

Abstract
Quantitative LC-MALDI is an underrepresented method, especially in large-scale experiments. The additional fractionation step that is needed for most MALDI-TOF-TOF instruments, the comparatively long analysis time, and the very limited number of established software tools for the data analysis render LC-MALDI a niche application for large quantitative analyses beside the widespread LC–electrospray ionization workflows.
Here, we used LC-MALDI in a relative quantification analysis of Staphylococcus aureus for the first time on a proteome-wide scale. Samples were analyzed in parallel with an LTQ-Orbitrap, which allowed cross-validation with a well-established workflow. With nearly 850 proteins identified in the cytosolic fraction and quantitative data for more than 550 proteins obtained with the MASCOT Distiller software, we were able to prove that LC-MALDI is able to process highly complex samples. The good correlation of quantities determined via this method and the LTQ-Orbitrap workflow confirmed the high reliability of our LC-MALDI approach for global quantification analysis.
Because the existing literature reports differences for MALDI and electrospray ionization preferences and the respective experimental work was limited by technical or methodological constraints, we systematically compared biochemical attributes of peptides identified with either instrument. This genome-wide, comprehensive study revealed biases toward certain peptide properties for both MALDI-TOF-TOF- and LTQ-Orbitrap-based approaches. These biases are based on almost 13,000 peptides and result in a general complementarity of the two approaches that should be exploited in future experiments.
One-dimensional gel-based liquid chromatography mass spectrometry (GeLC-MS)1 is a well-established technique in life science. In combination with in vivo labeling approaches such as stable isotope labeling by amino acids in cell culture (SILAC) (1) or 15N labeling (2), it allows the relative quantification of large numbers of proteins in a complex sample. Mass spectrometry (MS) measurements in such workflows are predominantly performed with electrospray ionization (ESI)-based mass spectrometers. Online coupling of a liquid chromatography (LC) system with fast MS spectra acquisition and high-mass-accuracy ESI instruments in conjunction with fractionation on both protein and peptide levels allows the analysis of very complex samples in a relatively short period of time (3). The identification and quantification of data can be done in an automatic or semi-automatic manner with a variety of well-established software packages (4).
In proteomic research, matrix-assisted laser desorption ionization time-of-flight tandem mass spectrometry (MALDI-TOF-TOF) is mainly used for the analysis of non-complex protein samples that do not require fractionation on the peptide level by means of liquid chromatography. In particular, the analysis of single protein spots resulting from the two-dimensional PAGE separation of complex samples is primarily carried out with this MS technique, as it allows the fast and reliable analysis of a high number of low-complex samples (5).
In most LC-MALDI workflows, the LC system and the MALDI instrument are coupled offline with a fractionation step in between. Online measurement of the LC eluate is not appropriate for most MALDI systems, as the mass analyzer is a closed vacuum chamber and sample insertion is based on a lock chamber, which precludes the direct injection of an LC run. Also, the measurement speed of most MALDI instruments is too low to analyze samples of mid and high complexity online, when large numbers of peptides elute in a short time frame. The comparatively low throughput in terms of single spectrum acquisition and the restricted online coupling for most instruments are generally circumvented by the offline coupling of an LC system and a MALDI-TOF-TOF instrument through a fractionation system (6). The decoupling from the chromatographic process makes MS measurements independent of the instrument's scan cycle time. The only restriction is the sample consumption in the ionization process. Besides counteracting excessively long cycle times of the mass spectrometer, the offline coupling also enables multiple measurements of the same LC run and therefore allows the selective analysis of single precursor ions after a first analysis of the data (7).
LC-MALDI for qualitative analysis generally ranks behind the widespread LC-ESI approaches. This is mainly because the additional fractionation step leads to longer analysis times. The discrepancy in usage is even bigger for relative quantification workflows. LC-MALDI is rarely employed for the analysis of in vivo labeled samples, especially in large-scale experiments. Even though it has been shown that shot-to-shot intensity variation, which is a general drawback for quantitative MALDI analysis, can be overcome with a suitable experimental setup (8), the lack of established software tools for the analysis of these data is evident and hampers the application of LC-MALDI in large-scale experiments.
Here, we describe for the first time a global analysis of in vivo 15N labeled samples with a GeLC-MS/MS workflow carried out with a MALDI-TOF-TOF instrument. In this workflow, proteins were prefractionated on a one-dimensional SDS gel and tryptically digested, and the resulting peptide mixtures were separated by means of reversed-phase LC. The LC eluate was then fractionated, which allowed offline coupling with a MALDI-TOF-TOF instrument. Data were analyzed with the Mascot Distiller software package. The same samples were also measured with an LTQ-Orbitrap as described in a paper by Hessling et al.2 This second data set from the very same samples allowed cross-validation with a well-established workflow.
We proved that LC-MALDI is an appropriate option for the quantitative analysis of in vivo labeled samples on a proteome-wide scale. We identified nearly 850 proteins and quantified more than 550 proteins within a reasonable analysis time. The resulting protein ratios correlate well with existing LTQ-Orbitrap data and should encourage groups equipped with a MALDI-TOF-TOF instrument to perform large-scale quantitative proteomic experiments.
The measurement of the same samples with two mass analyzers, one using ESI and the other using MALDI, also allowed the investigation of possible biases of one or the other mass analyzer toward peptides with certain physicochemical characteristics. These ionization preferences principally open opportunities with both technical and biological potential for deeper analysis of proteomes and increased sequence coverage in general, but they also could allow the exploration specifically of detectable peptides and/or proteins that could not be found with a particular ionization technique. Existing comparative studies in this field are few so far. All of them are limited by technical or methodological constraints of their time. Investigations were mainly hampered by restricted technical opportunities in the past, leading to exemplary use of samples of low complexity (9, 10), the application of divergent sample preparations such as different LC systems for peptide fractionation (9), and a scale in terms of identification numbers (10, 11) that is too small to enable general conclusions. The large amount of data and the avoidance of any technical variations in the present study allowed the most comprehensive comparison of ESI- and MALDI-generated data to date and revealed physicochemical biases in the detection of peptides, which confirms the generally complementary nature of the two ionization techniques.
MATERIALS AND METHODS
Sample Preparation and Chromatography
The same samples analyzed in this study via an LC-MALDI workflow had already been analyzed in an LTQ-Orbitrap (Thermo Fisher Scientific, Bremen, Germany) by Hessling et al.2 In that work, S. aureus COL (12) was grown under agitation at 37 °C in Luria-Bertani medium (Invitrogen, Wiesbaden, Germany). At an A540 of 0.5, vancomycin (Roth, Karlsruhe, Germany) was added to a concentration of 4.5 mg/l. This led to a decreased growth rate about 30 min after the addition of the antibiotic. Cells were harvested 100 min after stress induction at an A540 of about 1.4. Unstressed control samples were grown in Luria-Bertani medium and harvested during exponential growth at an A540 of 1.4 as well. The cultivations were carried out in triplicate to obtain three biological replicates.
The 15N-labeled standard used for quantification was the same as that used by Hessling et al.2 and was added to every sample before mass spectrometry measurement (13). This standard was a combined pool of vancomycin-stressed cells and exponentially growing cells grown in a 15N-enriched Bioexpress medium (Cambridge Isotope Laboratories, Andover, MA) that was supplemented with 5 g/l glucose. The standard was mixed with equal amounts of vancomycin-stressed and unstressed samples grown in Luria-Bertani medium as early during sample preparation as possible. Any protein loss during the cell lysis, digestion, and measurement was accounted for by equally affecting the respective 15N-labeled protein.
Harvested cultures were centrifuged for 15 min at 7000g at 4 °C. Pelleted cells were then washed in TBS (50 mm Tris, 150 mm NaCl, pH 8.0) and resuspended in 50 mm triethylammonium bicarbonate buffer. 500 μl of glass beads with a 0.1-mm diameter were added, and cells were disrupted using a Precellys 24 homogenizer (Peq Lab, Erlangen, Germany) for 30 s at 6800 rpm. Cell debris and glass beads were separated from the proteins via centrifugation for 10 min at 4 °C and 21,500g. A second centrifugation step of 30 min at 4 °C and 21,500g removed insoluble and aggregated proteins to obtain the cytosolic fraction of soluble proteins. The protein concentration was determined with the Roti-Nanoquant protein assay (Roth, Karlsruhe, Germany) according to the manufacturer's instructions, and the same protein amount of 15N-labeled standard was added. The sample was analyzed using the GeLC-MS workflow described by Otto et al. (5). The proteins were separated via one-dimensional SDS-PAGE; the gel was cut into 12 pieces, which were tryptically digested at 37 °C overnight. The resulting peptide mixtures were separated via reversed-phase column chromatography (Waters BEH 1.7 μm, 100 μm inner diameter × 100 mm, Waters Corporation, Milford, MA) operated on a nanoACQUITY-UPLC (Waters Corporation, Milford, MA). Peptides were first concentrated and desalted on a trapping column (Waters nanoACQUITY UPLC column, Symmetry C18, 5 μm, 180 μm inner diameter × 20 mm, Waters Corporation, Milford, MA) for 3 min at a flow rate of 1 ml/min with 99% (v/v) buffer A (0.1% (v/v) acetic acid). Subsequently, the peptides were eluted and separated with a nonlinear 80-min gradient from 5% to 60% (v/v) acetonitrile in 0.1% (v/v) acetic acid at a constant flow rate of 400 nl/min.
The membrane samples, which were only qualitatively analyzed to investigate possible detection preferences linked to the peptides' hydrophobicity, were also the same as used by Hessling et al.2 The membrane shaving protocol was carried out as described by Wolff et al. (14). Briefly, the soluble loops of the membranes were digested first with Proteinase K, and this was followed by chymotryptic digestion of transmembrane domains. The peptide-containing solution was loaded on a nanoACQUITY UPLC System (Waters Corporation) equipped with an analytical column (nanoACQUITY UPLC column, BEH130 C18, 1.7 μm, 100 μm × 100 mm; Waters Corporation) operated at 60 °C at 400 nl/min. Peptides were loaded directly on the column, and after being washed for 30 min with 99% (v/v) buffer A (0.1% (v/v) acetic acid), the peptides were eluted in a 5-h linear gradient from 5% to 90% (v/v) buffer B (90% (v/v) acetonitrile in 0.1% (v/v) acetic acid).
MS Analysis
MALDI-TOF-TOF Measurements
For MALDI-TOF-TOF measurements, the LC was coupled online with a Probot Microfraction Collector (Dionex GmbH, Idstein, Germany). The LC eluate was mixed online via a T-split with matrix solution used for MALDI analyses (3.3 mg/ml α-cyano-4-hydroxycinnamic acid in 50% (v/v) acetonitrile and 0.5% (v/v) trifluoroacetic acid; flow rate of 1.6 μl/min) and spotted onto a MALDI target plate, with a spot collection time of 15 s. Spotted targets were subsequently subjected to the 5800-MALDI-TOF-TOF analyzer and measured using TOF/TOF™ Series Explorer™ Software V4.1.0 (AB Sciex, Foster City, CA). MS spectra were recorded in a mass range from 700 to 4000 Da with a focus mass of 1700 Da. For one main spectrum, 15 subspectra with 200 shots per subspectrum were accumulated using a random search pattern and continuous stage movement.
Up to 20 precursor ions per spot were selected for MS/MS measurement using a job-wide interpretation method, with a fraction-to-fraction precursor mass tolerance of 200 ppm. For one main MS/MS spectrum, up to 25 subspectra with 250 shots per subspectrum were accumulated. The DynamicExitTM Algorithm was enabled using the highest threshold settings. These settings resulted in about 25,000 to 30,000 MS and MS/MS spectra per biological replicate with an MS analysis time of 20 to 30 h.
ESI-LTQ-Orbitrap Measurements
For LTQ-Orbitrap measurements, the LC system was coupled online with the mass analyzer. Analyses were performed as described by Hessling et al.2
Data Analysis
MALDI Data Analysis Using Mascot
MzML files were extracted from the instrument's internal Oracle database using MS Data Converter Beta version 1.2 (AB Sciex, Foster City, CA), processed with the default MALDI-TOF-TOF processing options, and searched with the Mascot search engine (V2.2, Matrix Science, London, UK) using Mascot Distiller version 2.4.2. All files belonging to one sample were processed together in a discrete multi-file project and searched against an S. aureus COL target-decoy protein sequence database. This database was composed of all protein sequences of S. aureus COL extracted from the National Center for Biotechnology Information bacteria genomes (www.ncbi.nlm.nih.gov/sites/entrez?Db=%20genome&Cmd=Retrieve&dopt=Protein%20%C3%BETable&list_uids=610). A set of the reversed sequences created by Bioworks Browser 3.2 EF2, as well as common contaminants such as keratin, was appended, resulting in 5864 database entries in total. Search parameters were as follows: enzyme type, trypsin, allowing two missed cleavage sites; peptide tolerance, 150 ppm; tolerance for fragment ions, 0.5 Da; variable modifications, oxidation of methionine (15.99 Da) and carbamidomethylation of cysteine (57.02 Da). 15N quantification was enabled, and only singly charged peptides were taken into account. For identification, at least two peptides per protein had to exceed the Mascot identity or homology threshold, using a p value threshold of 0.01. The protein false-positive rate was calculated for each analysis according to Peng et al. (15) and never exceeded 0.5% on the protein or peptide level.
Quantification was performed with the Quantification Toolbox of Mascot Distiller. Peptides needed to pass the following quality thresholds: correlation threshold, 0.9; fraction threshold, 0.8; and standard error threshold, 0.19. Threshold values were defined through repeated processing of datasets of technical replicates to find the most suitable conditions for generating the highest number of quantifiable peptides with sufficient reproducibility (data not shown). The resulting reproducibility values in the experiment are shown in the “Results” section. Because files of gel bands belonging to the same sample were processed independently, Mascot Distiller could yield more than one quantification value per peptide. Using Excel (Microsoft Corporation, Redmond, WA), an intensity-weighted average of each peptide was calculated. The median of the quantification values of all peptides belonging to the same protein determined the protein quantification value. Protein quantification results were median-centered, and ratios were log 2-transformed.
MALDI Data Analysis Using Sequest
Protein identification using Sequest and DTASelect, which was used only for the direct comparison of MALDI and ESI data, was done as described for LTQ-Orbitrap data by Hessling et al.2 using the same mzML files as for the Mascot analysis.
MzML files were searched with SEQUEST version v28 (rev.12) (Thermo Fisher Scientific) against an S. aureus COL target-decoy protein sequence database. This database was composed of all protein sequences of S. aureus COL extracted from the National Center for Biotechnology Information bacteria genomes (www.ncbi.nlm.nih.gov/sites/entrez?Db=%20genome&Cmd=Retrieve&dopt=Protein%20%C3%BETable&list_uids=610). A set of the reversed sequences created by BioworksBrowser 3.2 EF2, as well as common contaminants such as keratin, was appended. The searches were performed in two iterations. First, for the membrane shaving approach, the following search parameters were applied: enzyme type, none; peptide tolerance, 150 ppm; tolerance for fragment ions, 0.5 Da; b- and y-ion series; oxidation of methionine (15.99 Da) and carbamidomethylation (57.02 Da) of cysteine considered as variable modifications with a maximum of three modifications per peptide.
For MS analysis of the cytosolic samples, the following search parameters were used: enzyme type, trypsin, allowing two missed cleavage sites; peptide tolerance, 150 ppm; tolerance for fragment ions, 0.5 Da; b- and y-ion series; variable modification, oxidation of methionine (15.99 Da) and carbamidomethylation of cysteine (57.02 Da); maximum of three modifications per peptide. In the second iteration, the mass shift of all amino acids completely labeled with 15N-nitrogen was taken into account in the search parameters. For cytosolic samples, the resulting *.dta and *.out files were assembled and filtered using DTASelect (version 2.0.25) with the following parameters: -y 2 (only fully tryptic peptides) -c 2 (lowest accepted charge state) -C 4 (highest accepted charge state) -here (include only IDs in the current directory) -decoy Reverse_ (prefix that identifies decoy hits in the database) -p 2 (minimum of two peptides per protein) -t 2 (purge duplicate spectra on basis of XCorr) -u (include only loci with uniquely matching peptides) -MC 2 (maximum number of missed cleavage sites is two) -i 0.3 (30% as lowest proportion of fragment ions observed) -fp 0.005 (target false-positive rate of 0.005). The SEQUEST search results for the membrane shaving samples were probabilistically validated with Scaffold V3.4.8 (Proteome Software, Portland, OR) applying 95% protein and peptide identification probability filters and a minimum of two identified peptides per protein. The protein false-positive rate was calculated for each analysis according to Peng et al. (15) and never exceeded 0.5% on the protein or peptide level.
LTQ-Orbitrap Data Analysis
Data generated by LTQ-Orbitrap analysis were searched and quantified as described by Hessling et al.2
RESULTS AND DISCUSSION
General Identification of Proteins
Analysis of the six different cytosolic samples (three exponentially grown biological replicates and three vancomycin-stressed biological replicates) resulted in the identification of 848 proteins in total. More than 50% of these proteins could be found in at least five of these six samples (Fig. 1). A maximum of 697 proteins could be identified in one sample. The false-positive rate was checked for each replicate according to Peng et al. (15) and never exceeded 0.5% on the protein or peptide level. The large number of proteins found in the majority of all analyzed samples proves the consistency and robustness of the method. Analysis of the same samples with an LTQ-Orbitrap resulted in the identification of 1165 proteins with comparable filter criteria and false discovery rates (Hessling et al.2); 60% of them were detected in at least five out of six samples. The Venn diagram in Fig. 2A shows the very high overlap of identifications for the two instruments on the protein level. The number of 39 proteins exclusively identified with MALDI-TOF-TOF mass spectrometry is low, but despite the low number of additional proteins, our dataset of 848 proteins identified on a MALDI instrument is large enough to allow meaningful quantitative investigations in a highly complex sample. Therefore, the almost invariable use of MALDI instruments in proteomic research for non-complex samples seems to underestimate the potential of this technique.
Fig. 1.

Frequency of protein identifications. The proportion of proteins that could be identified with MALDI-TOF-TOF in one to six out of the six analyzed samples (three exponentially grown replicates and three vancomycin-stressed replicates) is displayed. More than 50% of all 848 proteins were detected in at least five out of six samples (the 15N standard that was spiked into all samples theoretically contained all proteins present in the two different physiological conditions).
Fig. 2.
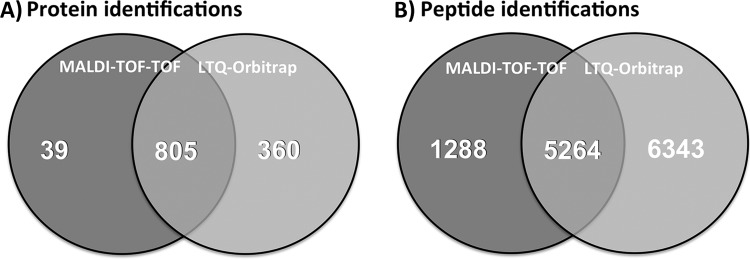
Venn diagrams of (A) identified proteins and (B) identified peptides. The numbers over dark gray areas represent identifications in the GeLC-MALDI-TOF-TOF workflow. The numbers over light gray areas represent identifications made via GeLC-LTQ-Orbitrap from the same samples. Numbers in the intersection areas relate to proteins that were identified with either method.
Quantification of Proteins
About half of all peptides identified via MALDI-TOF-TOF analysis could also be quantified on the basis of the corresponding 15N-labeled peptide of the spiked-in standard using the Mascot Distiller software. The proportion of identified peptides that could also be quantified via this method (quantification efficiency) is between 49% and 51% for the six different samples. In total, quantitative data for 554 proteins could be obtained, with nearly 50% of them in at least five out of six samples. Up to 415 proteins could be quantified in a single biological replicate.
The quantification efficiency for peptides identified with the LTQ-Orbitrap instrument and processed using Census software was around 80% for the different samples. This led to quantitative data for 1100 proteins; up to 972 proteins could be quantified in a single biological replicate. The significantly higher quantification efficiency for the LTQ-Orbitrap data might have been caused by the different data-analysis software packages, but quantification of LTQ-Orbitrap-generated data with MALDI Distiller delivered similar quantification efficiencies as with Census (80% to 90%). We suggest that the main reason for the lower quantification efficiency might be the lower number of MS1 scans per time frame of the LC run. As described in “Materials and Methods,” one fraction collected with the LC Probot represents 15 s of the LC run. In the LTQ-Orbitrap analysis, cycle times between two consecutive MS1 scans were less than 2 s. These highly time-resolved data enable a better statistical analysis of a peptide's elution peak. The higher the number of MS data points in such an elution peak is, the more accurate data processing in the quantification process will be, and therefore immanent statistical thresholds for trustworthy quantification will be less frequently exceeded, leading to higher quantification efficiency. A comparable time resolution for LC-MALDI-generated data is not achievable, as the analysis time would increase unreasonably. MS1 scans on a MALDI-TOF-TOF instrument take much more time than on the LTQ-Orbitrap, as one main MS1 spectrum is an average of multiple subspectra recorded on different locations of a spot. This is especially necessary in quantitative analyses in order to overcome the MALDI-specific problem of poor reproducibility of signal intensities (8).
14N/15N ratios for the same peptides of the different biological replicates analyzed with MALDI-TOF-TOF were in good correlation, as illustrated in Fig. 3A, in which peptide ratios of two exponentially growing biological replicates are plotted against each other. Calculated trend lines for these plots of biological replicates showed slopes near to 1 and an average coefficient of determination of 0.71. Given that these replicates include a biological variance, the correlation is high on technical level. The cross-validation of MALDI-TOF-TOF-generated peptide ratios with the LTQ-Orbitrap-generated data from the same sample is shown in Fig. 3B. The very good correlation of our data that were processed with Mascot Distiller with data generated by a well-established quantification workflow (16) that used the Census software (17) proves the general reliability of the new workflow.
Fig. 3.

Scatter plots for (A) two replicates analyzed with MALDI-TOF-TOF and (B) the same sample analyzed with MALDI-TOF-TOF and with LTQ-Orbitrap. A, log2 ratios of two exponentially grown biological replicates against the internal standard, both analyzed with MALDI-TOF-TOF, are plotted against each other. B, log2 ratios of one exponentially grown biological replicate against the internal standard, one analyzed with MALDI-TOF-TOF and the other with LTQ-Orbitrap, are plotted against each other. Linear trend lines (black) with the linear equation (y) and coefficient of determination (R2) were calculated.
There have been reports about the non-quantitative nature of MALDI-TOF-TOF mass spectrometry that refer to its reproducibility problems in term of spot-to-spot variability (18–21). This is mainly caused by inhomogeneous samples due to so-called hot spot formation in the crystallization process (22). Also, the ion suppression effect in the MALDI process hampers quantitative analysis with this ionization technique (23). But our results show that by averaging enough laser shots over a large sample area and using isotopically labeled peptides as internal standards, which were proposed strategies for overcoming the MALDI-specific problems in quantification (8), we were indeed able to obtain reproducible and reliable data (Fig. 3). Thus we converted these theoretical considerations into a viable experimental strategy.
Sensitivity of MALDI and ESI
The number of identified peptides was 6552 in the MALDI-TOF-TOF approach and 11,607 in the LTQ-Orbitrap analysis. Even though the absolute numbers point at a considerably higher sensitivity for the LTQ-Orbitrap in our analysis setup, the Venn diagram of peptide-level data shows considerably less overlap than that for the protein level (Fig. 2). This indicates that the two mass spectrometry techniques are complementary. In our case, an additional measurement with MALDI-TOF-TOF did not increase the number of identified proteins significantly, but it would enhance the sequence coverage of the identified proteins. The sample preparation and fractionation parameters were chosen not only to yield good sensitivity, but also to allow sample analysis in an appropriate time frame and direct comparison to the LTQ-Orbitrap analysis. Reducing sample collection times of the LC run fractionation would be advantageous for peptide detection but would be accompanied by an extended analysis time. With given parameters, the analysis time for one sample was between 20 and 30 h and thus was similar to that in the LTQ-Orbitrap analysis.
Previous studies comparing MALDI and ESI instruments with respect to their sensitivity in proteomic analysis have come up with different results, but the detection levels of the two compared instruments mostly did not vary as strongly as in the present study (3, 9, 11, 24, 25). Here we have to point out that the speed of technological development in the two ionization branches has varied in recent years. Faster development of new MALDI generations is highly desired in order to close this technological gap. We used state-of-the-art mass spectrometers for both ionization techniques, and we suggest that the amount of effort recently put into the development of LC-MS optimized ESI-MS/MS instruments resulted in the higher sensitivity of the ESI instrument in our study. But we also have to stress that we used one of the most sensitive ESI instruments available for the comparison. Several other types of ESI instruments commonly used in laboratories do not reach this high level of sensitivity and acquisition speed. With such instruments, the proportion of proteins and peptides specifically detected with ESI is much smaller, and the portion of MALDI-specific peptides and proteins is considerably higher (data not shown). Therefore, the MALDI approach might especially be taken into consideration by those laboratories not always equipped with the most modern generation of ESI instruments.
Detection Preferences of Both Ionization Methods
Despite this general disadvantage regarding the sensitivity of existing MALDI instruments relative to the most modern ESI instruments, the complementary nature of the two ionization techniques that was recognized in the few studies conducted in the past (9, 10) is still apparent. A strong indication of this is the observed discrepancy between very few exclusively identified proteins and a comparatively high number of exclusively identified peptides in the MALDI-MS-based data. This means that different peptides originating from the same protein, and therefore present in the same amounts, were specifically detected with either MS instrument. This observation may be linked to a preferential detection of peptides based on their biochemical properties.
The analysis of the same samples separated with the same chromatographic systems and parameters but two different mass spectrometry instruments allowed us to investigate whether peptides with certain biochemical features are preferentially detected by either instrument. Existing literature asserts different biases of MALDI or ESI ionization toward certain peptide properties. There are common hypotheses for both ionization methods (e.g. higher sensitivity of MALDI instruments toward more basic peptides) (26), but these findings are based on the analysis of peptides with different biochemical properties on the same instrument and therefore have only limited value for detailed conclusions about ionization preferences. Different systematic studies carried out on either single instrument type revealed that MALDI and ESI both preferentially ionize peptides with a large proportion of hydrophobic amino acids (27, 28). Moreover, these single-instrument analyses were restricted to small sets of synthetic (27) or small peptides (28), limiting the statistical inference for general conclusions with respect to ionization preferences. Additionally, these analyses of low-complex samples miss the large differences in peptide abundance and frequently occurring suppression effects that large-scale proteome studies have to deal with. Only a global, comparative analysis of the same samples on both instrument types allows one to determine whether peptides with certain characteristics are generally hard to access via mass spectrometry or whether changing the ionization technique would lead to an increase in sensitivity for such peptides. But comparative proteomic studies of MALDI and ESI instruments that directly compare both ionization techniques have been rare so far and generally are too small in terms of identification numbers to allow a statistically relevant analysis of identified peptides (9–11).
Our large dataset of nearly 13,000 identified peptides, of which more than 5200 could be detected with both mass analyzers, overcomes these restrictions and allows a statistically meaningful comparison of an ESI and a MALDI instrument regarding their biases in the ionization of peptides. In order to assign occurring differences between the two datasets to the different mass analyzers distinctively, we wanted to harmonize not only sample preparation, but also data processing for both datasets as far as possible. We performed the same database search for the three exponentially grown replicates analyzed with MALDI-TOF-TOF as we did in a previous study for the LTQ-Orbitrap-generated data, which means that data were searched with SEQUEST algorithm and afterward filtered with DTASelect to reach a target false-discovery rate of 0.5%. These results were compared with the corresponding LTQ-Orbitrap data.
To emphasize differences between the two detection techniques, we focused on peptides that were preferentially identified by only one mass analyzer. A preferentially identified peptide was specified as a peptide that was detected in all three replicates with one mass analyzer and at most in one replicate with the other mass analyzer, or in two out of the three replicates with one mass analyzer but in no replicate with the other. A comparison of the relative abundance of the different amino acids showed significant differences between the two datasets generated with either MALDI-TOF-TOF or LTQ-Orbitrap.
Composition-dependent Preferences of MALDI Ionization
An inspection of the data presented in Fig. 4 revealed that peptides containing an arginine or an aromatic amino acid were preferably detected by MALDI-TOF-TOF, whereas the presence of a lysine resulted in preferential detection with the LTQ-Orbitrap. About 65% of all MALDI-identified peptides shown in Fig. 2B ended with a C-terminal arginine, in contrast to only 24% for peptides identified with the LTQ-Orbitrap. The relative abundance of arginine is almost three times greater in MALDI-specific peptides than in Orbitrap-specific peptides (Fig. 4). The preferential MALDI ionization of tryptic peptides containing arginine was also observed by Krause and co-workers (29), as well as in different ionization comparison studies (9, 30). A more recent study by Dupré and co-workers seems to contradict these observations by noting higher quality MS/MS spectra from ESI instruments when analyzing arginine-containing peptides, whereas MALDI produced MS/MS spectra of higher quality when analyzing arginine-free peptides (31). This study was based on only 15 synthetically designed Lys-N proteolytic peptides and is not corroborated by our large dataset analysis of tryptic peptides.
Fig. 4.

Relative abundance of amino acids in peptides preferentially identified by either instrument. The proportion of all 20 proteinogenic amino acids is shown for peptides preferentially identified by either MALDI-TOF-TOF analysis (dark gray) or LTQ-Orbitrap analysis (light gray).
The high proportion of the aromatic amino acids tyrosine, tryptophan, and phenylalanine that we observed in our MALDI-detected peptides has been recognized before in limited datasets of particular groups of proteins (9) or synthetic peptides (32) and might be attributable to the beneficial photo excitation of these moieties during ionization (26). Additionally, the larger proportion of the secondary amino acid proline in our study in MALDI-identified peptides is noticeable. Baumgart and coworkers reported a single-instrument analysis in which MALDI generally preferred peptides with a large proportion of proline, arginine, phenylalanine, and leucine (27). Except for leucine, we can now support these findings in direct comparison to an ESI instrument (Fig. 4).
Our observed identification frequencies (Fig. 4) open interesting opportunities for research on MALDI-preferred peptides. All amino acids appreciably increased in MALDI analysis—arginine, tyrosine, tryptophan, phenylalanine, and proline—are amino acids of low occurrence in organisms. One of the rarest amino acids in S. aureus, tryptophan, with an average frequency of ∼1% in proteins, could be detected in ESI-specific peptides with a much lower rate of 0.2% only, whereas MALDI-specific peptides reflected its natural occurrence. An enhanced excitation in the MALDI process correlates to reported features of the indole moiety of tryptophan in fluorescence; its strong fluorescence masks the fluorescence of any other aromatic amino acid. But the amino acids containing a benzene moiety, tyrosine and phenylalanine, are even more overrepresented in MALDI-specific peptides, at around 5% and 6%. Their natural occurrence is between 4% and 4.5%, and they are underrepresented in ESI-specific peptides, at 3% or less. The non-aromatic ring moiety of proline seems to influence ionization efficiency in MALDI positively, too. Proline occurs in the ESI-specific data at little more than 3%, which fits its natural occurrence in S. aureus. In the MALDI analyses, proline was overrepresented with a proportion of nearly 5%. Proline-containing peptides might be of special interest in certain proteomics projects because of its ability to compromise secondary structures of proteins. Therefore, specifically structural peptides might be easier to ionize, as several structural proteins have higher proline concentrations. Altogether, these MALDI-specific preferences for amino acids of low occurrence should be used in future experiments targeting peptides containing these amino acids.
The amino acid of lowest occurrence in S. aureus, with a natural frequency below 1%, is cysteine. Cysteine was generally discriminated in our experimental setup and is too scarce to be considered in statistically reliable conclusions. This might be strongly linked to the first step of separation via one-dimensional SDS-PAGE, which could prevent the elution of insufficiently alkylated peptides. But we find it remarkable that this amino acid of lowest occurrence, which was probably discriminated in an early step of the sample preparation, could be found in MALDI with a distinguishable proportion, whereas in ESI it was almost undetectable (Fig. 4). This should be explored in a more suitable experimental setup in future.
Composition-dependent Preferences of ESI
Besides the already mentioned overrepresentation of lysine, there were some more amino acids with a significantly higher proportion in the peptides preferentially detected with the LTQ-Orbitrap in our data (Fig. 4). These amino acids often share certain biochemical properties. The aliphatic amino acids alanine, valine, leucine, and isoleucine all showed a moderately higher proportion in the LTQ-Orbitrap-identified peptides. This is true to a larger extent for methionine, which is not strictly aliphatic because of its sulfur-containing side chain but which shares chemical attributes of this group. Proline was the only exception in the class of aliphatic amino acids, as it occurred far more often in peptides specifically identified via MALDI. As already mentioned, both ionization techniques separately have been reported to favor hydrophobic over hydrophilic peptides (27, 28). The direct comparison previously revealed a higher preference of ESI toward aliphatic peptides in the particular group of DNA-binding proteins (9), which correlates well with our genome-wide observations. A positive effect of MALDI in terms of peptide ionization caused by branched amino acids that was noticed by Valero et al. (32) cannot be confirmed by our study, which shows moderate preferences of ESI for these amino acids.
Amino acids with a carboxyl-group-containing side chain (aspartic acid and glutamic acid) and two amino acids with a hydroxyl-group-containing side chain (serine and threonine) could be found in higher proportions in preferentially LTQ-Orbitrap-identified peptides (Fig. 4). The third proteinogenic, hydroxyl-containing amino acid, tyrosine, occurred more often in MALDI-specific data, as already mentioned. The strong bias of MALDI for aromatic residues seems to superimpose the relatively weak effect of the hydroxyl group in this amino acid. A preference of ESI was described for hydroxyl-group-containing peptides (9), but not for carboxyl-containing peptides that could be observed in our study for the first time.
Biochemical Properties of Preferentially Identified Peptides
The differential preferences of the two mass analyzers for certain amino acid classes correlate with differences in the biochemical properties of the peptides preferentially identified with one of the two mass spectrometers. The comparison of the isoelectric points (pI), calculated with ExPASy's Compute pI/Mw program, showed a higher proportion of peptides with lower pI values for the LTQ-Orbitrap-detected peptides and with higher pI values for the MALDI-TOF-TOF-based peptides (Fig. 5A). The average pI of all exclusively identified peptides (6.01) was higher in the MALDI-TOF-TOF- than in the LTQ-Orbitrap-obtained data (5.28). The bias of MALDI for peptides that contain a large proportion of basic amino acids becomes even more evident when comparing average pIs of all amino acids of the identified peptides. In Fig. 5B, the distribution curve of this average amino acid pI based on MALDI-TOF-TOF data is clearly shifted to more basic values. If aspartic and glutamic acid, which additionally carry acidic carboxyl groups, are omitted from this calculation, this shift is preserved (data not shown). Therefore, we can conclude that the shift displays a general phenomenon independent of the preference of the Orbitrap for peptides containing aspartic and glutamic acid.
Fig. 5.

Basicity of identified peptides. Relative occurrence of peptides in a certain pI range is shown for peptides preferentially identified with MALDI-TOF-TOF (dark gray) and LTQ-Orbitrap (light gray). In A, the frequency of peptides is shown according to the peptides' pI; in B, the frequency is according to the average pI of the amino acids of the peptides.
We also calculated the grand average of hydropathy (GRAVY) of the identified peptides according to Kyte and Doolittle (33). Positive GRAVY values indicate hydrophobic peptides (the higher the GRAVY, the stronger the hydrophobicity), whereas negative values indicate hydrophilic peptides (the lower the GRAVY, the stronger the hydrophilic character). The distribution of cytosolic peptides according to their GRAVY values is shown in Fig. 6A. The obtained overlap of the two distribution curves seems to contradict the supposed bias of ESI-MS for peptides containing more hydrophobic amino acids. To further investigate a possible preference for hydrophobic peptides of either ionization technique, we additionally analyzed the membrane shaving fraction, purified and analyzed via LTQ-Orbitrap by Hessling et al.,2 with MALDI-TOF-TOF. The membrane shaving fraction especially contains the hydrophobic membrane-spanning domains of peptides (14) and had never been analyzed before with MALDI-MS. In the preparation of these fractions, cell membranes are spun down via ultracentrifugation and digested with Proteinase K to deplete the soluble loops of membrane-associated proteins. In a consecutive step, the transmembrane domains are digested by chymotrypsin and are then accessible in mass spectrometry workflows. We compiled distribution curves of these more hydrophobic peptides according to their GRAVY values as well (Fig. 6B). The distribution curve of the preferentially LTQ-Orbitrap-identified peptides was clearly shifted to higher GRAVY values relative to MALDI-preferred peptides, supporting the reported bias of ESI for hydrophobic peptides (9) and the preference of ESI-MS for peptides containing aliphatic amino acids. The observed effects of single aliphatic amino acids were comparatively low, and we believe that the high proportion of aliphatic amino acids in the membrane shaving fraction and the preferential detection of peptides containing these amino acids by ESI-MS resulted in the considerably shifted distribution in this fraction.
Fig. 6.

Hydrophobicity of identified peptides. The relative occurrence of peptides in a certain GRAVY range is shown for peptides preferentially identified with MALDI-TOF-TOF (dark gray) and LTQ-Orbitrap (light gray). A is based on peptides identified in the cytosolic samples. B is based on peptides identified in the membrane shaving fraction.
An additional discriminating peptide attribute for detection by MALDI and ESI instruments was reported to be the peptide mass. Lasaosa and coworkers found a higher percentage of peptides with masses below 1400 Da in their LC-MALDI approach and a larger proportion of peptides with masses higher than 1400 Da in their LC-ESI analysis (11). In contrast, Stapels and Barofsky (9) and Seymour et al. (34) did not find any dependence on molecular mass. A discriminating effect of peptide mass could not be observed in our analysis, either (Fig. 7). Distribution curves of peptides detected with either instrument type showed variation, indeed, but a significant, mass-related bias could not be detected, supporting the observations of Stapels and Barofsky (9) and Seymour and coworkers (34).
Fig. 7.

Molecular weight of identified peptides. The relative occurrence of peptides in a certain molecular weight range is shown for peptides preferentially identified with MALDI-TOF-TOF (dark gray) and LTQ-Orbitrap (light gray).
Finally, we have to point out that highly sophisticated instruments always differ in more than one single component. The two instruments used for this study vary in far more than their ionization method (e.g. different mass analyzers and fragmentation methods), and these technical distinctions might have contributed to our observed peptide preferences. However, the overlapping of results of our analysis with those from prior comparative studies using completely different instrument types (but always an ESI and a MALDI instrument) strongly indicate primarily ionization-based results.
CONCLUSION
We were able to prove that the combination of global quantitative LC-MS analysis with MALDI-TOF-TOF instruments is feasible. Modern instrumentation and software solutions can be used to collect high-quality data in a reasonable time. Although the comparison with a state-of-the-art ESI instrument showed discrepancies in terms of identification numbers and quantification efficiency, it also revealed a complementary nature of the ionization techniques.
Most observed biases in our study, such as the tendency of MALDI to prefer peptides containing arginine, proline, and aromatic amino acids or of ESI to favor peptides containing hydrophobic amino acids, support earlier findings, but many contrary observations were made in the past as well. The technical or methodological constraints of earlier studies that aimed at a better understanding of peptide ionization preferences resulted in such uncertainties and partial contradictions. Single-instrument analysis, samples of low complexity, the application of divergent sample preparations such as different LC systems for peptide fractionation, and samples that were too small in terms of identification numbers hampered significant interpretation in most existing studies, and as a result minor effects might be overrepresented in the literature. Using a systematic approach, we tried to resolve this situation by undertaking a genome-wide analysis of highly complex samples and a detailed comparison of ionization preferences of almost 13,000 peptides on a scale unparalleled to date. The few existing studies also comparing MALDI and ESI peptide ionization used technically highly different instruments. Observed preferences consistent with earlier studies can therefore be attributed to the two applied ionization techniques. Contributions of other technical discrepancies between these two instrument types cannot be completely excluded, but such effects are not obvious.
We observed complementary results in terms of preferences of either technique for certain amino acids and biochemical properties of peptides. For practical purposes, these tendencies can be utilized, for example, in studies targeting proline-containing structural peptides and proteins or peptides specifically containing aromatic amino acids and arginine, or in the selective application of MALDI and ESI instruments to increase sequence coverage.
Supplementary Material
Acknowledgments
We thank Holger Kock for critically reading the manuscript.
Footnotes
* This work was supported by grants from the DFG (SFB/Transregio 34) and EU (BaSysBio LSHG-CT-2006-037469 and BaSynthec FP7-244093).
2 Hessling, B., Herbst, F.-A., Bonn, F., Otto, A., Rappen, G.-M., Bernhardt, J., Hecker, M., and Becher, D. Global proteome analysis of vancomycin stress in Staphylococcus aureus. Accepted for publication in the International Journal of Medical Microbiology.
1 The abbreviations used are:
- ESI
- electrospray ionization
- GeLC-MS
- one-dimensional gel liquid chromatography mass spectrometry
- GRAVY
- grand average of hydropathy
- S. aureus
- Staphylococcus aureus.
REFERENCES
- 1. Ong S.-E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 2. Oda Y., Huang K., Cross F. R., Cowburn D., Chait B. T. (1999) Accurate quantitation of protein expression and site-specific phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 96, 6591–6596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kuzyk M. A., Ohlund L. B., Elliott M. H., Smith D., Qian H., Delaney A., Hunter C. L., Borchers C. H. (2009) A comparison of MS/MS-based, stable-isotope-labeled, quantitation performance on ESI-quadrupole TOF and MALDI-TOF/TOF mass spectrometers. Proteomics 9, 3328–3340 [DOI] [PubMed] [Google Scholar]
- 4. Lau K. W., Jones A. R., Swainston N., Siepen J. A., Hubbard S. J. (2007) Capture and analysis of quantitative proteomic data. Proteomics 7, 2787–2799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Otto A., Bernhardt J., Hecker M., Becher D. (2012) Global relative and absolute quantitation in microbial proteomics. Curr. Opin. Microbiol. 15, 364–372 [DOI] [PubMed] [Google Scholar]
- 6. Mueller D. R., Voshol H., Waldt A., Wiedmann B., Van Oostrum J. (2007) LC-MALDI MS and MS/MS—an efficient tool in proteome analysis. Subcell. Biochem. 43, 355–380 [DOI] [PubMed] [Google Scholar]
- 7. Chen H., Rejtar T., Andreev V., Moskovets E., Karger B. L. (2005) Enhanced characterization of complex proteomic samples using LC-MALDI MS/MS: exclusion of redundant peptides from MS/MS analysis in replicate runs. Anal. Chem. 77, 7816–7825 [DOI] [PubMed] [Google Scholar]
- 8. Szájli E., Fehér T., Medzihradszky K. F. (2008) Investigating the quantitative nature of MALDI-TOF MS. Mol. Cell. Proteomics 7, 2410–2418 [DOI] [PubMed] [Google Scholar]
- 9. Stapels M. D., Barofsky D. F. (2004) Complementary use of MALDI and ESI for the HPLC-MS/MS analysis of DNA-binding proteins. Anal. Chem. 76, 5423–5430 [DOI] [PubMed] [Google Scholar]
- 10. Bodnar W. M., Blackburn R. K., Krise J. M., Moseley M. A. (2003) Exploiting the complementary nature of LC/MALDI/MS/MS and LC/ESI/MS/MS for increased proteome coverage. J. Am. Soc. Mass Spectrom. 14, 971–979 [DOI] [PubMed] [Google Scholar]
- 11. Lasaosa M., Delmotte N., Huber C. G., Melchior K., Heinzle E., Tholey A. (2009) A 2D reversed-phase x ion-pair reversed-phase HPLC-MALDI TOF/TOF-MS approach for shotgun proteome analysis. Anal. Bioanal. Chem. 393, 1245–1256 [DOI] [PubMed] [Google Scholar]
- 12. Shafer W. M., Iandolo J. J. (1979) Genetics of staphylococcal enterotoxin B in methicillin-resistant isolates of Staphylococcus aureus. Infect. Immun. 25, 902–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. MacCoss M. J., Wu C. C., Liu H., Sadygov R., Yates J. R. (2003) A correlation algorithm for the automated quantitative analysis of shotgun proteomics data. Anal. Chem. 75, 6912–6921 [DOI] [PubMed] [Google Scholar]
- 14. Wolff S., Hahne H., Hecker M., Becher D. (2008) Complementary analysis of the vegetative membrane proteome of the human pathogen Staphylococcus aureus. Mol. Cell. Proteomics 7, 1460–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peng J., Elias J. E., Thoreen C. C., Licklider L. J., Gygi S. P. (2003) Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J. Proteome Res. 2, 43–50 [DOI] [PubMed] [Google Scholar]
- 16. Otto A., Bernhardt J., Meyer H., Schaffer M., Herbst F.-A., Siebourg J., Mäder U., Lalk M., Hecker M., Becher D. (2010) Systems-wide temporal proteomic profiling in glucose-starved Bacillus subtilis. Nat. Commun. 1, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park S. K., Venable J. D., Xu T., Yates J. R. (2008) A quantitative analysis software tool for mass spectrometry-based proteomics. Nat. Methods 5, 319–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tholey A., Zabet-Moghaddam M., Heinzle E. (2006) Quantification of peptides for the monitoring of protease-catalyzed reactions by matrix-assisted laser desorption/ionization mass spectrometry using ionic liquid matrixes. Anal. Chem. 78, 291–297 [DOI] [PubMed] [Google Scholar]
- 19. Bucknall M., Fung K. Y. C., Duncan M. W. (2002) Practical quantitative biomedical applications of MALDI-TOF mass spectrometry. J. Am. Soc. Mass Spectrom. 13, 1015–1027 [DOI] [PubMed] [Google Scholar]
- 20. Dekker L. J., Dalebout J. C., Siccama I., Jenster G., Sillevis Smitt P. A., Luider T. M. (2005) A new method to analyze matrix-assisted laser desorption/ionization time-of-flight peptide profiling mass spectra. Rapid Commun. Mass Spectrom. 19, 865–870 [DOI] [PubMed] [Google Scholar]
- 21. Tholey A., Heinzle E. (2006) Ionic (liquid) matrices for matrix-assisted laser desorption/ionization mass spectrometry-applications and perspectives. Anal. Bioanal. Chem. 386, 24–37 [DOI] [PubMed] [Google Scholar]
- 22. Garden R. W., Sweedler J. V. (2000) Heterogeneity within MALDI samples as revealed by mass spectrometric imaging. Anal. Chem. 72, 30–36 [DOI] [PubMed] [Google Scholar]
- 23. Knochenmuss R., Dubois F., Dale M. J., Zenobi R. (1996) The matrix suppression effect and ionization mechanisms in matrix-assisted laser desorption/ionization. Rapid Commun. Mass Spectrom. 10, 871–877 [Google Scholar]
- 24. Hattan S. J., Marchese J., Khainovski N., Martin S., Juhasz P. (2005) Comparative study of [three] LC-MALDI workflows for the analysis of complex proteomic samples. J. Proteome Res. 4, 1931–1941 [DOI] [PubMed] [Google Scholar]
- 25. Stapels M. D., Cho J.-C., Giovannoni S. J., Barofsky D. F. (2004) Proteomic analysis of novel marine bacteria using MALDI and ESI mass spectrometry. J. Biomol. Tech. 15, 191–198 [PMC free article] [PubMed] [Google Scholar]
- 26. Burkitt W. I., Giannakopulos A. E., Sideridou F., Bashir S., Derrick P. J. (2003) Discrimination effects in MALDI-MS of mixtures of peptides—analysis of the proteome. Aust. J. Chem. 56, 369–377 [Google Scholar]
- 27. Baumgart S., Lindner Y., Kühne R., Oberemm A., Wenschuh H., Krause E. (2004) The contributions of specific amino acid side chains to signal intensities of peptides in matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun. Mass Spectrom. 18, 863–868 [DOI] [PubMed] [Google Scholar]
- 28. Cech N. B., Enke C. G. (2000) Relating electrospray ionization response to nonpolar character of small peptides. Anal. Chem. 72, 2717–2723 [DOI] [PubMed] [Google Scholar]
- 29. Krause E., Wenschuh H., Jungblut P. R. (1999) The dominance of arginine-containing peptides in MALDI-derived tryptic mass fingerprints of proteins. Anal. Chem. 71, 4160–4165 [DOI] [PubMed] [Google Scholar]
- 30. Medzihradszky K. F., Leffler H., Baldwin M. A., Burlingame A. L. (2001) Protein identification by in-gel digestion, high-performance liquid chromatography, and mass spectrometry: peptide analysis by complementary ionization techniques. J. Am. Soc. Mass Spectrom. 12, 215–221 [DOI] [PubMed] [Google Scholar]
- 31. Dupré M., Cantel S., Verdié P., Martinez J., Enjalbal C. (2011) Sequencing Lys-N proteolytic peptides by ESI and MALDI tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 22, 265–279 [DOI] [PubMed] [Google Scholar]
- 32. Valero M.-L., Giralt E., Andreu D. (1999) An investigation of residue-specific contributions to peptide desorption in MALDI-TOF mass spectrometry. Lett. Pept. Sci. 6, 109–115 [Google Scholar]
- 33. Kyte J., Doolittle R. F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 34. Seymour S., Booy A., Gundry R., Van Eyk J., Hunter C. (2010) Assessing the Complementarities of MALDI and ESI for Protein Identification in Complex Mixtures, Technical Note, AB SCIEX, 1–5, Foster City, CA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.