistant prostate cancer (CRPC): a single-centre experience. Ann Oncol. 2009;20:27–33. doi: 10.1093/annonc/mdn544. [DOI] [PubMed] [Google Scholar]
- 95.Danila DC, Morris MJ, de Bono JS, et al. Phase II multicenter study of abiraterone acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. J Clin Oncol. 2010;28:1496–501. doi: 10.1200/JCO.2009.25.9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Scher HI, Heller G, Molina A, et al. Evaluation of circulating tumor cell (CTC) enumeration as an efficacy response biomarker of overall survival (OS) in metastatic castration-resistant prostate cancer (mCRPC): Planned final analysis (FA) of COU-AA-301, a randomized double-blind, placebo-controlled phase III study of abiraterone acetate (AA) plus low-dose prednisone (P) post docetaxel. ASCO Annual Meeting; June 3–7, 2011; Chicago, IL. p. Abstract LBA4517. [Google Scholar]
- 97.Buyse M, Molenberghs G. Criteria for the validation of surrogate endpoints in randomized experiments. Biometrics. 1998;54:1014–29. [PubMed] [Google Scholar]
- 98.Burzykowski T, Molenberghs G, Buyse M, et al. Validation of surrogate endpoints in multiple randomized clinical trials with failure-time endpoints. Appl Stat. 2001;50:405–22. [Google Scholar]
- 99.Shaffer DR, Leversha MA, Danila DC, et al. Circulating tumor cell analysis in patients with progressive castration-resistant prostate cancer. Clin Cancer Res. 2007;13:2023–29. doi: 10.1158/1078-0432.CCR-06-2701. [DOI] [PubMed] [Google Scholar]