

Abstract
Vascular dementia ranks as the second leading cause of dementia in the United States. However, its underlying pathophysiological mechanism is not fully understood and no effective treatment is available. The purpose of the current study was to evaluate long-term cognitive deficits induced by transient middle cerebral artery occlusion (tMCAO) in rats and to investigate the underlying mechanism. Sprague-Dawley rats were subjected to tMCAO or sham surgery. Behavior tests for locomotor activity and cognitive function were conducted at 7 or 30 days after stroke. Hippocampal long term potentiation (LTP) and involvement of GABAergic neurotransmission were evaluated at 30 days after sham surgery or stroke. Immunohistochemistry and Western blot analyses were conducted to determine the effect of tMCAO on cell signaling in the hippocampus. Transient MCAO induced a progressive deficiency in spatial performance. At 30 days after stroke, no neuron loss or synaptic marker change in the hippocampus were observed. LTP in both sides of the hippocampus was reduced at 30 days after stroke. This LTP impairment was prevented by blocking GABAA receptors. In addition, ERK activity was significantly reduced in both sides of the hippocampus. In summary, we identified a progressive decline in spatial learning and memory after ischemic stroke that correlates with suppression of hippocampal LTP, elevation of GABAergic neurotransmission, and inhibition of ERK activation. Our results indicate that the attenuation of GABAergic activity or enhancement of ERK/MAPK activation in the hippocampus might be potential therapeutic approaches to prevent or attenuate cognitive impairment after ischemic stroke.
Keywords: stroke, long term potentiation, hippocampus, vascular dementia, cognition, GABA, ERK
Introduction
Stroke ranks as the fourth leading cause of death in the United States, with ischemic stroke comprising more than 80% of total stroke. The stroke patients must not only survive the acute stages of infarction, but they must then cope with the ongoing neurological impairment. Stroke is the most common cause of permanent disability among people in the United States and is associated with a high incidence of deficits in both sensorimotor function as well as cognitive ability (Phipps, 1991). Epidemiological studies have shown that the prevalence of dementia in ischemic stroke patients is nine-fold higher than controls at 3 months (Tatemichi et al., 1992), and 4-12 times higher than in controls 4 years after a lacuna infarct (Loeb et al., 1992). Many of these dementias developed progressively after stroke, and thus cannot be interpreted as direct consequence of the primary ischemic damage (Tatemichi et al., 1994).
Middle cerebral artery occlusion (MCAO) in rodents is considered to be a convenient, reproducible, and reliable model of cerebral ischemia in humans. MCAO typically results in extensive damage in the cortex and caudate putamen, and sensorimotor behavior impairment has been extensively studied in ischemic stroke models. Spontaneous partial or complete recovery of sensorimotor function has been reported consistently over time after ischemic stroke (DeVries et al., 2001; Markgraf et al., 1992; Yonemori et al., 1999). On the other hand, the assessment of the cognitive impairment after MCAO has yielded conflicting results (Bingham et al., 2012; Bland et al., 2000; DeVries et al., 2001). Characterization of long-term cognitive outcome after stroke will be critical for discovery of therapeutic approaches and evaluating efficacies of potential therapeutic agents.
In the present study, the progression of impaired spatial learning and memory performance after ischemic stroke was evaluated in a rat model of transient focal cerebral ischemia. Then, potential underlying mechanisms of the progressive impairments were studied in the hippocampus, a brain region recognized to play a vital role in spatial information processing and memory formation. Our study demonstrated a progressive decline in capacity to acquire spatial learning after ischemic stroke, which could be attributed to functional changes in the hippocampus distal to the primary infarct area.
Materials and Methods
Animal and experimental procedures
All procedures were approved by the IACUC of UNTHSC. Transient middle cerebral artery occlusion (tMCAO) was induced in adult male Sprague-Dawley rats (225 to 250 g, Charles River, 2-3 months old) as described previously (Liu et al., 2010). Rats were assigned to one of three groups randomly: (1) sham control, (2) 7 days post tMCAO and (3) 30 days post tMCAO. The left middle cerebral artery was occluded by a 3-0 monofilament suture for 1 hour. Sham surgery was performed as per tMCAO with the exception that no suture was introduced into the internal carotid artery. Separate groups of the rats were allowed to recover from surgery for different time periods. Laboratory staff performing behavioral assessments and biochemical/LTP measurements had no knowledge of the group assignments at the time of testing/assay.
Behavioral assessments
Spontaneous locomotor activity and spatial learning and memory tests were conducted in separate groups of the rats beginning either at 7 days or 30 days after tMCAO. Spontaneous locomotor activity was measured using a Digiscan apparatus (Omnitech Electronics) as described previously (Forster and Lal, 1991). For each of the testing sessions the rats were placed in clear acrylic chambers for a period of 60 minutes. The apparatus recorded the distance traveled in cm (i.e., forward locomotion) and the time spent in a 36 × 36 cm zone in the center of the chamber. After the first test (Test 1), the rats were re-tested at 37, 52, and 97 days after tMCAO.
Spatial learning/memory was tested using a Morris water maze as described previously (Sumien et al., 2006). During a pre-training phase, the rats were allowed to swim from one end of a straight alley that was submerged partially in the water maze tank, and climb to a hidden platform at the other end, where they remained for 10 s before being returned to a holding cage. A black tent over the tank prevented rats from exposure to spatial cues in the room during this phase. The pre-training sessions were conducted during the morning and afternoon of 2 consecutive days; they each consisted of 5 trials conducted at 10-min intervals. The black tent and alley were removed during 4 subsequent sessions (spatial acquisition phase) during which rats swam in the open tank from one of 4 different starting positions until they located the platform, which remained in a fixed position just below the water surface. Efficiency in reaching the platform on each trial was considered in terms of the path length and swim speed as assessed using a contrast video tracking system. Spatial acquisition sessions were conducted during the morning and afternoon of 2 consecutive days and consisted of 5 trials separated by 10 min. Sessions conducted on days 3 and 4 began with a probe trial during which the platform was lowered for 30 s, and memory performance was considered proportional to the time spent in a 20-cm annulus around the platform location. The probe trials were followed by 5 trials in which the platform was available in its location from the training phase. On the afternoon of day 4, the platform was moved to a new location, and the efficiency of each rat to learn the new position was measured subsequently on five trials. Training to the new location was continued on day 5 for 5 trials, and this was followed by a probe trial (as described above) 10 min following the last trial.
A test of visually-cued learning in the Morris water maze was conducted following the test for spatial learning and memory. During this phase, the location of the platform was shifted on each trial but the location was identified by a triangular flag (5 cm each side, 11 cm2) that was raised above the surface of the water. Visible platform sessions consisting of 5 trials at 10-min intervals were administered in the morning and afternoon of two consecutive days. Each trial began from a different starting point in the tank.
Electrophysiological analysis
Rats were sacrificed at 30 days after tMCAO or sham surgeries. Transverse hippocampal slices (350 μm thickness) were prepared and incubated in oxygenated artificial CSF. LTP of CA3-CA1 synapses from two bisected hippocampal slices of same animal were recorded simultaneously in two stations. Theta burst stimulus (TBS) protocol was used to induce LTP. For the experiment using GABAA receptor antagonist, picrotoxin (60 μM) was applied to the bath 10 min before TBS and continuously perfused throughout the recordings. The field excitatory postsynaptic potentiation (fEPSP) slopes were calculated with Clampfit 10.2, and all the slope values were normalized against the average slope over the 10 min before the tetanus. Magnitude of LTP was expressed with average value of normalized fEPSP slopes from 50 to 60 min following the tetanus.
Neuropathological and Western blot analysis
Immunohistochemistry of NeuN (Santa Cruz) was conducted in paraffin-embedded brain slices from sham and stroke rats sacrificed at 30 days after stroke as described previously (Li et al., 2011). Immunohistochemistry of pERK was conducted using frozen sections. For Western blot analyses, brains were harvested at 30 days after tMCAO or sham surgeries to evaluate expression of synaptic markers: NR2b, GluR4, PSD95 and GABAA receptor γ2 subunit (Santa Cruz) in the hippocampus. Band intensity was normalized to actin for quantitative analysis. Activation of Mitogen-activated protein (MAP) kinases (MAPK) pathway was evaluated by Western blots: phospho-P38 (pP38), P38, phospho-JNK (pJNK), JNK, phospho-ERK (pERK) and total ERK (Cell Signaling).
Statistical Analysis
Data were expressed as mean ± SEM. Student's t-test (paired or unpaired) or one or two-way ANOVA with Student–Newman–Keuls multiple comparison test was used to determine statistical significance (p<0.05). Spatial learning and memory data were considered in 3-way analyses of variance, with Trials and Days as within-groups factors, and Treatment as a between groups factor. Planned individual comparisons of treatment groups with control on specific trials were performed using single degree of freedom F tests within the 3-way interaction.
Results
Progression of functional impairment after ischemic stroke
A significant increase of spontaneous locomotor activity was observed for rats tested 30 days after tMCAO (Figure 1A). There was an increase in the mean of time spending within the center zone of the chamber for tMCAO groups when tested at 7 and 30 days after stroke, that was not statistically significant (p>0.05) (Figure 1B, test 1). During a retest at 52 and 97 days after tMCAO (Figure 1B, test 2), the time spent in the center zone by these rats was significantly increased (Figure 1B).
Figure 1.
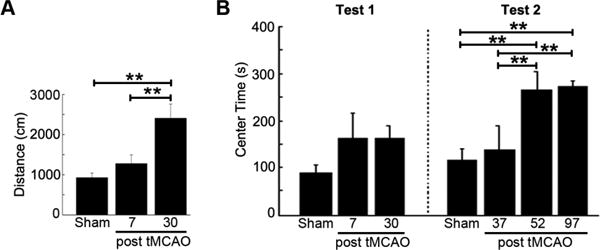
Increase of spontaneous locomotor activity after tMCAO. A. Movement of the rat in open field test for locomotor activity (** p<0.01). B. Increase of center time at 52 and 97 days after tMCAO. The animals were first tested (test 1) at 7 or 30 days after tMCAO in 7-day (n=13) or 30-day (n=15) group, respectively. Then, they were repeatedly tested (test 2) at 37, 52, and 97 days after tMCAO or sham surgery (sham) (** p<0.01).
By the end of the pre-training phase, all rats in the sham control and 7- and 30-days post tMCAO groups could swim efficiently and climb on to the platform using the straight alley. By the end of the spatial acquisition phase on day 2, all rats had also learned to navigate the water maze and reach the platform efficiently from different starting positions in the open tank (Figure 2A and B). However, groups that had received experimental stroke showed less efficient navigation when compared to the sham control group during the first five trials of acquisition and had poorer retention during the first trial of session 2. This deficit was more severe in the 30-days post tMCAO group than in the 7-day post tMCAO group and, additionally, the 30-days post tMCAO rats showed a deficit in probe trial performance (Figure 2C) and in learning the new platform position. Analyses of variance conducted on the path length data for acquisition and reversal phases yielded significant Trial × Day × Treatment interactions in support of the above observations, which suggested a time-dependent increase in deficient spatial performance in the water maze following tMCAO.
Figure 2.
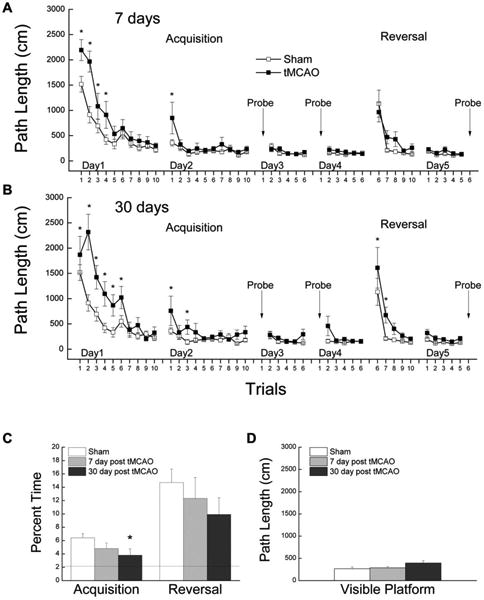
Ischemic stroke induced progressive spatial learning/memory impairment. A. Path length ± S.E.M as a function of trials during the spatial learning and reversal phases of Morris water maze for separate groups of rats tested 7 days following tMCAO (n=13) vs. sham control (n=19). Down arrows indicate probe trials (* p<0.05 vs. sham control using pooled error term from ANOVA). B. Path length ± S.E.M as a function of trials for separate groups of rats tested 30 days following tMCAO (n=15) as compared with the same sham control (n=19). C. Percent time spent in an annulus 20-cm around the platform location during probe trials during acquisition and reversal phases. The dotted line represents chance level of probe performance (* p<0.05 vs. sham control). D. Path length of the rats during their last session of visible platform.
The test of visually cued learning conducted following the spatial acquisition and reversal phases failed to suggest a significant effect of Treatment group on the ability of the rats to locate a platform tagged by a flag (p=0.146), suggesting no obvious visual deficits had developed following tMCAO (Figure 2D). Furthermore, the spatial learning and memory deficits were also not likely due to motor impairments as there was no effect of treatment group on path independent swim speed during pre-training, acquisition, or reversal (data not shown).
Hippocampal LTP is reduced after ischemic stroke
We measured the input-output relationship by stimulation of Schaffer collaterals at increasing current intensities (30-100 μA). Input-output curves for control rats and stroke rats at 30 days after tMCAO were not statistically different for a given stimulation current (Figure 3A, B). The average slopes of fEPSP from both sides of the hippocampus evoked by 100 μA stimulus are 0.903 ± 0.106 mV/ms in sham (n=19), and 0.875 ± 0.100 mV/ms in tMCAO animals (n=24), respectively (p>0.05). Moreover, no difference in input-output curves was observed between the ipsilateral and contralateral hippocampus from stroke animals (Figure 3A). We compared paired-pulse facilitation (PPF), a calcium-dependent form of presynaptic plasticity. Similar to input-output relation, we observed no difference in PPF between control and tMCAO groups across a range of inter-stimulus intervals (Figure 3B). These data suggest that general synaptic transmission and presynaptic plasticity are not altered in the CA1 area after tMCAO.
Figure 3.
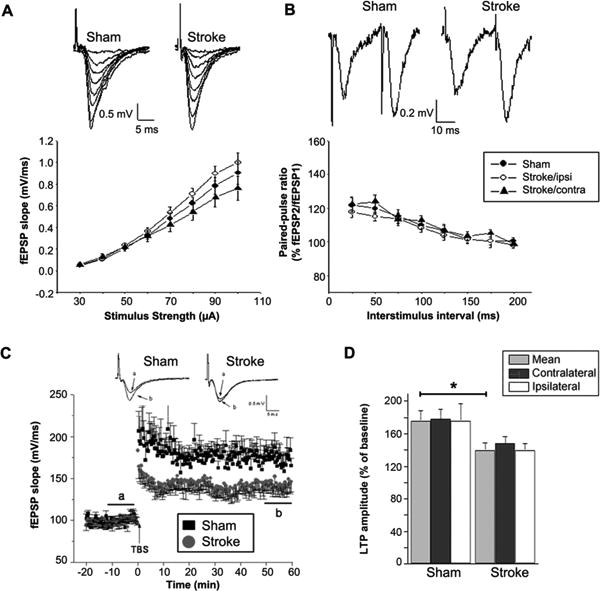
Ischemic stroke decreased hippocampal LTP in both hemispheres at 30 days after tMCAO. A. Input-output relationship shows the basic synaptic property of hippocampus slices from sham control and stroke rats at 30 days after tMCAO. B. Paired-pulse facilitation. The ratios of the EPSPs amplitude evoked by second stimulus to that by the first stimulus (EPSP2/EPSP1) are plotted against interpulse intervals. C. Time course of the fEPSP slopes in sham control (n=11) and stroke rats (n=14). Representative traces of fEPSPs recorded before (a) and after (b) TBS in hippocampal slices from sham control or stroke rats. D. LTP amplitude was indiscriminately reduced in both sides of the hippocampus at 30 day after tMCAO (*p<0.05, stroke vs sham control).
We investigated hippocampal LTP in rats sacrificed at 30 days after tMCAO. Hippocampal LTP in rats at 30 days after tMCAO (146 ± 8.1%, n=14) was significantly reduced compared with that in sham controls (176 ± 12%, n=11, p<0.05) (Figure 3C). Moreover, no significant difference was observed in LTP between the ipsilateral and contralateral hippocampus at 30 days after tMCAO (137 ± 11%, ipsilateral, n=9; 148 ± 8.8% contralateral, n=10; p>0.05). Taken together, these data indicate that tMCAO reduces LTP in both sides of the hippocampus without affecting synaptic transmission and presynaptic plasticity.
LTP impairment after stroke is mediated by GABAA receptors
We evaluated GABAergic neurotransmission and its potential role in the suppressed hippocampal LTP after ischemic stroke. In the presence of 60 μM picrotoxin, hippocampal LTP of the rats at 30 days after tMCAO was significantly increased (from 146 ± 8.1% to 179 ± 9.7%, p<0.05), and was not significantly different from that of sham controls (190 ± 13% in sham, n=7; 179 ± 9.7% in tMCAO, n=11. p>0.05) (Figure 4A). In addition, the magnitude of LTP increase in picorotoxin-treated slices was much greater in tMCAO group (33%, from 146 ± 8.1% to 179 ± 9.7%) than that in the sham control animals (14%, from 176 ± 11% to 190 ± 13%). However, no significant difference in the expression of GABAA receptor γ2 subunit was found with Western blot in the hippocampus between stroke and sham control animals (Figure 4B, C). These data indicate that functional enhancement in GABAergic neurotransmission might be involved in LTP inhibition in the hippocampus after tMCAO.
Figure 4.
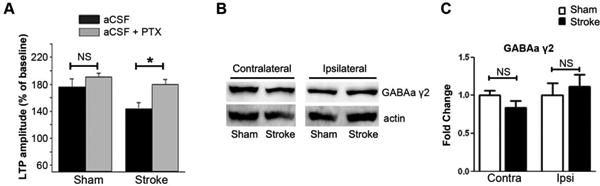
Inhibition of GABAA receptor function attenuated hippocampal LTP impairment after tMACO. A. LTP impairment after stroke is attenuated by picrotoxin (PTX) (*p<0.05, NS: not significant). Representative Western blots (B) and quantitative analysis (C) of GABAA γ2 subunit expression in the hippocampus of sham control and stroke rats at 30 days after tMCAO (n=5).
Transient MCAO reduces ERK activity in the hippocampus
Obvious lesions in the cortex and striatum were observed in all stroke rats after tMCAO. H&E staining and immunohistochemistry of NeuN indicated that there was no obvious neuron loss in the hippocampus at 30 days after tMCAO. TNUEL staining indicated that there was no obvious apoptosis in the hippocampus at 10 days and 30 days after tMCAO, while there was apparent apoptosis in the lesions area of the cortex at 10 days after tMCAO (Figure 5A). Ischemic damage was also observed in the anterior part of the entorhinal cortex (Figure 5A).
Figure 5.
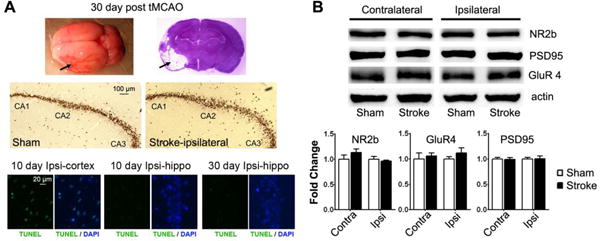
Neuropathological analyses of the hippocampus at 30 days after tMCAO. A. Upper panel: whole brain and coronal section H&E staining at 30 days after tMCAO (arrows indicate the infarct region). Middle panel: NeuN staining of the hippocampus (sham control and ipsilateral hippocampus of rats after tMCAO). Lower panel: TUNEL staining (green color) of ipsilateral cortex and hippocampus at 10 or 30 days after tMCAO. Nucleus were stained with DAPI in blue. B. Western blots and quantitative analysis of NR2b, GluR4 and PSD95 in the contralateral and ipsilateral hippocampus of sham control and stroke rats at 30 days after tMCAO (n=5).
We evaluated expression of NMDA receptor subunit NR2b, AMPA receptor subunit GluR4, and postsynaptic marker PSD95 with Western blot analyses. Quantitative analyses indicated no significant change in the expression of these markers at either ipsilateral or contralateral hippocampus compared with sham controls (Figure 5B).
We investigated the MAPK pathways in the hippocampus at 30 days after tMCAO. No obvious changes of JNK and P38 pathways were observed in rats at 30 days after tMCAO compared to sham controls (Figure 6A). On the other hand, a significant decrease in ERK activation was found in both sides of the hippocampus at 30 days after tMCAO compared with control rats (n=5, p <0.01) (Figure 6B, 6C). Consistently, immunohistochemistry indicated a loss of pERK in neurons at both side of hippocampus at 30 days after tMCAO (Figure 6C).
Figure 6.
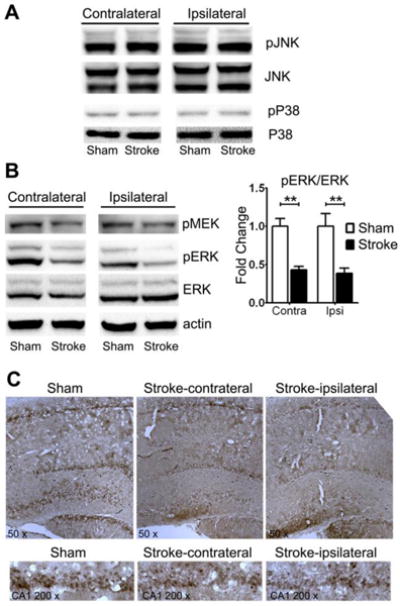
Reduction of ERK/MAPK activation in the ipsilateral and contralateral hippocampus at 30 days after tMCAO. A. Western blots show no change in p38 and JNK activation in the hippocampus. B. Depicted are representative Western blots of pMEK, pERK, and ERK in the hippocampus of sham control and stroke rats, indicating decreased phosphorylation of MEK and ERK. (** p<0.01, n=5). C. Representative immunohistochemistry of pERK in the hippocampus of sham control and stroke rats.
Discussion
Ischemic stroke is associated with a high incidence of sensorimotor and cognitive dysfunction. Both sensorimotor and cognitive impairments have been demonstrated in rats after MCAO. Sensorimotor impairment has been extensive studied in rats after MCAO. Spontaneous partial or complete recovery of sensorimotor function has been frequently reported over time after ischemic stroke in this model (DeVries et al., 2001; Karhunen et al., 2003; Markgraf et al., 1994; Markgraf et al., 1997; Roof et al., 2001; Yonemori et al., 1999). On the other hand, a progressive cognitive function impairment was indicated (Yang et al., 2006).
Traditional concepts of vascular dementia postulate that cognitive decline in patients with ischemic stroke results from stroke alone when a large volume of brain is affected by the infarct, which overcomes the brain's compensatory mechanisms. In addition, vascular dementia is not always related with large infarct (Roman et al., 1993). Clinically asymptomic vascular brain injury might also cause cognitive impairment (DeCarli, 2013). As many dementias develop progressively after stroke, an outcome that is difficult to interpret as a direct deficit induced by the primary cerebral damage. Rather, the progressive course of dementia suggests a progressive degenerative disorder (Tatemichi et al., 1994; Yang et al., 2006). The experimental protocols and indices of cognitive performance used in previous studies of experimental stroke injury have varied considerably, and this may account for significant inconsistency in the reported outcomes (Bingham et al., 2012). However, deficient performance of rats in the Morris water maze and other spatial tasks, which is a reliable outcome following hippocampal damage (Winocur and Moscovitch, 1990), would appear to be a consistent outcome over time after MCAO (Bingham et al., 2012; DeVries et al., 2001; Karhunen et al., 2003). In the present study, rats showed a progressive impairment of spatial learning and memory after ischemic stroke, which might be best described as initially a delayed acquisition, followed later by an impaired rate of learning and impaired retention which could be detected in the absence of visual or sensorimotor impairment. Neuropsychiatric symptoms are a common accompaniment of dementia (Lyketsos et al., 2002). In concordance with the progressive cognitive deficit after transient cerebral ischemia, the spontaneous locomotor activity showed a delayed increase in center zone time after transient focal cerebral ischemia, suggesting a progressive anxiolytic effect induced by ischemic stroke.
The current study was focused on the cognitive function after stroke. Nonetheless, our spontaneous locomotor activity test does not show a progressive decrease of motor activity after stroke. Instead, a progressive increase of spontaneous motor activity was observed after stroke. In addition, the swim speed analysis indicated no difference between the animals tested at 7 and 30 days after stroke (Supplement Figure 1). All these data suggests that the motor function recovery is unlike paralleled with the progressive cognitive decline after stroke.
Given the extensive neuronal network within and between each hemisphere, it might not be surprise that focal cerebral ischemia could trigger prolonged global reaction. Increasing evidence has indicated occlusion of a cerebral artery could induce both hemodynamic and cellular changes beyond the ischemic territory (Liu et al., 2012). It has been well known that the focal cerebral ischemia could induce stem cell proliferation in the subventricular and dentate gyrus, areas beyond the ischemic territory. The activation of adenosine monophosphate-activated protein kinase (AMPK), a critical energy sensor for energy metabolism, was found not only in the ischemic territory but also in non-ischemic hemisphere within 24 hours after transient focal cerebral ischemia induced by MCAO (McCullough et al., 2005). It was demonstrated that occlusion of a cerebral artery could induce hemodynamic change beyond the ischemic territory (Winters et al., 2012). Hippocampus plays a vital role in information processing, memory formation, and subsequent regulation of behavior (Lynch, 2004). MCAO induces very mild cerebral blood flow reduction in hippocampus in rats since it is supplied by posterior cerebral artery (Tanaka et al., 2000). Cognitive deficits after MCAO have been reported without neuron loss in the hippocampus (Okada et al., 1995; Wahl et al., 1992). In the current study, neither neuron loss nor reduction of synaptic markers was observed in the hippocampus at 30 days after tMCAO. Consistently, our electrophysiological study indicated that basic synaptic functions in the hippocampus were not affected by tMCAO as no difference was found in the input/output curve and the paired-pulse facilitation between stroke and sham controls. On the other hand, a significant reduction of LTP was observed at both sides of the hippocampus at 30 days after tMCAO.
Lesion of unilateral entorhinal cortex induced cognitive deficits as tested in Morris Water Maze in male rats while female rat didn't show any deficits (Roof et al., 1993). In our tMCAO model, partial lesion of the anterior part of the entorhinal cortex in the ipsilateral cortex was observed, while most part of the entorhinal cortex was saved. This partial lesion might have contributed to the cognitive deficits in water maze test. However, inhibition of the LTP in both hippocampi could have contributed more to the cognitive deficits in our tMCAO model (Bourtchuladze et al., 1994).
The GABAA receptor mediates most inhibition and plays a critical role in synaptic plasticity and learning behaviors. GABA acting at GABAA receptors limits postsynaptic depolarization during LTP induction and thus modifies LTP and learning. The involvement of GABAergic inhibitory interneurons during LTP induction is well documented (Kullmann and Lamsa, 2011). Our studies suggest that GABAergic neurotransmission is enhanced in the hippocampus after stroke as pharmacological blockade of GABAA receptors prevents LTP impairment after stroke. Our finding is in agreement with the observation that GABA function is enhanced in the brain area adjacent to stroke damage (Clarkson et al., 2010). Increased GABAergic neurotransmission in the hippocampus might also explain the anxiolytic effect induced by tMCAO (Gonzalez et al., 1998).
Activation of MAPKs, including ERK, JNK and p38, have been observed in the ischemic area immediately after MCAO (Irving and Bamford, 2002). In present study, we found that ERK, but not JNK and p38, activation was reduced in both sides of the hippocampus at 30 days after ischemic stroke. Activation of ERK pathway has been reported to be essential for LTP induction in the hippocampus (Davis et al., 2000) (Lynch, 2004). In addition, the ERK pathway has been implicated to negatively modulate GABAA receptor (Bell-Horner et al., 2006). We predict that the reduction of ERK activity could enhance GABAergic inhibition, hence contribute to the suppression of hippocampal LTP and cognitive impairment after ischemic stroke.
In current tMCAO model, GABAergic signaling and ERK pathway activity were affected in both ipsilateral and contralateral hippocampus. It has been found that, after MCAO, nitric oxide was released to cerebral spinal fluid (CSF) (Kader et al., 1993), which could lead to ERK inhibition (Estrada et al., 1997) and activation of GABAA receptor (Castel and Vaudry, 2001). GABAergic signaling can be regulated through different mechanisms, such as receptor expression, trafficking, activity modulation, GABA synthesis, release and recycling (Macdonald and Olsen, 1994). ERK pathway activity can also be inhibited by modulating different molecules of the pathway. Extensive experiments are needed to identify the underlying mechanisms. Nonetheless, our study indicated that GABA and ERK signaling might be potential targets to prevent vascular dementia induced by ischemic stroke. Alpha 5 subunit containing GABAA receptor subtype is expressed predominantly in the hippocampus and can be antagonized specifically by benzodiazepine inverse agonist L655,708 (Atack et al., 2006), which has been shown to promote cognitive performance and reduce motor function deficits after experimental stroke in rodent (Clarkson et al., 2010). Intraventricular administration of EGF has been shown to promote neurongenesis after stroke (Teramoto et al., 2003), and reduce cognitive deficits after brain trauma (Sun et al., 2010). We speculate that these two methods might be capable to attenuate cognitive deficits after stroke.
In conclusion, our current study identifies a progressive impairment of spatial learning/memory performance and a concurrent reduction of hippocampus LTP after ischemic stroke in an experimental MCAO model. Our results suggest further that elevated GABAergic inhibition and decreased ERK activation in the hippocampus may constitute the underlying mechanisms, and that attenuation of GABAA receptor activity and/or enhancement of ERK/MAPK signaling in the hippocampus might be potential therapeutic targets to prevent or attenuate cognitive function impairment after ischemic stroke.
Supplementary Material
Highlights.
tMCAO induced a progressive decline in spatial learning and memory.
tMCAO induced bilateral hippocampal LTP suppression.
tMCAO increased hippocampal GABAergic neurotransmission.
tMCAO decreased hippocampal ERK activation.
Acknowledgments
Sources of Fundings: National Institutes of Health grants R01NS054687 (SY), R01NS054651 (SY), P01AG022550 (JWS), UNT Health Science Center Faculty Seed Grant (RQH), National Natural Science Foundation of China Grant 81228009 (SY, FY).
Footnotes
Disclosures: none.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atack JR, et al. L-655,708 enhances cognition in rats but is not proconvulsant at a dose selective for alpha5-containing GABAA receptors. Neuropharmacology. 2006;51:1023–9. doi: 10.1016/j.neuropharm.2006.04.018. [DOI] [PubMed] [Google Scholar]
- Bell-Horner CL, et al. ERK/MAPK pathway regulates GABAA receptors. J Neurobiol. 2006;66:1467–74. doi: 10.1002/neu.20327. [DOI] [PubMed] [Google Scholar]
- Bingham D, et al. Watermaze performance after middle cerebral artery occlusion in the rat: the role of sensorimotor versus memory impairments. J Cereb Blood Flow Meta. 2012;32:989–99. doi: 10.1038/jcbfm.2012.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland ST, et al. Early exclusive use of the affected forelimb after moderate transient focal ischemia in rats : functional and anatomic outcome. Stroke. 2000;31:1144–52. doi: 10.1161/01.str.31.5.1144. [DOI] [PubMed] [Google Scholar]
- Bourtchuladze R, et al. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- Castel H, Vaudry H. Nitric oxide directly activates GABA(A) receptor function through a cGMP/protein kinase-independent pathway in frog pituitary melanotrophs. J Neuroendocrinol. 2001;13:695–705. doi: 10.1046/j.1365-2826.2001.00683.x. [DOI] [PubMed] [Google Scholar]
- Clarkson AN, et al. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature. 2010;468:305–9. doi: 10.1038/nature09511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, et al. The MAPK/ERK cascade targets both Elk-1 and cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J Neurosci. 2000;20:4563–72. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCarli C. Clinically asymptomatic vascular brain injury: a potent cause of cognitive impairment among older individuals. J Alzheimers Dis. 2013;33(Suppl 1):S417–26. doi: 10.3233/JAD-2012-129004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries AC, et al. Cognitive and behavioral assessment in experimental stroke research: will it prove useful? Neurosci Biobehav Rev. 2001;25:325–42. doi: 10.1016/s0149-7634(01)00017-3. [DOI] [PubMed] [Google Scholar]
- Estrada C, et al. Nitric oxide reversibly inhibits the epidermal growth factor receptor tyrosine kinase. Biochem J. 1997;326(Pt 2):369–76. doi: 10.1042/bj3260369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster MJ, Lal H. Neurobehavioral biomarkers of aging: influence of genotype and dietary restriction. Biomed Environ Sci. 1991;4:144–65. [PubMed] [Google Scholar]
- Gonzalez LE, et al. Stimulation of benzodiazepine receptors in the dorsal hippocampus and median raphe reveals differential GABAergic control in two animal tests of anxiety. Eur J Neurosci. 1998;10:3673–80. doi: 10.1046/j.1460-9568.1998.00375.x. [DOI] [PubMed] [Google Scholar]
- Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab. 2002;22:631–47. doi: 10.1097/00004647-200206000-00001. [DOI] [PubMed] [Google Scholar]
- Kader A, et al. Nitric oxide production during focal cerebral ischemia in rats. Stroke. 1993;24:1709–16. doi: 10.1161/01.str.24.11.1709. [DOI] [PubMed] [Google Scholar]
- Karhunen H, et al. Long-term functional consequences of transient occlusion of the middle cerebral artery in rats: a 1-year follow-up of the development of epileptogenesis and memory impairment in relation to sensorimotor deficits. Epilepsy Res. 2003;54:1–10. doi: 10.1016/s0920-1211(03)00034-2. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Lamsa KP. LTP and LTD in cortical GABAergic interneurons: emerging rules and roles. Neuropharmacology. 2011;60:712–9. doi: 10.1016/j.neuropharm.2010.12.020. [DOI] [PubMed] [Google Scholar]
- Li W, et al. Regulation of matrix metalloproteinase 2 by oligomeric amyloid beta protein. Brain Res. 2011;1387:141–8. doi: 10.1016/j.brainres.2011.02.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, et al. Combination therapy of 17beta-estradiol and recombinant tissue plasminogen activator for experimental ischemic stroke. J Pharmacol Exp Ther. 2010;332:1006–12. doi: 10.1124/jpet.109.160937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, et al. Neuroprotection targeting ischemic penumbra and beyond for the treatment of ischemic stroke. Neurol Res. 2012;34:331–7. doi: 10.1179/1743132812Y.0000000020. [DOI] [PubMed] [Google Scholar]
- Loeb C, et al. Dementia associated with lacunar infarction. Stroke. 1992;23:1225–9. doi: 10.1161/01.str.23.9.1225. [DOI] [PubMed] [Google Scholar]
- Lyketsos CG, et al. Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: results from the cardiovascular health study. JAMA. 2002;288:1475–83. doi: 10.1001/jama.288.12.1475. [DOI] [PubMed] [Google Scholar]
- Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84:87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Olsen RW. GABAA receptor channels. Annu Rev Neurosci. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- Markgraf CG, et al. Sensorimotor and cognitive consequences of middle cerebral artery occlusion in rats. Brain Res. 1992;575:238–46. doi: 10.1016/0006-8993(92)90085-n. [DOI] [PubMed] [Google Scholar]
- Markgraf CG, et al. Recovery of sensorimotor function after distal middle cerebral artery photothrombotic occlusion in rats. Stroke. 1994;25:153–9. doi: 10.1161/01.str.25.1.153. [DOI] [PubMed] [Google Scholar]
- Markgraf CG, et al. Behavioral recovery patterns in rats receiving the NMDA receptor antagonist MDL 100,453 immediately post-stroke. Pharmacol Biochem Behav. 1997;56:391–7. doi: 10.1016/s0091-3057(96)00231-6. [DOI] [PubMed] [Google Scholar]
- McCullough LD, et al. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280:20493–502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- Okada M, et al. Long-term spatial cognitive impairment after middle cerebral artery occlusion in rats: no involvement of the hippocampus. J Cereb Blood Flow Metab. 1995;15:1012–21. doi: 10.1038/jcbfm.1995.127. [DOI] [PubMed] [Google Scholar]
- Phipps MA. Assessment of neurologic deficits in stroke. Acute-care and rehabilitation implications. Nurs Clin North Am. 1991;26:957–70. [PubMed] [Google Scholar]
- Roman GC, et al. Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology. 1993;43:250–60. doi: 10.1212/wnl.43.2.250. [DOI] [PubMed] [Google Scholar]
- Roof RL, et al. A comparison of long-term functional outcome after 2 middle cerebral artery occlusion models in rats. Stroke. 2001;32:2648–57. doi: 10.1161/hs1101.097397. [DOI] [PubMed] [Google Scholar]
- Roof RL, et al. Gender-specific impairment on Morris water maze task after entorhinal cortex lesion. Behav Brain Res. 1993;57:47–51. doi: 10.1016/0166-4328(93)90060-4. [DOI] [PubMed] [Google Scholar]
- Sumien N, et al. Profiling psychomotor and cognitive aging in four-way cross mice. Age (Dordr) 2006;28:265–82. doi: 10.1007/s11357-006-9015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, et al. The effect of epidermal growth factor in the injured brain after trauma in rats. J Neurotrauma. 2010;27:923–38. doi: 10.1089/neu.2009.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, et al. Persistent CREB phosphorylation with protection of hippocampal CA1 pyramidal neurons following temporary occlusion of the middle cerebral artery in the rat. Exp Neurol. 2000;161:462–71. doi: 10.1006/exnr.1999.7313. [DOI] [PubMed] [Google Scholar]
- Tatemichi TK, et al. Dementia after stroke: baseline frequency, risks, and clinical features in a hospitalized cohort. Neurology. 1992;42:1185–93. doi: 10.1212/wnl.42.6.1185. [DOI] [PubMed] [Google Scholar]
- Tatemichi TK, et al. Risk of dementia after stroke in a hospitalized cohort: results of a longitudinal study. Neurology. 1994;44:1885–91. doi: 10.1212/wnl.44.10.1885. [DOI] [PubMed] [Google Scholar]
- Teramoto T, et al. EGF amplifies the replacement of parvalbumin-expressing striatal interneurons after ischemia. J Clin Invest. 2003;111:1125–32. doi: 10.1172/JCI17170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl F, et al. Neurological and behavioral outcomes of focal cerebral ischemia in rats. Stroke. 1992;23:267–72. doi: 10.1161/01.str.23.2.267. [DOI] [PubMed] [Google Scholar]
- Winocur G, Moscovitch M. Hippocampal and prefrontal cortex contributions to learning and memory: analysis of lesion and aging effects on maze learning in rats. Behav Neurosci. 1990;104:544–51. doi: 10.1037//0735-7044.104.4.544. [DOI] [PubMed] [Google Scholar]
- Winters A, et al. Transient Focal Cerebral Ischemia Induces Long-Term Cerebral Vasculature Dysfunction in a Rodent Experimental Stroke Model. Transl Stroke Res. 2012;3:279–285. doi: 10.1007/s12975-012-0148-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, et al. Endovascular middle cerebral artery occlusion in rats as a model for studying vascular dementia. Age (Dordr) 2006;28:297–307. doi: 10.1007/s11357-006-9026-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemori F, et al. Spatial cognitive performance after chronic focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1999;19:483–94. doi: 10.1097/00004647-199905000-00002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.