

Abstract
Current strategies to study N-glycoproteins in complex samples are often discrete, focusing on either N-glycans or N-glycosites enriched by sugar-based techniques. In this study we report a simple and rapid sample preparation platform, the GlycoFilter, which allows a comprehensive characterization of N-glycans, N-glycosites, and proteins in a single workflow. Both PNGase F catalyzed de-N-glycosylation and trypsin digestions are accelerated by microwave irradiation and performed sequentially in a single spin filter. Both N-glycans and peptides (including de-N-glycosylated peptides) are separately collected by filtration. The condition to effectively collect complex and heterogeneous N-glycans was established on model glycoproteins, bovine ribonuclease B, bovine fetuin, and human serum IgG. With this platform, the N-glycome, N-glycoproteome and proteome of human urine and plasma were characterized. Overall, a total of 865 and 295 N-glycosites were identified from three pairs of urine and plasma samples, respectively. Many sites were defined unambiguously as partially occupied by the detection of their nonsugar-modified peptides (128 from urine and 61 from plasma), demonstrating that partial occupancy of N-glycosylation occurs frequently. Given the likely high prevalence and variability of partial occupancy, glycoprotein quantification based exclusively on deglycosylated peptides may lead to inaccurate quantification.
N-glycosylation is one of the most abundant post-translational modifications of proteins. It is estimated that more than 50% of human proteins are N-glycosylated (1). This type of modification is critical to many fundamental biologic and pathologic processes such as: structural modulation of proteins, cell-cell signaling and interactions, pathogen-host recognition, and tumor progression (2, 3). Inherently, N-glycans are extremely heterogeneous, and subtle variations in the composition or structure may induce dramatic biological consequences (4, 5). Because of their heterogeneity, N-glycans typically need to be released from the parent glycoproteins to be accurately characterized or quantified (3, 6).
Identifying the sugar-modified position (glycosite) in a glycoprotein is also critical to understanding the biological role of N-glycosylation (7). Current methods that determine glycosites often use sugar-based enrichment techniques, such as hydrazide chemistry (8) or lectin affinity (9). The extracted glycoproteins or glycopeptides are subjected to de-N-glycosylation, and the deglycosylated peptides are then sequenced by liquid chromatography-tandem MS (LC-MS/MS) to characterize the previously glycosylated sites with a standard bottom-up proteomic approach (10). Filtration has been previously applied to collect peptides (FASP) (11) and deglycosylated peptides after lectin-enrichment (N-glyco FASP) (12). Although these enrichment techniques can identify low-abundant glycosites because of the enrichment selectivity (13), typically they are not feasible for characterization of the N-glycome, because glycans are oxidized and altered when coupled to the hydrazide groups (8), and the selective affinity of the lectin usually biases the N-glycome.
N-glycosylation is often considered “irreversible” once a glycoprotein is exported into the extracellular matrix. The addition of a dolichol-linked N-glycan precursor (Glc3Man9GlcNAc2) onto a nascent peptide is an enzyme-catalyzed and nontemplate driven process (2). However, the likelihood and efficiency of this addition are impacted by many factors including: (1) the concentration and activity of oligosaccharyltransferase (2, 14), (2) the availability of dolichol-linked N-glycan precursor (2, 14), (3) the length of time the glycosylated region is unfolded during passage across the endoplasmic reticulum membrane (2, 14), (4) the accessibility of the glycosite, which is greatly impacted by neighboring amino acids (15), and (5) the conformation of a glycosylated protein (correctly folded or not) (14). Because of these variables, the majority of the common N-glycosylation consensus motif (Asn-XXX-Ser/Thr, in which XXX is any amino acid except proline) in human proteins are not actually modified by a sugar chain (15).
Furthermore, a glycosite may be partially occupied (PO),1 a state in which both the glycosylated (sugar-modified asparagine) and nonglycosylated (nonsugar-modified asparagine) forms coexist. For example, human corticosteroid-binding globulin, a major plasma glycoprotein with six N-glycosites, has variable degrees in occupancy among its six glycosites (ranging from 70 to 99.5%) that also seem to change with pregnancy (16). Although the biological implications of partial occupancy in N-glycosylation are not well understood, to date, there are no well-defined strategies that can readily identify PO glycosites, particularly in a complex mixture. Current sugar-based enrichment methodologies alone are typically incapable of determining whether a particular glycosite is partially occupied or not, because the nonglycosylated peptides are typically removed.
Here we demonstrate a simple, rapid but comprehensive sample preparation platform, the GlycoFilter, which collects N-glycans and peptides separately in a single spin filter device. We demonstrate that glycans, including large acidic glycans, can be effectively separated and captured using a simple shift in pH combined with filtration. Although the lectin-based enrichment method of N-glyco FASP also uses a filtration principle to identify lectin-specific glycosites (12), this platform enables efficient downstream characterization of the N-glycans, N-glycosites, and the remaining proteome of a simple or complex biological sample. Furthermore, the GlycoFilter has the additional nonbiased capability to identify PO N-glycosites using a standard LC-MS/MS approach.
EXPERIMENTAL PROCEDURES
Materials and Reagents
The standard glycoproteins, bovine ribonuclease B, bovine fetuin, and human IgG (from serum) were obtained from Sigma-Aldrich (St. Louis, MO). PNGase F (glycerol free) was purchased from New England Biolab. (Ipswich, MA). Sequencing grade trypsin was obtained from Promega (Madison, WI). The Viva Spin 2 series of spin filters (10K and 30K MWCO, 2 ml volume, Polyethersulfone-type membrane) were purchased from Sartorius Stedium Biotech (Aubagne, France). H218O (98%) was obtained from Rotem (Arava, Israel). The centrifugation was performed in a fixed-angle rotor of a bench-top centrifuge 5804R (Eppendorf). The default spinning period was 20 min at 10,000 × g, unless otherwise specified.
Urine and Plasma Preprocessing
Urine and plasma samples were obtained from three healthy volunteers under an Institutional Review Board-approved procedure. The processing of urine samples and the depletion of albumin in urine were performed according to the reported one-step protocol (17). Twelve to 15 ml of urine from each donor was depleted of albumin yielding 600 μg of depleted urine protein. These aliquots were divided into two for the outlined studies. Plasma was obtained by the centrifugation of whole blood (10 min × 3000 × g). Forty microliters of plasma were depleted of the seven most abundant plasma proteins by MARS 7 spin column (Agilent, Santa Clara, CA) according to the vendor's protocol, yielding ∼350 μg of depleted plasma proteins. The concentration of depleted urine and plasma samples were measured by the Bradford assay in triplicate.
The GlycoFilter Platform Workflow
Preprocessing
The samples were dissolved in 8 M urea/0.2 M Tris-HCl buffer (pH 8.5), and transferred into the sample chamber of a filter device (10 kDa molecular weight cutoff (MWCO) for ribonuclease B and fetuin, and 30 kDa MWCO for urine and plasma). The proteins were reduced by dithiothreitol (25 mm final concentration, 45 min at room temperature) and alkylated by iodoacetamide (30 mm final concentration, 45 min at room temperature in a dark environment). The low-molecular-weight molecules (reagents, salts, buffers, etc.) were removed by repeated centrifugation with 0.5 ml of 50 mm ammonium bicarbonate buffer in H218O (×4). Specifically for urine samples, additional centrifugations were conducted with 0.5 ml of 50 mm ammonium bicarbonate buffer in H216O (×6) before H218O-based buffer washing.
De-N-glycosylation
Additional 50 mm ammonium bicarbonate buffer in H218O was added into the sample to cover the membrane of the sample chamber. PNGase F (∼1 μl of enzyme per 200 μg of protein) was introduced into the solution, and the de-N-glycosylation was performed by a 20-min domestic microwave protocol (18). The device was cooled in an ice bath after the de-N-glycosylation. For complex urine and plasma samples, this step was performed twice with fresh PNGase F. The released N-glycans were eluted into the collecting chamber of the filter device by repeated centrifugation with 0.5 ml of pure water (×1) and 0.5 ml of ice-cold 0.1% formic acid (×3) successively. All the flow-through fractions were combined, and dried completely in a speed-vacuum.
Proteolytic Digestion
The sample was adjusted back to basic pH by repeated centrifugation with 0.5 ml of 50 mm ammonium bicarbonate in normal H216O (×2). Additional 50 mm ammonium bicarbonate buffer in normal H216O was added to cover the membrane of the filter. The first aliquot of trypsin was introduced to the sample solution (trypsin: protein = 1:50 by weight). The digestion was performed by a 6 min domestic microwave protocol (19). After cooling in an ice bath, the sample was subjected to a second digestion with fresh trypsin. The tryptic peptides were eluted into the collection chamber of the filter device by repeated centrifugation with 0.5 ml of 50 mm ammonium bicarbonate solution (×3). All the peptide fractions were combined, and stored at −20 °C.
Isoelectric Focusing of Peptides
The tryptic peptides from all urine and plasma samples were focused into 24 fractions using a 3100 OFFGEL fractionator (Agilent, Santa Clara, CA) as described previously (17). Briefly, the 24 cm, pH 3–10 IPG DryStrips (GE healthcare) were rehydrated for 20 min with the IPG buffer pH 3–10. Samples were dissolved in 3.6 ml of IPG buffer, and equally distributed into each well. Mineral oil was added to the both ends to prevent drying. Focusing was performed according to the preset program up to 50 kV h with maximum current of 50 μA. Fractions were collected from each well. An additional 100 μl of 0.1% formic acid was added to each well, and extracted after 10 min. The extracted peptides were combined and dried completely in a speed-vacuum.
Mass Spectrometric Analyses of Permethylated N-Glycans
Without further purification, the collected glycans were permethylated (20). In cases in which < 10 μg of glycoproteins were used, a solid-phase permethylation procedure was conducted to avoid sample loss (21). Under selective cases, de-N-glycosylated proteins were recovered from the sample chamber of the filter with 50 mm ammonium bicarbonate buffer, and further processed onto C18 SPE cartridge (Sep-Pak Vac, Waters, Milford, MA). The flow-through fraction (2% acetonitrile/0.1 trifluoroacetic acid) from the C18 SPE was collected, dried completely, and permethylated, to detect any remaining N-glycans in the protein solution. The matrix solution was prepared by dissolving 10 mg of 2,5-dihydroxybenzoic acid (DHB) in a volume of 1 ml of 50% methanol containing 1 mm sodium acetate. Glycans were spotted directly onto a stainless steel MALDI plate and mixed with an equal volume of matrix solution (0.5–1 μl). MALDI-MS was carried out on an MDS SCIEX 4800 (Applied Biosystems, Carlsbad, CA) using the interactive mode. The external calibration was performed using the ProteoMass Peptide MALDI-MS calibration kit (Sigma-Aldrich, St. Louis, MO). MS data were processed using Data Explorer 4.9 (Applied Biosystems, Carlsbad, CA). Glycan samples were also analyzed on a LTQ-Orbitrap XL mass spectrometer (Thermo Scientific, Waltham, MA) coupled with a TriVersa Nanomate (Advion, Ithaca, CA). MS profiles were generated by the high mass accuracy Orbitrap, and MS/MS fragmentation spectra were obtained by collision-induced dissociation in the linear ion trap (LTQ). As for human urinary and plasma N-glycans, each peak was assigned a putative topology, based on its m/z value and plausible biosynthetic pathway. No further verifications were conducted.
Mass Spectrometric Analyses of Peptides
The OFFGEL fractionated tryptic peptides were further desalted with Strong Cation Toptips (Poly Sulfoethyl A, Catalog # TT2SSA) by the vendor's protocol (Glygen, Columbia, MD). Briefly, dried peptides were dissolved in 50 μl of binding solution (0.1% formic acid and 20% acetonitrile in water), and applied onto the Toptips. The peptide samples were further washed with 150 μl of the binding solution before eluting with 150 μl of releasing solution (5% ammonium hydroxide and 30% methanol in water). The eluted peptides were dried completely and stored for mass spectrometric analysis. The peptides were analyzed by an LTQ-Orbitrap XL mass spectrometer (Thermo Scientific, Waltham, MA) connected to an autosampler and nanoflow HPLC pump (Eksigent, Dublin, CA). The reversed phase columns were packed in-house by using Magic C18 particles (3 μm, 200 Å; Michrom Bioresource), and PicoTip Emitters (New Objective). The peptides were eluted with a 60 min linear gradient (0–35% ACN with 0.2% formic acid), and data acquired in a data dependent mode, fragmenting the seven most abundant peaks by CID, with dynamic exclusion for 60 s. All precursor scans were performed in the Orbitrap, and MS/MS spectra were obtained from the low resolution linear ion trap. Buffer A was 0.2% formic acid, buffer B was acetonitrile and 0.2% formic acid, and loading buffer was 5% formic acid with 5% acetonitrile.
Database Searching and Validation
The 200 most intense fragment ions of each raw product ion spectrum were used to generate .mgf files using the peak-list-generating software ProteoWizard (2.2.2881, released on 2011–7-25). Searches were performed against the UniProtKB/Swiss-Prot database (Homo sapiens, released in 2010_07) containing both forward and reverse protein sequences (40566 total entries, of which 20283 were targets) using the Mascot search engine (v 2.2.1) (22). One miscleavage per peptide was allowed and mass tolerances were 10 ppm for precursor and 0.8 Da for fragment ions. The search included fixed modification of carbamidomethylation on cysteine, and variable modifications: oxidation (Met), Gln → pyro-Glu (N-terminal Gln), Glu → pyro-Glu (N-terminal Glu), and deamidation (O16) on both asparagine and glutamine, and O18-incorporated deamidation on asparagine. The FDR analysis used the R implementation of a flexible mixture model as described by Choi et al. to calculate global and local peptide-level false discovery rates (23). The false discovery rate (FDR) was controlled at 1% at the peptide level by searching the same data set against a decoy database, and a minimum of two unique peptides per protein was required for the identification of proteins. Peptides were grouped to all proteins that contain them by graphical analysis. A protein group was treated as a single entity with one representative protein (accession number). Other proteins that share all or some of the exact same set of peptides are separately listed (See the supplementary excel tables for the protein identifications). The representative protein is always the identification with the most peptide matches among the group members. In the case of multiple choices, the one with the lowest accession number is chosen as the representative.
A deglycopeptide was defined by two criteria: (1) the peptide sequence possessed the common N-glycosylation consensus motif (Asn-XXX-Ser/Thr, in which XXX is any amino acid except proline); and (2) the specific asparagine residue within that motif was identified as the 18O-incorporated deamidation derivative (N, +2.9982 Da) (9). Glycoproteins in this study were defined as those proteins identified by at least one unique deglycopeptide. All annotated MS/MS spectra to identify the deglycopeptides and PO sites of each individual sample were included in the supplementary material.
RESULTS
The GlycoFilter Platform
The workflow of the GlycoFilter platform comprises three sequential steps: preprocessing, de-N-glycosylation, and proteolysis, all of which are performed in a single spin filter device (Fig. 1). During the preprocessing step, the sample is reduced and alkylated in the sample chamber of the filter. This is followed by the first round of filtration to remove excessive reagents and other interfering small molecules. The de-N-glycosylation is catalyzed by PNGase F in a H218O environment. With subsequent acidification, the second round of filtration collects the released N-glycans. The remaining de-N-glycosylated proteins undergo proteolysis with trypsin, and the tryptic peptides are collected by the third round of filtration. Both enzyme reactions are accelerated by a domestic microwave (18, 19), which is a cost-effective technique that significantly reduces the overall processing time. The novel feature of this platform is the GlycoFilter's ability to sequentially collect N-glycans and peptides (both deglycosylated and nonglycosylated). The eluates are then used to characterize N-glycome, N-glycosites, and proteome via standard techniques. The GlycoFilter platform was evaluated in three distinct aspects: (1) the separation efficiency of released N-glycans by filtration, as filtration is not an established method to separate glycans from proteins; (2) the general impact on proteomic performance of complex biological samples after PNGase F catalyzed de-N-glycosylation; and (3) the identification of N-glycosites and PO N-glycosites.
Fig. 1.
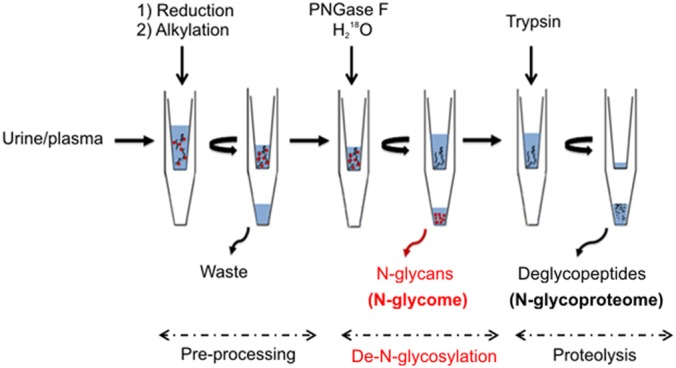
The experimental workflow of the GlycoFilter platform. The entire platform comprises three consecutive steps: preprocessing, de-N-glycosylation, and proteolysis. Each step involves separation by high-speed centrifugation and filtration. During the initial preprocessing step, the proteins are reduced and alkylated, and excess reagents are removed by the first round of filtration. The de-N-glycosylation is catalyzed by PNGase F in an H218O environment to label the de-N-glycosylation site in the protein backbone with one atom of 18O. The released N-glycans are collected by the second round of filtration. The proteolysis is catalyzed by trypsin, and the tryptic peptides are collected by the third round of filtration. Both enzymatic reactions can be accelerated by the assistance of a domestic microwave, resulting in a dramatic decrease of the overall processing time.
The Separation Efficiency of N-Glycans by Filtration
Although N-glycans are much smaller than most proteins, the separation of N-glycans from proteins based on their size difference has rarely been used (24). The initial goal was to test the feasibility of a commercial filter device (10 kDa MWCO) to separate released N-glycans from model glycoproteins, bovine ribonuclease B, bovine fetuin, and human IgG from serum. Decreasing amounts of ribonuclease B (10, 5, and 1 μg) were separately deglycosylated, and their N-glycans were collected by filtration. Even at very low amounts of glycoprotein (1 μg of ribonuclease B is approximately equivalent to 75 picomole of sugar), all five known high-mannose type of N-glycans were unambiguously detected by MALDI-MS (Fig. 2A) with an intensity pattern analogous to larger amounts of starting material (supplemental Fig. S1) and other reports (18). This initial experiment demonstrated the capability of filtration to separate hydrophilic sugar molecules from proteins.
Fig. 2.

The MALDI-MS spectra of permethylated N-glycans released from bovine ribonuclease B (A), bovine fetuin (B), and urine sample U1 (C), collected by the GlycoFilter platform. The Neu5Gc peaks in (B) are annotated with an*. The compositions of the two largest glycans (m/z 4324.9 and 4412.9) in (B) were further confirmed by high resolution ESI-MS and MS/MS fragmentation spectra (Supplemental Fig. S3). All peaks in U1 (C) were assigned a putative topology based on the N-glycosylation biosynthetic pathway and their m/z values. All peaks were single sodium adduct and the monoisotopic peak was annotated.
Bovine fetuin was then evaluated because it has large complex-type multi-antennary N-glycans containing several N-acetylneuraminic acid residues (Neu5Ac) (25), which may be difficult to separate by filtration. Initially, the large N-glycans with more than three Neu5Ac residues could not be efficiently eluted by filtration (supplemental Fig. S2). We theorized that the basic solution used for de-N-glycosylation causes deprotonation of the carboxyl group in sialic acid, which may lead to strong ionic interactions with positively charged groups in proteins and thereby prevent the elution of the acidic N-glycans by filtration. By acidifying to a lower pH (3–4), we effectively and efficiently collected all reported N-glycans of fetuin with a similar intensity pattern as previously reported (21) (Fig. 2B). Notably, additional minor species containing N-glycolylneuraminic acid residue (Neu5Gc) (4), which were not detected by other means, were also observed (Fig. 2B). Two unreported N-glycans at m/z 4324.9 and 4412.9 were also detected, both containing five and four Neu5Ac residues, respectively (Fig. 2B). Their sugar compositions were further confirmed by high accuracy mass spectrometry (MS) and MS/MS fragmentation (supplemental Fig. S3). Overall, acidification appeared to significantly improve the ability to separate and collect large and acidic N-glycans from proteins using filtration.
Because glycan analysis is a regulatory requirement for therapeutic glycoproteins, IgG from human serum was also tested for their compatibility on this platform. As expected, common IgG-type N-glycans (26), G0F, G1F, and G2F, are the most abundant peaks in the MALDI-MS spectrum (supplemental Fig. S4), and the overall intensity pattern was similar to a previous multigroup collaborative report (27), demonstrating that the GlycoFilter might be applicable on the analysis of therapeutic glycoproteins.
Human Urine N-Glycome
To test the capabilities of the GlycoFilter platform on complex samples, urine samples from three healthy individuals were obtained (U1, U2, and U3). The N-glycan profile of each sample was assessed by MALDI-MS. The MALDI-MS profile of U1 had the widest range of glycan peaks ranging from m/z 1345.8 (a fucosylated core structure) to m/z 4587.3 (a complex-type tetra-antennary structure with four Neu5Ac residues based on their molecular weights) (Fig. 2C). Acidic glycans dominated the profile of U1, with only a few neutral compositions smaller than 2500 Da. Further profiling by high accuracy MS detected a total of 87 distinct sugar compositions, of which many were low abundance species and 58 contained Neu5Ac residues (supplemental Table S1). In contrast, the N-glycan profiles of U2 and U3 had fewer acidic species, but many more neutral and fucosylated compositions (supplemental Table S2 and S3). The mass range of the detection was similar in all urine samples, in that several glycans larger than 4000 Da were detected in all three samples, suggesting that the variability in urinary N-glycans compositions may be more likely from differences in the individuals, as opposed to variability in the separation method.
To check whether urinary N-glycans were completely separated by the acidic elution, an additional urine sample was obtained from the donor of U1. After de-N-glycosylation in the filter, eight acidic elutions were conducted. The initial three and the remaining five elutions were respectively combined. The deglycosylated proteins were recovered from the sample chamber, and passed through a C18 SPE cartridge. The flow-through fractions of C18 SPE were collected to detect any remaining N-glycans in the proteins solution. As shown in their MALDI-MS (Supplemental Fig. S5), no glycans were detected from the later acidic elutions (#4–8) or the flow-through fraction of C18 SPE, further demonstrating the effectiveness of the GlycoFilter to separate glycans from proteins.
The Impact of PNGase F-Catalyzed De-N-Glycosylation on Proteomic Performance
The impact of PNGase F catalyzed de-N-glycosylation on the urinary proteome was assessed by dividing each U1, U2, and U3 samples into two equivalent aliquots. Each aliquot was processed in parallel using the GlycoFilter platform (Fig. 1), except for the absence of PNGase F in one aliquot (Control); and the addition of PNGase F for de-N-glycosylation in the other aliquot (+PNGase F) (supplemental Fig. S6). Both aliquots were processed in an H218O environment to identify the deglycosylated sites in +PNGase F samples; and to determine the degree of chemical deamidation in the Control.
Table I and Fig. 3 present the comparisons of the three pairs of urinary proteomes (+PNGase F versus Control). Overall, +PNGase F aliquots in all three pairs consistently displayed similar or slightly better proteomic performance at the peptide level (average gain of 713 peptides per urine sample) as compared with the Control (Fig. 3A). The majority of deglycopeptides (>90%) were uniquely found in the deglycosylated samples, whereas the majority of glycoproteins (> 85%) were also identified in Control samples, implying that other peptides were used to identify these glycoproteins in Control samples (Fig. 3A). Considering there is some degree of inherent variation in LC-MS/MS sampling, the differences may be even more dramatic than those we observed.
Table I. A comparison of the urinary proteomes (U1, U2, and U3) with and without de-N-glycosylation (+PNGase F vs. Control) (supplementary Fig. S6). Proteins were identified by a minimum two peptides per protein and a peptide false discovery rate of ≤1%. The 18O-incorporated peptides were defined as peptides containing deamidated asparagines that were incorporated with one 18O-atom (18O-incorporated aspartic acid). Deglycopeptides were assigned by meeting two criteria: (1) the peptide sequence possessed the N-glycosylation consensus motif Asn-xxx-Ser/Thr (where XXX is any amino acid except proline), and (2) the specific asparagine within the motif was deamidated by one 18O-atom. Deglycopeptides identified in Control samples demonstrated the degree of chemical deamidation and were used to calculate the possible false assignment rate of deglycopeptides in +PNGase F samples. Glycoproteins were defined as the proteins containing at least one deglycopeptide.
| PNGase F | Proteins |
Peptides |
18O-Incorporated peptides |
Deglycopeptides |
Glycoproteins |
||||
|---|---|---|---|---|---|---|---|---|---|
| + | − | + | − | + | − | + | − | + | |
| U1 | 1051 | 981 | 7039 | 6087 | 556 | 39 | 505 | 5 | 287 |
| U2 | 1057 | 990 | 6668 | 6151 | 599 | 18 | 573 | 1 | 314 |
| U3 | 1149 | 1104 | 7639 | 6968 | 697 | 23 | 656 | 1 | 356 |
Fig. 3.

The comparisons of three different urinary proteomes (U1, U2, and U3) with and without PNGase F (+PNGase F versus Control, supplemental Fig. S6) to determine the impact of upfront de-N-glycosylation on overall proteomic performance. A, A Venn-Diagram comparison between +PNGase F and Control at the peptide and the protein levels. The numbers in parentheses indicate the deglycopeptides or glycoproteins identified. In B and C the x axis indicates the log2 value of the ratio of unique peptide counts for either glycoproteins (B) or other proteins (C) identified in common between +PNGase F and Control. The y axis designated the number of proteins within that specific ratio.
Approximately 70% of the overall identified proteins were co-identified in both +PNGase F and Control samples. These co-identified proteins (Fig. 3A) were split into two distinct groups: glycoproteins and other proteins. The log2 ratio of unique peptide counts was calculated for each co-identified protein. A total of 2692 ratios derived from 832 glycoproteins and 1860 other proteins were generated (supplemental Table S4). Plotting the unique peptide count ratio against their frequency of occurrence demonstrated that the vast majority of co-identified glycoproteins had a log2 ratio ≥0.4 indicating a more than 30% increase in unique peptides because of the impact of de-N-glycosylation (Fig. 3B). In fact, 144 of the 832 co-identified glycoproteins had a log2 ratio ≥1, indicating that more than a twofold increase in protein backbone coverage (+PNGase F versus Control) was achieved. On the other hand, the frequency of other proteins had an almost Gaussian distribution around x = 0 indicating a negligible effect on the identification of unique peptides of other proteins with the addition of PNGase F (Fig. 3C). Overall, the upfront de-N-glycosylation appeared to enhance the peptide coverage of glycoproteins without a sacrifice in the coverage of other proteins.
Chemical Deamidation
Because spontaneous chemical deamidation of asparagine will also yield an aspartic acid identical to that from de-N-glycosylation, it was necessary to determine the degree of concurrent chemical deamidation in the H218O environment, as this may cause false assignments of deglycopeptides (28). To measure the degree of chemical deamidation, the Control was also identically processed in H218O without the addition of PNGase F (supplemental Fig. S6). A total of 1852 nonredundant peptides were identified with an 18O-incorporated aspartic acid in the three +PNGase F samples (Table I). As expected, the majority of them (1734) resided on the N-glycosylation consensus motifs indicating that these aspartic acid residues were formed as a result of PNGase F treatment (Table I). In contrast, only 80 peptides were identified to contain an 18O-incorporated aspartic acid in three Control samples, of which seven were located within the consensus motif, indicating that the combined criteria detailed under “Experimental Procedures” to determine a deglycopeptide is effective to minimize the false assignment of deglycopeptides from chemical deamidation (Table I).
Identifying Partially Occupied (PO) Glycosites
Of the 1734 deglycopeptides, a total of 865 nonredundant N-glycosites were identified in the three urine samples, with more than 50% not previously reported as glycosites according to the most recent UniProtKB/Swiss-Prot database (May 2012). In addition to compositional and structural variations of a glycan at a particular glycosite (3), a glycosite may be completely modified or only partially modified by a glycan. By using the GlycoFilter platform to collect nonglycosylated and deglycosylated peptides, a glycosite can be unambiguously determined as PO if both the deglycopeptide (18O-incorporated aspartic acid) and nonglycosylated peptide (asparagine) were detected. For example, when testing bovine fetuin, we detected the known partially occupied site by identifying both the nonglycosylated and deglycosylated peptides (Asn-176, via the tryptic sequence: VVHAVEVALATFNAESNGSLQLVEISR). In the more complex body fluid samples there were many examples of PO. For example, the unique sequence “DLDMFINASK” within the glycoprotein attractin contained a PO glycosite (Asn-1198) in which both the deglycopeptide and nonglycosylated peptide were identified by their distinct MS/MS spectra (Figs. 4A and 4B). A 3 Da mass difference in several fragment ions determined the exact position of asparagine and 18O-incorporated aspartic acid residues, respectively. Even glycoproteins with a single glycosite were found to be PO. For example, the nonglycosylated form of complement factor H related protein 2, which contains a single N-glycosylation consensus motif, was detected in all urine samples (Table II) indicating that N-glycans may not be crucial to regulate a glycoprotein's extracellular destination (2). Using this strategy, 128 glycosites, of which 101 had been previously defined as glycosites, were found to be PO in urine (supplemental Table S5). The PO status of these known glycosites could not have been determined without the detections of their nonglycosylated counterparts.
Fig. 4.
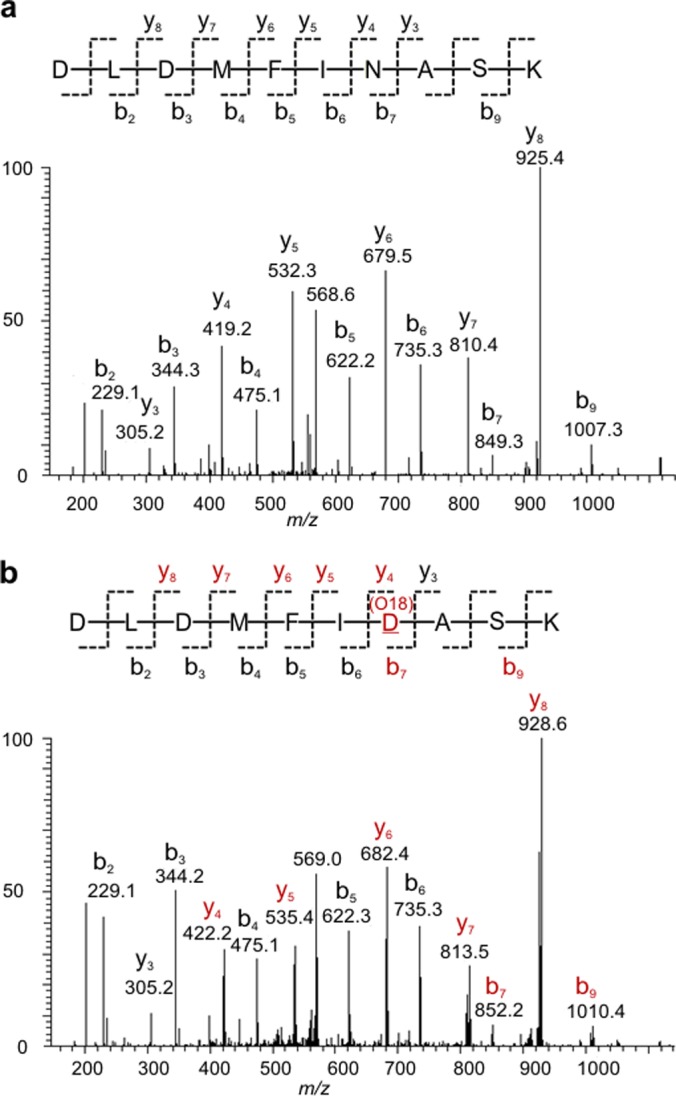
Two distinct MS/MS spectra of the peptide sequence “DLDMFINASK” of the glycoprotein attractin (O75882, ATRN_HUMAN) confirmed a PO glycosite at asparagine-1198. A, The position of the nonsugar-modified asparagine within the sequence was identified by the y4/b7 ions (the precursor ion: m/z 577.2818 2+). B, The position of de-N-glycosylated asparagine (18O-incorporated aspartic acid) was identified by the 3 Da mass shift of a series of b- and y- ions (red highlight) (the precursor ion: m/z 578.7756 2+)The nomenclature of peptide fragment ions was based on previous report (45).
Table II. Examples of selected glycoproteins identified in urine samples (U1, U2, and U3) with PO on specific glycosites. The N-glycosite consensus sequences with the specific sugar-modified asparagine (highlighted as bold) are listed. Glycosites reported and annotated in UniProtKB/Swiss-Prot database are noted as (+). Those not confirmed as glycosites in UniProtKB/Swiss-Prot database are noted as (−). G: glycosite was detected in the deglycopeptide (18O-incorporated aspartic acid) only; N: glycosite was detected in the nonglycosylated peptide only; P: the glycosite was detected in both nonglycosylated and deglycosylated peptides; and (−): the glycosite was not detected.
| UniProt Accession # | Protein name | Glycosite # | Consensus Sequence | UniProt Status | U1 | U2 | U3 |
|---|---|---|---|---|---|---|---|
| Q03591 | Complement factor H-related protein 1 | 126 | NNISS | + | N | N | P |
| P01133 | Pro-epidermal growth factor | 104 | LNGSR | − | G | G | - |
| 148 | GNNSH | − | G | G | G | ||
| 324 | GNCSS | − | P | G | P | ||
| 404 | RNVSE | − | - | P | G | ||
| 596 | ENISQ | − | P | P | - | ||
| 815 | DNITE | − | - | G | G | ||
| 926 | ENASC | − | - | G | - | ||
| P01042 | Kininogen-1 | 48 | QNQSN | + | P | P | P |
| 169 | NNNTQ | + | P | G | P | ||
| 205 | TNCSK | + | P | P | P | ||
| 294 | NNATF | + | P | P | P | ||
| O75882 | Attractin | 264 | SNSSD | + | - | G | G |
| 300 | CNSSD | + | - | G | G | ||
| 325 | ANQSF | − | - | G | G | ||
| 416 | GNVTN | + | G | G | G | ||
| 428 | HNESW | + | G | - | G | ||
| 575 | HNDTS | + | G | - | - | ||
| 731 | RNHSC | + | P | G | P | ||
| 914 | LNGSV | − | G | G | G | ||
| 923 | ANHSA | − | G | G | G | ||
| 1043 | LNSSM | + | - | G | G | ||
| 1054 | YNWSF | − | G | G | G | ||
| 1073 | INQSI | − | N | G | - | ||
| 1082 | ENLTT | − | P | G | G | ||
| 1198 | INASK | + | N | P | P |
If a specific glycoprotein contains multiple glycosites, each glycosite may display a distinct occupancy pattern as compared with other sites. For example, pro-epidermal growth factor has a total of nine N-glycosylation consensus motifs. Of the seven glycosites that were identified in this study, three of the glycosites were found to be PO (Asn-324, 404, and 596) (Table II). The glycoprotein kininogen-1 has four glycosites (Asn-48, 169, 205, and 294) that have been frequently detected by common sugar-based enrichment techniques (8, 13, 29), but in this study, all four sites were found to be PO (Table II). As another example, the heavily glycosylated glycoprotein attractin has 25 consensus motifs, of which 12 have been previously defined as glycosites. In this study, a total of 14 glycosites were identified, of which six were newly determined glycosites (Table II). The nonglycosylated forms were detected for four glycosites (Asn-731, 1073, 1082, and 1198), implying a propensity for nonglycosylation and PO at particular glycosites. These and many other identified glycoproteins (supplemental Table S5) clearly demonstrate that different sites within the same glycoprotein may have a dramatically different pattern in occupancy and that the occupancy of each individual glycosite may vary among samples.
Human Plasma
To further test the technical capabilities of the GlycoFilter, three plasma samples from the same urine donors were also processed. The MALDI-MS profiles of plasma N-glycans were highly similar among three samples (supplemental Fig. S7), indicating that there may be minimal variation of the N-glycome in normal plasma. Alternatively, the lack of variation in the N-glycans could be attributed to the well-known fact that plasma is dominated by a number of constant and highly abundant glycoproteins (30). In this study an average of 316 proteins and 240 deglycopeptides were identified per plasma sample (supplemental Table S6). A total of 295 nonredundant glycosites (supplemental Table S7), including 61 PO sites, were identified from three plasma samples, further indicating the frequent occurrence of partial occupancy in N-glycosylation.
DISCUSSION
In this report, we describe the GlycoFilter as a rapid and widely applicable sample preparation platform for N-glycoproteins that can separately collect both N-glycans and peptides (nonglycosylated and deglycosylated) using a single spin filter device. This platform allows the characterizations of the N-glycome, the former N-glycosites, the PO N-glycosites, and the proteome of a simple mixture or a complex sample such as urine and plasma. The simple nature of the GlycoFilter may also improve the depth of characterization of the glycans and the deglycosylated protein backbone of purified proteins, such as during the analysis of therapeutic glycoproteins. As demonstrated on urine samples, the upfront de-N-glycosylation in H218O environment not only improved the coverage of identified glycoproteins, but also effectively minimized the false assignment of deglycopeptides because of the chemical deamidation. The GlycoFilter platform provides a strategy that allows for the identification of PO in the N-glycoproteome, which is difficult using sugar-based enrichment methods, because of the inherent selectively of the enrichment. In this report we purposefully did not attempt to determine the natural variations of the N-glycome or N-glycoproteome of normal urine and plasma; however, by specifically interrogating biologic replicates of urine and plasma, we demonstrated the technical feasibility of the GlycoFilter on complex samples.
Technical Merits for Glycomics
The GlycoFilter platform has numerous technical merits. In general, processing samples in a single filter minimizes sample transfer, facilitates sample clean up, and permits multiplexing. The GlycoFilter is fully compatible with current glycomics strategies to profile glycans such as reductive derivatization with fluorescent reagents (33) (data not shown) or permethylation (27). To demonstrate its advantage for glycomics research, urine was tested because it is a complex protein analyte (34), which contains significant amounts of chemically heterogeneous metabolites and salts (17). This analytical issue may be one of the reasons that there are few dedicated reports on the urinary N-glycome as compared with extensive glycomics studies on plasma. Our previous efforts to obtain urinary N-glycans using a series of common affinity techniques such as: C18, porous graphitized carbon and hydrophilic interaction chromatography (35), yielded few glycan peaks in MALDI-MS (unpublished data). By using the GlycoFilter, urinary N-glycans were obtained without the need for further purification, highlighting the GlycoFilter's technical advantage in glycomics studies, particularly on complex mixtures. If a universal O-glycanase were available in the future, separation of O-linked glycans could also potentially be integrated into the GlycoFilter platform. Current chemical releasing methods, such as the reductive β-elimination (32), might result in the degradation of protein backbone, and may not be compatible with this platform.
Comparison to the Existing N-Glyco FASP Protocol
Although filtration has previously been applied to collect peptides (FASP) (11) and deglycopeptides using lectin enrichment (N-glyco FASP) (12), filtration has not been reportedly used to separate large and complex N-glycans from deglycosylated proteins. Even though the GlycoFilter is also a filter-based sample preparation method, the distinctive order of steps and buffers of the GlycoFilter protocol exploit the inherent size and chemical properties of N-glycans to allow for efficient separation of the glycome from the proteome. As demonstrated in this study, large N-glycans containing two or more sialic acid residues are difficult to elute without proper pH adjustment (supplemental Figs. S2 and S5). In comparison to current glycomic protocols, the GlycoFilter provides purified glycans that do not require further manipulation. In contrast, the lectin-enrichment based N-glyco FASP protocol (12, 36) would require additional steps to release and capture the glycome after enrichment. The captured glycome would presumably be a reflection of the choice of lectin(s). The GlycoFilter is designed to qualitatively detect PO glycosites because of the identification of the nonglycosylated peptides. Because no method can specifically enrich the nonglycosylated counterparts of deglycopeptides, the detection of PO glycosites is a function of the limitations of MS. The current sugar-based strategies, including N-glyco FASP, may have an inherent sensitivity to detect low abundant sugar-occupied glycosites. Presumably, the PO sites might potentially be identified by the sugar-based enrichment strategies if the nonbinding flow-through is captured and analyzed; however, the detection of nonglycosylated counterparts of deglycopeptides would be also confined by the limitation of MS.
The Implication of the Frequency of PO Glycosites
Current sugar-based enrichment techniques have been shown to significantly enhance the capability to detect low-abundance sugar-occupied glycosites (8–10, 37), however detecting the naturally occurring nonsugar-occupied counterparts of these glycosites requires a secondary experiment. Although not the primary objective of this study, a large number of partially occupied glycosites were identified in healthy individuals. This suggests that incomplete occupancy on an N-glycosite is a common occurrence (supplemental Table S5 and S7). The ability to detect a larger number of the nonglycosylated glycopeptides in a complex mixture is more likely a function of the inherent limitations of data-dependent acquisition mode of LC-MS/MS (38), and other analytical variables such as: fractionation, LC gradient, or the depletion of abundant proteins. We presume that in complex mixtures the actual occurrence of partial occupancy may be more frequent than what was detected. Maximizing the MS variables for a deeper or a directed interrogation would improve this detection, as there are no specific enrichment methods for the nonglycosylated glycopeptides.
Because a glycoprotein may have PO glycosites, or the degree of occupancy of a specific glycosite may change with pathological conditions, such as type I congenital disorders of glycosylation (39, 40), quantifying a glycoprotein using only the sugar-occupied portion may lead to inaccurate quantification. Accordingly, many have reported that changes or variations in protein expression and glycosylation occupancy may not always be analogous or parallel (41, 42). Even though a recent approach proposed analyzing the change of site-specific occupancy between samples using only hydrazide enriched deglycopeptides (43), without a corresponding measurement of the sample specific protein expression this technique may result in incomplete measurements. For example, a twofold increase in site-specific occupancy would be masked by a twofold decrease in protein expression. This concept is similar to protein phosphorylation as it is well known that the stoichiometry of phosphorylation of a site is variable among samples (44). Analysis becomes even more complex with glycoproteins with multiple sites, which may be partially occupied to different degrees. The GlycoFilter platform may improve the quantification of a glycoprotein because all deglycopeptides, nonglycosylated glycopeptides and other peptides (not originating from glycosylated regions) are collected and can be quantified simultaneously by any peptide-based quantification techniques (38). Concomitantly, the identification of a partially occupied glycosite may facilitate the selection of the ideal peptides for quantifying a glycoprotein in a more directed manner.
The Quantification of Occupancy
Unfortunately, unlike phosphopeptides and nonphosphopeptides, which are chemically identical after phosphatase treatment (44), the conversion of sugar-linked asparagine to aspartic acid by PNGase F alters the original peptide chemical identity, leading to different ionization responses in mass spectrometry. Therefore, it is inaccurate to measure the degree of occupancy of a partially occupied glycosite by directly comparing the intensity ratio of deglycopeptides and nonglycosylated peptides in MS. With the advent of the GlycoFilter technology, we expect more and more PO glycosites will be reported and documented in the future. With this knowledge, the degree of occupancy of the glycosite of interest could be more accurately measured by integrating the GlycoFilter platform into a directed mass spectrometry workflow with isotopic internal deglycopeptides and/or nonglycosylated peptide standards (38, 39).
Supplementary Material
Acknowledgments
We thank all members of the Proteomics Center at Boston Children's Hospital for their assistance. We particularly thank Dr. Ali Vaezzadeh, Dr. Judith Steen, and Dr. Hanno Steen for their suggestions on this project. We thank Peter Warren for his assistance on the bioinformatics analysis. We also thank the Department of Urology, Boston Children's Hospital for their continued support.
Footnotes
* This work was supported by the US National Institutes of Health Grants DK077836 (R.S.L.) and DK077836S1 (R.S.L.).
 This article contains supplemental Figs. S1 to S7 and Tables S1 to S7.
This article contains supplemental Figs. S1 to S7 and Tables S1 to S7.
1 The abbreviations used are:
- PO
- partially occupied
- N-Glycosite
- N-glycosylation site
- Deglycopeptide
- de-N-glycosylated peptide
- MWCO
- molecular weight cutoff.
REFERENCES
- 1. Apweiler R., Hermjakob H., Sharon N. (1999) On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys.. Acta 1473, 4–8 [DOI] [PubMed] [Google Scholar]
- 2. Stanley P., Schachter H., Taniguchi N. (2009) N-Glycans. In: Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E., eds. Essentials of Glycobiology, 2nd Ed., Cold Spring Harbor; (NY) [PubMed] [Google Scholar]
- 3. Mariño K., Bones J., Kattla J. J., Rudd P. M. (2010) A systematic approach to protein glycosylation analysis: a path through the maze. Nat. Chem. Biol. 6, 713–723 [DOI] [PubMed] [Google Scholar]
- 4. Ghaderi D., Taylor R. E., Padler-Karavani V., Diaz S., Varki A. (2010) Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nat. Biotechnol. 28, 863–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bosques C. J., Collins B. E., Meador J. W., 3rd, Sarvaiya H., Murphy J. L., Dellorusso G., Bulik D. A., Hsu I. H., Washburn N., Sipsey S. F., Myette J. R., Raman R., Shriver Z., Sasisekharan R., Venkataraman G. (2010) Chinese hamster ovary cells can produce galactose-alpha-1,3-galactose antigens on proteins. Nat. Biotechnol. 28, 1153–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hart G. W., Copeland R. J. (2010) Glycomics hits the big time. Cell 143, 672–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Steentoft C., Vakhrushev S. Y., Vester-Christensen M. B., Schjoldager K. T., Kong Y., Bennett E. P., Mandel U., Wandall H., Levery S. B., Clausen H. (2011) Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat. Methods 8, 977–982 [DOI] [PubMed] [Google Scholar]
- 8. Zhang H., Li X. J., Martin D. B., Aebersold R. (2003) Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol. 21, 660–666 [DOI] [PubMed] [Google Scholar]
- 9. Kaji H., Saito H., Yamauchi Y., Shinkawa T., Taoka M., Hirabayashi J., Kasai K., Takahashi N., Isobe T. (2003) Lectin affinity capture, isotope-coded tagging and mass spectrometry to identify N-linked glycoproteins. Nat. Biotechnol. 21, 667–672 [DOI] [PubMed] [Google Scholar]
- 10. Pan S., Chen R., Aebersold R., Brentnall T. A. (2011) Mass spectrometry based glycoproteomics–from a proteomics perspective. Mol. Cell. Proteomics 10, R110.003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wiśniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 12. Zielinska D. F., Gnad F., Wiśniewski J. R., Mann M. (2010) Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 141, 897–907 [DOI] [PubMed] [Google Scholar]
- 13. Liu T., Qian W. J., Gritsenko M. A., Camp D. G., 2nd, Monroe M. E., Moore R. J., Smith R. D. (2005) Human plasma N-glycoproteome analysis by immunoaffinity subtraction, hydrazide chemistry, and mass spectrometry. J. Proteome Res. 4, 2070–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Walsh C. T., Garneau-Tsodikova S., Gatto G. J., Jr. (2005) Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem. Int. Ed. Engl. 44, 7342–7372 [DOI] [PubMed] [Google Scholar]
- 15. Petrescu A. J., Milac A. L., Petrescu S. M., Dwek R. A., Wormald M. R. (2004) Statistical analysis of the protein environment of N-glycosylation sites: implications for occupancy, structure, and folding. Glycobiology 14, 103–114 [DOI] [PubMed] [Google Scholar]
- 16. Sumer-Bayraktar Z., Kolarich D., Campbell M. P., Ali S., Packer N. H., Thaysen-Andersen M. (2011) N-glycans modulate the function of human corticosteroid-binding globulin. Mol. Cell. Proteomics 10, M111.009100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vaezzadeh A. R., Briscoe A. C., Steen H., Lee R. S. (2010) One-step sample concentration, purification, and albumin depletion method for urinary proteomics. J. Proteome Res. 9, 6082–6089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou H., Briscoe A. C., Froehlich J. W., Lee R. S. (2012) PNGase F catalyzes de-N-glycosylation in a domestic microwave. Anal. Biochem. 427, 33–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vaezzadeh A. R., Deshusses J. M., Waridel P., François P., Zimmermann-Ivol C. G., Lescuyer P., Schrenzel J., Hochstrasser D. F. (2010) Accelerated digestion for high-throughput proteomics analysis of whole bacterial proteomes. J. Microbiol. Methods 80, 56–62 [DOI] [PubMed] [Google Scholar]
- 20. Ciucanu I., Costello C. E. (2003) Elimination of oxidative degradation during the per-O-methylation of carbohydrates. J. Am. Chem. Soc. 125, 16213–16219 [DOI] [PubMed] [Google Scholar]
- 21. Kang P., Mechref Y., Klouckova I., Novotny M. V. (2005) Solid-phase permethylation of glycans for mass spectrometric analysis. Rapid Commun. Mass Spectrom. 19, 3421–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 23. Choi H., Ghosh D., Nesvizhskii A. I. (2008) Statistical validation of peptide identifications in large-scale proteomics using the target-decoy database search strategy and flexible mixture modeling. J. Proteome Res. 7, 286–292 [DOI] [PubMed] [Google Scholar]
- 24. Qin H., Zhao L., Li R., Wu R., Zou H. (2011) Size-selective enrichment of N-linked glycans using highly ordered mesoporous carbon material and detection by MALDI-TOF MS. Anal. Chem. 83, 7721–7728 [DOI] [PubMed] [Google Scholar]
- 25. Green E. D., Adelt G., Baenziger J. U., Wilson S., Van Halbeek H. (1988) The asparagine-linked oligosaccharides on bovine fetuin. Structural analysis of N-glycanase-released oligosaccharides by 500-megahertz 1H NMR spectroscopy. J. Biol. Chem. 263, 18253–18268 [PubMed] [Google Scholar]
- 26. Huhn C., Selman M. H., Ruhaak L. R., Deelder A. M., Wuhrer M. (2009) IgG glycosylation analysis. Proteomics 9, 882–913 [DOI] [PubMed] [Google Scholar]
- 27. Wada Y., Azadi P., Costello C. E., Dell A., Dwek R. A., Geyer H., Geyer R., Kakehi K., Karlsson N. G., Kato K., Kawasaki N., Khoo K. H., Kim S., Kondo A., Lattova E., Mechref Y., Miyoshi E., Nakamura K., Narimatsu H., Novotny M. V., Packer N. H., Perreault H., Peter-Katalinic J., Pohlentz G., Reinhold V. N., Rudd P. M., Suzuki A., Taniguchi N. (2007) Comparison of the methods for profiling glycoprotein glycans–HUPO Human Disease Glycomics/Proteome Initiative multi-institutional study. Glycobiology 17, 411–422 [DOI] [PubMed] [Google Scholar]
- 28. Palmisano G., Melo-Braga M. N., Engholm-Keller K., Parker B. L., Larsen M. R. (2012) Chemical deamidation: a common pitfall in large-scale N-linked glycoproteomic mass spectrometry-based analyses. J. Proteome Res. 11, 1949–1957 [DOI] [PubMed] [Google Scholar]
- 29. Chen R., Jiang X., Sun D., Han G., Wang F., Ye M., Wang L., Zou H. (2009) Glycoproteomics analysis of human liver tissue by combination of multiple enzyme digestion and hydrazide chemistry. J. Proteome Res. 8, 651–661 [DOI] [PubMed] [Google Scholar]
- 30. Tu C., Rudnick P. A., Martinez M. Y., Cheek K. L., Stein S. E., Slebos R. J., Liebler D. C. (2010) Depletion of abundant plasma proteins and limitations of plasma proteomics. J. Proteome Res. 9, 4982–4991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Addona T. A., Shi X., Keshishian H., Mani D. R., Burgess M., Gillette M. A., Clauser K. R., Shen D., Lewis G. D., Farrell L. A., Fifer M. A., Sabatine M. S., Gerszten R. E., Carr S. A. (2011) A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat. Biotechnol. 29, 635–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carlson D. M. (1968) Structures and immunochemical properties of oligosaccharides isolated from pig submaxillary mucins. J. Biol. Chem. 243, 616–626 [PubMed] [Google Scholar]
- 33. Xia B., Kawar Z. S., Ju T., Alvarez R. A., Sachdev G. P., Cummings R. D. (2005) Versatile fluorescent derivatization of glycans for glycomic analysis. Nat. Methods 2, 845–850 [DOI] [PubMed] [Google Scholar]
- 34. Halim A., Nilsson J., Ruetschi U., Hesse C., Larson G. (2012) Human urinary glycoproteomics; attachment site specific analysis of N- and O-linked glycosylations by CID and ECD. Mol. Cell. Proteomics 11, M111.013649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ruhaak L. R., Zauner G., Huhn C., Bruggink C., Deelder A. M., Wuhrer M. (2010) Glycan labeling strategies and their use in identification and quantification. Anal. Bioanal. Chem. 397, 3457–3481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boersema P. J., Geiger T., Wisniewski J. R., Mann M. (2013) Quantification of the N-glycosylated Secretome by Super-SILAC During Breast Cancer Progression and in Human Blood Samples. Mol. Cell. Proteomics 12, 158–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wada Y., Gu J., Okamoto N., Inui K. (1994) Diagnosis of carbohydrate-deficient glycoprotein syndrome by matrix-assisted laser desorption time-of-flight mass spectrometry. Biol. Mass Spectrom. 23, 108–109 [DOI] [PubMed] [Google Scholar]
- 38. Domon B., Aebersold R. (2010) Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721 [DOI] [PubMed] [Google Scholar]
- 39. Hülsmeier A. J., Paesold-Burda P., Hennet T. (2007) N-glycosylation site occupancy in serum glycoproteins using multiple reaction monitoring liquid chromatography-mass spectrometry. Mol. Cell. Proteomics 6, 2132–2138 [DOI] [PubMed] [Google Scholar]
- 40. Sturiale L., Barone R., Garozzo D. (2011) The impact of mass spectrometry in the diagnosis of congenital disorders of glycosylation. J. Inherited Metabolic Dis. 34, 891–899 [DOI] [PubMed] [Google Scholar]
- 41. Liu Z., Cao J., He Y., Qiao L., Xu C., Lu H., Yang P. (2010) Tandem 18O stable isotope labeling for quantification of N-glycoproteome. J. Proteome Res. 9, 227–236 [DOI] [PubMed] [Google Scholar]
- 42. Ueda K., Takami S., Saichi N., Daigo Y., Ishikawa N., Kohno N., Katsumata M., Yamane A., Ota M., Sato T. A., Nakamura Y., Nakagawa H. (2010) Development of serum glycoproteomic profiling technique; simultaneous identification of glycosylation sites and site-specific quantification of glycan structure changes. Mol. Cell. Proteomics 9, 1819–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pan S., Tamura Y., Chen R., May D., McIntosh M. W., Brentnall T. A. (2012) Large-scale quantitative glycoproteomics analysis of site-specific glycosylation occupancy. Mol. Biosyst. 8, 2850–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu R., Haas W., Dephoure N., Huttlin E. L., Zhai B., Sowa M. E., Gygi S. P. (2011) A large-scale method to measure absolute protein phosphorylation stoichiometries. Nat. Methods 8, 677–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Roepstorff P., Fohlman J. (1984) Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed. Mass Spectrom. 11, 601. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.