

Abstract
Protein kinases are implicated in multiple diseases such as cancer, diabetes, cardiovascular diseases, and central nervous system disorders. Identification of kinase substrates is critical to dissecting signaling pathways and to understanding disease pathologies. However, methods and techniques used to identify bona fide kinase substrates have remained elusive. Here we describe a proteomic strategy suitable for identifying kinase specificity and direct substrates in high throughput. This approach includes an in vitro kinase assay-based substrate screening and an endogenous kinase dependent phosphorylation profiling. In the in vitro kinase reaction route, a pool of formerly phosphorylated proteins is directly extracted from whole cell extracts, dephosphorylated by phosphatase treatment, after which the kinase of interest is added. Quantitative proteomics identifies the rephosphorylated proteins as direct substrates in vitro. In parallel, the in vivo quantitative phosphoproteomics is performed in which cells are treated with or without the kinase inhibitor. Together, proteins phosphorylated in vitro overlapping with the kinase-dependent phosphoproteome in vivo represents the physiological direct substrates in high confidence. The protein kinase assay-linked phosphoproteomics was applied to identify 25 candidate substrates of the protein-tyrosine kinase SYK, including a number of known substrates and many novel substrates in human B cells. These shed light on possible new roles for SYK in multiple important signaling pathways. The results demonstrate that this integrated proteomic approach can provide an efficient strategy to screen direct substrates for protein tyrosine kinases.
Protein phosphorylation plays a pivotal role in regulating biological events such as protein–protein interactions, signal transduction, subcellular localization, and apoptosis (1). Deregulation of kinase-substrate interactions often leads to disease states such as human malignancies, diabetes, and immune disorders (2). Although a number of kinases are being targeted to develop new drugs, our understanding of the precise relationships between protein kinases and their direct substrates is incomplete for the majority of protein kinases (3). Thus, mapping kinase–substrate relationships is essential for the understanding of biological signaling networks and the discovery and development of drugs for targeted therapies (4). Toward this goal, various in vitro kinase assays using synthetic peptide libraries (5), phage expression libraries (6), protein arrays (7–9), or cell extracts (10, 11) have been explored for the screening of kinase substrates.
Besides classical biochemical and genetic methods, mass spectrometry-based high throughput approaches have become increasingly attractive because they are capable of sequencing proteins and localizing phosphorylation sites at the same time. Mass spectrometry-based proteomic methods have been extensively applied to kinase-substrate interaction mapping (12) and global phosphorylation profiling (13–15). Although thousands of phosphorylation events can be inspected simultaneously (16, 17), large-scale phosphoproteomics does not typically reveal direct relationships between protein kinases and their substrates.
Recently, several mass spectrometry-based proteomic strategies have been introduced for identifying elusive kinase substrates (7, 18, 19). Taking advantage of recent advances of high speed and high-resolution mass spectrometry, these methods used purified, active kinases to phosphorylate cell extracts in vitro, followed by mass spectrometric analysis to identify phosphoproteins. These approaches commonly face the major challenge of distinguishing phosphorylation events triggered by the kinase reaction from background signals introduced by endogenous kinase activities (20). To dissect the phosphorylation cascade, Shokat and colleagues developed an approach named Analog-Sensitive Kinase Allele (ASKA)1 (21). In their approach, a kinase is engineered to accept a bulky-ATP analog exclusively so that direct phosphorylation caused by the analog-sensitive target kinase can be differentiated from that of wild type kinases. As a result, indirect effects caused by contaminating kinases during the in vitro kinase assay are largely eliminated. ASKA has recently been coupled with quantitative proteomics, termed Quantitative Identification of Kinase Substrates (QIKS) (12), to identify substrate proteins of Mek1. Recently, one extension of the ASKA technique is for the analog ATP to carry a γ-thiophosphate group so that in vitro thiophosphorylated proteins can be isolated for mass spectrometric detection (22–24). In addition to ASKA, radioisotope labeling using [γ-32P]ATP (10), using concentrated purified kinase (25), inactivating endogenous kinase activity by an additional heating step (11), and quantitative proteomics (26, 27) are alternative means aimed to address the same issues. All of these methods, however, have been limited to the identification of in vitro kinase substrates.
To bridge the gap between in vitro phosphorylation and physiological phosphorylation events, we have recently introduced an integrated strategy termed Kinase Assay-Linked Phosphoproteomics (KALIP) (28). By combining in vitro kinase assays with in vivo phosphoproteomics, this method was demonstrated to have exceptional sensitivity for high confidence identification of direct kinase substrates. The main drawback for the KALIP approach is that the kinase reaction is performed at the peptide stage to eliminate any problems related to contamination by endogenous kinases. However, the KALIP method may not be effective for kinases that require a priming phosphorylation event (i.e. a previous phosphorylation, on substrate or kinase, has effect on following phosphorylation) (29), additional interacting surfaces (30), or a docking site on the protein (31). For example, basophilic kinases require multiple basic resides for phosphorylation and tryptic digestion will abolish these motifs, which are needed for effective kinase reactions.
We address the shortcoming by introducing an alternative strategy termed Protein Kinase Assay-Linked Phosphoproteomics (proKALIP). The major difference between this method and the previous KALIP method is the utilization of protein extracts instead of digested peptides as the substrate pool. The major issue is how to reduce potential interference by endogenous kinase activities. One effective solution is to use a generic kinase inhibitor, 5′-(4-fluorosulfonylbenzoyl)adenosine (FSBA), which was widely used for covalent labeling of kinases (32, 33), kinase isolation (34), kinase activity exploration (35, 36), and more recently kinase substrate identification by Kothary and co-workers (37). However, an extra step is required to effectively remove the inhibitor before the kinase reaction, which may decrease the sensitivity. ProKALIP addresses the issue by carrying out the kinase reaction using formerly in vivo phosphorylated proteins as candidates. This step efficiently improves the sensitivity and specificity of the in vitro kinase reaction. Coupled with in vivo phosphoproteomics, proKALIP has gained a high sensitivity and provided physiologically relevant substrates with high confidence.
To demonstrate the proKALIP strategy, the protein-tyrosine kinase SYK was used as our target kinase. SYK is known to play a crucial role in the adaptive immune response, particularly in B cells, by facilitating the antigen induced B-cell receptor (BCR) signaling pathways and modulating cellular responses to oxidative stress in a receptor-independent manner (38, 39). SYK also has diverse biological functions such as innate immune recognition, osteoclast maturation, cellular adhesion, platelet activation, and vascular development (38). In addition, the expression of SYK is highly correlated to tumorigenesis by promoting cell–cell adhesion and inhibiting the motility, growth, and invasiveness of certain cancer cells (40). In this study, we attempt to identify bona fide substrates of SYK in human B cells using the proKALIP approach and demonstrate the specificity and sensitivity of this strategy.
EXPERIMENTAL PROCEDURES
Cell Culture
Human DG-75 B lymphoma cells (ATCC) were grown in RPMI 1640 media (Sigma) supplemented with 10% heat inactivated fetal bovine serum (FBS), 1 mm sodium pyruvate, 100 μg/ml streptomycin, 100 IU/ml penicillin, and 0.05 mm 2-mercaptoethanol in 5% CO2 at 37 °C. For stable isotope labeling of amino acids in cell culture (SILAC) experiments, cells were grown in SILAC RPMI-1040 media (Sigma) supplemented with 10% dialyzed inactivated FBS (Sigma), 1 mm sodium pyruvate, 100 μg/ml streptomycin, 100 IU/ml penicillin, and 0.05 mm 2-mercaptoethanol, and either l-Lysine and l-Arginine for “light” samples, or 13C6-Arginine and 13C6-Lysine (Isotec) for “heavy” samples. Complete incorporation of the “heavy” amino acids was confirmed by mass spectrometry analysis with the cell lysate after at least six passages. Light and heavy cells were normalized based on the cell number for each experiment. For the pervanadate treatment, cells were stimulated with 20 mm sodium pervanadate for 15 min at 37 °C. For the IgM stimulation, cells were incubated with 50 μl/ml anti-IgM antibody (Rockland, Rockland, ME) for 15 min at 4 °C. The cells were then washed with PBS, collected, and frozen at −80 °C for further use.
Enrichment of Phosphotyrosine-containing Proteins
Cells were lysed in buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 5 mm EDTA, 1% Nonidet P-40, 1 mm sodium orthovanadate, 1× phosphatase inhibitor mixture (Sigma), and 10 mm sodium fluoride for 20 min on ice. The cell debris was cleared by centrifugation at 16,000 × g for 10 min. The supernatant containing soluble proteins was collected. The cell lysate was incubated with anti-phosphotyrosine antibodies (PT66 and PY20) conjugated to agarose beads overnight at 4 °C with agitation. The beads were washed twice with 500 μl of the lysis buffer and twice with water. To inhibit endogenous kinases, the beads were incubated with 1 mm 5′-(4-Fluorosulfonylbenzoyl)adenosine (FSBA) with 10% dimethyl sulfoxide in Tris-HCl, pH 7.5 at 30 °C for 1 h. Phosphoproteins were eluted with 100 mm triethylamine twice. The eluents were combined and dried down to 10% of original volume under vacuum.
In Vitro Kinase Reaction
Samples of phosphoproteins were re-suspended in 200 μl of phosphatase buffer (Roche). 2U of phosphatase (Roche) was added and incubated at 37 °C for 1 h. The phosphatase was deactivated by heating at 75 °C for 5 min. Samples were incubated in buffer containing 300 ng SYK (Sigma), 5 mm MgCl2, and 1 mm ATP at 30 °C for 30 min. The reaction was quenched by 8 m urea with 5 mm dithiothreitol at 37 °C for 1 h. Proteins were alkylated in 15 mm iodoacetamide for 1 h in the dark at room temperature and then digested with proteomics-grade trypsin at a 1:50 ratio overnight at 37 °C. For reciprocal SILAC experiments, the light and heavy samples were treated with or without kinase respectively, and then they were pooled in equal amounts after the reaction.
Phosphopeptide Enrichment
The tryptic peptides were first desalted using a Sep-pak C18 column (Waters) and dried. Next, the peptide mixture was re-suspended in 100 μl of loading buffer (100 mm glycolic acid, 1% trifluroacetic acid, 50% acetonitrile) to which 5 nmol of the PolyMAC-Ti reagent was added (41). The mixture was then incubated for 5 min. Two-hundred microliters of 300 mm HEPES, pH 7.7, was added to the mixture to achieve a final pH of 6.3. The solution was transferred to a spin column (Boca Scientific) containing Affi-Gel Hydrazide beads (Bio-Rad, Hercules, CA) to capture the PolyMAC-Ti dendrimers. The column was gently agitated for 10 min and then centrifuged at 2,300 × g for 30 s to collect the unbound flow-through. The beads were washed once with 200 μl loading buffer, twice with a mixture of 100 mm acetic acid, 1% trifluoroacetic acid, and 80% acetonitrile, and once with water. The phosphopeptides were eluted from dendrimers by incubating the beads twice with 100 μl of 400 mm ammonium hydroxide for 5 min. The eluates were collected and dried under vacuum.
Mass Spectrometric Data Acquisition
Peptide samples were dissolved in 8 μl of 0.1% formic acid and injected into an Eksigent NanoLC Ultra 2D HPLC system. The reverse phase chromatography was performed using an in-house C18 capillary column packed with 5 μm C18 Magic beads resin (Michrom; 75 μm i.d. and 12 cm bed length). The mobile phase buffer consisted of 0.1% formic acid in ultra-pure water with an eluting buffer of 0.1% formic acid (Buffer A) in 100% CH3CN (Buffer B) run over a linear gradient (2–35% Buffer B, 60 min) with a flow rate of 300 nl/min. The electrospray ionization emitter tip was generated on the prepacked column with a laser puller (Model P-2000, Sutter Instrument Co.). The Eksigent Ultra2D HPLC system was coupled online with a high-resolution hybrid duel-cell linear ion trap Orbitrap mass spectrometer (LTQ-Orbitrap Velos; Thermo Fisher). The mass spectrometer was operated in the data-dependent mode in which a full-scan MS (from m/z 300–1700 with the resolution of 60,000 at m/z 400) was followed by 20 CID MS/MS scans of the most abundant ions. Ions with the charge state of +1 were excluded. The dynamic exclusion time was set to 60 s after two fragmentations.
Database Search and Quantitation
The LTQ-Orbitrap raw files were searched directly against the Homo sapiens database with no redundant entries (93,289 entries; human International Protein Index (IPI) v.3.83) using the SEQUEST algorithm on Proteome Discoverer (Version 1.3; Thermo Fisher). Peptide precursor mass tolerance was set to 10 ppm, and MS/MS tolerance was set to 0.8 Da. Search criteria included a static modification of +57.0214 Da on cysteine residues, a dynamic modification of +15.9949 Da on oxidized methionine, and a dynamic modification of +79.996 Da on phosphorylated serine, threonine, and tyrosine residues. Searches were performed with full tryptic digestion and allowed a maximum of two missed cleavages on peptides analyzed by the sequence database. False discovery rates (FDR) were set to 1% for each analysis. Proteome Discoverer generated a reverse “decoy” database from the same protein database, and any peptide passing the initial filtering parameters from this decoy database was defined as a false positive. The minimum cross-correlation factor (Xcorr) filter was re-adjusted for each charge state separately to optimally meet the predetermined 1% FDR based on the number of random false-positives matched with the reversed “decoy” database. Thus, each data set had its own passing parameters. The number of unique phosphopeptides and nonphosphopeptides were then manually counted and compared. Phosphorylation site localization from collision-induced dissociation (CID) mass spectra was determined by PhosphoRS scores (42). For phosphopeptides with ambiguous phosphorylation sites, only one phosphorylation site with the highest score was selected for further data interpretation. For SILAC experiments, a dynamic modification of +6.020 Da was added on Arginine and Lysine in addition to the above parameters. The default template for SILAC 2plex (Arg6, Lys6) in Proteome Discoverer was used for quantification, and the Light/Heavy ratio of each peptide was reported in the supplemental data.
Data Analysis
Motif-X was used for predicting the specificity of kinases according to identified phosphosites. Parameters were set to peptide length = 13, occurrence = 10, and significance p value less than 0.00001. For the pathway analysis, the proteins showing increased phosphorylation with SILAC ratio more than 2 were extracted. Those proteins were submitted to Ingenuity Pathway Analysis (IPA) (Ingenuity Systems) for the kinase-substrate functional annotation.
Immunoprecipitation and Western Blotting Experiments
Cells were collected and lysed in buffer containing 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 5 mm EDTA, 1% Nonidet P-40, 1 mm sodium orthovanadate, 1× phosphatase inhibitor mixture (Sigma), 10 mm sodium fluoride, 1× Mini Complete protease inhibitor mixture (Roche) for 20 min on ice. Samples were cleared of debris and normalized based on the protein concentration. Then 1 mg of lysate was pre-incubated with 20 μl Protein A/G agarose beads (Thermo) for 20 min at 4 °C to remove nonspecific binding proteins, before further incubation with 10 μg of antibodies for 4 h at 4 °C. Antibodies were anti-β-tubulin and anti-UBA1 rabbit polyclonal antibody from Cell Signaling and anti-STIP1 mouse monoclonal antibody from Abcam. The samples were then incubated with 20 μl of Protein A/G agarose beads again for capturing antibody-antigen complexes for 4 h at 4 °C. The beads were washed and bound proteins were eluted by boiling the beads in SDS loading buffer with 50 mm DTT for 5 min. The eluents were separated on a 12% SDS-polyacrylamide gel and transferred onto a polyvinylidene difluoride membrane. The membranes were probed using antibodies against proteins of interest. To detect phosphorylation, the membranes were stripped and re-probed using 4G10 anti-phosphotyrosine antibody (Millipore).
RESULTS
The KALIP Strategy for Direct Kinase Substrate Identifications
Traditional proteomic strategies using kinase assays with whole cell extracts only identify potential kinase substrates in vitro. In addition, most of these strategies face interferences from endogenous kinase activity and background phosphorylation in cell lysates. In contrast, kinase assays with a peptide mixture can circumvent the problem of endogenous kinase interference. However, the loss of substrate structure decreases kinase specificity for substrates and may lead to a high false positive rate of substrate identification.
We devised here the proKALIP strategy that combines a newly designed kinase reaction carried out in vitro and classic kinase-modulated phosphoproteomics in vivo (Fig. 1) to identify direct kinase substrates with high confidence. In our strategy, we introduced a critical step by generating a pool of proteins derived from intact kinase substrates as candidates. This was achieved by treating cells with a phosphatase inhibitor to enhance the overall level of protein phosphorylation. Phosphoproteins were then purified using antibody-based immunochromatography. At the same time the endogenous kinases were inhibited by pre-incubating with the generic, irreversible kinase inhibitor, FSBA. This step efficiently eliminated the problem associated with endogenous kinase contamination. The excess FSBA was washed away so that it did not suppress the activity of the exogenous kinase in the following step. The addition of a phosphatase subsequently removed phosphate groups from the phosphoproteins to generate a pool of highly relevant substrate candidates for the following in vitro kinase reaction. After enzymatic digestion, phosphopeptides were enriched using Polymer-based Metal ion Affinity Capture (PolyMAC) (41), followed by mass spectrometric analysis to identify peptide sequences and phosphorylation sites. Because the phosphatase may not completely remove all background endogenous phosphorylation, the SILAC method is applied to quantify the change in phosphorylation after the kinase reaction. Cells were differentially labeled at the beginning of the procedure to generate two sets of protein lysates (Fig. 1, left panel). These two samples were treated with or without kinase respectively during the kinase reaction and then pooled for MS analysis. This procedure generated a list of in vitro direct substrates of the target kinase.
Fig. 1.

Methodology of proKALIP to identify kinase substrates. proKALIP combines in vitro kinase reactions and in vivo phosphoproteomics. In the in vitro kinase reaction, substrate proteins are isolated from cell lysates through affinity purification, dephosphorylated by alkaline phosphatase, and rephosphorylated by kinase of interest. After an in vitro kinase reaction, phosphopeptides are further enriched and analyzed by mass spectrometry for sequencing and site identification. Theoretical substrate has a higher intensity in kinase+ sample compared with the control in SILAC experiments. In in vivo phosphoproteomics, kinase dependent phosphorylation events are identified by comparing two phosphoproteomes with kinase perturbations. Genuine substrates are the phosphopeptides present within both data sets from in vitro kinase reaction and in vivo phosphoproteomics.
Because in vitro kinase reactions typically display a degree of promiscuity (43), we further incorporated endogenous phosphoproteomic analysis from the previous study (28) using cells whose kinase of interest was either active or inhibited (Fig. 1, right panel). In brief, two pools of identical cells were treated with the SYK inhibitor and dimethyl sulfoxide respectively. The total protein lysates were trypsin digested and the tyrosine phosphorylated peptides in the whole lysates were immunoprecipitated by the anti-phosphotyrosine antibody. A further purification of the phosphopeptides was performed using PolyMAC (41) followed by the LC-MS analysis.
On one hand, the phosphorylated proteins generated by the kinase reaction in vitro include both direct kinase substrates and potentially artificial candidates caused by the loss of physiological regulatory mechanisms under in vitro conditions. On the other hand, endogenous phosphoproteomics data illustrate direct kinase substrates as well as proteins phosphorylated downstream in the cascade. Therefore, the overlap of in vitro and in vivo candidates should represent the proteins with the highest probability of being genuine direct kinase substrates among those phosphorylated proteins detected. The total number of direct kinase substrates depends on the specificity and activity of the kinase as well as the actual cell types that are used.
Inhibition of Endogenous Kinases by the Generic Kinase Inhibitor
One major challenge for in vitro kinase reactions with whole cell extracts is how to distinguish direct kinase phosphorylation from background phosphorylation caused by endogenous kinase activities. We have employed several methods in proKALIP, including the use of a high concentration of purified kinase (10, 25) and quantification of phosphorylation before and after the kinase reaction (44). In addition, we utilized FSBA, an ATP analog cross-linker that can competitively block the ATP binding pocket by targeting the conserved lysine residue in the catalytic domain to inhibit endogenous kinase activities (37). FSBA was incubated with the phosphotyrosine proteins capturing on the beads before the elution step. The excess FSBA was washed away so that it did not suppress the activity of the exogenous kinase in the following step. We examined the efficiency of FSBA inhibition by comparing kinase reactions with and without FSBA treatment based on quantitative measurements. SILAC quantitation revealed that the overall phosphorylation of the FSBA+ sample was indeed lower than that of the FSBA- sample (Fig. 2, supplemental Tables S1, S2). Under kinase reaction conditions without any exogenous kinase, the phosphorylation detected by MS is considered to be residual background phosphorylation after the dephosphorylation and residual endogenous kinase activities. In our experiments, the low level of phosphorylation indicated that the prior dephosphorylation step was highly efficient. Additionally, the contrast between FSBA- and FSBA+ samples indicated that autophosphorylation of endogenous kinases was existed (Fig. 2A, supplemental Table S3). When SYK was introduced under the kinase reaction conditions, the level of phosphorylation was noticeably higher in the FSBA- sample than in the FSBA+ sample, indicating prior treatment of kinase inhibitors such as FSBA is necessary to inhibit any downstream kinase activities triggered by the addition of active SYK in vitro (Fig. 2B, supplemental Table S4). By using a generic kinase inhibitor to diminish the influence of endogenous kinases, we propose that proKALIP should have a lower false positive rate than traditional cell lysate-based in vitro kinase screening methods.
Fig. 2.
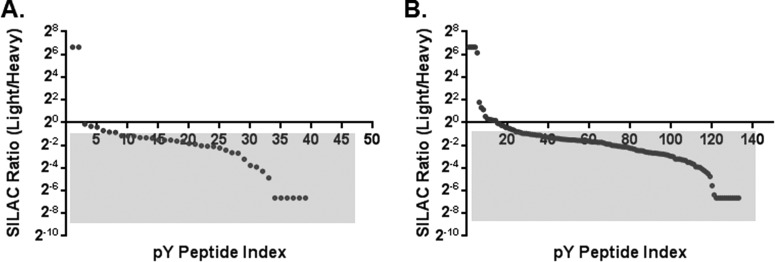
Evaluation of FSBA inhibition under in vitro kinase reaction condition. A, Kinase assay with no exogenous kinase and FSBA was added in the light fraction. B, Kinase assay with the exogenous SYK and FSBA was added in the light fraction.
Identification of Direct SYK Substrates
We applied the proKALIP strategy to examine potential substrates for SYK in human B cells (the step-by-step scheme is illustrated in supplemental Fig. S1). Human DG75 B cells were first treated with pervanadate, a generic protein-tyrosine phosphatase inhibitor, to elevate global tyrosine phosphorylation levels in the cells. Tyrosine phosphorylated proteins were isolated using a mixture of immobilized antibodies (PT66 and PY20, Sigma) against the phosphotyrosine residue. Phosphate groups were then removed using an alkaline phosphatase, which was subsequently inactivated by pulse heating. The collection of candidate substrate proteins from heavy and light isotope labeled SILAC cells were then incubated with or without purified active SYK, respectively, in the kinase reaction buffer. The reactions were quenched and then samples were pooled, trypsin digested, enriched using PolyMAC, and analyzed by mass spectrometry. The experimental conditions for phosphatase treatment, subsequent phosphatase removal, and kinase activity were optimized as described previously (28). In each step, changes in phosphotyrosine content were examined by Western blotting (supplemental Fig. S2).
To improve the quantitative measurement, we further carried out reciprocal SILAC experiments by reversing labeling of samples for the kinase reaction. A total of 463 unique tyrosine phosphorylated peptides representing 226 phosphoproteins and 315 phosphotyrosine sites (supplemental Table S5) were quantified in a sample that was derived from 8 mg DG75 whole cell extract (4 mg for each isotope labeled cell lysate). A large elevation in phosphorylation in samples treated with SYK kinase was clearly observed when comparing the two isotope labeled samples (Figs. 3A, 3B). However, there was residual phosphorylation detected in the phosphatase+/kinase- sample, demonstrating the necessity of quantitation to differentiate SYK catalyzed phosphorylation events from the background. The forward and reverse SILAC results were normalized based on the median ratios to avoid potential bias caused by the introduction of purified SYK only in one isotopic form. Likely because of the large variation of kinase preference toward substrates, the ratios were observed to span a broad range. In case some physiological substrates with low phosphorylation efficiency by SYK in vitro might be lost when overlapping with in vivo data set, we did not apply a statistical cutoff for in vitro substrates. Instead, tyrosine phosphosites with higher phosphorylation in intensity after the kinase reaction were considered as significant (red region in Fig. 3C) and their corresponding proteins were considered to be in vitro substrates. In total, from those phosphotyrosine sites that had increased phosphorylation level by SYK, 84 unique phosphorylation sites can be extracted from reciprocal SILAC (supplemental Table S6, supplemental Fig. S3).
Fig. 3.

Ratio distributions of phosphopeptide intensity before and after kinase reaction using reciprocal SILAC quantifications. A, SYK was added in the heavy fraction, whereas light fraction was the control. B, SYK was added in the light fraction, whereas heavy fraction was the control. C, Dot-plot depicting the overlap of the reciprocal data sets of tyrosine phosphorylation sites identified using in vitro kinase reaction.
The sites on substrates that are phosphorylated by protein kinases are dependent, in part, on the surrounding sequence of amino acids as well as protein structure. Compared with protein microarray screening, proKALIP provides a convenient and efficient method to determine kinase specificity by using an extensive collection of potential kinase substrates in an intact cell. Among the phosphorylation sites with increased phosphorylation in vitro (supplemental Table S6), two major classes of consensus sequences for SYK substrates were identified using Motif-X (45) (Fig. 4A). A clear enrichment of acidic residues surrounding the phosphorylated tyrosine in the consensus sequences was consistent with reported motifs determined by phage-display library screening and sequence alignment of SYK substrates (46). This result demonstrates the specificity of our approach in extracting the primary sequence preferences for tyrosine kinases.
Fig. 4.

A, Consensus sequence analysis of in vitro kinase phosphorylation for SYK specificity. Upper panel, frequency plot of all amino acids flanking the phosphotyrosine site. Lower panel, significantly enriched motif from SYK substrates using Motif-X; B, Venn diagram illustrating the overlap of tyrosine phosphorylation sites identified by in vitro kinase reaction and in vivo phosphoproteomics in DG75 cells. C, Categories of biological processes for identified SYK substrates.
Although the list of phosphoproteins generated by an in vitro kinase assay provides important clues for identification of actual protein substrates, specificity of kinases in vitro can be compromised because of high concentrations of the exogenous kinase and a loss of physiological context. To identify bona fide substrates, we compared the in vitro results with the in vivo SYK dependent phosphotyrosine events (supplemental Table S7). The in vivo data set was retrieved from our previous study SYK (28), in which the phosphotyrosine proteomes of DG75 cells that were treated with or without the SYK inhibitor piceatannol. The differential phosphotyrosine proteins were identified by mass spectrometry (Technical details in experimental procedure). The unique tyrosine phosphopeptides identified in the untreated cells but not in cells treated with the SYK inhibitor are considered to be SYK dependent phosphorylation events in vivo, which include SYK's direct substrates and downstream signaling molecules as well. This resulted in 37 tyrosine phosphorylation sites representing 25 unique proteins in both in vivo and in vitro data sets as genuine candidates of SYK direct substrates with the highest confidence (Fig. 4B, Table I).
Table I. Substrate candidates of SYK kinase identified in both reciprocal in vitro and in vivo methods.
| Accessions | Gene name | Protein name | Phosphorylation sites |
|---|---|---|---|
| P52565 | ARHGDIA | Rho GDP dissociation inhibitor (GDI) alpha | Y156 |
| Q9P0L7 | BTK | Bruton agammaglobulinemia tyrosine kinase | Y551 |
| Q9UJU6 | DBNL | Drebrin-like | Y162 |
| Q53H88 | DCTN2 | Dynactin 2 (p50) | Y91 |
| Q14318 | FKBP8 | FK506 binding protein 8, 38kDa | Y351 |
| Q8N6F7 | GCET2 | Germinal center expressed transcript 2 | Y148 |
| Q6GVN4 | HCA59 | Hepatocellular carcinoma-associated antigen 59 | Y68 |
| P14317 | HCLS1 | Hematopoietic cell-specific Lyn substrate 1 | Y103, Y140, Y222 |
| Q9BW71 | HIRIP3 | HIRA interacting protein 3 | Y111 |
| Q86X89 | MAP1B | Microtubule-associated protein 1B | Y1044, Y1062, Y1337, Y1410, Y1889, Y1921, Y1938, Y1940, Y1957, Y2025 |
| P23511 | NFYA | Nuclear transcription factor Y, alpha | Y237 |
| Q9NWQ8 | PAG1 | Phosphoprotein associated with glycosphingolipid microdomains 1 | Y163, Y417 |
| Q9NWS0 | PIH1D1 | PIH1 domain containing 1 | Y194 |
| P16885 | PLCG2 | Phospholipase C, gamma 2 (phosphatidylinositol-specific) | Y759 |
| Q06124 | PTPN11 | Protein tyrosine phosphatase, non-receptor type 11 | Y580 |
| Q9NQC3 | RTN4 | Reticulon 4 | Y718 |
| Q53HM6 | SF3A3 | Splicing factor 3a, subunit 3, 60kDa | Y479 |
| Q9BSV4 | SFPQ | Splicing factor proline/glutamine-rich | Y488 |
| O00193 | SMAP | Small acidic protein | Y84 |
| P31948 | STIP1 | Stress-induced-phosphoprotein 1 | Y354 |
| Q99426 | TBCB | Tubulin folding cofactor B | Y98 |
| Q15785 | TOMM34 | Translocase of outer mitochondrial membrane 34 | Y54 |
| Q5JP53 | TUBB | Tubulin, beta class I | Y33 |
| Q96HX0 | TUBB4B | Tubulin, beta 4B class IVb | Y50 |
| Q5JRS2 | UBA1 | Ubiquitin-like modifier activating enzyme 1 | Y106 |
Validating Novel SYK Substrates In Vitro and In Vivo
Among the 25 candidate proteins shown in both in vivo and in vitro lists (supplemental Table S8), four were known substrates of SYK with known tyrosine phosphorylation sites confirmed by traditional biochemical approaches in B cells (proKALIP column in Table II). Note that novel phosphotyrosine sites were also identified on previously characterized substrates such as germinal center B-cell-expressed transcript 2 protein (GCET2) and hematopoietic lineage cell-specific protein (HS1). At the same time, many previously unreported substrates were identified as well. To better understand the SYK phosphorylation, the identified SYK substrates are functionally grouped into four biological processes: immune cell responses, cancer development, gene regulation, and regulation of cell morphology (Fig. 4C). The proteins involved in immune cell signaling include multiple well-known BCR pathway enzymes such as Bruton's tyrosine kinase (BTK); phospholipase C-γ 2 (PLCG2); phosphoprotein associated with glycosphingolipid-enriched microdomains 1 (PAG1)), and GCET2. Among them, BTK, PLCG2 and GCET2 were known to be SYK substrates, whereas PAG1 has not been reported so far.
Table II. Comparison of peptide and protein level KALIP for known SYK substrates identification. Abbreviations: BTK, bruton tyrosine kinase; PLCG2, 1- phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-2; GCET2, germinal center B-cell-expressed transcript 2 protein; HPK1, mitogen-activated protein kinase kinase kinase kinase 1; BLNK, B-cell linker protein; LAT2, linker for activation of T-cells family member 2; HS1, hematopoietic lineage cell-specific protein; TUBA, tubulin alpha.
| Substrate | Phosphopeptides | Peptide KALIP | Protein KALIP | Ratio(SYK+/SYK-) | References |
|---|---|---|---|---|---|
| BTK | RYVLDDEyTSSVGSK | - | Y | 2.5 | (58) |
| PLCG2 | MyVDPSEINPSMPQR | - | Y | 3 | (59) |
| GCET2 | GNSAEEYyENVPCKA | Y | Y | Quantified other sites | (60) |
| HPK1 | LSESSDDDyDDVDIP | Y | - | - | (55) |
| BLNK | LLEDEADyVVPVEDN | Y | - | - | (57) |
| LAT2 | EDEESEDyQNSASIHWQR | Y | - | - | (56) |
| HS1 | EPEPENDyEDVEEMDR | Y | Y | Quantified other sites | (61) |
| TUBA | MAALEKDYEEVGVDS | Y | - | Quantified other sites | (62) |
To further investigate novel roles of SYK in enriched biological processes other than immune response, we selected three candidates for biochemical validation: β-tubulin (TUBB) in the cell morphology network, ubiquitin activating enzyme (UBA1) in the cell death and cancer development pathway, and stress induced protein 1 (STIP1) involved in gene regulation. Each protein was immunoprecipitated from DG75 cell lysates and then incubated with SYK under the kinase reaction conditions. Analyses of the reaction products by Western blotting with a pan anti-phosphotyrosine antibody confirmed that all three proteins served as direct substrates for SYK (Fig. 5). Such a high true hit rate suggests the low false positive rate of proKALIP for substrate discovery. To check whether TUBB, UBA1, and STIP1 could be phosphorylated by SYK in cells, we monitored changes In their phosphorylation in vivo by treating DG75 cells with anti-IgM antibody (47), which activates SYK through aggregation of the B-cell antigen receptor (BCR). Receptor-stimulated changes in tyrosine phosphorylation were detected in all three substrates (Fig. 5). Together with their direct phosphorylation by SYK in vitro, those substrates are likely involved in phosphorylation signaling in the downstream BCR pathway.
Fig. 5.
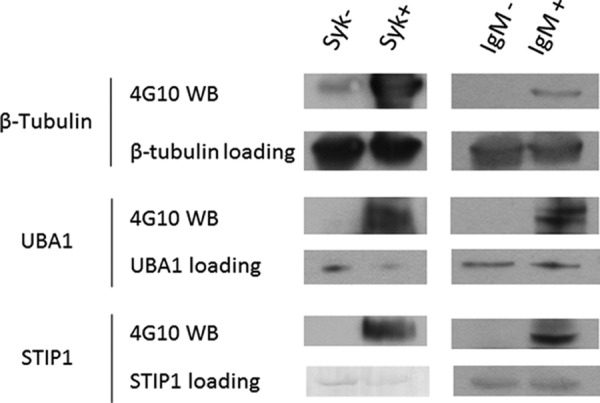
Western blotting for confirmation of SYK substrates through in vitro kinase assay and in vivo phosphorylation changes in response to BCR stimulation in DG-75 cells. For in vitro kinase assay, individual proteins were immunoprecipitated from cell lysates. The immune complex was then incubated with in a kinase reaction containing purified SYK kinase. For BCR stimulation, DG75 B cells were treated without (IgM-) or with (IgM+) anti-IgM antibodies. Individual proteins were directly isolated from cell lysates. The reaction mixtures were separated by SDS-PAGE and blotted with antigen antibodies and anti-phosphotyrosine antibody (4G10).
To our knowledge, this is the first report other than large-scale phosphoproteomic screenings indicating that β-tubulin phosphorylation is directly related to SYK. However, it is not surprising because many known substrates for SYK, including some tubulin proteins, are also involved in cellular organization. For example, α-tubulin has been reported to be phosphorylated by SYK on a tyrosine located at the C terminus (48, 49). Several other substrates such as MAP1B, ARHGDIA, and TBCB are also associated with this pathway, demonstrating a probable role for SYK in cell organization networks.
Ubiquitin-activating enzyme 1 (UBA1) phosphorylation is intriguing because UBA1, the principal E1 ligase, is essential for all ubiquitination pathways. SYK, itself, is modified by ubiquitination (50). The phosphorylation of UBA1 suggests possible correlation between SYK activation and protein ubiquitination in B cells. The effect of phosphorylation on the activity of UBA1 is currently under investigation. The identification of several direct SYK substrates involved in the ubiquitination pathway may provide insightful information in further understanding the regulation of ubiquitination in response to B-cell activation.
Stress induced protein 1 (STIP1) and SYK are predicted to be interacting proteins based on the Human Protein-Protein Interaction Prediction database (PIPs). Phosphorylation of Y354 on STIP1 has been characterized by a large-scale tyrosine phosphorylation study (51). This phosphorylation was also shown to be dependent on SYK in phosphoproteomic studies of MDA-MB-231 (41) and DG75 cells (28). Our in vitro kinase assay confirmed that Y354 was indeed the direct phosphorylation site of SYK. Besides, it is known that STIP1 promotes the association between 70 kDa heat shock cognate protein (HSC70) and heat shock protein 90 (HSP90) (52). HSC70 and HSP90 can be phosphorylated by SYK both in vitro and in vivo (28). Our results indicates potentially important role of STIP1 and associated proteins' phosphorylation by SYK in signaling networks that could be further examined.
DISCUSSION
Although thousands of phosphorylation events can be detected in a single mass spectrometry experiment, identifying direct kinase substrates in a physiological environment remains a daunting task, even with modern quantitative phosphoproteomics. To screen for direct substrates, several groups have used cell lysates to identify proteins that can be phosphorylated in vitro by specific protein kinases. By linking the in vitro kinase assays and quantitative phosphoproteomics together, proKALIP offers an efficient strategy for identifying bona fide kinase substrates.
Primarily, proKALIP distinguishes itself from other strategies by enriching phosphoproteins at an early stage. Several benefits are gained by this pre-enrichment step. First, the collection of substrates is derived from formerly tyrosine phosphorylated proteins. This extra step efficiently reduces the basal phosphorylation that may interfere with the kinase reaction and result in high false positives. Second, application of the effective phosphatase inhibitor pervanadate elevates phosphorylation levels, allowing isolation of a large number of phosphoproteins and increasing the overall sensitivity of the strategy. Third, the generic alkaline phosphatase has higher efficacy with purified phosphotyrosine proteome than with crude cell lysates. Pre-enrichment helps phosphotyrosine proteins to be specifically dephosphorylated instead of using large amounts of phosphatase to dephosphorylate all the phosphorylation, including high level of serine and threonine phosphorylated proteins that would not be used as SYK substrates. Fourth, the enrichment yields the proteins that have been actually phosphorylated within the intact cell instead of the proteins that may be exclusively phosphorylated in vitro. Fifth, the resin-captured phosphoproteome is compatible with multiple pretreatments before the kinase reaction. For example, to eliminate endogenous kinase activity in our study, a generic kinase inhibitor was incubated with phosphoproteins captured on the anti-phosphotyrosine antibody conjugated beads. The inhibitor was then easily washed away, so that it did not influence the activity of the target exogenous kinase in the following step. In summary, by promoting the target kinase reaction and suppressing background phosphorylation, our pre-enrichment resulted in high sensitivity and specificity with low false positive rate.
The proKALIP method performs the kinase reaction at the protein level instead of at the peptide level as previously described (28). Reactions carried out at the peptide level (KALIP), efficiently eliminates problems related to contamination with endogenous kinases. However, a loss of substrate structure during the kinase reaction by using enzymatically digested peptides may introduce false-positives for some types of kinases whose specificity is not exclusively based on the primary sequences of their substrates. Moreover, abolishing certain motifs may yield false-negative results. For example, trypsin digestion following on lysine or arginine is not compatible with substrate recognition by basophilic kinases. These drawbacks may result in certain limits to the KALIP method. To address this issue, proKALIP preserves the substrate structure that may serve a critical role in the kinase selectivity and activity. Because some kinases use secondary (noncatalytic domain) contacts with their substrates (30, 53), keeping full-length native substrates will likely give a higher degree of success in many in vitro kinase screenings.
Overlapping in vitro and in vivo candidates leads to the identification of direct kinase substrates with low false discovery rates. The percentage of overlapping proteins in the in vitro and in vivo data is largely dependent on the individual kinase in the study. A kinase high in the upstream signaling network can lead to an extensive list of phosphoproteins in the in vivo analysis because of numerous downstream signaling events. On the other hand, if the kinase is a downstream signaling molecule, then the list can be quite small. In the present study, for example, because SYK is an upstream kinase in the BCR signaling pathway, a large number of SYK-dependent tyrosine phosphorylation sites were identified in vivo. However, only a small portion is caused by direct phosphorylation by SYK.
The strategy to overlap the in vitro and in vivo data may lead to the exclusion of true kinase substrates. This possibility was assessed by close examination of the in vitro substrates list and the in vivo list. As we mentioned before, the intercept of in vitro and in vivo lists gave 4 known substrate proteins of SYK. When we further examined the proteins exclusively identified in either in vitro or in vivo list, we only found two more known substrates, alpha-tubulin and 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-1 (PLCG1), in the in vivo list, implying the limitation of false negative rate was probably not because of the overlapping of in vitro and in vivo lists in the proKALIP strategy. Moreover, the failure of discovering alpha-tubulin is because of the technical limitation of SILAC based quantitation that the C-terminal phosphopeptide has no Lys or Arg residue for isotope labeling. This limitation should be possibly circumvented by other quantitation methods such as label-free quantification. The failure to identify other known substrates in B cells in our strategy is likely because of the fact that some substrates may have low expression level or low tyrosine phosphorylation stoichiometry challenging for mass spectrometry detection. In the previous KALIP report, we observed the distinct substrates pools of SYK when using two cell lines, DG75 B cell and MDA-MB-231 breast cancer cell respectively, as the starting materials (28). Together, we hypothesized that the sensitivity of proKALIP to exhaust all possible substrates can be possibly improved by using more samples for both in vivo phosphoproteomics and the in vitro kinase assay.
Comparing the SYK substrates identified from the protein level and the peptide level phosphorylation methods (Table II), there are some common tyrosine phosphorylation sites confirmed by both approaches as well as a number of sites identified by the KALIP and proKALIP separately. In addition to different experimental settings, there are potential factors that lead to the distinction. First, proKALIP takes substrate structure into account whereas the peptide level in vitro kinase assays only examine the kinase specificity based on primary sequence. Overall, the enrichment of the consensus SYK recognition motif in proKALIP at the protein level is not as significant as KALIP at the peptide level. For example, the three novel substrates we confirmed in this study were uniquely identified in B cells by proKALIP, but not in KALIP. Among them, STIP1 and TUBB do not exhibit a conventional acidic SYK recognition motif flanking the phosphotyrosine residue, suggesting a possible higher order mechanism other than the primary sequence recognition for their recognition and phosphorylation by SYK. One potential limitation in the proKALIP strategy is the use of some harsh sample processing conditions, especially the pulse heating step that may alter proteins' nature conformation. In addition, although the FSBA treatment step is employed, complete inhibition of all endogenous kinases may remain difficult to accomplish. Furthermore, one technical challenge of proKALIP for studying the serine/threonine kinases is the pre-enrichment of phosphoproteins. Currently, there are no highly efficient anti-phosphoserine and anti-phosphothreonine antibodies available for immunoprecipitation. To further examine the sensitivity of proKALIP, we interrogated our list of substrates with a previously reported B-lymphocyte protein expression repertoire (54). Mitogen-activated protein kinase kinase kinase kinase 1 (55), Linker for activation of T-cell family member 2 (56) and B-cell linker protein (57), three of the known substrates that were detected by KALIP but missed in proKALIP screening, has relatively low expression level (the normalization expression value are missing for the first two and 0.00093 for the last one). Taken together, KALIP appears advantageous to discover low abundance kinase substrates, whereas proKALIP may be superior to identify the substrates phosphorylated by kinases based on the tertiary structure, according to the motif analysis discussed in the previous session. However, these two methods are both powerful by virtue of the total number of known SYK substrates identified and the similar number of known substrates uniquely identified with either method. Thus, these two strategies complement each other to yield a more complete listing of kinase-specific substrates.
CONCLUSION
Here we illustrate an integrated proteomic strategy for screening kinase substrates and defining the specificity of protein kinases. In our study, large numbers of known and novel SYK substrates with well characterized consensus sequences were identified, supporting the hypothesis that SYK plays complex roles in multiple signaling pathways that are essential for the regulation of cell growth. In addition, a more comprehensive kinase substrate repertoire will provide invaluable information for the discovery of new pathways for specific kinases. Thus, the proteomic strategy used here, combined with other existing approaches for the identification of direct kinase substrates, can be a powerful tool to shed light on complex signaling networks.
Supplementary Material
Footnotes
* This was supported in part by an NSF CAREER award CHE-0645020 (W.A.T.), and by National Institutes of Health grants GM088317 (WAT), CA115465 (RLG and WAT), and CA037372 (RLG).
 This article contains supplemental Figs. S1 to S3 and Tables S1 to S8.
This article contains supplemental Figs. S1 to S3 and Tables S1 to S8.
1 The abbreviations used are:
- ASKA
- Analog-Sensitive Kinase Allele
- BCR
- B-cell receptor
- FDR
- False discovery rate
- FSBA
- 5′-(4-Fluorosulfonylbenzoyl)adenosine
- GO
- Gene ontology
- IPA
- Ingenuity Pathway Analysis
- KALIP
- Kinase Assay-Linked Phosphoproteomics
- PolyMAC
- Polymer-based Metal-ion Affinity Capture
- proKALIP
- Protein Kinase Assay Linked with Phosphoproteomics
- QIKS
- Quantitative Identification of Kinase Substrates
- SILAC
- Stable isotope labeling by amino acids in cell culture.
REFERENCES
- 1. Hunter T. (2000) Signaling–2000 and beyond. Cell 100, 113–127 [DOI] [PubMed] [Google Scholar]
- 2. Blume-Jensen P., Hunter T. (2001) Oncogenic kinase signalling. Nature 411, 355–365 [DOI] [PubMed] [Google Scholar]
- 3. Bensimon A., Heck A. J., Aebersold R. (2012) Mass spectrometry-based proteomics and network biology. Annu. Rev. Biochem. 81, 379–405 [DOI] [PubMed] [Google Scholar]
- 4. Cohen P. (2002) Protein kinases–the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 1, 309–315 [DOI] [PubMed] [Google Scholar]
- 5. Yaffe M. B., Leparc G. G., Lai J., Obata T., Volinia S., Cantley L. C. (2001) A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nat. Biotechnol. 19, 348–353 [DOI] [PubMed] [Google Scholar]
- 6. Fukunaga R., Hunter T. (1997) MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J. 16, 1921–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Amanchy R., Zhong J., Molina H., Chaerkady R., Iwahori A., Kalume D. E., Grønborg M., Joore J., Cope L., Pandey A. (2008) Identification of c-Src tyrosine kinase substrates using mass spectrometry and peptide microarrays. J. Proteome Res. 7, 3900–3910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mok J., Im H., Snyder M. (2009) Global identification of protein kinase substrates by protein microarray analysis. Nat. Protoc. 4, 1820–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lam K. S., Liu R., Miyamoto S., Lehman A. L., Tuscano J. M. (2003) Applications of one-bead one-compound combinatorial libraries and chemical microarrays in signal transduction research. Acc. Chem. Res. 36, 370–377 [DOI] [PubMed] [Google Scholar]
- 10. Knebel A., Morrice N., Cohen P. (2001) A novel method to identify protein kinase substrates: eEF2 kinase is phosphorylated and inhibited by SAPK4/p38delta. EMBO J. 20, 4360–4369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Troiani S., Uggeri M., Moll J., Isacchi A., Kalisz H. M., Rusconi L., Valsasina B. (2005) Searching for biomarkers of Aurora-A kinase activity: identification of in vitro substrates through a modified KESTREL approach. J. Proteome Res. 4, 1296–1303 [DOI] [PubMed] [Google Scholar]
- 12. Morandell S., Grosstessner-Hain K., Roitinger E., Hudecz O., Lindhorst T., Teis D., Wrulich O. A., Mazanek M., Taus T., Ueberall F., Mechtler K., Huber L. A. (2010) QIKS–Quantitative identification of kinase substrates. Proteomics 10, 2015–2025 [DOI] [PubMed] [Google Scholar]
- 13. Ficarro S. B., McCleland M. L., Stukenberg P. T., Burke D. J., Ross M. M., Shabanowitz J., Hunt D. F., White F. M. (2002) Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 20, 301–305 [DOI] [PubMed] [Google Scholar]
- 14. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 15. Karisch R., Fernandez M., Taylor P., Virtanen C., St-Germain J. R., Jin L. L., Harris I. S., Mori J., Mak T. W., Senis Y. A., Östman A., Moran M. F., Neel B. G. (2011) Global proteomic assessment of the classical protein-tyrosine phosphatome and “Redoxome”. Cell 146, 826–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villén J., Haas W., Sowa M. E., Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bodenmiller B., Aebersold R. (2010) Quantitative analysis of protein phosphorylation on a system-wide scale by mass spectrometry-based proteomics. Methods Enzymol. 470, 317–334 [DOI] [PubMed] [Google Scholar]
- 18. Huang S. Y., Tsai M. L., Chen G. Y., Wu C. J., Chen S. H. (2007) A systematic MS-based approach for identifying in vitro substrates of PKA and PKG in rat uteri. J. Proteome Res. 6, 2674–2684 [DOI] [PubMed] [Google Scholar]
- 19. Coba M. P., Pocklington A. J., Collins M. O., Kopanitsa M. V., Uren R. T., Swamy S., Croning M. D., Choudhary J. S., Grant S. G. (2009) Neurotransmitters drive combinatorial multistate postsynaptic density networks. Sci. Signal. 2, ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kettenbach A. N., Schweppe D. K., Faherty B. K., Pechenick D., Pletnev A. A., Gerber S. A. (2011) Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci. Signal. 4, rs5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shah K., Liu Y., Deirmengian C., Shokat K. M. (1997) Engineering unnatural nucleotide specificity for Rous sarcoma virus tyrosine kinase to uniquely label its direct substrates. Proc. Natl. Acad. Sci. U.S.A. 94, 3565–3570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Allen J. J., Li M., Brinkworth C. S., Paulson J. L., Wang D., Hübner A., Chou W. H., Davis R. J., Burlingame A. L., Messing R. O., Katayama C. D., Hedrick S. M., Shokat K. M. (2007) A semisynthetic epitope for kinase substrates. Nat. Methods 4, 511–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blethrow J. D., Glavy J. S., Morgan D. O., Shokat K. M. (2008) Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc. Natl. Acad. Sci. U.S.A. 105, 1442–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chi Y., Welcker M., Hizli A. A., Posakony J. J., Aebersold R., Clurman B. E. (2008) Identification of CDK2 substrates in human cell lysates. Genome Biol. 9, R149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cohen P., Knebel A. (2006) KESTREL: a powerful method for identifying the physiological substrates of protein kinases. Biochem. J. 393, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grosstessner-Hain K., Hegemann B., Novatchkova M., Rameseder J., Joughin B. A., Hudecz O., Roitinger E., Pichler P., Kraut N., Yaffe M. B., Peters J. M., Mechtler K. (2011) Quantitative phospho-proteomics to investigate the polo-like kinase 1-dependent phospho-proteome. Mol. Cell. Proteomics 10, M111/008540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bodenmiller B., Wanka S., Kraft C., Urban J., Campbell D., Pedrioli P. G., Gerrits B., Picotti P., Lam H., Vitek O., Brusniak M. Y., Roschitzki B., Zhang C., Shokat K. M., Schlapbach R., Colman-Lerner A., Nolan G. P., Nesvizhskii A. I., Peter M., Loewith R., von Mering C., Aebersold R. (2010) Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci. Signal. 3, rs4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xue L., Wang W. H., Iliuk A., Hu L., Galan J. A., Yu S., Hans M., Geahlen R. L., Tao W. A. (2012) Sensitive kinase assay linked with phosphoproteomics for identifying direct kinase substrates. Proc. Natl. Acad. Sci. U.S.A. 109, 5615–5620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goldberg G. S., Alexander D. B., Pellicena P., Zhang Z. Y., Tsuda H., Miller W. T. (2003) Src phosphorylates Cas on tyrosine 253 to promote migration of transformed cells. J. Biol. Chem. 278, 46533–46540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burkhard K. A., Chen F., Shapiro P. (2011) Quantitative analysis of ERK2 interactions with substrate proteins: roles for kinase docking domains and activity in determining binding affinity. J. Biol. Chem. 286, 2477–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Levinson N. M., Seeliger M. A., Cole P. A., Kuriyan J. (2008) Structural basis for the recognition of c-Src by its inactivator Csk. Cell 134, 124–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khandekar S. S., Feng B., Yi T., Chen S., Laping N., Bramson N. (2005) A liquid chromatography/mass spectrometry-based method for the selection of ATP competitive kinase inhibitors. J. Biomol. Screen 10, 447–455 [DOI] [PubMed] [Google Scholar]
- 33. Sim C. H., Lio D. S., Mok S. S., Masters C. L., Hill A. F., Culvenor J. G., Cheng H. C. (2006) C-terminal truncation and Parkinson's disease-associated mutations down-regulate the protein serine/threonine kinase activity of PTEN-induced kinase-1. Hum. Mol. Genet. 15, 3251–3262 [DOI] [PubMed] [Google Scholar]
- 34. Feige J. J., Cochet C., Pirollet F., Chambaz E. M. (1983) Identification of the catalytic subunit of an oligomeric casein kinase (G type). Affinity labeling of the nucleotide site using 5′-[p-(fluorosulfonyl)benzoyl]adenosine. Biochemistry 22, 1452–1459 [DOI] [PubMed] [Google Scholar]
- 35. Renzone G., Salzano A. M., Arena S., D'Ambrosio C., Scaloni A. (2006) Selective ion tracing and MSn analysis of peptide digests from FSBA-treated kinases for the analysis of protein ATP-binding sites. J. Proteome Res. 5, 2019–2024 [DOI] [PubMed] [Google Scholar]
- 36. Moore L. L., Fulton A. M., Harrison M. L., Geahlen R. L. (2004) Anti-sulfonylbenzoate antibodies as a tool for the detection of nucleotide-binding proteins for functional proteomics. J. Proteome Res. 3, 1184–1190 [DOI] [PubMed] [Google Scholar]
- 37. Knight J. D., Tian R., Lee R. E., Wang F., Beauvais A., Zou H., Megeney L. A., Gingras A. C., Pawson T., Figeys D., Kothary R. (2012) A novel whole-cell lysate kinase assay identifies substrates of the p38 MAPK in differentiating myoblasts. Skelet. Muscle 2, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mocsai A., Ruland J., Tybulewicz V. L. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat. Rev. Immunol. 10, 387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Geahlen R. L. (2009) Syk and pTyr'd: Signaling through the B cell antigen receptor. Biochim. Biophys. Acta 1793, 1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhou Q., Geahlen R. L. (2009) The protein-tyrosine kinase Syk interacts with TRAF-interacting protein TRIP in breast epithelial cells. Oncogene 28, 1348–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iliuk A. B., Martin V. A., Alicie B. M., Geahlen R. L., Tao W. A. (2010) In-depth analyses of kinase-dependent tyrosine phosphoproteomes based on metal ion-functionalized soluble nanopolymers. Mol. Cell. Proteomics 9, 2162–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taus T., Kocher T., Pichler P., Paschke C., Schmidt A., Henrich C., Mechtler K. (2011) Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 10, 5354–5362 [DOI] [PubMed] [Google Scholar]
- 43. Manning B. D., Cantley L. C. (2002) Hitting the target: emerging technologies in the search for kinase substrates. Sci. STKE 2002, pe49. [DOI] [PubMed] [Google Scholar]
- 44. Kettenbach A. N., Rush J., Gerber S. A. (2011) Absolute quantification of protein and post-translational modification abundance with stable isotope-labeled synthetic peptides. Nat. Protoc. 6, 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schwartz D., Gygi S. P. (2005) An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 23, 1391–1398 [DOI] [PubMed] [Google Scholar]
- 46. Schmitz R., Baumann G., Gram H. (1996) Catalytic specificity of phosphotyrosine kinases Blk, Lyn, c-Src and Syk as assessed by phage display. J. Mol. Biol. 260, 664–677 [DOI] [PubMed] [Google Scholar]
- 47. Harwood N. E., Batista F. D. Early events in B cell activation. Annu. Rev. Immunol. 28, 185–210 [DOI] [PubMed] [Google Scholar]
- 48. Faruki S., Geahlen R. L., Asai D. J. (2000) Syk-dependent phosphorylation of microtubules in activated B-lymphocytes. J. Cell Sci. 113 (Pt 14), 2557–2565 [DOI] [PubMed] [Google Scholar]
- 49. Fernandez J. A., Keshvara L. M., Peters J. D., Furlong M. T., Harrison M. L., Geahlen R. L. (1999) Phosphorylation- and activation-independent association of the tyrosine kinase Syk and the tyrosine kinase substrates Cbl and Vav with tubulin in B-cells. J. Biol. Chem. 274, 1401–1406 [DOI] [PubMed] [Google Scholar]
- 50. Dangelmaier C. A., Quinter P. G., Jin J., Tsygankov A. Y., Kunapuli S. P., Daniel J. L. (2005) Rapid ubiquitination of Syk following GPVI activation in platelets. Blood 105, 3918–3924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rush J., Moritz A., Lee K. A., Guo A., Goss V. L., Spek E. J., Zhang H., Zha X. M., Polakiewicz R. D., Comb M. J. (2005) Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 23, 94–101 [DOI] [PubMed] [Google Scholar]
- 52. Longshaw V. M., Chapple J. P., Balda M. S., Cheetham M. E., Blatch G. L. (2004) Nuclear translocation of the Hsp70/Hsp90 organizing protein mSTI1 is regulated by cell cycle kinases. J. Cell Sci. 117, 701–710 [DOI] [PubMed] [Google Scholar]
- 53. Moarefi I., LaFevre-Bernt M., Sicheri F., Huse M., Lee C. H., Kuriyan J., Miller W. T. (1997) Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature 385, 650–653 [DOI] [PubMed] [Google Scholar]
- 54. Shiio Y., Eisenman R. N., Yi E. C., Donohoe S., Goodlett D. R., Aebersold R. (2003) Quantitative proteomic analysis of chromatin-associated factors. J. Am. Soc. Mass Spectrom. 14, 696–703 [DOI] [PubMed] [Google Scholar]
- 55. Tsuji S., Okamoto M., Yamada K., Okamoto N., Goitsuka R., Arnold R., Kiefer F., Kitamura D. (2001) B cell adaptor containing src homology 2 domain (BASH) links B cell receptor signaling to the activation of hematopoietic progenitor kinase 1. J. Exp. Med. 194, 529–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang W., Sloan-Lancaster J., Kitchen J., Trible R. P., Samelson L. E. (1998) LAT: The ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 92, 83–92 [DOI] [PubMed] [Google Scholar]
- 57. Fu C., Turck C. W., Kurosaki T., Chan A. C. (1998) BLNK: a central linker protein in B cell activation. Immunity 9, 93–103 [DOI] [PubMed] [Google Scholar]
- 58. Baba Y., Hashimoto S., Matsushita M., Watanabe D., Kishimoto T., Kurosaki T., Tsukada S. (2001) BLNK mediates Syk-dependent Btk activation. Proc. Natl. Acad. Sci. U.S.A. 98, 2582–2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kurosaki T., Tsukada S. (2000) BLNK: connecting Syk and Btk to calcium signals. Immunity 12, 1–5 [DOI] [PubMed] [Google Scholar]
- 60. Pan Z., Shen Y., Ge B., Du C., McKeithan T., Chan W. C. (2007) Studies of a germinal centre B-cell expressed gene, GCET2, suggest its role as a membrane associated adapter protein. Br. J. Haematol. 137, 578–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brunati A. M., Deana R., Folda A., Massimino M. L., Marin O., Ledro S., Pinna L. A., Donella-Deana A. (2005) Thrombin-induced tyrosine phosphorylation of HS1 in human platelets is sequentially catalyzed by Syk and Lyn tyrosine kinases and associated with the cellular migration of the protein. J. Biol. Chem. 280, 21029–21035 [DOI] [PubMed] [Google Scholar]
- 62. Peters J. D., Furlong M. T., Asai D. J., Harrison M. L., Geahlen R. L. (1996) Syk, activated by cross-linking the B-cell antigen receptor, localizes to the cytosol where it interacts with and phosphorylates alpha-tubulin on tyrosine. J. Biol. Chem. 271, 4755–4762 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.