

Diatoms frustules are created under the control of biomolecules (silaffins, silacidins and long-chain polyamines) at close to physiologic conditions.[1–4] The mechanism of biosilica formation was traditionally based on the ability of zwitterionic water-soluble proteins to create macromolecular assemblies for silica polymerization.[5–7] Recent discoveries of water-insoluble collagen matrices within certain sponge biosilica spicules[8], chitin-based scaffolds in sponge and diatom biosilica formations[9,10], as well as cingulins within diatom girdle bands[4], revive the use of insoluble biomimetic organic templates for morphogenesis of non-porous silica structures. The use of fibrillar collagen as templates for biosilica synthesis was unsuccessful in the past as only extrafibrillar silica deposition was observed.[11,12] Intrafibrillar mineralization of collagen has important implications from a biophysical perspective.[13] Here, we report a collagen biosilicification scheme based on fusion of stabilized polysilicic acid into a fluidic precursor phase upon their infiltration into polyamine-enriched collagen. The latter serves as a template and catalyst for polymerization of the precursor phase into silica that faithfully reproduces the collagen tertiary architecture. Our findings provide a new concept in biosilica materials synthesis which does not require phosphate supplements.
Type-I collagen has been widely used for hybrid biomaterials synthesis because of its biocompatibility.[14,15] Nevertheless, the lack of mechanical resistance of highly porous, non-mineralized collagen matrices limits their application as stress-bearing scaffolds for bone repair.[5] The potential stimulating effect of silicic acid on osteogenesis[16,17]provides the incentive to produce silicified collagen matrices for bone regeneration applications. Previous studies have explored methods to produce silica-collagen hybrid materials by simultaneous self-assembly of collagen and silica. However, the kinetics of collagen self-assembly and silica polymerization are not fully compatible.[18] Other studies that utilized fibrillar collagen as templates produced silica particles in close vicinity of the collagen fibrils but not within the fibrils, which limits the strength of the biohybrid materials.[12] Silica-collagen hybrids may also be potentially usedfor preparation of non-biological functional materials;[19,20] however, the fundamental prerequisite is that collagen fibrils should be fully infiltrated with a high intrafibrillar silica content.
During diatom cell wall synthesis, silicic acid is stabilized and prevented from spontaneous polymerization into silica.[21] Silicic acid gels rapidly in vitro at pH 5.5 without stabilization (Supporting Information S2, Figure S2-A and S3, Figure S3-A). Thus, we used choline chloride to stabilize silicic acid hydrolyzed from tetraethyl orthosilicate.[22] Based on assessment of gelling times, 72 mM choline chloride is required to stabilize 1.5% silicic acid (pH 5.5) for 72 h before gelling (Supporting Information S2, Figure S2-A). This provided sufficient time for the choline-stabilized silicic acid (Ch-SA) to infiltrate type-I collagen sponges. The mean particle diameter of Ch-SA was ~9.7 nm based on dynamic light scattering measurements (Supporting Information S2, Figures S2-B and S2-C).
A series of control experiments was initially performed using unstained sections prepared for transmission electron microscopy (TEM) to determine the optimal conditions for silicification of collagen sponges with Ch-SA (Supporting Information 3, Figures S3 A-E). By embracing the model of cingulin proteins as water-insoluble templates in the silicification of diatom girdle bands,[4] successful intrafibrillar silicification of collagen was achieved in 4 days after immersing polyallylamine (PAH)-enriched collagen sponges (treatment with 6.67×10−4 M PAH for 4 h) in 1.5% Ch-SA. Unlike hydrated non-silicified collagen sponges that collapsed after drying, the silicified sponges retained their original shapes after dehydration (Supporting Information S3, Figure S3-F). This collagen silicification scheme does not involve the use of phosphates to induce phase separation of PAH into macromolecular assemblies.[23] Despite identification of extrafibrillar silica particles by scanning electron microscopy (SEM), the silicified collagen sponges remained highly porous (Supporting Information S3). The well-packed manifestation of the silicified collagen fibrils was attributed to their extensive intrafibrillar silicification. Under TEM, electron-dense minerals were identified within the fibrils that reproduced the cross banding and microfibrillar architecture of fibrillar collagen (Figure 1). The presence of silicon within the collagen fibrils was confirmed using scanning transmission electron microscopy-energy dispersed X-ray analysis (STEM-EDX; Figure 2). Examination of the initial stage of silicification at 24 h identified a continuous electron-dense phase that was formed by fusion of Ch-SA nanoparticles after their infiltration into the PAH-enriched collagen fibrils (Supporting Information S4). Micro-computed tomography of the silicifiedcollagen sponges showed that silicification occurred throughout the entire sponge (Supporting Information S5).
Figure 1. Unstained TEM images of silicified PAH-enriched collagen.

a, A leaflet from a silicified collagen sponge showing electron-dense minerals inside collagen fibrils (bar = 1 mm). b, Intrafibrillar electron-dense minerals replicate the cross banding and microfibrillar architecture of fibrillar collagen (bar = 100 nm). Inset: selected area electron diffraction reveals the amorphous nature of the infiltrated minerals. c, A heavily silicified collagen fibril showing continuous intrafibrillar mineral strands (bar = 50 nm). d, Cross section showing mineral infiltration from the surface to the center of the fibrils (bar = 50 nm).
Figure 2. STEM-EDX of an unstained leaflet within a silicified PAH-enriched collagen sponge.

Darkfield image shows mineral deposition within the collagen fibrils (bar = 500 nm). The latter contain predominantly silicon and oxygen.
Silicified PAH-enriched collagen sponges were characterized using different analytical techniques. Fourier transform-infrared spectroscopy was used to identify peaks associated with hydrated silica (Supporting Information S6). As both electron diffraction (Figure 1b, inset) and powder X-ray diffraction of the silicified sponges showed that the infiltrated mineral phase was amorphous, we sintered the silicified sponges in atmospheric air to 1000 °C and observed different crystalline silica phases (predominantly tridymite and cristabolite) after sintering (Supporting Information S7). This confirms that the mineral phase prior to sintering is amorphous silica. Using 29Si cross polarization-magic angle spinning nuclear magnetic resonance spectroscopy (CP-MAS NMR), three different Q species were delineated from the NMR spectrum that provide information on the connectivity of the silica network: Q4 (bulk siloxane), Q3 (single silanol) and Q2 (geminal silanol) (Figure 3a).[24] The Q4:Q3:Q2 intensity ratio (1:0.76:0.19) of the biosilicified collagen matrix is comparable to the ratio identified from the diatom Coscinodiscus granii (1:0.71:0.05),[23] indicating that they have a similar degree of silica condensation. When 13C CP-MAS NMR was performed on the silicified collagen sponges, we observed an amino acid profile that is characteristic of collagen (Figure 3b).[24] High resolution thermogravimetric analysis showed that the mineral content in the silicified PAH-enriched collagen sponges was 57.2% (Figure 4a). Although this mineral content is lower than that of mineralized bone (ca. 60–70 wt%)[25], one has to take into account the lower molecular weight of silica (60.1) versus that of hydroxyapatite (502.3). Changes in derivative weight with temperature further revealed weight loss-rate profiles (Figure 4b) characteristic of collagen[26] and hydrated silica.[27]
Figure 3. Solid-state NMR of silicified PAH-enriched collagen sponges.

a, 29Si CP-MAS spectrum. The broad peak between -78 to -125 ppm was deconvoluted to reveal Q4 (siloxane) at ~110 ppm, Q3 (single silanol) at ~100 ppm and Q2 (geminal silanol) at ~91 ppm. b, 13C CP-MAS spectrum. Signals assigned to glycine (Gly), alanine (Ala), proline (Pro) and hydroxyproline (Hyp) correspond to those identified from fibrillar collagen.[24]
Figure 4. TGA of non-silicified vs silicified PAH-enriched collagen sponges.

a, Plot of weight loss vs temperature showing a 57.2 wt% increase in mineral content after silicification. b, Plot of derivative weight vs temperature. For the non-silicified sponge, the two peaks at 49.6 °C and 64.7 °C are caused by dissociation of water from the primary and secondary hydration compartments of type I collagen. The two additional peaks observed at high temperatures are attributed to decomposition (289.7 °C) and combustion of the organic matrix (752 °C).[26] For the silicified sponge, three desorption processes can be observed: physisorbed water (51.6 °C), hydrogen-bonded structural water (289.7 °C) and water released by condensation of silanol to siloxane (547.8 °C).[27]
The mechanical properties of silicified collagen sponges were investigated by examining their compressive stress-strain responses (Figure 5). The tangent modulus of the silicified sponges based on responses from 0–5% strain is 599.8 ± 166.0 kPa, which is ~48,000 times higher than that of non-silicified sponges (0.013 ± 0.004 kPa). For comparison, the elastic modulus of human trabecular bone is highly dependent on density and range from ~10 MPa to over 10 GPa.[28] The modulus of toughness of the silicified collagen sponges (165.3 ± 5.0 kPa) is ~1500 times higher than the value obtained for non-silicified sponges (0.11 ± 0.002 kPa). The modulus of toughness of trabecular bone with density = 0.3 kg/cm3 is 1.2 MPa.[29] Although both the stiffness and toughness of the silicified collagen matrices are lower than human bone, one has to consider that fiber leaflets of a collagen sponge are much thinner (< 5 μm; Figure 2, Supporting Information S3) than natural bone trabeculae (mean trabecular thickness 140–172 μm).[30,31] Collagen fibrils within fiber leaflets of collagen sponges are also somewhat isolated and are not able to reinforce one another, as in bone. Moreover, collagen sponges lack the different levels of hierarchy that are seen in bone. As silicified collagen sponges demonstrated improved stress-strain relationships over the highly compliant non-silicified sponge, they are potentially useful as porous scaffolds for bone repair in regions with minimal to moderate load-bearing requirements.
Figure 5. Compressive stress-strain responses of silicified PAH-enriched vs non-silicified, hydrated collagen sponges.
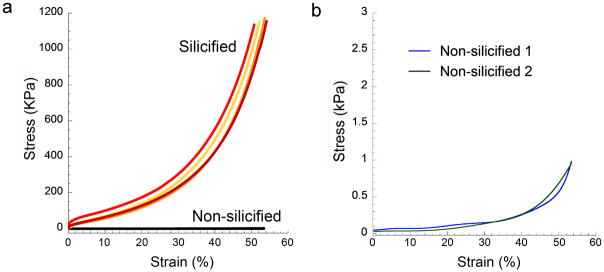
a, Stress-strain responses were recorded only to the maximum capacity of the load cell and did not represent failure of the silicified sponges. b, For the non-silicified sponges, the stress axis is scaled down for better visualization.
To verify that intrafibrillar silicification of collagen is not a “staining artifact” (sections were examined unstained), we sintered silicified collagen sponges to completely remove the organic components (Supporting Information S8). Appearance of non-porous fiber cores indicates that collagen fibrils were heavily silicified with amorphous silica prior tosintering. To verify that silicification of reconstituted collagen sponges can be reproduced in natural collagen, we silicified PAH-enriched rat tail tendon collagen in 1.5% Ch-SA and obtained the same results (Supporting Information S9). Electron tomography and 3-D visualization of silicified rat tail tendon collagen are included in Supporting Information S9.
Our findings provide a new concept in biosilica materials synthesis that does not require phosphate supplements. Figure 6 is a schematic depicting the hypothetical events that occur in this biosilicification scheme. It is possible that PAH treatment of collagen fibrils results in phase separation of the PAH into macromolecular assemblies within the microfibrillar milieu of the fibrils. As Ch-SA nanoparticles infiltrate the collagen fibril, they fuse to produce a liquid-like silica precursor phase resembling the polymer-induced liquid precursors in calcium carbonate and calcium phosphate mineralization of collagen[32,33], that eventually fills the gap zones and microfibrillar spaces. An alternative mechanism is that PAH covers the collagen sponge surface, with is positive charge attracting the negatively charged silica species to facilitate silicification of the PAH-coated sponges. Either mechanism results in the PAH-enriched collagen serving as a template for polymerization of the silica precursor phase. Complete infiltration of the water-filled spaces by the liquid-like silica precursor phase results in replication of the cross banding patterns and microfibrillar architecture of the collagen fibril.
Figure 6.

A schematic of a biosilicification scheme based on the use of a polyamine-enriched collagen template.
Experimental Section
Methods summary can be found in Supporting Information S1. A flow chart summarizing the experimental design can be found in Supporting Information Figure S1.
Supplementary Material
Acknowledgments
This work was supported by grant R21 DE019213 from NIDCR (PI. Franklin Tay), the PSRP and ESA awards from the Georgia Health Sciences University and grantNSFC 81130078 from Nature Science Foundation of China (PI. Ji-hua Chen). We thank R. Smith (Electron Microscopy Core Unit, Georgia Health Sciences University, USA) for performing electron diffractions, F. Chan (Electron Microscopy Unit, The University of Hong Kong, China) for performing STEM-EDX and electron tomography and M. Burnside for secretarial support.
References
- 1.Kröger N, Deutzmann R, Sumper M. Science. 1999;286:1129–1132. doi: 10.1126/science.286.5442.1129. [DOI] [PubMed] [Google Scholar]
- 2.Kröger N, Lorenz S, Brunner E, Sumper M. Science. 2002;29:584–586. doi: 10.1126/science.1076221. [DOI] [PubMed] [Google Scholar]
- 3.Wenzl S, Hett R, Richthammer P, Sumper M. Angew Chem. 2008;120:1753–1756. doi: 10.1002/anie.200704994. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:1729–1732. doi: 10.1002/anie.200704994. [DOI] [PubMed] [Google Scholar]
- 4.Scheffel A, Poulsen N, Shian S, Kröger N. Proc Natl Acad Sci USA. 2011;108:3175–3180. doi: 10.1073/pnas.1012842108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cha JN, Stucky GD, Morse DE, Deming TJ. Nature. 2000;403:289–292. doi: 10.1038/35002038. [DOI] [PubMed] [Google Scholar]
- 6.Brott LL, Naik RR, Pikas DJ, Kirkpatrick SM, Tomlin DW, Whitlock PW, Clarson SJ, Stone MO. Nature. 2001;13:291–293. doi: 10.1038/35095031. [DOI] [PubMed] [Google Scholar]
- 7.Sumper M, Brunner E. Adv Func Mater. 2006;16:17–27. [Google Scholar]
- 8.Ehrlich H, et al. Nat Chem. 2010;2:1084–1088. doi: 10.1038/nchem.899. [DOI] [PubMed] [Google Scholar]
- 9.Ehrlich H, Krautter M, Hanke T, Simon P, Knieb C, Heinemann S, Worch H. J Exp Zool B Mol Dev Evol. 2007;308:473–483. doi: 10.1002/jez.b.21174. [DOI] [PubMed] [Google Scholar]
- 10.Brunner E, Richthammer P, Ehrlich H, Paasch S, Simon P, Ueberlein S, vanPée KH. Angew Chem. 2009;121:9904–9907. doi: 10.1002/anie.200905028. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:9724–9727. doi: 10.1002/anie.200905028. [DOI] [PubMed] [Google Scholar]
- 11.Heinemann S, Heinemann C, Ehrlich H, Meyer M, Baltzer H, Worch H, Hanke T. Adv Eng Mater. 2007;9:1061–1068. [Google Scholar]
- 12.Desimone MF, Hélary C, Rietveld IB, Bataille I, Mosser G, Giraud-Guille MM, Livage J, Coradin T. Acta Biomater. 2010;6:3998–4004. doi: 10.1016/j.actbio.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 13.Gupta HS. Proc Natl Acad Sci USA. 2006;103:17741–17746. doi: 10.1073/pnas.0604237103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puxkandl R, Zizak I, Paris O, Keckes J, Tesch W, Bernstorff S, Purslow P, Fratzl P. Philos Trans R Soc Lond B Biol Sci. 2002;357:191–197. doi: 10.1098/rstb.2001.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glowacki J, Mizuno S. Biopolymers. 2008;89:338–344. doi: 10.1002/bip.20871. [DOI] [PubMed] [Google Scholar]
- 16.Reffitt DM, Ogston N, Jugdaohsingh R, Cheung HF, Evans BA, Thompson RP, Powell JJ, Hampson GN. Bone. 2003;32:127–135. doi: 10.1016/s8756-3282(02)00950-x. [DOI] [PubMed] [Google Scholar]
- 17.Hoppe A, Güldal NS, Boccaccini AR. Biomaterials. 2011;32:2757–2574. doi: 10.1016/j.biomaterials.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Eglin D, Coradin T, GiraudGuille MM, Helary C, Livage J. Biomed Mater Engin. 2005;15:43–50. [PubMed] [Google Scholar]
- 19.Shin Y, Wang C, Exarhos GJ. Adv Mater. 2005;17:73–77. [Google Scholar]
- 20.Bao Z, et al. Nature. 2007;446:172–175. doi: 10.1038/nature05570. (Supporting Information S10; S25) [DOI] [PubMed] [Google Scholar]
- 21.Hildebrand M, Dahlin K, Volcani BE. Mol Gen Genet. 1999;260:480–486. doi: 10.1007/s004380050920. [DOI] [PubMed] [Google Scholar]
- 22.Bronder SR. Stabilizing orthosilicic acid comprising preparation and biological preparation. 5,922,360 United States Patent Office. 1999
- 23.Lutz K, Gröger C, Sumper M, Brunner E. Phys Chem Chem Phys. 2005;7:2812–2815. doi: 10.1039/b505945c. [DOI] [PubMed] [Google Scholar]
- 24.Saitô H, Tabeta R, Shoji A, Ozaki T, Ando I, Miyata T. Biopolymers. 1984;23:2279–2297. doi: 10.1002/bip.360231111. [DOI] [PubMed] [Google Scholar]
- 25.Weiner S, Traub W, Wagner HD. J Struct Biol. 1999;126:241–255. doi: 10.1006/jsbi.1999.4107. [DOI] [PubMed] [Google Scholar]
- 26.Lim JJ, Shamos MH. Biopolymers. 1974;13:1791–1907. doi: 10.1002/bip.1974.360130912. [DOI] [PubMed] [Google Scholar]
- 27.Odlyha M, Scott RPW, Simpson CF. J Thermal Anal. 1993;40:1197–1212. [Google Scholar]
- 28.Carter DR, Hayes WC. J Bone Joint Surg Am. 1977;59:954–962. [PubMed] [Google Scholar]
- 29.Ashman RB, Rho JY. J Biomech. 1988;21:177–181. doi: 10.1016/0021-9290(88)90167-4. [DOI] [PubMed] [Google Scholar]
- 30.Parfitt AM, Mathews CHE, Villanueva AR, Kleerekoper M, Frame B, Rao DS. J Clin Invest. 1983;72:1396–1409. doi: 10.1172/JCI111096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parkinson IH, Forbes D, Sutton-Smith P, Fazzalari NL. J Osteoporos. 2010:641578. doi: 10.4061/2010/641578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olszta MJ, Douglas EP, Gower LB. Calcif Tissue Int. 2003;72:583–591. doi: 10.1007/s00223-002-1032-7. [DOI] [PubMed] [Google Scholar]
- 33.Jee SS, Thula TT, Gower LB. Acta Biomater. 2010;6:3676–3686. doi: 10.1016/j.actbio.2010.03.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.