

Abstract
Bcl2-associated athanogene 3 (BAG3), a member of the BAG family of co-chaperones, plays a critical role in regulating apoptosis, development, cell motility, autophagy, and tumor metastasis and in mediating cell adaptive responses to stressful stimuli. BAG3 carries a BAG domain, a WW domain, and a proline-rich repeat (PXXP), all of which mediate binding to different partners. To elucidate BAG3's interaction network at the molecular level, we employed quantitative immunoprecipitation combined with knockdown and human proteome microarrays to comprehensively profile the BAG3 interactome in humans. We identified a total of 382 BAG3-interacting proteins with diverse functions, including transferase activity, nucleic acid binding, transcription factors, proteases, and chaperones, suggesting that BAG3 is a critical regulator of diverse cellular functions. In addition, we characterized interactions between BAG3 and some of its newly identified partners in greater detail. In particular, bioinformatic analysis revealed that the BAG3 interactome is strongly enriched in proteins functioning within the proteasome-ubiquitination process and that compose the proteasome complex itself, suggesting that a critical biological function of BAG3 is associated with the proteasome. Functional studies demonstrated that BAG3 indeed interacts with the proteasome and modulates its activity, sustaining cell survival and underlying resistance to therapy through the down-modulation of apoptosis. Taken as a whole, this study expands our knowledge of the BAG3 interactome, provides a valuable resource for understanding how BAG3 affects different cellular functions, and demonstrates that biologically relevant data can be harvested using this kind of integrated approach.
Bcl2-associated athanogene 3 (BAG3)1 is a member of the human BAG family of molecular co-chaperone proteins (1). BAG proteins share a common C-terminal BAG domain that mediates interactions with the ATPase domain of heat shock protein (Hsp) 70 (1), but they have divergent N termini that confer different properties and functions to each member (2). For example, in addition to the BAG domain, BAG3 contains a proline-rich (PXXP) region and an N-terminal WW domain (1) that mediate binding to partners other than Hsp70. Moreover, in addition to the BAG domain, BAG3 possesses one of the longest C-terminal sequences among the known BAG proteins. These multifaceted structural characteristics underlie the ability of BAG3 to modulate biological processes such as apoptosis, development, cytoskeleton organization, and autophagy and to mediate cell adaptive responses to stressful stimuli (3).
As a chaperone, BAG3 seems to influence cell survival by interacting with different molecular partners and thus activating multiple pathways. For example, BAG3 can potently suppress cell apoptosis via its interaction with Hsp70, a chaperone protein able to modulate apoptosis by interfering with cytochrome c release, apoptosome formation, and other events in the death process (4, 5). In addition, BAG3 binds PLC-gamma (6) and Bcl-2, with which it synergizes to prevent cell death (7). Other cellular signaling molecules reported to be regulated by BAG3 include Raf-1, CDK-4, EGFR, MMP-2, Hsps B8 and B6, and focal adhesion kinase (3). Notably, recent reports have shown additional functions of this chaperone in the regulation of autophagy (8, 9), virus replication (10, 11), and NF-κB activity (12). Moreover, in a previous study, we demonstrated that the up-regulation of BAG3 may be part of the cellular response that SARS-CoV relies on for replication (13).
Despite the multifunctional roles of this stress- and survival-related protein, little is known about the molecular mechanisms underlying these functions. Given that most proteins are part of one or more protein complexes, knowledge of protein–protein interactions is essential in order for one to understand the nature of protein-mediated biological processes. Some of the interacting partners of BAG3 are known, but the diverse functions identified for BAG3 suggest that this protein might have an even greater assortment of binding partners. Thus, the identification and characterization of the entire set of BAG3 interaction partners is critical for a comprehensive understanding of its biological functions. To this end, we have performed a global analysis of BAG3 interaction partners using the quantitative immunoprecipitation combined with knockdown (QUICK) and proteome microarray methods.
The QUICK method, recently developed by Mann and colleagues (14), is a relatively new method for identifying interactions between proteins at endogenous levels under physiological conditions via a combination of stable isotope labeling with amino acids in cell culture (SILAC), RNA interference (RNAi)-induced knockdown, co-immunoprecipitation, and quantitative MS. This highly sensitive and accurate approach for protein–protein interaction analysis has been applied to identify the interaction partners of β-catenin and Cbl (14), 14-3–3ζ (15), Lrrk2 (16), and Stat3 (17).
Proteome microarrays, also known as protein chips, are constructed by spotting hundreds to thousands of individually purified proteins at a high density on a solid surface (18, 19). A major breakthrough in this technology came from a study by Zhu et al. in which a proteome-wide protein array consisting of 5,800 unique yeast proteins was constructed and used to identify calmodulin- and phospholipid-binding proteins (20). Since then, proteome microarrays have been employed to probe various types of protein biochemical properties, enabling high-throughput screening of thousands of proteins in a single experiment (21–23). Recently, a human proteome microarray comprising 17,000 purified, full-length proteins was constructed and used to identify highly specific monoclonal antibodies (24). Proteome microarrays provide information about direct biochemical and physical interactions among biomolecules and have been successfully used for the identification of protein interaction partners (20, 25, 26). They have become one of the most powerful multiplexed detection platforms for addressing diverse biological questions in a high-throughput fashion.
Although both QUICK and proteome microarrays have been successfully used for the identification of protein–protein interactions, each approach detects and reports these interactions in a distinct manner. For example, the proteome microarrays detect binary interactions, whereas the QUICK method purifies and identifies protein complexes. The QUICK method uses untagged endogenous bait proteins to pull down prey proteins followed by mass spectrometric detection of proteins, whereas the proteome microarray method relies on separate protein complementation schemes to ultimately report on whether a protein pair is interacting. In addition, the QUICK method identifies interactions at approximately physiological cellular protein concentrations, whereas the concentrations of the interacting partners in the proteome microarray screens are not necessarily comparable to those found in the native environment. However, the protein concentrations used are less diverse on the proteome microarray, and this may facilitate the identification of BAG3 interacting proteins expressed at low levels under physiological conditions. The QUICK method is presently not a high-throughput method, whereas the proteome microarray is an outstandingly flexible tool for high-throughput studies. Therefore, although each approach has distinct advantages, disadvantages, and limitations, the combination of these complementary tools provides a powerful methodology for proteomic investigations.
In this study, we employed both QUICK and proteome microarray methods to decipher the human BAG3 interactome. Our results confirm that BAG3 indeed interacts with many more proteins than previously thought and suggest a number of different functions for BAG3, which can now be further explored. This study also demonstrates that the complementary information obtained by combining the QUICK method and proteome microarrays can provide a comprehensive portrait of the protein–protein interactions with any protein of interest.
EXPERIMENTAL PROCEDURES
Cell Culture
Cells from human cervical cancer cell line HeLa, lung cancer cell line A549, and multiple myeloma cell line U266 were purchased from the American Type Culture Collection (Rockville, MD). HeLa and A549 cells were routinely maintained in Dulbecco modified Eagle's medium (DMEM) (Sigma-Aldrich, Saint Louis, MO) supplemented with 1% penicillin/streptomycin and 10% fetal bovine serum (FBS) (Sigma-Aldrich). All cells were grown at 37 °C in a humidified incubator with an atmosphere of 5% CO2.
QUICK Identification of BAG3 Interacting Proteins
SILAC Labeling and RNA Interference
The BAG3 interaction proteins in HeLa cells were identified using the QUICK approach. SILAC labeling was performed essentially as described elsewhere (15). A SILAC Protein Quantitation Kit (Pierce Biotechnology, Rockford, IL) was used according to the manufacturer's instructions. In brief, HeLa cells were grown in SILAC DMEM (Pierce) containing 10% v/v dialyzed FBS and 0.1 mg/ml heavy [13C6] or light [12C6] l-lysine. To ensure full incorporation of heavy- and light-labeled amino acids, cells were grown for at least six cell doublings prior to treatment. For BAG3 RNA interference, small interfering RNAs (siRNAs) specific for BAG3 (siBAG3: 5′-GCAUGCCAGAAACCACUCA-3′) or negative control siRNA (siNC: 5′-UUCUCCGAACGUGUCACGUTT-3′) were purchased from Shanghai GenePharma (Shanghai, China). Heavy [13C6]-labeled HeLa cells were transfected with siBAG3 using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. A control experiment was performed by transfecting light [12C6]-labeled HeLa cells with negative control siNC. A549 cells were transfected using the same protocol. Changes in BAG3 protein levels were determined via Western blotting.
In Vivo Formaldehyde Cross-linking
72 h post-transfection, cells were harvested, washed with PBS, and incubated in 1.0% w/v paraformaldehyde in PBS for 10 min at 37 °C. To stop the cross-linking reaction, 1.25 m glycine was added to a final concentration of 125 mm for 5 min at room temperature. Cells were harvested, washed three times with ice-cold washing buffer (10 μm Tris-HCl, 250 μm sucrose, pH 7.0), and transferred to a clean 1.5-ml Eppendorf tube. Cells were lysed with RIPA lysis buffer, and cellular debris was removed via centrifugation for 30 min at 13,200 × g at 4 °C. Supernatants were collected, and protein concentrations were measured in duplicate using a BCA protein assay kit (Pierce).
Antibody Coupling and Immunoprecipitation
In order to prevent interference from an excess of antibodies with mass spectrometric detection, the BAG3 antibody was coupled to Protein A/G Sepharose (GE Healthcare). Protein A/G Sepharose was mixed with antibody in 0.1 m borate buffer, pH 8.2, for 30 min at room temperature with gentle shaking, after which the Sepharose beads were washed with excess borate buffer. The Sepharose was washed with 0.2 m triethanolamine, pH 8.2, and then resuspended in 20 volumes of dimethyl pimelimidate dihydrochloride (Pierce) freshly made up in 0.2 m triethanolamine, readjusting the pH to 8.2. The mixture was agitated gently at room temperature for 1 h, and the reaction was stopped by centrifuging the beads (500 × g for 1 min) and resuspending in an equal volume of ethanolamine (pH 8.2) of the same molarity as the dimethyl pimelimidate dihydrochloride. After 5 min, the cross-linked beads were centrifuged and washed three times with lysis buffer. Prior to immunoprecipitation, 2 mg of protein from light siNC transfected HeLa cells was mixed with 2 mg of protein from heavy siBAG3 transfected HeLa cells and precleared with 0.2 ml Protein A/G Sepharose for 2 h. Protein A-Sepharose beads coupled with antibodies were mixed with precleared mixtures and rotated for 18 h at 4 °C. The beads were washed twice with 2 ml of lysis buffer containing 0.1% Triton X-100 and twice with 2 ml of lysis buffer without detergent. After complete removal of the supernatant with a microliter syringe, immunoprecipitated proteins were eluted with 3 × 200 μl of 5% acetic acid pH 3 and dried in a speed-vacuum. Laemlli sample buffer, pH 7.4 (10 mm Tris, 10% v/v glycerol, 2% w/v SDS, 5 mm EDTA, 0.02% bromphenol blue, and 6% β-mercaptoethanol), was added and boiled for 20 min at 95 °C. Proteins were then separated on a 10% SDS-PAGE gel and visualized with Coomassie Blue staining.
Protein Separation and In-gel Digestion
Protein bands were excised from the SDS-PAGE gel and cut into 15 sections for in-gel tryptic digestion. The excised sections were chopped into small particles, washed in water, and completely destained using 100 mm ammonium bicarbonate in 50% acetonitrile. A reduction step was performed by adding 100 μl of 10 mm DTT at 37 °C for 1 h. The proteins were alkylated by the addition of 100 μl of 50 mm iodoacetamide and allowed to react in the dark at room temperature for 1 h. Gel sections were washed first in water and then in acetonitrile, and finally they were dried using a SpeedVac (Thermo Fisher Scientific, Waltham, MA) for 30 min. Digestion was carried out using 20 μg/ml sequencing-grade modified trypsin (Promega) in 50 mm ammonium bicarbonate. Sufficient trypsin solution was added to swell the gel pieces, which were kept at 4 °C for 45 min and then incubated at 37 °C overnight. The supernatants were transferred into a 200-μl microcentrifuge tube, and the gels were extracted once with extraction buffer (67% acetonitrile containing 2.5% trifluoroacetic acid). The peptide extract and the supernatant of the gel slices were combined and then completely dried in the SpeedVac.
Mass Spectrometry and Protein Identification and Quantification
Dried peptides were reconstituted in 5% acetonitrile/0.1% formic acid. LC-MS/MS analyses were performed on an LTQ-Orbitrap mass spectrometer (Thermo Fisher) equipped with an Agilent 1100 high-performance liquid chromatography (HPLC) pump (Agilent Technologies) and a Famos autosampler (LC Packings). The peptide mixtures were loaded onto a C18 column (100 μm inner diameter, 10 cm long, 5 μm resin, from Michrom Bioresources, Auburn, CA) using an autosampler. Peptides were eluted with a 0%–35% gradient (Buffer A, 0.1% formic acid and 5% acetonitrile; Buffer B, 0.1% formic acid and 95% acetonitrile) over 90 min and detected online in an LTQ-Orbitrap using a data-dependent method (27). All samples were analyzed in triplicate. The general mass spectrometric conditions were as follows: spray voltage, 1.80 kV; no sheath and auxiliary gas flow; ion transfer tube temperature, 200 °C. Ion selection thresholds were 1,000 counts for MS2 and 500 counts for MS3. An activation q = 0.25 and an activation time of 30 ms were applied in MS2 acquisitions. The mass spectrometers were operated in positive ion mode, employing a data-dependent automatic switch between MS and MS2 acquisition modes. For each cycle, one full MS scan of the five most intense ions in the Orbitrap at 1 × 106 AGC target was followed by five MS2 scans in the LTQ at 5,000 AGC target. Selected ions were excluded from further selection for 90 s. The maximum ion accumulation time was 500 ms for full MS scans and 100 ms for MS2 scans. All MS/MS spectra were collected using normalized collision energy (a setting of 35%), an isolation window of 3 m/z, and one micro-scan. The application of mass spectrometer scan functions and HPLC solvent gradients was controlled by an XCalibur data system (Thermo Fisher Scientific, Waltham, MA). The acquired raw files were processed using BioWorks 3.3.1 (Thermo Finnigan, San Jose, CA), and the MS/MS spectra were searched using the SEQUEST search engine (version 28.13, Thermo Fisher Scientific, Waltham, MA) against a catenated database combining the International Protein Index human database (version 3.63, 84,118 sequences), the reversed sequences of all sequences in the International Protein Index human database, and 262 commonly observed contaminants. The following search criteria were employed: full tryptic specificity was required; two missed cleavages were allowed; parent ion mass tolerance = 20 ppm; mass tolerance for fragment ion = 0.5 Da; minimum peptide length of six residues; Cys (+57.0215 Da, carbamidomethylation) set as a fixed modification; Met (+15.9949 Da, oxidation) and Lys (+6.0201 Da, SILAC heavy amino acid) considered as variable modifications. Peptides were accepted if their Xcorr was greater than or equal to 1.9, 2.5, 3.5, or 4.5 for +1, +2, +3, and +4 charged ions, respectively, and if their ΔCn was at least 0.1. The global false discovery rate for both peptides and proteins was set to 0.01. If the peptide charge and peptide sequence were the same, we selected only the peptide with the highest Xcorr value. The selected peptides were converted to dtaselect-filter.txt by the dtaselect-filter file format and quantified by Census (28). Only proteins with a minimum of two quantifiable peptides were included in our final dataset. The quantification was based on three independent QUICK experiments, and only proteins identified and quantified in at least two of three experiments were considered for further analysis. Census normalized protein ratios were utilized for the downstream analyses, and a p value of 0.05 was selected as the threshold for significant enrichment of BAG3 interaction proteins. Only those proteins with p ≤ 0.05 were considered as BAG3-interacting proteins.
Proteome Microarray-based BAG3 Interactome Screen
Human Proteome Microarray Construction
N-terminal GST-tagged human proteins (∼17,000) were purified from 6 ml yeast culture as described elsewhere (24). Briefly, yeast transformants were grown at 30 °C and inoculated into SC-ura/raffinose media. The cultures were grown overnight and induced at an A600 of 0.6 to 0.8 to a final concentration of 2% galactose. Cells were then harvested via centrifugation after 6 h. The cells were lysed via shaking with glass beads, and the supernatant was incubated with glutathione-Sepharose (GE Healthcare). The beads were washed and the proteins were eluted with 40 mm glutathione. The purified proteins were aliquoted into 384-well plates and printed onto Fullmoon slides (Fullmoon Biosystems, Sunnyvale, CA) using a 48-pin contact microarrayer (ChipWriter Pro, Bio-Rad, Hercules, CA). Each protein was spotted in duplicate.
Probe BAG3 on Human Proteome Microarray
Proteome microarrays were blocked with blocking buffer (1% BSA in 0.1% Tween 20, TBST) for 1 h at room temperature with gentle agitation. BAG3 protein (Abnova, Taipei, Taiwan) was diluted to 0.01 μg/μl in blocking buffer and incubated on the blocked proteome microarray at room temperature for 1 h. The microarrays were washed three times for 5 min each time with TBST, incubated with 1:1,000 diluted BAG3 antibody (Abcam Inc., Cambridge, MA) in blocking buffer for 1 h at room temperature, and then washed three more times (5-min washes) in TBST. The microarrays were further incubated with a Cy5 conjugated second antibody at 1:1,000 dilution (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) for 1 h at room temperature and underwent three more 5-min washes. The microarrays were spun dry at 1,500 rpm for 3 min and subjected to scanning with a Genepix 4200A (Axon Instruments, Sunnyvale, CA) in order for results to be visualized and recorded. A GenePix Pro 6.0 was used for data analysis.
Proteome Microarray Data Processing
The data were extracted by GenePix Pro 6.0 from the microarray images. To generate the candidate list of BAG3 binding proteins, the signal-to-noise ratios (SNR = F635 median/B635 median) were first calculated for all the spots. The SNR of a protein was averaged from two duplicated spots. The SNRs of all the proteins were calculated for microarrays with or without BAG3 as SNR(+) or SNR(−), respectively. Another index, the fold change, was defined as (Signal(+) − Background(+))/(Signal(−) − Background(−)) and was generated for all the proteins. To determine the final candidates, the cutoff was set as SNR(+) ≥ 2 and fold change ≥ 3.
Western Blotting Analysis
Protein extracts (50 μg) prepared with RIPA lysis buffer were resolved on a 10% SDS-PAGE gel and transferred to an Immobilon-P PVDF transfer membrane (Millipore, Bedford, MA) via electroblotting. After blocking with 5% nonfat milk, the membranes were probed with rabbit monoclonal anti-Bag1, goat polyclonal anti-Bag2, rabbit monoclonal anti-BAG3, rabbit polyclonal anti-Bag4, rabbit polyclonal anti-Bag5 (Abcam Inc., Cambridge, MA), mouse anti-Actin monoclonal (Santa Cruz Biotechnology, Santa Cruz, CA), and mouse anti-ubiquitin monoclonal antibodies (P4D1, Cell Signaling). Blots were then incubated with a streptavidin alkaline phosphatase-conjugate secondary antibody (Amersham Biosciences, Piscataway, NJ) for 1 h at room temperature. The signal was visualized with ECL detection reagent (GE Healthcare).
Bioinformatics Analysis of the BAG3 Interactome
The BAG3 interactome was classified based on the PANTHER (Protein Analysis through Evolutionary Relationships) system, which is a unique resource that classifies genes and proteins according to their functions (29). Functional diversity among proteins present in two datasets was analyzed as previously described (30). The relative distribution of all proteins from Homo sapiens, as annotated according to the PANTHER functional classes, is shown by the line labeled “Expected” in Fig. 3. It denotes the relative fraction (coverage) of all human proteins that belong to a given category. The deviations of the QUICK and proteome microarray datasets from this distribution are shown by different-colored bars and values in Fig. 3.
Fig. 3.
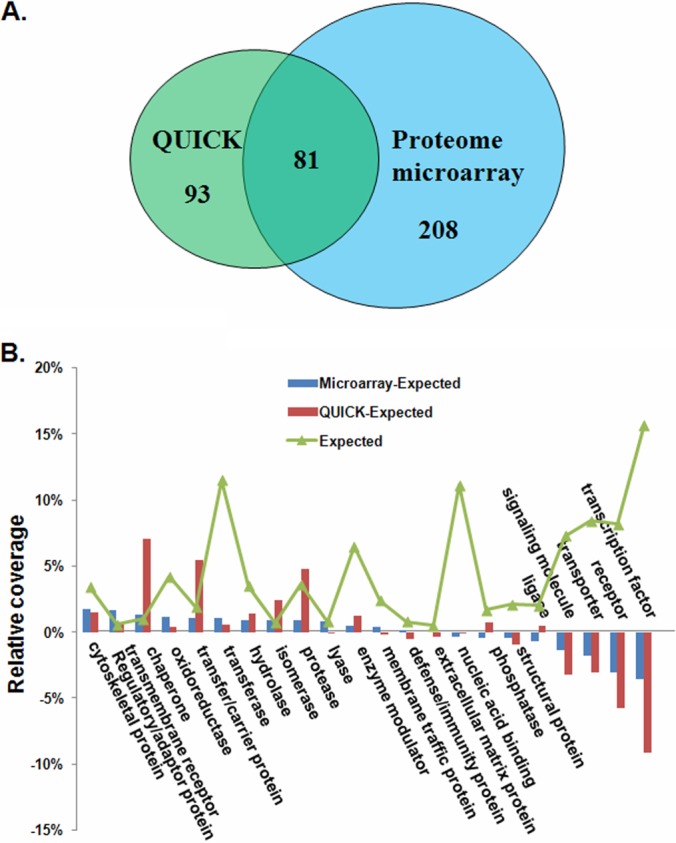
Comparison of BAG3 interacting proteins identified via the QUICK and proteome microarray methods. A, Venn diagrams showing the degree of overlap between the QUICK and proteome microarray data sets. B, functional diversity among proteins present in two datasets. The relative distribution of all proteins from Homo sapiens, annotated according to the PANTHER functional classes, is shown by the line labeled “Expected.” This denotes the relative fraction (coverage) of all human proteins that belong to a given category. Deviations of the QUICK and proteome microarray datasets from this distribution are shown by different colored bars; for example, proteins labeled “transcription factor” are relatively underrepresented in the two datasets.
To determine whether any types of proteins were overrepresented, enrichment analysis of Gene Ontology (GO) terms (31), Kyoto Encyclopedia of Genes and Genomes pathways (32), and Pfam domain families (33) was performed using the Web-accessible program DAVID 6.7 (34, 35). The default human proteome was used as the background list. The significance of the enrichments was statistically evaluated with a modified Fisher's exact test (EASE score), and a p value for each term was calculated by applying a Benjamini-Hochberg false discovery rate correction (34, 35). For GO term enrichments, the GO FAT annotation available in DAVID was used. GO FAT is a subset of the GO term set created by filtering out the broadest ontology terms so as to not overshadow more specific ones. The enrichment of GO biological process terms was also analyzed using Cytoscape and its Plugin BiNGO 2.3 (36), using the complete GO term set and a hypergeometric statistical test with Benjamini-Hochberg false discovery rate correction. The GO categories GO SLim Generic assignment, the distribution of cellular components, molecular functions, and biological processes of the BAG3 interactome were analyzed.
The BAG3 interactome was searched against the STRING database, version 8 (37), for protein–protein interactions. Only interactions between proteins belonging to the BAG3 interactome dataset were selected. STRING defines a metric called a “confidence score” to define interaction confidence; we fetched all interactions for our BAG3 interactome dataset that had a confidence score of ≥0.4 (medium confidence). A network of protein–protein interactions was generated in this manner and then visualized by Cytoscape v2.8.1. This BAG3 interaction network was further analyzed for densely connected regions using a graph theoretic clustering algorithm, “Molecular Complex Detection” (MCODE) (38). MCODE is part of the plug-in toolkit in Cytoscape (39). The top four high-ranking modules were selected for further analysis and rendering.
Co-immunoprecipitation and Western Blotting
Cell lysates were prepared as described above. Co-immunoprecipitation and Western blotting were performed essentially as described earlier (15). Antibodies used in this study and their sources were as follows: 14-3–3ζ, autophagy related 3 (ATG3), and proteasome subunit, beta type 1 (PSMB1) antibodies (Santa Cruz Biotechnology, Santa Cruz, CA); mitogen-activated protein kinase 7 (MAPK7) and heat shock transcription factor 1 (HSF1) antibodies (Cell Signaling, Danvers, MA); and BAG3 and Hsp70 antibodies (Abcam, Inc., Cambridge, MA).
Surface Plasmon Resonance Analysis
Surface plasmon resonance (SPR) experiments were carried out at 25 °C using a BIAcore 3000 instrument (BIAcore AB, Uppsala, Sweden). SPR buffers, regeneration solutions, and sensor chips were purchased from GE Healthcare. Chemicals were from Sigma (St. Louis, MO) unless otherwise stated. Purchased proteins were BAG3 (Abnova, Taipei, Taiwan), 14-3–3ζ, Hsp70, MAPK7, ATG3, PSMB1, HSF1, and ENO1 (OriGene Technologies, Rockville, MD). The surface of the CM5 chip was activated following a standard 1-ethyl-3(3-dimethylaminopropyl)-carbodiimide hydrochloride/N-hydroxyl-succinimide amine coupling Biacore protocol. For the immobilization of proteins, the BAG3 protein was injected in 100 μl of 10 mm sodium acetate (flow rate: 10 μl/min). After immobilization, each surface was blocked by 1 m ethanolamine at pH 8.5 for 6 min. For interaction measurements, recombinant human proteins were dissolved according to the manufacturer's instructions (OriGene Technologies, Rockville, MD). Various concentrations of proteins were injected over this interaction surface for 5 min in a buffer containing 150 mm NaCl, 5 mm CaCl2, 0.005% (v/v) Tween 20, and 20 mm Hepes (pH 7.4) with a flow rate of 5 μl/min. The sensor surface was regenerated between sample injections by washing with 6 m guanidine-HCl for 0.5 min at a flow rate of 50 μl/min. An equivalent volume of each protein sample (analyte) was injected over the chip surface with no protein immobilized to serve as a blank phase for background subtraction. ENO1, which was identified as a background protein in this study, was injected as a negative control using the same conditions. All experiments were repeated at least three times. The kinetic parameters (ka, association rate constant; kd, dissociation rate constant; KD = kd/ka = equilibrium dissociation constant) for each interaction were determined by globally fitting the experimental data using BIAevaluation software 4.1 (GE Healthcare).
Plasmid Constructs and Transient Transfection
A cDNA encoding human BAG3 was generated via PCR from a human brain cDNA library (Invitrogen, Carlsbad, CA) and subcloned into the EcoRI/XhoI sites of the eukaryotic expression plasmid pcDNA3 tagged with a Flag epitope. HeLa or A549 cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
Proteasome Activity Assay
Cytosolic extracts (without protease inhibitors) were used to measure proteasome activity using a 20S proteasome assay kit (Chemicon, Temecula, CA) following the manufacturer's instructions. The proteasome has three distinct peptidase activities: chymotryptic, tryptic, and caspase-like activities (40). The specific proteasome activities were measured as described previously, with a few modifications (41, 42). Treated or untreated HeLa and A549 cells were washed with PBS and pelleted via centrifugation. Cell pellets were lysed via repeated freeze-thaw cycles in 0.25 m sucrose, 25 mm Hepes (pH 7.8), 10 mm MgCl2, 1 mm EDTA, and 1 mm dithiothreitol. Lysates were centrifuged at 12,000 × g for 10 min, and supernatants were collected. Protein concentrations were measured in duplicate using a BCA protein assay kit (Pierce). Cell lysates were diluted with proteolysis assay buffer (50 mm Hepes (pH 7.8), 20 mm KCl, 5 mm MgCl2, and 1 mm dithiothreitol) to a final concentration of 0.5 mg/ml. The specific proteasome activity was measured via the addition of 170 μl proteolysis assay buffer and 30 μl Suc-LLVY-AMC (chymotryptic substrate; Biomol International, Plymouth Meeting, PA), Z-ARR-AMC (tryptic substrate; Calbiochem), or Z-LLE-AMC (caspase-like substrate; Biomol International) (1 mm stock solution in DMSO) to 100 μl of the diluted cell lysate. Assays of 26S proteasome complexes were carried out in the presence of 5 mm ATP, which stimulates 26S proteasome activity. 20S proteasome activity was measured in the presence of 0.02% SDS without ATP, which activates 20S proteasomes and partially inhibits 26S proteasomes. The mixture was incubated at 37 °C for 2 h. Following proteasome cleavage, the fluorescent group, 7-amido-4-methylcoumarin (AMC), was released and measured on a Perkin-Elmer fluorometer using an excitation wavelength of 380 nm and an emission wavelength of 460 nm. The data are plotted as mean ± standard error and are expressed as a percentage of the control, which was considered to be 100%. All samples were assayed in three independent experiments. The statistical significance of the data was tested using Student's t test for unpaired samples. Differences were considered to be significant if p ≤ 0.05 (*).
Detection of Apoptosis
Apoptosis was detected by using Annexin V/PI staining. In brief, cells were washed once in PBS and then stained with Annexin V-FITC and PI (2 mg/ml) according to the manufacturer's instructions. Samples were acquired on a FACScan flow cytometer (Becton Dickinson, San Jose, CA) and analyzed with WinMDI 2.8. All samples were assayed in three independent experiments. The statistical significance of the data was tested using Student's t test for unpaired samples. Differences were considered to be significant if p ≤ 0.05 (*).
RESULTS
QUICK Identification of BAG3 Interacting Proteins
We identified BAG3 interacting proteins first using the QUICK approach, a SILAC-based quantitative strategy to capture endogenous protein–protein interactions (Fig. 1A). A prerequisite for the QUICK assay is the availability of RNAi cells exhibiting reduced expression of the protein of interest. BAG3 gene silencing was achieved by transfecting HeLa cells with BAG3 siRNA (siBAG3). BAG3 knockdown efficiency was monitored by means of Western blotting using a BAG3-specific antibody. Levels of BAG3 proteins were markedly reduced in siBAG3-treated HeLa cells realative to HeLa cells transfected with negative control siRNA (siNC) (Fig. 1B). The depletion of intracellular levels of BAG3 showed no appreciable effect on the expression of the other BAG isoforms or on the internal control, actin (Fig. 1B). Proteins were immunoprecipitated, and peptides from pooled eluates were subjected to LC-MS/MS. As expected, the level of BAG3 was 4.89-fold lower in the knock-down treatment than in the control (supplemental Table S1). From among the quantified proteins, we include here only the results for those proteins that could be quantified in two or more experiments. Using the criteria described in “Experimental Procedures,” we identified a total of 174 proteins as potential BAG3 interaction partners (supplemental Table S1). Twenty-five proteins had p values greater than 0.05, indicating that under our experimental conditions, they are likely to interact nonspecifically with BAG3 and were therefore considered as background proteins (supplemental Table S2). Detailed information about the quantified proteins and the ratios for each peptide are summarized in supplemental Table S2.
Fig. 1.
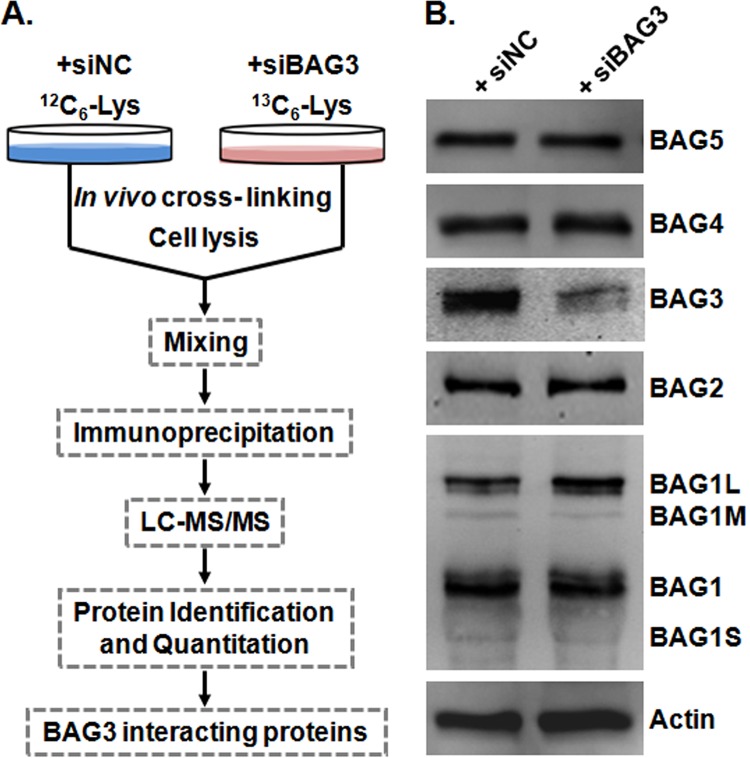
Identification of BAG3 interacting proteins with QUICK. A, overall workflow of the QUICK method. B, expression of BAG isoforms in BAG3 knockdown HeLa cells (siBAG3) and negative control cells (siNC). Relative to control cells, BAG3 knockdown HeLa cells showed diminished BAG3 protein levels, as expected. Depletion of BAG3 showed no obvious effect on the expression of the other BAG isoforms. Actin blotting was performed to ensure equal loading.
Identification of BAG3 Interacting Proteins Using a Human Proteome Microarray
We next used a human proteome microarray to identify putative BAG3 interacting proteins (Fig. 2A). As shown in Fig. 2B, a protein microarray composed of ∼17,000 individually purified human proteins as N-terminal GST fusions was incubated with affinity-purified human BAG3 protein. To detect binding events, we applied a BAG3-specific monoclonal antibody to the microarray and followed with incubation with a Cy5-labeled secondary antibody (Fig. 2B, left panel). Because anti-BAG3 and Cy5-labeled secondary antibodies can potentially recognize proteins other than BAG3 on the human proteome microarray, we also performed a control binding experiment in the absence of BAG3 on the microarray (Fig. 2B, right panel). Using stringent criteria as described in “Experimental Procedures,” 289 proteins were identified as potential BAG3 interacting proteins (supplemental Table S3), 11 of which are shown in Fig. 2C. To make an estimation of the false positive rate, we adopted a statistical model developed for estimating DNA microarray false positive rates by Wang et al. (43). Based on our experience, for proteome microarray experiments, the discordant rate of two replicated microarrays could be easily controlled under 10%. If we assume that the discordant rate of this study was 10%, then the false positive rate is (10%)2/(1 − 2*10% + (10%)2) = 1.23%.
Fig. 2.
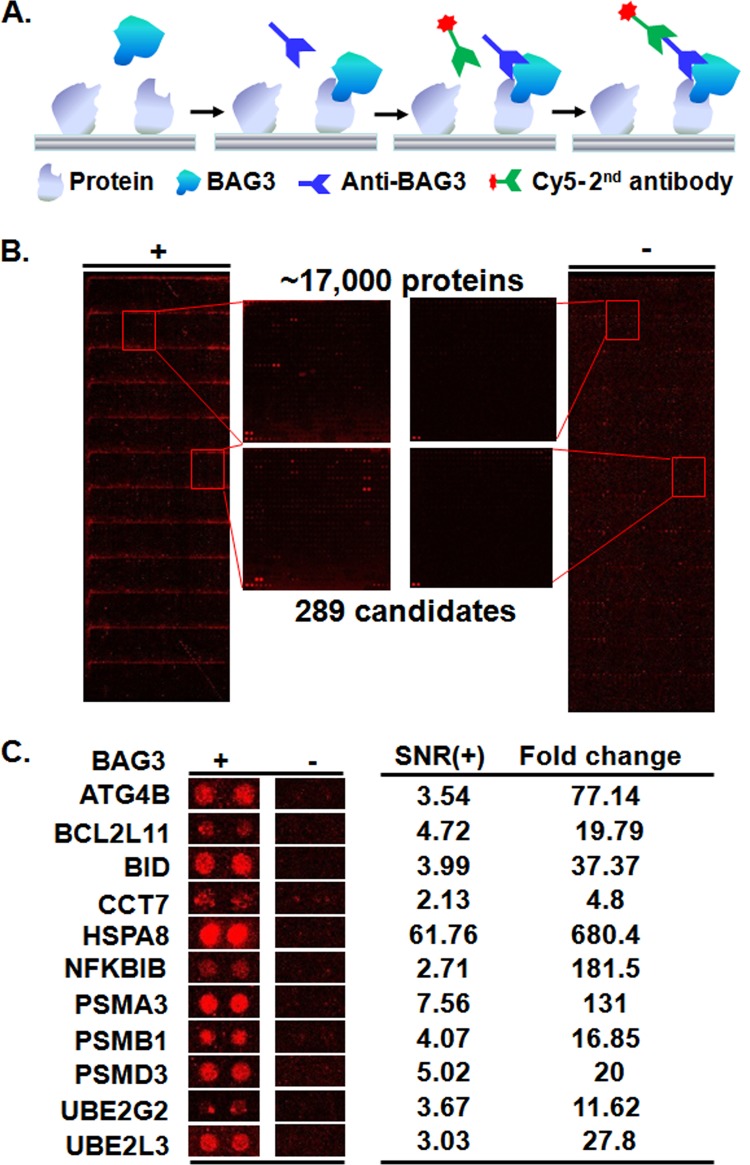
Identification of BAG3 interacting proteins with a human proteome microarray. A, workflow of the proteome microarray strategy for the identification of protein–protein interactions. B, human proteome microarrays were probed with BAG3 (left), followed by a BAG3 antibody and a Cy5-labeled secondary antibody. A control experiment was carried out without BAG3 (right). Many stronger signals can be observed from the microarray with BAG3 than from the microarray without BAG3. Enlarged areas from the same locations of the two micorarrays with and without BAG3 are illustrated in the middle. When the signals from the two microarrays were compared, 289 candidate BAG3 interacting proteins were identified. C, representative BAG3 interacting proteins.
Functional Diversity in the Two Datasets and Construction of the BAG3 Interactome
A comparison of the QUICK and proteome microarray results revealed that a substantial fraction of the proteins were shared between the two datasets. Of all 382 proteins identified as BAG3 interacting proteins, 81 (21.2%) were identified by both methods (Fig. 3A). Some of the known BAG3 interacting proteins, such as Hsp70 (5), sequestosome 1 (9), CCT (44), and HspB6 (45), were recovered in our datasets (supplemental Table S4), supporting the reliability of our data.
Although 20% of the proteins in the two datasets overlapped, each dataset contained a distinct group of proteins, and functional classification of these proteins permitted characterization of the different properties of the two datasets. Fig. 3B shows the relative distribution of functional classes for all Homo sapiens proteins in 22 PANTHER functional classes (29) (labeled “Expected”) (30) and the interacting proteins from the QUICK and proteome microarray datasets. Across all 22 PANTHER functional classes, we found the greatest deviations in the QUICK dataset to be associated with an overrepresentation in the chaperone, transfer/carrier protein, and protease categories and an underrepresentation of transcription factors and receptors. In the proteome microarray dataset, adaptor and cytoskeletal proteins were the two most overrepresented categories, and transcription factors and receptors were the most underrepresented. It is noteworthy that the transcription factor and receptor categories in the proteome microarray dataset were much less underrepresented than in QUICK (Fig. 3B). Therefore, there are large, clear differences between the two datasets, particularly for proteins with chaperone, transcription factor, or receptor functions. A plausible explanation for this difference is that transcription factors and receptors are generally expressed at low levels (46), making them hard to identify with QUICK, because the affinity purification step will most likely result in a loss of these proteins. However, this is not a problem with the protein microarray because each protein is overexpressed in yeast and individually purified. Similarly, proteins that function in organized assemblies, such as chaperones, would be preferentially included in the QUICK dataset, whereas the binary binding assay performed on the proteome microarrays is not designed to identify interactions involving more than two proteins. In order to take full advantage of the complementary nature of data obtained via the QUICK and proteome microarray methods, we combined the proteins identified by each method, giving a nonredundant list of 382 human proteins. This list, referred to as the BAG3 interactome (supplemental Table S4), was used in all subsequent analyses.
Categorization and GO Analysis of the BAG3 Interactome
To evaluate the biological relevance of the BAG3 interactome, the PANTHER classification system was used to classify these interacting proteins according to their functions and biological processes. The PANTHER classification analysis revealed that the interactome can be classified into 17 groups according to biological processes and 23 groups according to molecular functions (supplemental Fig. S1). The largest group within the BAG3 interactome is the transferase family (12%). Other significant functional categories include nucleic acid binding (10%), transcription factors (8%), enzyme modulators (8%), proteases (7%), and chaperones (6%). These results confirm that BAG3 is a critical regulator of diverse cellular functions across widespread biological processes (3). It should be noted that many of these proteins are multifunctional and are assigned to more than one functional class.
We performed a GO analysis to gain insights into the functional roles of BAG3, identifying enriched terms in the BAG3 interactome. First, the assignment of GO categories gave us an overview of the GO distribution (supplemental Fig. S2). Next, we performed GO biological process, molecular function, and cellular component analysis (Table I and supplemental Tables S5–S7). Consistent with the fact that BAG3 is a molecular chaperone regulator, 47 proteins were annotated in the “catabolic processes” term (GO term: 9057, p = 8.33 × 10−12). The biological process terms are presented as a graph in which direct links describe the hierarchy and relationships between terms (supplemental Fig. S3A). Colored nodes are those that are significantly overrepresented. Examination of the closest branch points of the overrepresented GO biological processes indicated that four major functional groups were strongly enriched in the BAG3 interactome, including two clusters of biological processes involved in the control of the proteasome-ubiquitination system and metabolic processes and two other clusters related to the regulation of cellular processes such as cell cycle and cell death (supplemental Fig. S3A). In the GO molecular function category, we found that the most overrepresented functions were involved in regulating enzyme activity, such as threonine-type endopeptidases and peptidases, glutathione transferases, and small protein-activating enzymes (supplemental Fig. S3B). For example, threonine-type endopeptidase and peptidase activities were significantly overrepresented with a p value of 2.17 × 10−9. In addition, functions including cytoskeletal protein binding, calcium-dependent phospholipid binding, proteasome activator activity, and coenzyme binding were also significant (p < 0.01). In the GO cellular component category, we found that most of the BAG3 interactome proteins identified were localized in non-membrane-bounded organelles (p = 8.83 × 10−4), the cytosol (p = 6.23 × 10−15), and the cytoskeleton (p = 2.99 × 10−4) (supplemental Fig. S3C). Notably, 21 BAG3 interacting proteins were attributed to the proteasome complex (p = 7.25 × 10−20), suggesting that that BAG3 might interact with the proteasome and regulate its activity.
Table I. GO terms enriched in the BAG3 interactome. The top five GO biological process, molecular function, and cellular component terms enriched in the BAG3 interactome are listed. A complete list can be found in supplementary Tables S5–S7.
| Term | Term | Description | Counta | %b | p valuec |
|---|---|---|---|---|---|
| Biological process | GO:0051443 | Positive regulation of ubiquitin-protein ligase activity | 24 | 7.16 | 1.07E-22 |
| GO:0051351 | Positive regulation of ligase activity | 24 | 7.16 | 3.21E-22 | |
| GO:0031398 | Positive regulation of protein ubiquitination | 25 | 7.46 | 5.37E-22 | |
| GO:0051437 | Positive regulation of ubiquitin-protein ligase activity during mitotic cell cycle | 23 | 6.87 | 1.37E-21 | |
| GO:0051438 | Regulation of ubiquitin-protein ligase activity | 24 | 7.16 | 1.78E-21 | |
| Molecular function | GO:0004298 | Threonine-type endopeptidase activity | 9 | 2.69 | 2.17E-09 |
| GO:0070003 | Threonine-type peptidase activity | 9 | 2.69 | 2.17E-09 | |
| GO:0004364 | Glutathione transferase activity | 7 | 2.09 | 1.74E-06 | |
| GO:0016765 | Transferase activity, transferring alkyl or aryl (other than methyl) groups | 8 | 2.39 | 4.64E-05 | |
| GO:0008092 | Cytoskeletal protein binding | 25 | 7.46 | 6.40E-05 | |
| Cellular component | GO:0000502 | Proteasome complex | 21 | 6.27 | 7.25E-20 |
| GO:0005829 | Cytosol | 70 | 20.9 | 6.23E-15 | |
| GO:0005839 | Proteasome core complex | 9 | 2.69 | 1.79E-09 | |
| GO:0048770 | Pigment granule | 12 | 3.58 | 1.09E-06 | |
| GO:0042470 | Melanosome | 12 | 3.58 | 1.09E-06 |
a Number of BAG3 interacting proteins.
b Percentage of mapped proteins associated with each term.
c Statistical significance of the difference between the fraction of BAG3 interacting proteins assigned to this GO term and the fraction of all proteins within the human protein set assigned to this GO term.
The subcellular localization and function of proteins are often dictated by their domains. We performed a similar analysis at the structural level by investigating the enrichment of protein domains in the BAG3 interactome. 93.4% of the BAG3 interacting proteins contain at least one Pfam-annotated domain; those domains that are significantly enriched in the BAG3 interactome are listed in Table II. Pfam domains associated with the proteasome and 14-3–3 proteins were most overrepresented in the BAG3 interactome (Table II). It is well established that Bag3 has a modular structure that contains many domains to mediate potential interactions with chaperones and/or other proteins (47). For example, it has been reported that the PXXP region of BAG3 can bind to Src homology 3 (SH3)-containing proteins (6). Consistent with these reports, nine BAG3 interactors are SH3-containing proteins, and SH3 domain families are enriched in the BAG3 interactome (p < 10−6) (Table II). Therefore, it is likely that these proteins may use the SH3 domain to interact with the PXXP region of BAG3.
Table II. Pfam domain families enriched in the BAG3 interactome.
| Term | Description | Counta | %b | p valuec |
|---|---|---|---|---|
| PF00227 | Proteasome | 9 | 2.58 | 1.47E-09 |
| PF00244 | 14-3–3 | 6 | 1.72 | 6.32E-08 |
| PF00018 | SH3 | 9 | 2.58 | 1.33E-07 |
| PF02798 | Glutathione S-transferase | 7 | 2.01 | 1.88E-06 |
| PF00191 | Annexin | 5 | 1.43 | 1.34E-04 |
| PF01023 | S-100 | 6 | 1.72 | 1.75E-04 |
| PF00899 | ThiF family | 4 | 1.15 | 2.62E-04 |
| PF00179 | Ubiquitin-conjugating enzyme | 5 | 1.43 | 8.66E-04 |
| PF02252 | PA28-beta | 3 | 0.86 | 1.18E-03 |
| PF02251 | PA28-alpha | 3 | 0.86 | 1.18E-03 |
| PF00179 | UQ-con | 6 | 1.72 | 1.29E-03 |
| PF00012 | HSP70 | 4 | 1.15 | 1.53E-03 |
| PF01370 | NAD dependent epimerase/dehydratase family | 3 | 0.86 | 7.84E-03 |
| PF00452 | Bcl-2 | 4 | 1.15 | 9.22E-03 |
| PF01370 | Epimerase | 3 | 0.86 | 1.03E-02 |
| PF00493 | MCM | 3 | 0.86 | 1.31E-02 |
| PF00012 | Hsp70 protein | 3 | 0.86 | 1.62E-02 |
| PF00004 | AAA | 5 | 1.43 | 1.65E-02 |
| PF00158 | Sigma54-activat | 3 | 0.86 | 1.95E-02 |
| PF00036 | EF hand | 5 | 1.43 | 2.41E-02 |
| PF10585 | Ubiquitin-activating enzyme active site | 2 | 0.57 | 3.96E-02 |
| PF10584 | Proteasome subunit A N-terminal signature | 2 | 0.57 | 3.96E-02 |
| PF01399 | PCI domain | 3 | 0.86 | 3.98E-02 |
| PF00515 | TPR-1 | 6 | 1.72 | 4.86E-02 |
a Number of BAG3 interacting proteins.
b Percentage of mapped proteins associated with each term.
c Statistical significance of the difference between the fraction of BAG3 interacting proteins assigned to this GO term and the fraction of all proteins within the human protein set assigned to this GO term.
Signaling Pathway and Protein Interaction Network Analysis
To determine whether BAG3 plays a role in signaling pathways, we superimposed the human Kyoto Encyclopedia of Genes and Genomes pathway database on the BAG3 interactome. As shown in supplemental Table S8, many identified BAG3 interacting proteins are involved in pathways responsible for the control of key physiological and pathological processes. In particular, 27 BAG3 partners were found to be involved in the regulation of protein degradation. Of these, 16 proteins are mapped to the proteasome, 8 to ubiquitin-mediated proteolysis, and 3 to the regulation of autophagy. Furthermore, we identified 12 BAG3 interacting proteins, including CHUK, HspA8, JUND, MAPK7, and MAPK11, which take part in MAPK signaling pathways involved in various cellular functions, including cell proliferation, differentiation, and migration (supplemental Table S8). In addition, five proteins, including Hsp90AA1, NFKBIB, and MAPK11, were assigned to the NOD-like receptor signaling pathway, a crucial pathway in the recognition of intracellular ligands. Furthermore, some of the proteins in our BAG3 interactome, such as Hsp90, MAPK11, CHUK, and 14-3–3, serve as key molecular switches in these signaling pathways. In summary, pathway analysis of the BAG3 interactome implied direct involvement of the BAG3 interactome in complex physiological and pathological systems such as the proteasome-ubiquitination system, immune responses, cancer, and neurodegenerative disorders.
To generate a BAG3 protein interaction network, we used the STRING database of physical and functional interactions (48). 283 (74%) of the identified BAG3 interacting proteins mapped to the BAG3 interaction network either directly or indirectly (Fig. 4A). Recent large-scale, high-throughput studies have established that most, if not all, cellular functions and processes are carried out by protein complexes rather than by individual proteins. In attempts to better understand biological processes, mapping interactions between protein complexes is as important as mapping interactions between individual proteins. To characterize protein complexes that associate with BAG3, therefore, we analyzed the BAG3 interactome for highly connected regions using MCODE (38) and determined the interconnectivity between these complexes and BAG3 by means of protein network analysis. As shown in Figs. 4B–4E, several complexes and cellular functions formed prominent, highly connected clusters. For example, many proteasome subunits are connected in a dense protein–protein interaction network that forms the densest cluster (Cluster 1), whereas BAG3 forms a complex with Hsp90AA1, HspA1A, HSF1, and ST13 (Cluster 4). It is therefore tempting to speculate that the physiological interaction between these two complexes might contribute to the cooperation and/or coordination of their functions in the control of numerous intracellular signaling and regulatory pathways.
Fig. 4.

Protein interaction network of the BAG3 interactome. A, the complete BAG3 interaction network obtained from STRING with a confidence score greater than 0.4. The network contains 283 nodes and 2,863 edges. B–E, the four top-ranked and tightly connected network clusters obtained with MCODE are color-coded and rendered as separate modules.
Validation of BAG3 Interacting Proteins by Reciprocal Immunoprecipitation
To validate some of the proteins identified via QUICK and proteome microarrays, co-immunoprecipitation experiments and Western blot analysis were performed. Based on how well their biological functions and importance were known, we selected two proteins identified only by QUICK, 14-3–3ζ and ATG3, and two proteins identified only by proteome microarrays, MAPK7 and HSF1, for validation. We also included two proteins, PSMB1 and HSP70, that were identified by both methods for validation. 14-3–3ζ belongs to the 14-3–3 protein family, which mediates signal transduction by binding to phosphoserine-containing proteins (49). ATG3, the yeast Apg3 homolog, is a protein-conjugating enzyme essential for autophagy (50). PSMB1 is the beta subunit of the highly ordered ring-shaped 20S core of the proteasome (51). MAPK7 plays a role in diverse cellular processes such as proliferation, differentiation, and cell survival (52). HSF1 is a DNA-binding protein that specifically binds heat shock promoter elements and activates transcription (53). Hsp70 was included as a positive control for the co-immunoprecipitation experiments because it was previously reported to physically interact with BAG3 (5). As shown in Fig. 5A, endogenously expressed 14-3–3ζ, ATG3, PSMB1, MAPK7, HSF1, and Hsp70 proteins could be readily detected in the BAG3 complex immunoprecipitated with anti-BAG3 antibodies. These interactions are specific, as no detectable signals were observed in IgG negative controls. Furthermore, reciprocal immunoprecipitation—Western blot analysis confirmed their specific interactions in cells (Figs. 5B–5G). Thus, compelling evidence shows that 14-3–3ζ, ATG3, PSMB1, MAPK7, HSF1, and Hsp70 proteins are bona fide BAG3 interacting proteins.
Fig. 5.

Validation of newly identified BAG3 interactors by co-immunoprecipitation. Immunoprecipitation assays of 14-3–3ζ, ATG3, Hsp70, PSMB1, MAPK7, HSF1, and BAG3 proteins were carried out as described under “Experimental Procedures.” A, immunoblotting demonstrated that BAG3 protein binds with 14-3–3ζ, ATG3, PSMB1, MAPK7, HSF1, and Hsp70 proteins. No BAG3-interacting bands were observed in the negative control (IgG). “Input” stands for the total cell lysate extracted from HeLa cells. Similarly, reverse immunoprecipitation assays confirmed the binding of (B) 14-3–3ζ, (C) ATG3, (D) PSMB1, (E) Hsp70, (F) MAPK7, and (G) HSF1 protein with BAG3.
Validation of BAG3 Interacting Proteins by SPR
As an independent approach, we employed SPR to determine the binding affinity of BAG3 interacting proteins. We included one protein, ENO1, which was identified as a background protein as a negative control for these SPR experiments. This in vitro assay system clearly showed that 14-3–3ζ, ATG3, PSMB1, MAPK7, HSF1, and Hsp70 interact with BAG3 immobilized on the SPR surface. As expected, we did not observe any appreciable interaction between ENO1 and BAG3 under the same conditions (Table III). Using the SPR data, we further determined the affinity constants (KD) for the interaction between immobilized BAG3 and 14-3–3ζ, ATG3, PSMB1, MAPK7, HSF1, and Hsp70 as 51, 170, 1,200, 1,100, 34, and 17 nm, respectively (Table III).
Table III. Binding affinities of newly identified BAG3 interacting proteins.
| Interacting proteins | ka (M−1 s−1)a | kd (s−1)b | KD (M) (kd/ka)c |
|---|---|---|---|
| 14-3–3ζ | (1.4 ± 0.4) × 106 | (7.1 ± 3.3) × 10−2 | (5.1 ± 2.2) × 10−8 |
| ATG3 | (4.2 ± 2.2) × 103 | (7.2 ± 1.8) × 10−4 | (1.7 ± 2.4) × 10−7 |
| PSMB1 | (3.8 ± 1.7) × 103 | (4.6 ± 0.6) × 10−3 | (1.2 ± 0.8) × 10−6 |
| HSP70 | (3.2 ± 1.5) × 104 | (5.3 ± 2.1) × 10−4 | (1.7 ± 1.3) × 10−8 |
| MAPK7 | (7.3 ± 3.4) × 104 | (8.1 ± 1.5) × 10−2 | (1.1 ± 1.6) × 10−6 |
| HSF1 | (9.6 ± 2.5) × 104 | (3.3 ± 1.2) × 10−3 | (3.4 ± 1.8) × 10−8 |
| ENO1 | NDd | NDd | NDd |
a ka: association rate.
b kd: dissociation rate.
c KD: kd/ka, dissociation constant.
d ND, not determined because of a low interaction signal.
BAG3 Regulates Proteasome Activity
GO analysis revealed that the most overrepresented biological process in the BAG3 interactome is involved in the control of the proteasome function, suggesting that BAG3 is a critical regulator of proteasome activity. To evaluate the role of BAG3 in regulating proteasome activity, we knocked down BAG3 using RNAi and examined its potential impact in HeLa cells. Western blotting using anti-BAG3 antibodies in RNAi-treated cells confirmed a significant decrease in the protein level of BAG3 relative to the negative control (Fig. 6A). Because previous studies have shown that BAG3 is induced by the proteasome inhibitor MG-132 at the transcriptional level, and because the induction of BAG3 by proteasome inhibition is anti-apoptotic (54, 55), we performed MG-132 treatment together with BAG3 knockdown. As shown in Fig. 6B, knockdown of BAG3 alone did not affect apoptosis in HeLa cells. However, in the BAG3-knockdown background, MG-132-induced apoptosis was significantly higher (49.5 ± 5.1%) than that in control cells treated with a control siRNA (36.1 ± 2.4%; p < 0.05). We further measured the effects of BAG3 knockdown on 20S proteasome activity in HeLa cells. As shown in Fig. 6C, knockdown of BAG3 resulted in a significant decrease in 20S proteasome activity (p < 0.05). In cells transfected with BAG3 siRNA, proteasome activity of MG-132-treated cells (42 ± 3.1%) was significantly lower than that in cells transfected with control siRNA (58 ± 6.4%; p < 0.05). Inhibition of proteasome activity should prevent the rapid degradation of ubiquitin-tagged proteins. Our results (Fig. 6D) show that BAG3 knockdown caused an accumulation of ubiquitin-tagged proteins in HeLa cells, as determined via Western blotting using anti-ubiquitin antibody. The 20 and 26S proteasomes have at least three distinct kinds of proteolytic activities, including chymotrypsin-, trypsin-, and caspase-like activities (40). To monitor specific protease activities of the 20 and 26S proteasomes, three fluorogenic substrates (Suc-LLVY-AMC for chymotrypsin-like, Z-ARR-AMC for trypsin-like, and Z-LLE-AMC for caspase-like activity) were incubated with cell lysates from treated or untreated cultures. As shown in Fig. 6E, knockdown of BAG3 resulted in a significant decrease in chymotrypsin-like activity of the 26S proteasome (p < 0.05). The caspase-like and trypsin-like activities of the 26S proteasome were slightly, but not significantly, less in BAG3 silenced HeLa cells. In cells transfected with BAG3 siRNA, chymotrypsin-like activity of MG-132 treated cells (43 ± 5.4%) was significantly lower than that in cells transfected with control siRNA (56 ± 4.5%) (p < 0.05), whereas the caspase-like and trypsin-like activities of the 26S proteasome were not significantly different. Similar to its effects on the 26S proteasome, BAG3 knockdown resulted in a significant (p < 0.05) decrease in the chymotrypsin-like activity of the 20S proteasome in HeLa cells with or without MG-132 treatment (Fig. 6F). The effects of BAG3 on the specific protease activities of the 20 and 26S proteasomes were further determined using additional A549 (human lung adenocarcinoma epithelial cell) cancer cells. Similar to its effects on apoptosis and the proteasome in HeLa cells, BAG3 knockdown resulted a significant (p < 0.05) increase in MG-132 sensitivity and decrease in the chymotrypsin-like activity of the 26S and 20S proteasome in A549 cells (supplemental Fig. S4).
Fig. 6.

The effect of knockdown of BAG3 on proteasome activity and MG-132 sensitivity in HeLa cells. A, knockdown of BAG3 in HeLa cells was confirmed by Western blotting. B, quantitative analysis of BAG3 knockdown on apoptosis in HeLa cells. Data are expressed as the mean ± S.D. of the fractions of apoptotic cells from at least three experiments. C, knockdown of BAG3 resulted in a marked decrease in proteasome activity. D, knockdown of BAG3 increased the ubiquitin-tagged proteins in HeLa cells. Western blotting was carried out with anti-ubiquitin antibody using total cell lysates extracted from HeLa cells. Actin served as a loading control. Effect of knockdown of BAG3 on the chymotryptic, tryptic, and caspase-like activities of the (E) 26S or (F) 20S proteasome in HeLa cells, as measured using fluorogenic substrates. Experiments were repeated a minimum of three times. *p < 0.05 when compared with the control group. 1: untreated HeLa cells; 2: HeLa cells treated with control siRNA; 3: HeLa cells treated with BAG3 siRNA; 4: HeLa cells treated with MG-132; 5: HeLa cells treated with control siRNA and MG-132; 6: HeLa cells treated with BAG3 siRNA and MG-132.
These results prompted us to investigate the effect of BAG3 overexpression on proteasome activity and apoptosis. BAG3 was transiently overexpressed in HeLa cells, as confirmed by Western blotting (Fig. 7A). As shown in Fig. 7B, overexpression of BAG3 reduced MG-132 sensitivity in HeLa cells relative to cells transfected with a control vector. Quantitative analysis revealed that the percentage of apoptotic cells when BAG3 was overexpressed together with MG-132 treatment (24.9% ± 3.1%) was significantly lower than that in the vector control (34.2% ± 3.4%; p < 0.05). When BAG3 was overexpressed, the 20S proteasome activity of MG-132 treated cells (77% ± 7.1%) was significantly higher (p < 0.01) than that of MG-132-treated cells transfected with a control vector (62% ± 5.4%) (Fig. 7C), and the accumulation of ubiquitin-tagged proteins in MG-132-treated HeLa cells was reduced (Fig. 7D). We further examined the specific effect of BAG3 on proteasome activity. As shown in Figs. 7E and 7F, the chymotrypsin-like activity of MG-132-treated cells (68% ± 4.2% for 26S and 71% ± 6.1% for 20S) was significantly higher (p < 0.05) than that of MG-132-treated cells transfected with a control vector (54% ± 5.4% for 26S and 58% ± 4.5% for 20S). In addition, similar effects of BAG3 overexpression on apoptosis and proteasome activities were also observed in A549 cells (supplemental Fig. S5).
Fig. 7.

The effect of overexpression of BAG3 on proteasome activity and MG-132 sensitivity in HeLa cells. A, overexpression of BAG3 in HeLa cells was confirmed by Western blotting. B, overexpression of BAG3 reduces MG-132 sensitivity in HeLa cells. C, overexpression of BAG3 resulted in a marked increase in proteasome activity in MG-132-treated cells. D, overexpression of BAG3 decreased the ubiquitin-tagged proteins in MG-132-treated HeLa cells. Effect of overexpression of BAG3 on the chymotryptic, tryptic, and caspase-like activities of the (E) 26S or (F) 20S proteasome in HeLa cells, as measured using fluorogenic substrates. Experiments were repeated a minimum of three times. *p < 0.05 when compared with the control group. 1: untreated HeLa cells; 2: HeLa cells treated with blank vector; 3: HeLa cells treated with BAG3 plasmid; 4: HeLa cells treated with MG-132; 5: HeLa cells treated with blank vector and MG-132; 6: HeLa cells treated with BAG3 plasmid and MG-132.
Together, these results suggest that BAG3 plays an important role in regulating proteasome activity in cancer cells. Our results also support the notion that the inhibition of BAG3 could in principle be a useful therapeutic strategy for the treatment of cancer.
DISCUSSION
BAG3 has a multifaceted ability to form complexes with many proteins; it is involved in a number of major biological processes including apoptosis, development, cytoskeleton organization, virus replication, and autophagy. Recently, we found that BAG3 plays an important role in SARS-CoV replication (13). However, the mechanisms underlying BAG3's functions remain somewhat enigmatic and controversial. A key step in understanding the physiological function of BAG3 is assigning BAG3 to appropriate biochemical pathways through the identification of its interacting partners. In this study, we performed a global analysis of the interactome of BAG3 using the QUICK and proteome microarray methods. Our results clearly demonstrate that datasets generated via QUICK and proteome microarrays form distinct, partially overlapping sets with different protein–protein interaction characteristics (Fig. 3). The differences between the QUICK and proteome microarray results were not unexpected, as many studies have shown that different proteomic methods possess distinct strengths and weaknesses and provide complementary types of information (56–58). Therefore, we consolidated these two datasets into a single set that was designated as the BAG3 interactome. This dataset should provide a starting point for more in-depth studies on the functions of BAG3.
Because our bioinformatics analysis indicated that the proteasome-ubiquitination process and proteasome complex were strongly enriched in the BAG3 interactome, we investigated their relationship in greater depth. We report for the first time that BAG3 interacts with the proteasome and regulates its activity. This agrees well with reports that proteasome-interacting partners dynamically associate with proteasomes and alter proteasome function (59–64). In this study, down-regulation of BAG3 was found to decrease proteasome activity and increase cell death in response to proteasome inhibition. This effect can be attributed to the fact that BAG3 inhibition suppresses a wide constellation of anti-apoptotic protective cellular pathways, thereby rendering cells more sensitive to various pro-apoptotic stimuli. This indicates that BAG3 might reduce the apoptotic response to proteasome inhibition, probably by maintaining proteasome activity. Proteasomes degrade a multitude of protein substrates in the cytosol and nucleus and are thus essential for many aspects of cellular function (65). Proteasome inhibitors represent a novel class of anticancer drugs with very promising clinical activity in multiple myeloma and possibly in various solid tumors (66). However, proteasome inhibitors also activate survival signals, and thus it seems plausible that the activation of survival signaling cascades might compromise their antitumoral efficacy (67). The synergistic effect between BAG3 inhibition and MG-132 observed here confirms the functional significance of BAG3 and suggests the possibility that combination treatments that include BAG3 inhibitors may augment clinical efficacy and overcome clinical refractoriness to proteasome inhibitors.
Notably, there were seven heat shock proteins (Hsp90AA1, Hsp90AB1, HspA1A, HspA1L, HspA2, HspA8, and HspB6) among the BAG3 interacting proteins (supplemental Table S4), two of which (Hsp90AA1 and HspA1A) have been predicted by MCODE (38) to form a complex with BAG3 (Fig. 4B). Heat shock proteins, known as molecular chaperones that assist in protein folding, are the products of several distinct gene families that are required for cell survival during stress and are named according to their molecular weights (68). Different classes of heat shock proteins play diverse roles in governing proper protein assembly, folding, and translocation (69). Among them, the Hsp70s control protein homeostasis in the cell during and following applied stress and, together with other chaperones, regulate physiological processes, including vesicular trafficking and signaling for growth, differentiation, and apoptosis (70). It is well established that BAG3 contains a conserved BAG domain able to interact with Hsp70 proteins and that the influence of BAG3 on cell survival is mediated at least in part by its binding to Hsp70 proteins (3, 71). Our data therefore support the idea that BAG3 interacts with both the proteasome and Hsp70 and raise the possibility that BAG3 might form a complex in vivo with Hsp70 and the proteasome. It is likely that the interaction of BAG3 with the proteasome and Hsp70 may regulate the stability and dynamics of targeted proteins. It is also possible that such interactions may be dynamically regulated by stimuli in coordination with the diverse functions of these proteins during cellular processes such as cell survival (stress response), proliferation, migration, and apoptosis. The interaction of BAG3 with the proteasome and Hsp70 might therefore represent a novel mechanism for maintaining the stability and dynamics of targeted proteins in cancer cells. Further work that combines individual knockouts of BAG3, Hsp70, and proteasome subunits with quantitative proteomics is ongoing to clarify the role of these proteins in cancer cells.
Seventeen of the BAG3 interacting proteins belong to the transfer/carrier protein class, including five ubiquitin-conjugating enzymes and two autophagy-related proteins (Atg3 and Atg7). The interaction of Atg3 with BAG3 was confirmed via reciprocal co-immunoprecipitation and SPR analysis (Fig. 5 and Table III). ATG genes are highly conserved among eukaryotes and are required for macroautophagy and its related processes. The discovery of ATG genes, originally in yeast and subsequently in multicellular organisms, has provided an important breakthrough in the understanding of autophagy and of its functions in physiology and disease (72). To date, more than 30 ATG genes have been identified. The corresponding gene products constitute the core machinery that coordinates specific steps in the autophagic pathway (73). It is well established that protein degradation through both the proteasome and autophagy pathways is regulated by BAG3 and its interacting proteins (3, 9). BAG3 participates, along with HspB8, a member of the HspB family of molecular chaperones, in the degradation of misfolded and aggregated proteins via autophagy (74, 75). BAG3 may act as a molecular switch to determine whether proteins are degraded in a proteasomal or autophagic manner (3, 9). Additionally, BAG3 was found to act in concert with the ubiquitin-binding protein sequestosome 1 to increase autophagic activity (3, 9). In this study, we have shown clearly that Atg3 and Atg7 are BAG3-associated proteins. This suggests that, through binding to Atg proteins, BAG3 may regulate the formation and maturation of autophagosomes and their fusion with lysosomes. Further experiments are necessary to establish the functional consequences and detailed mechanisms of these interactions, as well as to define further the role of BAG3 in autophagy signaling. These data, combined with data from previous studies, will provide a new window into the autophagy machinery and a possible new target for therapeutic intervention.
To our knowledge, this is the first study that has combined QUICK and proteome microarrays to obtain a comprehensive profile of protein–protein interactions. Both of these technologies have their own unique advantages and limitations, and thus combining the two provides a powerful approach for proteomic investigations. With BAG3 as an example, we have shown that QUICK and proteome microarrays are complementary approaches (Fig. 3A). In addition, well-validated BAG3 interacting proteins such as Hsp70 (5), sequestosome 1 (9), and HspB6 (45) were successfully identified via both of these approaches. The lists of BAG3 interacting proteins generated by the two approaches overlapped to a high degree; 47% (81/174) of the QUICK candidates were also identified by the proteome microarray. Furthermore, we constructed a very comprehensive interactome with 382 BAG3 interacting proteins by combining the independent results obtained using these two methodologies. This complementary strategy of using both QUICK and proteome microarrays could easily be adopted for interactome construction for other proteins of interest, such as transcriptional factors, the protein–protein interactions of which cannot theoretically be studied using traditional yeast two-hybrid assays.
The complete BAG3 interactome obtained using these complementary approaches provides an essential step toward our understanding of the system-wide role of BAG3 in the regulation of various cellular processes such as signal transduction, transcription, proteasome-ubiquitination, and autophagy. It is now important to further characterize the interactions of BAG3 with different binding partners in different physiological and pathophysiological contexts.
Supplementary Material
Acknowledgments
We thank Dr. Daniel Czajkowsky for critical reading and Dr. Joy Fleming for English editing.
Footnotes
* This work was supported by the State Key Development Program for Basic Research in China (Grant Nos. 2012CB518700 and 2010CB529205), the National High Technology Research and Development Program of China (Grant Nos. 2012AA020103 and 2012AA020203), the National Natural Science Foundation of China (Grant No. 31000388), and the Hundred Talents Program of the Chinese Academy of Sciences.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- AMC
- 7-amido-4-methylcoumarin
- ATG3
- autophagy related 3
- BAG
- Bcl2-associated athanogene
- GO
- Gene Ontology
- HSF
- heat shock transcription factor
- Hsp
- heat shock protein
- MAPK
- mitogen-activated protein kinase
- PSMB1
- proteasome subunit, beta type 1
- QUICK
- quantitative immunoprecipitation combined with knockdown
- RNAi
- RNA interference
- SILAC
- stable isotope labeling with amino acids in cell culture
- SPR
- surface plasmon resonance.
REFERENCES
- 1. Doong H., Vrailas A., Kohn E. C. (2002) What's in the “BAG”?—a functional domain analysis of the BAG-family proteins. Cancer Lett. 188, 25–32 [DOI] [PubMed] [Google Scholar]
- 2. Kabbage M., Dickman M. B. (2008) The BAG proteins: a ubiquitous family of chaperone regulators. Cell. Mol. Life Sci. 65, 1390–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rosati A., Graziano V., De Laurenzi V., Pascale M., Turco M. C. (2011) BAG3: a multifaceted protein that regulates major cell pathways. Cell Death Dis. 2, e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Doong H., Rizzo K., Fang S., Kulpa V., Weissman A. M., Kohn E. C. (2003) CAIR-1/BAG-3 abrogates heat shock protein-70 chaperone complex-mediated protein degradation: accumulation of poly-ubiquitinated Hsp90 client proteins. J. Biol. Chem. 278, 28490–28500 [DOI] [PubMed] [Google Scholar]
- 5. Hishiya A., Kitazawa T., Takayama S. (2010) BAG3 and Hsc70 interact with actin capping protein CapZ to maintain myofibrillar integrity under mechanical stress. Circ. Res. 107, 1220–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Doong H., Price J., Kim Y. S., Gasbarre C., Probst J., Liotta L. A., Blanchette J., Rizzo K., Kohn E. (2000) CAIR-1/BAG-3 forms an EGF-regulated ternary complex with phospholipase C-gamma and Hsp70/Hsc70. Oncogene 19, 4385–4395 [DOI] [PubMed] [Google Scholar]
- 7. Lee J. H., Takahashi T., Yasuhara N., Inazawa J., Kamada S., Tsujimoto Y. (1999) Bis, a Bcl-2-binding protein that synergizes with Bcl-2 in preventing cell death. Oncogene 18, 6183–6190 [DOI] [PubMed] [Google Scholar]
- 8. Behl C. (2011) BAG3 and friends: co-chaperones in selective autophagy during aging and disease. Autophagy 7, 795–798 [DOI] [PubMed] [Google Scholar]
- 9. Gamerdinger M., Hajieva P., Kaya A. M., Wolfrum U., Hartl F. U., Behl C. (2009) Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 28, 889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kyratsous C. A., Silverstein S. J. (2008) The co-chaperone BAG3 regulates Herpes Simplex Virus replication. Proc. Natl. Acad. Sci. U.S.A. 105, 20912–20917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kyratsous C. A., Silverstein S. J. (2007) BAG3, a host cochaperone, facilitates varicella-zoster virus replication. J. Virol. 81, 7491–7503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baud V., Karin M. (2009) Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 8, 33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang L., Zhang Z. P., Zhang X. E., Lin F. S., Ge F. (2010) Quantitative proteomics analysis reveals BAG3 as a potential target to suppress severe acute respiratory syndrome coronavirus replication. J. Virol. 84, 6050–6059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Selbach M., Mann M. (2006) Protein interaction screening by quantitative immunoprecipitation combined with knockdown (QUICK). Nat. Methods 3, 981–983 [DOI] [PubMed] [Google Scholar]
- 15. Ge F., Li W. L., Bi L. J., Tao S. C., Zhang Z. P., Zhang X. E. (2010) Identification of novel 14-3–3zeta interacting proteins by quantitative immunoprecipitation combined with knockdown (QUICK). J. Proteome Res. 9, 5848–5858 [DOI] [PubMed] [Google Scholar]
- 16. Meixner A., Boldt K., Van Troys M., Askenazi M., Gloeckner C. J., Bauer M., Marto J. A., Ampe C., Kinkl N., Ueffing M. (2011) A QUICK screen for Lrrk2 interaction partners—leucine-rich repeat kinase 2 is involved in actin cytoskeleton dynamics. Mol. Cell. Proteomics 10, M110.001172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zheng P., Zhong Q., Xiong Q., Yang M., Zhang J., Li C., Bi L. J., Ge F. (2012) QUICK identification and SPR validation of signal transducers and activators of transcription 3 (Stat3) interacting proteins. J. Proteomics 75, 1055–1066 [DOI] [PubMed] [Google Scholar]
- 18. Chen C. S., Korobkova E., Chen H., Zhu J., Jian X., Tao S. C., He C., Zhu H. (2008) A proteome chip approach reveals new DNA damage recognition activities in Escherichia coli. Nat. Methods 5, 69–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tao S. C., Chen C. S., Zhu H. (2007) Applications of protein microarray technology. Comb. Chem. High Throughput Screen. 10, 706–718 [DOI] [PubMed] [Google Scholar]
- 20. Zhu H., Bilgin M., Bangham R., Hall D., Casamayor A., Bertone P., Lan N., Jansen R., Bidlingmaier S., Houfek T., Mitchell T., Miller P., Dean R. A., Gerstein M., Snyder M. (2001) Global analysis of protein activities using proteome chips. Science 293, 2101–2105 [DOI] [PubMed] [Google Scholar]
- 21. Yang L., Guo S., Li Y., Zhou S., Tao S. (2011) Protein microarrays for systems biology. Acta Biochim. Biophys. Sin. (Shanghai) 43, 161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jeong J. S., Rho H. S., Zhu H. (2011) A functional protein microarray approach to characterizing posttranslational modifications on lysine residues. Methods Mol. Biol. 723, 213–223 [DOI] [PubMed] [Google Scholar]
- 23. Lin Y. Y., Lu J. Y., Zhang J., Walter W., Dang W., Wan J., Tao S. C., Qian J., Zhao Y., Boeke J. D., Berger S. L., Zhu H. (2009) Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell 136, 1073–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jeong J. S., Jiang L., Albino E., Marrero J., Rho H. S., Hu J., Hu S., Vera C., Bayron-Poueymiroy D., Rivera-Pacheco Z. A., Ramos L., Torres-Castro C., Qian J., Bonaventura J., Boeke J. D., Yap W. Y., Pino I., Eichinger D. J., Zhu H., Blackshaw S. (2012) Rapid identification of monospecific monoclonal antibodies using a human proteome microarray. Mol. Cell. Proteomics 11, O111.016253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schnack C., Danzer K. M., Hengerer B., Gillardon F. (2008) Protein array analysis of oligomerization-induced changes in alpha-synuclein protein-protein interactions points to an interference with Cdc42 effector proteins. Neuroscience 154, 1450–1457 [DOI] [PubMed] [Google Scholar]
- 26. Virok D. P., Simon D., Bozso Z., Rajko R., Datki Z., Balint E., Szegedi V., Janaky T., Penke B., Fulop L. (2011) Protein array based interactome analysis of amyloid-beta indicates an inhibition of protein translation. J. Proteome Res. 10, 1538–1547 [DOI] [PubMed] [Google Scholar]
- 27. Haas W., Faherty B. K., Gerber S. A., Elias J. E., Beausoleil S. A., Bakalarski C. E., Li X., Villen J., Gygi S. P. (2006) Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol. Cell. Proteomics 5, 1326–1337 [DOI] [PubMed] [Google Scholar]
- 28. Park S. K., Venable J. D., Xu T., Yates J. R., 3rd (2008) A quantitative analysis software tool for mass spectrometry-based proteomics. Nat. Methods 5, 319–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mi H., Guo N., Kejariwal A., Thomas P. D. (2007) PANTHER version 6: protein sequence and function evolution data with expanded representation of biological pathways. Nucleic Acids Res. 35, D247–D252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu X., Ivanic J., Memisevic V., Wallqvist A., Reifman J. (2011) Categorizing biases in high-confidence high-throughput protein-protein interaction data sets. Mol. Cell. Proteomics 10, M111.012500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., Sherlock G. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kanehisa M., Goto S. (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Finn R. D., Tate J., Mistry J., Coggill P. C., Sammut S. J., Hotz H. R., Ceric G., Forslund K., Eddy S. R., Sonnhammer E. L., Bateman A. (2008) The Pfam protein families database. Nucleic Acids Res. 36, D281–D288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 35. Huang da W., Sherman B. T., Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maere S., Heymans K., Kuiper M. (2005) BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449 [DOI] [PubMed] [Google Scholar]
- 37. Jensen L. J., Kuhn M., Stark M., Chaffron S., Creevey C., Muller J., Doerks T., Julien P., Roth A., Simonovic M., Bork P., von Mering C. (2009) STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 37, D412–D416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bader G. D., Hogue C. W. (2003) An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Adams J. (2003) The proteasome: structure, function, and role in the cell. Cancer Treat. Rev. 29 Suppl 1, 3–9 [DOI] [PubMed] [Google Scholar]
- 41. Glas R., Bogyo M., McMaster J. S., Gaczynska M., Ploegh H. L. (1998) A proteolytic system that compensates for loss of proteasome function. Nature 392, 618–622 [DOI] [PubMed] [Google Scholar]
- 42. Grune T., Reinheckel T., Joshi M., Davies K. J. (1995) Proteolysis in cultured liver epithelial cells during oxidative stress. Role of the multicatalytic proteinase complex, proteasome. J. Biol. Chem. 270, 2344–2351 [DOI] [PubMed] [Google Scholar]
- 43. Wang X., Hessner M. J., Wu Y., Pati N., Ghosh S. (2003) Quantitative quality control in microarray experiments and the application in data filtering, normalization and false positive rate prediction. Bioinformatics 19, 1341–1347 [DOI] [PubMed] [Google Scholar]
- 44. Fontanella B., Birolo L., Infusini G., Cirulli C., Marzullo L., Pucci P., Turco M. C., Tosco A. (2010) The co-chaperone BAG3 interacts with the cytosolic chaperonin CCT: new hints for actin folding. Int. J. Biochem. Cell Biol. 42, 641–650 [DOI] [PubMed] [Google Scholar]
- 45. Fuchs M., Poirier D. J., Seguin S. J., Lambert H., Carra S., Charette S. J., Landry J. (2010) Identification of the key structural motifs involved in HspB8/HspB6-Bag3 interaction. Biochem. J. 425, 245–255 [DOI] [PubMed] [Google Scholar]
- 46. Jiang D., Jia Y., Jarrett H. W. (2011) Transcription factor proteomics: identification by a novel gel mobility shift-three-dimensional electrophoresis method coupled with southwestern blot and high-performance liquid chromatography-electrospray-mass spectrometry analysis. J. Chromatogr. A 1218, 7003–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McCollum A. K., Casagrande G., Kohn E. C. (2010) Caught in the middle: the role of Bag3 in disease. Biochem. J. 425, e1–e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. von Mering C., Huynen M., Jaeggi D., Schmidt S., Bork P., Snel B. (2003) STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 31, 258–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Matta A., Siu K. M., Ralhan R. (2012) 14-3–3 zeta as novel molecular target for cancer therapy. Expert Opin. Ther. Targets epub pubmed/22512284 [DOI] [PubMed] [Google Scholar]
- 50. Besteiro S., Brooks C. F., Striepen B., Dubremetz J. F. (2011) Autophagy protein Atg3 is essential for maintaining mitochondrial integrity and for normal intracellular development of Toxoplasma gondii tachyzoites. PLoS Pathog. 7, e1002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Keutgens A., Zhang X., Shostak K., Robert I., Olivier S., Vanderplasschen A., Chapelle J. P., Viatour P., Merville M. P., Bex F., Gothot A., Chariot A. (2010) BCL-3 degradation involves its polyubiquitination through a FBW7-independent pathway and its binding to the proteasome subunit PSMB1. J. Biol. Chem. 285, 25831–25840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sebolt-Leopold J. S., Herrera R. (2004) Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat. Rev. Cancer 4, 937–947 [DOI] [PubMed] [Google Scholar]
- 53. Whitesell L., Lindquist S. (2009) Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin. Ther. Targets 13, 469–478 [DOI] [PubMed] [Google Scholar]
- 54. Du Z. X., Zhang H. Y., Meng X., Gao Y. Y., Zou R. L., Liu B. Q., Guan Y., Wang H. Q. (2009) Proteasome inhibitor MG132 induces BAG3 expression through activation of heat shock factor 1. J. Cell. Physiol. 218, 631–637 [DOI] [PubMed] [Google Scholar]
- 55. Wang H. Q., Liu H. M., Zhang H. Y., Guan Y., Du Z. X. (2008) Transcriptional upregulation of BAG3 upon proteasome inhibition. Biochem. Biophys. Res. Commun. 365, 381–385 [DOI] [PubMed] [Google Scholar]
- 56. Ge F., Bi L. J., Tao S. C., Xu X. D., Zhang Z. P., Kitazato K., Zhang X. E. (2011) Proteomic analysis of multiple myeloma: current status and future perspectives. Proteomics Clin. Appl. 5, 30–37 [DOI] [PubMed] [Google Scholar]
- 57. Elliott M. H., Smith D. S., Parker C. E., Borchers C. (2009) Current trends in quantitative proteomics. J. Mass Spectrom. 44, 1637–1660 [DOI] [PubMed] [Google Scholar]
- 58. Yu X., Ivanic J., Memisevic V., Wallqvist A., Reifman J. (2011) Categorizing biases in high-confidence high-throughput protein-protein interaction data sets. Mol. Cell. Proteomics, M111.012500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Guerrero C., Tagwerker C., Kaiser P., Huang L. (2006) An integrated mass spectrometry-based proteomic approach: quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Mol. Cell. Proteomics 5, 366–378 [DOI] [PubMed] [Google Scholar]
- 60. Zong C., Gomes A. V., Drews O., Li X., Young G. W., Berhane B., Qiao X., French S. W., Bardag-Gorce F., Ping P. (2006) Regulation of murine cardiac 20S proteasomes: role of associating partners. Circ. Res. 99, 372–380 [DOI] [PubMed] [Google Scholar]
- 61. Bousquet-Dubouch M. P., Baudelet E., Guerin F., Matondo M., Uttenweiler-Joseph S., Burlet-Schiltz O., Monsarrat B. (2009) Affinity purification strategy to capture human endogenous proteasome complexes diversity and to identify proteasome-interacting proteins. Mol. Cell. Proteomics 8, 1150–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Huang L., Burlingame A. L. (2005) Comprehensive mass spectrometric analysis of the 20S proteasome complex. Methods Enzymol. 405, 187–236 [DOI] [PubMed] [Google Scholar]
- 63. Aiken C. T., Kaake R. M., Wang X., Huang L. (2011) Oxidative stress-mediated regulation of proteasome complexes. Mol. Cell. Proteomics 10, R110.006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guerrero C., Milenkovic T., Przulj N., Kaiser P., Huang L. (2008) Characterization of the proteasome interaction network using a QTAX-based tag-team strategy and protein interaction network analysis. Proc. Natl. Acad. Sci. U.S.A. 105, 13333–13338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kwak J., Workman J. L., Lee D. (2011) The proteasome and its regulatory roles in gene expression. Biochim. Biophys. Acta 1809, 88–96 [DOI] [PubMed] [Google Scholar]
- 66. Driscoll J. J., Minter A., Driscoll D. A., Burris J. K. (2011) The ubiquitin+proteasome protein degradation pathway as a therapeutic strategy in the treatment of solid tumor malignancies. Anticancer Agents Med. Chem. 11, 242–246 [DOI] [PubMed] [Google Scholar]
- 67. Ruschak A. M., Slassi M., Kay L. E., Schimmer A. D. (2011) Novel proteasome inhibitors to overcome bortezomib resistance. J. Natl. Cancer Inst. 103, 1007–1017 [DOI] [PubMed] [Google Scholar]
- 68. Calderwood S. K., Khaleque M. A., Sawyer D. B., Ciocca D. R. (2006) Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem. Sci. 31, 164–172 [DOI] [PubMed] [Google Scholar]
- 69. Richter K., Haslbeck M., Buchner J. (2010) The heat shock response: life on the verge of death. Mol. Cell 40, 253–266 [DOI] [PubMed] [Google Scholar]
- 70. Lanneau D., Brunet M., Frisan E., Solary E., Fontenay M., Garrido C. (2008) Heat shock proteins: essential proteins for apoptosis regulation. J. Cell. Mol. Med. 12, 743–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gentilella A., Khalili K. (2011) BAG3 expression in glioblastoma cells promotes accumulation of ubiquitinated clients in an Hsp70-dependent manner. J. Biol. Chem. 286, 9205–9215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yang Z., Klionsky D. J. (2010) Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12, 814–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Xie Z., Klionsky D. J. (2007) Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 9, 1102–1109 [DOI] [PubMed] [Google Scholar]
- 74. Carra S. (2009) The stress-inducible HspB8-Bag3 complex induces the eIF2alpha kinase pathway: implications for protein quality control and viral factory degradation? Autophagy 5, 428–429 [DOI] [PubMed] [Google Scholar]
- 75. Carra S., Brunsting J. F., Lambert H., Landry J., Kampinga H. H. (2009) HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2{alpha} phosphorylation. J. Biol. Chem. 284, 5523–5532 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.