

Abstract
The endothelial glycocalyx is a dynamic layer of macromolecules at the luminal surface of vascular endothelium that is involved in fluid homeostasis and regulation. Its role in vascular permeability and edema formation is emerging but is still not well understood. In this special article, we highlight key concepts of endothelial dysfunction with regards to the glycocalyx and provide new insights into the glycocalyx as a mediator of processes central to the development of pulmonary edema and lung injury.
Introduction
The endothelial glycocalyx (EG) is a complex and multicomponent layer of macromolecules at the luminal surface of vascular endothelium. This concept was proposed more than 70 years ago and its composition is well studied as detailed in two reviews;1,2 however, its role in mechanisms of endothelial protection and injury and subsequent clinical implications have just recently become evident. The EG consists of a variety of endothelial membrane-bound molecules, including glycoproteins and proteoglycans, that provide the basis for plasma-endothelial cell interaction. EG structure, though well characterized in vitro, is poorly defined in vivo because of its dynamically changing composition by self-assembly and enzymatic degradation or shear-dependent shedding of its elements. Its major constituents are hyaluronic acid and the negatively charged heparan sulphate proteoglycans. Together with glycosaminoglycans (GAG) and plasma proteins, the EG layer as a whole forms the endothelial surface layer which acts as a barrier to circulating cells and large molecules. Considerable prognostic and therapeutic promise lies with the emergence of the EG as a key mediator of endothelial dysfunction in pathogenic states, particularly with regard to vascular permeability and edema formation. Several studies have demonstrated the role of the EG in plasma/interstitial fluid balance and solute exchange,3,4,5 mechanotransduction that couples intravascular pressure and shear stress (i.e., biomechanical forces) to endothelial cell responses (i.e., biochemical signals),6 and the inflammatory response cascade via physical blockade of neutrophils to the endothelial cell surface.7,8,9 This review explores the emerging evidence for the role of the EG in vascular permeability, examines evidence for modulation by the EG of inflammatory processes that lead to edema formation, and provides insight into the role of the EG in the development of pulmonary edema and lung injury. The concept of the glycocalyx as a mechanotransducer of pathophysiologic signals in the pathogenesis of lung injury after pulmonary resection surgery is also explored.
The Starling Equation and Pulmonary Edema
Our understanding of vascular permeability as well as plasma/interstitial fluid movement and edema formation has changed with recognition of and insight into the EG, a meshwork of proteins and soluble components that forms a major barrier to water and plasma protein exchange. The fundamental principle guiding microvascular filtration and transcapillary fluid shifts was proposed in 1896 by Starling;10 however, this traditional model has been revised given our current, more sophisticated view of the endothelial barrier and its dynamic components. Starling initially devised a series of experiments showing that fluid movement across the walls of capillaries (and postcapillary venules) is passive and dependent on pressure gradients across the endothelium. He suggested that fluid filtration is a balance between opposing hydrostatic and colloid (protein) osmotic pressures. Since hydrostatic pressure decreases along a capillary, it follows that filtration occurs along the arterial end of capillaries and reabsorption at the venous end of capillaries, though this model has been challenged in more recent years.11,12 Not until decades later did Starling’s initial observations become expressed in mathematical format,13,14 known as the Starling equation:
This equation depicts the volume filtration rate (Jv) per unit area of capillary wall (A) as a balance of hydrostatic pressure forces and oncotic pressure forces existing in the capillaries and interstitium, respectively. The hydraulic conductance (Lp) and reflection coefficient (σ) are considered constants; ΔP is the difference between local capillary blood pressure (Pc) and interstitial fluid hydrostatic pressure (Pi) whereas Δπ is the difference between the osmotic pressure exerted by the macromolecules in plasma (πc) and interstitial fluid (πi).
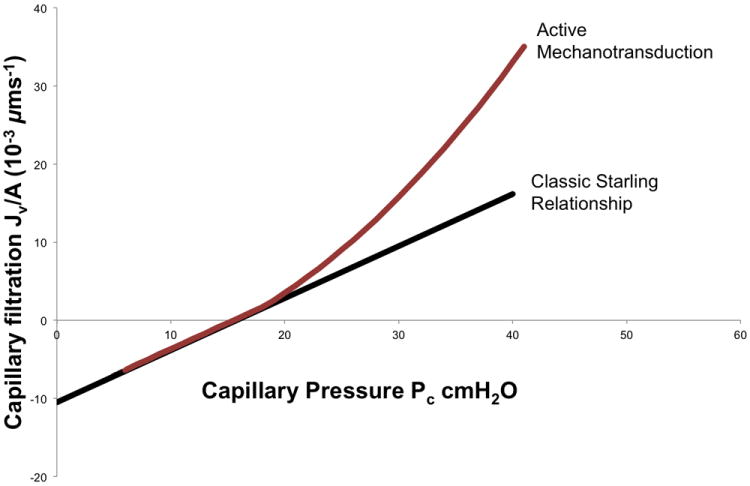
According to the Starling equation, there are several considerations by which interstitial pulmonary edema might develop: increased capillary hydrostatic pressure, increased capillary permeability to fluid or plasma proteins, decreased plasma oncotic pressure, and increased negative pressure within the interstitial space. Increased pulmonary vascular permeability is a hallmark of lung injury; in fact, it is an essential provision of noncardiogenic pulmonary edema formation in which disruption of the alveolocapillary membrane occurs and alveoli are flooded with protein-rich fluid. As in other tissues, it has traditionally been accepted that fluid balance in the lungs obeys the Starling equation15; newer revisions of this classical model, however, have been described115,16,17 to incorporate the glycocalyx into microvascular fluid exchange and suggest that fluid movement between capillary endothelium and surrounding tissues may not follow a simple Starling model. In addition, the lungs may be more resistant to edema formation than other organs18 with varying permeability (Lp) in different vascular segments. Research has demonstrated in isolated lung models19 that Lp is segmentally distributed in the pulmonary microvasculature and varies with peak inflation pressures. In addition, applying the concept that Lp might vary with physiologic or pathophysiologic changes has directed investigators towards a more complicated model of transcapillary permeability with the idea that fluid flux is not simply and passively governed by transcapillary pressure differences. In fact, evidence has accumulated in vivo20,21,22,23,24,25 (isolated animal lung models) and in vitro26 that transendothelial hydrostatic pressure alters vascular permeability and that the pressure versus Lp relationship displays nonlinear dynamics as evidenced in lung endothelial cells. That is, hydrostatic pressure alters vascular permeability in a nonlinear fashion (Figure 1). This deviation from the classic Starling relationship served as the impetus for investigations into cell mechanotransduction and the elucidation of non-Starling mechanisms of barrier regulation that involve the glycocalyx.26,27
Figure 1.
Transcapillary fluid flux: Classic Starling relationship versus active mechanotransduction. Classic Starling principle (black line) predicts a linear relationship between capillary pressure (cm H2O) and transcapillary fluid flux (μm/sec). The x-intercept is the point where plasma oncotic pressure equals capillary hydrostatic pressure, resulting in zero net fluid flux. During active endothelial mechanotransduction (maroon line), capillary hydrostatic pressure activates signaling pathways that increase endothelial permeability resulting in nonlinear dynamics and a higher water flux than would be predicted by the summation of net Starling forces. Classic Starling forces can predict fluid flux over short time intervals (seconds to minutes) while mechanotransduction pathways are time-dependent, requiring 10-20 minutes to manifest changes in endothelial permeability.
The Glycocalyx and Vascular Permeability
A little more than fifty years ago, evidence suggested that vasoactive substances could initiate edema formation by inducing the formation of gaps between endothelial cells.28 It is now recognized that junctions between adjacent endothelial cells are modified in response to inflammatory mediators. However, fluid movement across the pulmonary endothelium into the interstitium and subsequently the alveolar air spaces is generally thought to occur by a paracellular pathway;29 electron microscopy studies have demonstrated the formation of such paracellular gaps at active inflammatory sites within the vasculature.30 One early hypothesis of pressure-induced permeability changes in endothelial microvasculature was the concept of the “stretched pore phenomenon”31,32 whereby elevated vascular pressure increased the size of endothelial pores that led to macromolecule passage. This model has since been refuted.21 Other models using cultured endothelial monolayers have suggested that endothelial contraction is a principal mechanism leading to increased vascular permeability.33
Though the first major consideration of the EG to vascular barrier competence was suggested in the early 1980s,34 its exact role in this regard has been poorly defined until more recently. More recent evidence has led to a better understanding of the functional significance of the EG in regulating vascular barrier function and permeability, transmission of shear stress to endothelial cells, and modulation of inflammatory cell-cell interactions.2 EG structure is thought to influence transport of water and proteins through breaks in the tight junction of endothelial cells but may not influence fluid flux through larger junctional openings.35
The EG consists mainly of endothelial-bound glycoproteins and proteoglycans including the syndecan and glypican families that carry negatively charged GAG side chains. The proteoglycans consist of a core protein to which one or more GAG side chains are linked. The core protein groups span the cell membrane (syndecans) or insert into the membrane with a GPI anchor (glypicans). There are five types of GAG chains, and these linear disaccharide polymers have variable lengths. The most abundant of these are heparan sulfate, chondroitin sulfate, and hyaluronan for which many of the EG’s biophysical properties are ascribed.36 Heparan sulfate serves as a dominant structural component in the glycocalyx whereas chondroitin sulfate and hyaluronan contribute significantly to vascular permeability.37 Hyaluronan is the only GAG that is not covalently linked to other proteins and has no negatively charged sulfate groups. It is important to emphasize, however, that the glycocalyx is part of a dynamic equilibrium with soluble components in plasma (lipids and proteins) as well which contribute to barrier regulation. These components form (in vivo) what has been termed the endothelial surface layer38 (ESL) in which molecules are continually being replaced without a distinct boundary between the EG and plasma. It is this dynamic structure that forms the physiological oncotic gradient at the vascular barrier. Under physiologic conditions, the glycocalyx structure is fairly stable with its components representing a balance between synthesis of new glycans and shear-dependent shedding of existing ones. This ESL concept has led to the generation of new models of fluid homeostasis including the low filtration-low resorption model.39 It has been suggested that the intact ESL in rat mesenteric microvessels creates an inwardly directed oncotic gradient due to a protein-free space beneath the protein-rich ESL which in turn opposes the hydrostatic gradient.11 In this way, new flow of ultrafiltrate to the interstitial space is limited. Because endothelium is continuous along the vessel wall, back-diffusion of colloid from the interstitial space is prevented, serving to maintain an oncotic gradient across the ESL.
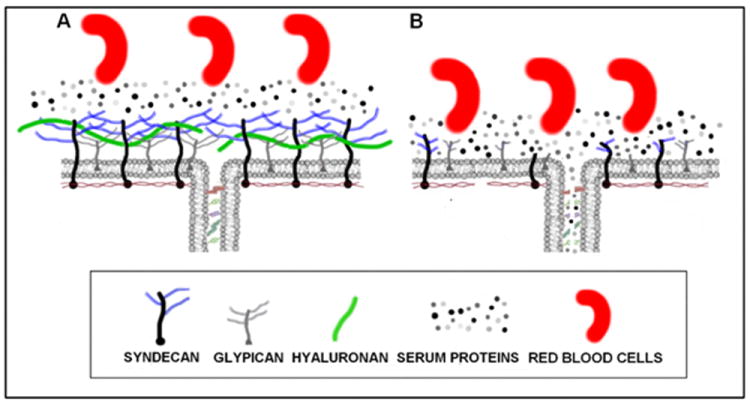
The EG has been recognized to be an important determinant in vascular homeostasis and permeability.3,40 It has been shown to have a role as a barrier to exchange of macromolecules34 and leukocyte-endothelial adhesion41 as well as a repository for factors and enzymes such as antithrombin III42 and superoxide dismutase.43 Membrane-bound glycoproteins and proteoglycans with associated GAG side chains form a cross-linked mesh and serve with other luminal soluble proteins to create a red blood cell exclusion zone (Figure 2) which is decreased when the glycocalyx is degraded.44 As demonstrated in rat mesenteric arteries using fluorescently labeled dextrans,45 the EG limits passage of larger molecules with increasing permeability for smaller ones. Partial degradation (enzymatic removal) and loss of glycocalyx integrity in rat myocardial capillaries has been shown to lead to edema.4 In one early study, Adamson demonstrated that partial digestion of the endothelial surface with pronase more than doubled hydraulic permeability within the capillary wall.46 In addition, neutralization of the highly negatively charged components of the glycocalyx (many of the GAG chains are highly sulfated) leads to increased albumin uptake in cultured endothelial cells47 and increased permeability to dextrans in rat mesenteric arteries.5 However, such studies have been limited to cultured cell lines or experimental animal models with few directly linking changes in glycocalyx structure to changes in endothelial permeability.
Figure 2.
The glycocalyx is a complex layer of protetoglycans, glycosaminoglycans and glycolipids on the endothelial surface. (A) An intact glycocalyx limits water and protein flux into the cell-cell junction by forming a molecular filter over the junctional orifice. The glycocalyx also creates a scaffolding upon which serum proteins accumulate and form the immobile plasma layer directly adjacent to the vessel wall. Collectively, the glycocalyx and protein layer create the red blood cell exclusion zone used to determine the functional thickness of the glycocalyx. (B) During inflammation, proteases degrade the glycocalyx and endothelial cells shed constituents through cell-associated sheddases. Loss of the glycocalyx scaffolding eliminates the immobile plasma layer. Breakdown of the glycocalyx is associated with increased vascular permeability due to loss of the junctional barrier and opening of the intracellular junction, as evidence by increased water and protein flux through the junction. Note the protein-free space under the glycocalyx (left panel) that may significantly affect Starling forces across the cell-cell junction (see text for detail).
The Glycocalyx and Mechanotransduction
The pulmonary microvasculature is sensitive to a variety of injurious stimuli including excessive mechanical forces during ventilation and increased pulmonary vascular pressures, ischemia-reperfusion injury, and inflammatory mediators.48 Endothelial perturbations from such stimuli are important and are likely to be significant in the pathophysiology of lung injury.49 Furthermore, mechanistic approaches designed to understand vascular barrier function and permeability reveal the complexity of these processes.33 Investigations50 have shown that pressure-induced vascular permeability changes may be induced by cellular signaling. In this way, the endothelial response to pressure changes may play a pivotal role in the pathogenesis of microvascular injury in lung injury. Using a cultured bovine aortic endothelial cell model, one group of researchers has demonstrated that endothelial albumin permeability is shear dependent, i.e., influenced by laminar shear stress of blood at the vascular wall.51 This same group has also shown that specific components of the glycocalyx, heparan sulfate proteoglycans, are involved in mechanotransduction and permeability in whole animal lung models.25 It is thought that these specific proteoglycans are the most common endothelial cell surface GAG, comprising 50% to 90% of the total amount of proteoglycans present in the EG.38 Additional evidence52 demonstrates that heparan sulfates are key factors in inflammatory cationic peptide-induced signaling that links lung endothelial cytoskeletal changes and subsequent barrier dysfunction as measured by transendothelial electrical resistance, thus suggesting a role for specific EG components in inflammatory mediation. The syndecans (the largest group of heparan sulfate proteoglycans on the endothelial cell surface and the only proteoglycan that spans the cell membrane and penetrates the cytoplasm) appear to be mediators for these signaling events, though the mechanisms remain poorly defined. A common final pathway for many inflammatory stimuli is the activation of an endothelial-specific myosin-light chain kinase and the cascading activation of signaling that results in actin stress fiber formation and endothelial cell contraction that opens cell-cell junctions, resulting in enhanced permeability.29 However, heparan sulfate-directed and syndecan-mediated signaling may act through novel mechanisms to induce changes in permeability.
Mechanotransduction, the transmission of force (shear stress) to the cellular surface, and subsequent cell response by signaling mechanisms is central to pathophysiologic alterations in endothelial permeability to fluid and proteins. Increasing evidence suggests that the EG plays a critical role in these processes. As an interface between flowing blood and cellular elements, it is in a position to serve as a transducer of hemodynamic signals and is mechanically and functionally linked to the cell membrane and cytoskeleton. Its components have been implicated in cytoskeletal organization and rearrangement during shear stress and the subsequent activation of downstream effects influencing permeability and flow. As mentioned previously, syndecans in the glycocalyx are linked to cytoskeletal elements including actin via their cytoplasmic domains.35 Syndecan clustering and actin cytoskeletal reorganization have been shown to occur from heparan sulfate-directed signaling. Nitric oxide (NO), a vasodilator catalyzed by endothelial nitric oxide synthase (eNOS), is perhaps the most notable of vasoregulating agents whose production is altered by mechanotransduction. The production of NO is an early and rapid cellular response to changes in shear stress at the luminal surface; in addition, NO is an important signaling molecule in activated endothelial cells. As recently reviewed,35 shear stresses applied at the luminal surface of the endothelium increase Lp by stimulating NO release. Circumferential stretch of pulmonary endothelial cells which is affected by both microvascular perfusion pressures and alveolar ventilation pressures has been demonstrated to activate NO release.53 Evidence18,54 suggests that the glycocalyx, specifically heparan sulfate GAG chains in the glycocalyx, senses fluid shear stress and mediates enhanced NO production. That is, the glycocalyx is the mechanosensor for the NO response. Figure 3 provides a model for this mechanotransduction, highlighting shear stress-induced changes in the glycocalyx leading to cellular changes which ultimately activate eNOS and cause increased vascular permeability. That release of NO in endothelial cells has also been shown to be regulated by actin filaments55 provides a conceptual framework linking this pathway with mechanotransduction and cell surface EG components.
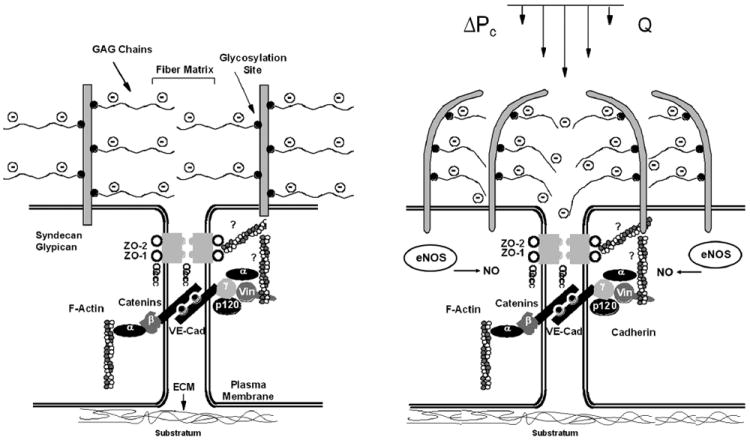
Figure 3.
Schematic illustrating the hypothesized role of the glycocalyx in lung vascular mechanotransduction. Left: during static conditions, the glycocalyx maintains barrier function over the intercellular junction. Right: during increased vascular pressure, the increased hydraulic flow through the glycocalyx deforms or stresses the glycosaminoglycan (GAG) fibers, which in turn activates endothelial nitric oxide synthase (eNOS) and leads to barrier dysfunction. ΔPc, change in capillary pressure; Q, flow; ZO-1 and ZO-2, zonula occludens-1 and -2; vin, vinculin; VE-Cad, vascular endothelial cadherin; ECM, extracellular matrix (Adapted from Dull R, Cluff M, Kingston J, Hill D, Chen H, Hoehne S, Malleske D, Kaur R. Lung heparan sulfates modulate Kfc during increased vascular pressure: evidence for glycocalyx-mediated mechanotransduction. Am J Physiol Lung Cell Mol Physiol 2012;302:L816-28).
Previous studies have demonstrated the role of eNOS and eNOS-derived NO in acute inflammation and local vascular permeability.56 Interestingly, depletion of heparan sulfate from the cell surface glycocalyx with the enzyme heparinase prevents production of NO. Both heparinase and hyaluronidase (a hyaluronan-digesting enzyme) have been demonstrated to block shear-induced NO production as well as increases in Lp, though chondroitinase (cleaving chondroitin sulfate) only partially inhibits the shear-Lp response. This suggests that glycocalyx perturbation may increase endothelial permeability via NO release but that degradation or destruction of the glycocalyx may lead to inflammatory and endothelial responses by mechanisms other than NO release such as increased leukocyte adhesion41 and barrier disruption creating protein and fluid extravasation. Of note, work in bovine pulmonary endothelium57 has shown particular cationic copolymers (which bind to the negatively charged glycocalyx) effectively block pressure-induced increases in Lp, suggesting that the glycocalyx may be a therapeutic target to enhance endothelial barrier function in pathological states such as lung injury.
The Glycocalyx and Lung Injury – A New Paradigm
Glycocalyx damage impairs a number of important endothelial cell functions leading to impaired mechanotransduction with changes in fluid shear stress,6,58 activation of coagulation pathways,59 adhesion of leukocytes7,59 and platelets60 to the endothelial cell surface, leakage of fluid and plasma proteins into the interstitium,46,61 and resultant tissue edema.62 Specific pathophysiological triggers that lead to damage of the glycocalyx are being actively investigated and remain poorly understood. As noted in an editorial,63 it is not entirely clear whether loss of glycocalyx integrity that causes endothelium dysfunction inevitably leads to disease at the tissue or organ level. Nonetheless, protection of the glycocalyx seems to be a promising goal in many clinical scenarios particularly since its degradation is so closely associated with the pathophysiology of inflammation, capillary leak, and edema formation in diverse injury and disease states including ischemia/reperfusion injury, hypoxia, inflammation, trauma, hypervolemia, atherosclerosis, diabetes, and hypertension.64
Pulmonary resection surgery imposes upon the lung and its vasculature a number of insults which place patients undergoing these procedures at increased risk for lung injury and pulmonary edema. The clinical severity of lung injury may vary, ranging from pneumonitis to the most severe form of lung injury, acute respiratory distress syndrome (ARDS). Acute lung injury after pulmonary resection has been variously described as postlung resection pulmonary edema, postpneumonectomy pulmonary edema (PPE), and low pressure or noncardiogenic pulmonary edema. The risk factors for and etiology of this phenomenon remain poorly defined. Importantly, while the incidence of lung injury using the 1992 American European Consensus Conference guidelines is relatively low, ranging from 1% to 7% after lobectomy and 4% to 7% after pneumonectomy, the mortality rate can be as high as 40 percent in those patients undergoing pneumonectomy.65 ARDS is indistinguishable from PPE and is characterized by inflammation and vascular injury with loss of endothelial integrity and resultant high protein edema.66 Postmortem studies in patients with PPE have demonstrated the classical features of ARDS seen from other causes.67 In addition, ARDS in its initial phase has the same histologic features as seen after lung resection.68 It is possible that damage or degradation of glycocalyx integrity by ischemia and inflammatory responses is a major mediator for edema formation in these settings. Though the pathogenesis of pulmonary edema and specific etiologies of lung injury after pulmonary resection are not well understood,69 ongoing investigations are improving our understanding of the complex factors involved in the pathophysiology of postlung resection lung injury and ARDS. Several studies have focused on the contribution of patient and surgical factors as well as anesthetic variables including one-lung ventilation,70,71,72 protective ventilation strategies,73,74 fluid management,75,76 and the role of putative protective pharmacologic agents.77
There is a paucity of data with specific regard to glycocalyx integrity and damage and lung injury. It is known that patients with lung injury have high pleural fluid protein levels, implicating increased endothelial permeability with endothelial injury.65 Likely etiologies include hemodynamic shear stress and increased pulmonary capillary pressure as well as cytokine release, oxidative stress, and ischemia-reperfusion injury during lung deflation and reinflation with one-lung ventilation. Evidence in animal models suggests that the glycocalyx is degraded in response to inflammation8,59 and ischemia-reperfusion injury8, among other pathophysiologic perturbations. Components of the glycocalyx are seen in bronchoalveolar lavage fluid of animals with lung injury78 consistent with the hypothesis that glycocalyx degradation leads to endothelial injury. Furthermore, IV administration of a major GAG component of the glycocalyx (hyaluronan) has been shown to be protective from sepsis- and intratracheally induced lung injury in rats.79 Other in vitro and in vivo animal models of inflammatory lung injury80 have implicated glycocalyx components in endothelial protection. A recent study of experimental sepsis81 provided results that sepsis-induced respiratory failure in humans was associated with higher plasma heparan sulfate degradation activity. These data suggest that major components of the glycocalyx may maintain pulmonary vascular integrity.
Lung Injury, Pulmonary Resection, and Pulmonary Edema
Pulmonary resection surgery is greatly facilitated by one-lung ventilation which results in blood flow redistribution to the dependent, ventilated lung by gravitational effects, hypoxic pulmonary vasoconstriction, and mechanical effects on the operative lung. After pulmonary resection or pneumonectomy, pulmonary flow within the remaining lung parenchyma increases which leads to an increase in pulmonary vascular resistance and pulmonary artery pressures resultant from flow to the previously restricted vascular bed.76,82,83 Fundamental to our understanding of postlung resection lung injury is the development of a low-pressure, high-protein-content pulmonary edema, an indication of injury to the endothelium. 69,84 Pulmonary endothelial cells are exposed to shear stress and higher transvascular pressures after pneumonectomy; these changes may induce alterations in cellular signaling pathways including the production of reactive oxygen species (ROS) and NO.58 Of significance, plasma markers of oxidative damage and lung injury in patients undergoing routine thoracic surgery were found to be largest in patients undergoing pneumonectomy, modest in lobectomy, and not significant in lesser (wedge, segment) resections.85 In addition, increased pulmonary blood flow and pressure are accompanied by an increase in endothelial permeability in pneumonectomy but not lobectomy patients,76 suggesting that the degree of vascular bed restriction or degree of resultant flow, pressure, and shear stresses are important factors in endothelial dysfunction or damage. The mechanisms by which alterations in pulmonary flow and pressure modulate inflammatory responses via vascular permeability changes remain elusive.
The increase in pulmonary blood flow that occurs after pulmonary resection is likely not a benign phenomenon. That increased cardiac output and pulmonary blood flow may be injurious to the pulmonary vasculature is suggested in animal models of experimental lung injury. These studies support the concept that increased pulmonary blood flow may induce lung injury or aggravate a preexisting injury state.86,87,88,89,90 In a canine oleic acid lung injury model,87 cardiac output increases induced by terbutaline exacerbated macromolecule leakage from pulmonary capillaries. This increased permeability may derive from a recruitment of “leaky” capillaries and an overall increase in the capillary exchange surface area whereas increased flow by itself may be a risk factor for alterations in capillary leakage. In a similar lung injury model,91 larger increases in capillary filtration coefficient (hydraulic conductivity Lp multiplied by filtration surface area) were demonstrated by increasing perfusate flow than by increasing pulmonary venous pressure, a finding that may relate to augmentation of Lp, an increase in the effective area of filtration, or both. An increase in Lp as a result of pulmonary overperfusion and increased shear stress is suggested by some92 but not all animal studies.93,94 Increased pulmonary endothelial permeability has been demonstrated in humans undergoing lung resection using technetium-99m-labeled albumin. The etiology of these endothelial changes is thought to be due to increased shear stress in the reduced vascular bed under constant cardiac output, thereby causing endothelial damage and increased permeability.
These studies suggest that augmentation of pulmonary blood flow in the context of preexisting injury increases biochemical, cellular, and histological features of lung injury. The extent of resection required to cause blood flow-induced injury is less certain. In a canine model of pulmonary resection95 a resection volume of 80% was required before the capillary filtration coefficient was increased. It remains unclear how this translates to patients undergoing pulmonary resection or how other co-morbidities and lung-related insults such as cigarette smoking or asthma may influence the susceptibility to flow-induced injury. Clearly this is an area that requires active investigation. However, as highlighted earlier,76 endothelial permeability is increased after pneumonectomy in humans, suggesting that pulmonary resection surgery itself constitutes significant subclinical injury. Furthermore, pneumonectomy has been determined to be an independent predictor of primary lung injury in a retrospective analysis96 of more than 800 pulmonary resection procedures.
Though the precise identity of initial subclinical endothelial injury in lung injury remains uncertain, it seems likely that inflammation is a component. Unilateral lung injury may induce a systemic inflammatory response affecting the uninjured lung.97,98 Once rendered susceptible by systemic or regional inflammation (first hit), the remaining lung parenchyma may be more susceptible to subsequent injurious stimuli (second hit). The effects of increased blood flow and pulmonary vascular pressures on lung injury generation or exacerbation may thereby be rendered more potent in the context of a preexisting inflammatory condition or injury to the contralateral lung. In a rat model,99 unilateral lung injury was induced and cardiac output increased (>70%) by dobutamine challenge demonstrating that the opposite (contralateral) lung exhibited markedly increased inflammatory cell counts (neutrophils and macrophages) in lavage fluid with increased edema and histopathologic features of lung injury. In this study, dobutamine caused an increase in both cardiac output and pulmonary arterial pressure; though increased pulmonary pressure has been proposed as a feature causing damage to the alveolocapillary membrane, it is not possible to discern if flow or pressure was the leading factor associated with contralateral lung injury. This study supports two important concepts: (1) augmentation of pulmonary blood flow and accompanying changes in pressure- and flow-related shear stresses elicit lung injury with accompanying cellular, biochemical, and histopathologic findings, and (2) a subtle inflammatory response in an uninjured lung increases the susceptibility to injury from subsequent insults.
Damage to the Glycocalyx
Though still incompletely understood, the EG plays a role in maintaining vascular integrity in at least two important ways. First, it serves as a passive barrier to the efflux of proteins and fluid from the capillary lumen. In this sense, it serves to augment the classical Starling forces preventing capillary fluid leakage and edema formation. Second, the EG is a dynamic structure functionally linked to the cell membrane and cytoskeleton and has been demonstrated51,53,55,100 to function as a mechanotransducer of pressure and shear stress within the vascular lumen, as discussed earlier. It has also been identified as a site of damage after ischemia / reperfusion injury.
Numerous studies101,102,103 have demonstrated the importance of oxygen radicals in reperfusion injury; more recent studies104,105 implicate the glycocalyx directly by such EG-bound proteins as xanthine oxidoreductase and superoxide dismutase, enzymes involved in endothelial oxidative stress. Animal models8,106 and human studies107,108 have also shown reperfusion-mediated glycocalyx degradation. In isolated animal heart models,109 a brief period of warm no-flow ischemia with subsequent reperfusion has been demonstrated to cause near-complete glycocalyx degradation with increased vascular permeability. We assume that similar shedding of the lung vascular glycocalyx occurs during ischemia reperfusion injury though such studies have yet to be performed. In adults undergoing aortic surgery,107 increased plasma levels of glycocalyx components as measured by immunoassays of syndecan-1 and heparan sulfate have been shown.
Advances in imaging techniques have enabled reproducible measurements of the EG in humans,110 providing evidence of glycocalyx damage during acute and chronic inflammatory states; one such inflammatory response is that elicited from a surgical insult such as pulmonary resection. This response may be sufficient to “prime” the pulmonary vasculature, rendering it more sensitive to subsequent insults that may lead to lung injury (mild-severe ARDS using revised Berlin Definition111). Such insults may include injurious modes of mechanical ventilation, oxidative stress, ischemia reperfusion injury, and increased fluid loads. During major thoracic surgery, additional mechanisms of injury may present during reperfusion of the remaining operative lung including oxidant stress involving ROS.112 The subsequent pressure- and flow-related increases within the pulmonary vasculature may then be mechanotransduced by the glycocalyx, leading to increased permeability and leukocyte adhesion. These effects appear to be an important response to increased pulmonary pressure and flow. That they are also dependent on the glycocalyx is supported by work demonstrating the GAG dependence of shear stress-induced alterations in hydraulic conductivity and attenuation of this response by inhibition of NO synthase and scavenging of ROS.55 This finding is consistent with the clinical observation of increased oxidative stress in patients undergoing pulmonary resection surgery. Thus, the activation of eNOS (and possibly ROS production) after pneumonectomy appears to be an important adaptive response to increased pulmonary pressure and flow, promoting compensatory vasodilation and pulmonary growth. Furthermore, it is dependent on an intact EG as evidenced by selective enzymatic removal of terminal sialic acids, heparan sulfate, or hyaluronic acid which blocks the effect of shear-induced NO production. A corollary of this adaptation, at least in exaggerated circumstances, may be the liberal increase in permeability accompanying pulmonary resection and lung injury.
Factors intrinsic and extrinsic to the endothelium may be associated with damage to the glycocalyx. Hydrostatic increases within the pulmonary microvasculature activate metabolic and cellular changes which are a “pro-inflammatory” endothelial cell phenotype. This includes neutrophil activation and adhesion, a critical step in endothelial injury and the pathogenesis of lung injury. Neutrophil adhesion to the altered endothelial cell is facilitated by augmented expression of adhesion glycoproteins including E-selectin113 which is also activated by pressure-mediated changes in endothelial phenotype. Inhibition of neutrophil adhesion protects against development of pulmonary vascular endothelial injury,114 and more recent evidence38 has shown that polycationic peptides used to mimic neutrophil-derived inflammatory proteins induce injury to endothelial cells mediated by and dependent upon the glycocalyx component heparan sulfate. These cellular adhesion processes as well as other as inflammatory mediators such as tumor necrosis factor-alpha59,115 have been demonstrated to cause degradation of the glycocalyx; the ultimate implications of these findings, or whether strategies aimed at glycocalyx protection offer therapeutic benefit, remain unclear.
The glycocalyx/mechanotransduction model of lung injury predicts that the successful blockade of pressure- or flow-induced signals transduced by the glycocalyx would lead to an attenuation of permeability and pressure-induced augmentation in hydraulic conductivity of the endothelial cell layer. Though research in this area remains in its infancy, it has already been demonstrated that the glycocalyx is an important target for such intervention. The use of a cationic copolymer57 rationally designed to integrate into the glycocalyx to augment passive glycocalyx barrier functions has been shown to decrease albumin permeability in pulmonary endothelial cells under normal and inflammatory conditions and to block pressure-induced augmentation in hydraulic conductivity. While such investigation may lead to the development of rational therapies for the prevention or treatment of lung injury, it also currently provides additional evidence for the role and importance of the glycocalyx in mediating pathologic processes integral in the development of lung injury.
Summary and Perspectives
Pulmonary endothelial dysfunction plays a major role in lung injury via alterations in barrier permeability, thus promoting pulmonary edema formation; the adjoining glycocalyx has recently emerged as a major endothelial element involved in the regulation of vascular integrity and fluid homeostasis. A glycocalyx mechanotransduction-mediated lung injury model predicts that activation of pressure- or flow-induced signals during pulmonary resection may lead to augmentation in endothelial cell hydraulic conductivity, i.e., capillary permeability, involved in the formation of pulmonary edema. These processes remain poorly understood. Further insights into lung microvasculature, the glycocalyx, and its associated role in mechanotransduction challenge our fundamental understanding of lung fluid balance. Our current knowledge of the glycocalyx and endothelial dysfunction supports its role in mediating pathologic processes integral in the development of pulmonary edema. These concepts may also lead to the development of rational therapies for the prevention or treatment of lung injury. For instance, given the compromised state of the glycocalyx under injurious conditions and evidence for its control of fluid flux into the interstitium, better defined conservative fluid management strategies may attenuate lung injury.
Preventing the shedding or degradation of endothelial glycocalyx constituents induced by inflammatory cytokines and plasma proteases may have substantial clinical benefits during thoracic surgery. Identifying the pathways that promote shedding and the proteases involved in glycocalyx-breakdown are just being learned in sepsis-induced lung injury81 and similar studies are needed to understand the complex relationship between mechanical forces, inflammatory signaling within the endothelium, and breakdown of the glycocalyx. In addition, development of therapeutic agents that modulate mechanotransduction and/or stabilize the glycocalyx might improve our ability to mitigate lung injury associated with disease states and pulmonary resection surgery. Indeed, the proof-in-concept for this idea has been verified in a basic science model of the lung capillary endothelium in which one group of researchers57,116 has designed, synthesized, and tested novel biomimetic polymers that bind avidly to the glycocalyx and enhance barrier function during basal states. The polymers also attenuate pressure and shear-stress-mediated changes in permeability, in part, by preventing NO production. Modulating endothelial barrier function in a clinically beneficial way, however, remains elusive.
Acknowledgments
Funding: The authors were not funded for the preparation of this manuscript.
Footnotes
-
Name: Stephen R. Collins, MDContribution:This author prepared the manuscript.Attestation: This author approves the final manuscript.
-
Name: Randal S. Blank, MD, PhDContribution: This author assisted in preparation of the manuscript.Attestation: This author approves the final manuscript.
-
Name: Lindy S. Deatherage, MDContribution: This author assisted in preparation of the manuscript.Attestation: This author approves the final manuscript.
-
Name: Randal O. Dull, MD, PhDContribution: This author assisted in preparation of the manuscript.Attestation: This author approves the final manuscript.
- This manuscript was handled by: Michael J. Murray, MD, PhD
The authors declare no conflicts of interest.
Contributor Information
Stephen R. Collins, Department of Anesthesiology, University of Virginia Health System, Charlottesville, Virginia.
Randal S. Blank, Department of Anesthesiology, University of Virginia Health System, Charlottesville, Virginia.
Lindy S. Deatherage, Department of Anesthesiology, University of Utah, Salt Lake City, Utah.
Randal O. Dull, Department of Anesthesiology and Bioengineering, University of Illinois at Chicago College of Medicine, Chicago, Illinois.
References
- 1.Reitsma S, Slaaf D, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch – Eur J Physiol. 2007;454:345–59. doi: 10.1007/s00424-007-0212-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng. 2007;9:121–67. doi: 10.1146/annurev.bioeng.9.060906.151959. [DOI] [PubMed] [Google Scholar]
- 3.Vink H, Duling BR. Capillary endothelial surface layer selectivity reduces plasma solute distribution volume. Am J Physiol Heart Circ Physiol. 2000;278:H285–89. doi: 10.1152/ajpheart.2000.278.1.H285. [DOI] [PubMed] [Google Scholar]
- 4.van den Berg BM, Vink H, Spaan JA. The endothelial glycocalyx protects against myocardial edema. Circ Res. 2003;92:592–94. doi: 10.1161/01.RES.0000065917.53950.75. [DOI] [PubMed] [Google Scholar]
- 5.Van Haaren PM, VanBavel E, Vink H, Spaan JA. Charge modification of the endothelial surface layer modulates the permeability barrier of isolated rat mesenteric small arteries. Am J Physiol Heart Circ Physiol. 2005;289:H2503–07. doi: 10.1152/ajpheart.00587.2005. [DOI] [PubMed] [Google Scholar]
- 6.Mochizuki S, Vink H, Hiramatsu O, Kajita T, Shigeto F, Spaan JA, Kajiya F. Role of hyaluronic acid in shear-induced endothelium-derived nitric oxide release. Am J Physiol Heart Circ Physiol. 2003;285:H722–26. doi: 10.1152/ajpheart.00691.2002. [DOI] [PubMed] [Google Scholar]
- 7.Constantinescu AA, Vink H, Spaan JA. Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler Thromb Vasc Biol. 2003;23:1541–47. doi: 10.1161/01.ATV.0000085630.24353.3D. [DOI] [PubMed] [Google Scholar]
- 8.Mulivor AW, Lipowsky HH. Inflammation- and ischemia-induced shedding of venular glycocalyx. Am J Physiol Heart Circ Physiol. 2004;286:H1672–80. doi: 10.1152/ajpheart.00832.2003. [DOI] [PubMed] [Google Scholar]
- 9.Lipowsky H. The endothelial glycocalyx as a barrier to leukocyte adhesion and its mediation by extracellular proteases. Annals Biomed Eng. 2012;40(4):840–48. doi: 10.1007/s10439-011-0427-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Starling EH. On the absorption of fluids from the connective tissue spaces. J Physiol. 1896;19:312–26. doi: 10.1113/jphysiol.1896.sp000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adamson RH, Lenz JF, Zhang X, Adamson GN, Weinbaum S, Curry FE. Oncotic pressures opposing filtration across non-fenestrated rat microvessels. J Physiol. 2004;557:889–907. doi: 10.1113/jphysiol.2003.058255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levick JR. Revision of the Starling principle: new views of tissue fluid balance. J Physiol. 2004;557:704. doi: 10.1113/jphysiol.2004.066118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Landis EM. Micro-injection studies of capillary permeability. The relation between capillary pressure and the rate at which fluid passes through the walls of single capillaries. Am J Physiol. 1927;82:217–38. [Google Scholar]
- 14.Pappenheimer JR, Soto-Rivera A. Effective osmotic pressure of the plasma proteins and other quantities associated with the capillary circulation in the hind-limbs of cats and dogs. Am J Physiol. 1948;152:471–91. doi: 10.1152/ajplegacy.1948.152.3.471. [DOI] [PubMed] [Google Scholar]
- 15.Michel CC. Starling: the formulation of his hypothesis of microvascular fluid exchange and its significance after 100 years. Exp Physiol. 1997;82:1–30. doi: 10.1113/expphysiol.1997.sp004000. [DOI] [PubMed] [Google Scholar]
- 16.Weinbaum S. 1997 Whitaker distinguished lecture: models to solve mysteries in biomechanics at the cellular level: a new view of fiber matrix layers. Ann Biomed Eng. 1998;26:627–43. doi: 10.1114/1.134. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Adamson RH, Curry FR, Weinbaum S. A 1-D model to explore the effects of tissue loading and tissue concentration gradients in the revised Starling principle. Am J Physiol Heart Circ Physiol. 2006;291:H2950–2964. doi: 10.1152/ajpheart.01160.2005. [DOI] [PubMed] [Google Scholar]
- 18.Effros RM, Parker JC. Pulmonary vascular heterogeneity and the Starling hypothesis. Microvasc Res. 2009;78:71–77. doi: 10.1016/j.mvr.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 19.Parker JC, Yoshikawa S. Vascular segmental permeabilities at high peak inflation pressure in isolated rat lungs. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1203–1209. doi: 10.1152/ajplung.00488.2001. [DOI] [PubMed] [Google Scholar]
- 20.Rippe B, Townsley M, Thigpen J, Parker JC, Korthuis RJ, Taylor AE. Effects of vascular pressure on the pulmonary microvasculature in isolated dog lungs. J Appl Physiol. 1984;57(1):233–39. doi: 10.1152/jappl.1984.57.1.233. [DOI] [PubMed] [Google Scholar]
- 21.Ehrhart IC, Hofman WF. Pressure-dependent increase in lung vascular permeability to water but not protein. J Appl Physiol. 1992;72(1):211–18. doi: 10.1152/jappl.1992.72.1.211. [DOI] [PubMed] [Google Scholar]
- 22.Ehrhart IC, McCloud LL, Organos SE, Catravas JD, Hofman WF. Effect of high blood flow on pulmonary vascular permeability to protein. J Appl Physiol. 1994;76(6):2342–47. doi: 10.1152/jappl.1994.76.6.2342. [DOI] [PubMed] [Google Scholar]
- 23.Parker JC, Ivey CL. Isoproterenol attenuates high vascular pressure-induced permeability increases in isolated rat lungs. J Appl Physiol. 1997;83:1962–67. doi: 10.1152/jappl.1997.83.6.1962. [DOI] [PubMed] [Google Scholar]
- 24.Parker RE, Roselli RJ, Harris TR, Brigham KL. Effects of graded increases in pulmonary vascular pressures on lung fluid balance in unanesthetized sheep. Circ Res. 1981;49:1164–72. doi: 10.1161/01.res.49.5.1164. [DOI] [PubMed] [Google Scholar]
- 25.Dull RO, Cluff M, Kingston J, Hill D, Chen H, Hoehne S, Malleske DT, Kaur R. Lung heparan sulfates modulate Kfc during increased vascular pressure: evidence for glycocalyx-mediated mechanotransduction. Am J Physiol Lung Cell Mol Physiol. 2012;302:L816–28. doi: 10.1152/ajplung.00080.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tarbell JM, Demaio L, Zaw MM. Effect of pressure on hydraulic conductivity of endothelial monolayers: role of endothelial cleft shear stress. J Appl Physiol. 1999;87:261–68. doi: 10.1152/jappl.1999.87.1.261. [DOI] [PubMed] [Google Scholar]
- 27.Dull RO, Mecham I, McJames S. Heparan sulfates mediate pressure-induced increase in lung endothelial hydraulic conductivity via nitric oxide/reactive oxygen species. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1452–58. doi: 10.1152/ajplung.00376.2006. [DOI] [PubMed] [Google Scholar]
- 28.Majno G, Palade GE. Studies on inflammation. The effect of histamine and serotonin on vascular permeability: an electron microscopic study. J Cell Biol. 1961;11:571–605. doi: 10.1083/jcb.11.3.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dudek S, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 30.Hirata A, Baluk P, Fujiwara T, McDonald DM. Localization of focal silver staining at endothelial gaps in inflamed venules examined by scanning electron microscopy. Am J Physiol Lung Cell Mol Physiol. 1995;269:L403–18. doi: 10.1152/ajplung.1995.269.3.L403. [DOI] [PubMed] [Google Scholar]
- 31.Shirley HH, Wolfram CG, Wasserman K, Mayerson HS. Capillary permeability to macromolecules: stretched pore phenomenon. Am J Physiol. 1957;190:189–93. doi: 10.1152/ajplegacy.1957.190.2.189. [DOI] [PubMed] [Google Scholar]
- 32.Wasserman K, Loeb L, Mayerson HS. Capillary permeability to macromolecules. Circ Res. 1955;3:594–603. doi: 10.1161/01.res.3.6.594. [DOI] [PubMed] [Google Scholar]
- 33.Curry FE, Noll T. Spotlight on microvascular permeability. Cardiovasc Res. 2010;87:195–97. doi: 10.1093/cvr/cvq188. [DOI] [PubMed] [Google Scholar]
- 34.Curry FE, Michel CC. A fiber matrix model of capillary permeability. Microvasc Res. 1980;20:96–99. doi: 10.1016/0026-2862(80)90024-2. [DOI] [PubMed] [Google Scholar]
- 35.Tarbell JM. Shear stress and the endothelial transport barrier. Cardiovasc Res. 2010;87:320–30. doi: 10.1093/cvr/cvq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Callaghan R, Job KM, Dull RO, Hlady V. Stiffness and heterogeneity of the pulmonary endothelial glycocalyx measured by atomic force microscopy. Am J Physiol Lung Cell Mol Physiol. 2011;301(3):L353–60. doi: 10.1152/ajplung.00342.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao L, Lipowsky HH. Composition of the endothelial glycocalyx and its relation to its thickness and diffusion of small solutes. Microvasc Res. 2010;80:394–401. doi: 10.1016/j.mvr.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pries AR, Secomb TW, Gaehtgens P. The endothelial surface layer. Pflügers Arch – Eur J Physiol. 2000;440:653–66. doi: 10.1007/s004240000307. [DOI] [PubMed] [Google Scholar]
- 39.Jacob M, Bruegger D, Rehm M, Stoeckelhuber M, Welsch U, Conzen P, Becker BF. The endothelial glycocalyx affords compatibility of Starling’s principle and high cardiac interstitial albumin levels. Cardiovasc Res. 2007;73:575–586. doi: 10.1016/j.cardiores.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 40.Henry CB, Duling BR. Permeation of the luminal capillary glycocalyx is determined by hyaluronan. Am J Physiol. 1999;277:H508–14. doi: 10.1152/ajpheart.1999.277.2.H508. [DOI] [PubMed] [Google Scholar]
- 41.Mulivor AW, Lipowsky HH. Role of glycocalyx in leukocyte-endothelial cell adhesion. Am J Physiol Heart Circ Physiol. 2002;283:H1282–91. doi: 10.1152/ajpheart.00117.2002. [DOI] [PubMed] [Google Scholar]
- 42.Shimada K, Kobayashi M, Kimura S, Nishinaga M, Takeuchi K, Ozawa T. Anticoagulant heparin-like glycosaminoglycans on endothelial cell surface. Jpn Circ. 1991;55:1016–21. doi: 10.1253/jcj.55.1016. [DOI] [PubMed] [Google Scholar]
- 43.Abrahamsson T, Brandt U, Marklund SL, Sjoqvist PO. Vascular bound recombinant extracellular superoxide dismutase type C protects against the detrimental effects of superoxide radicals on endothelium dependent arterial relaxation. Circ Res. 1992;70:264–71. doi: 10.1161/01.res.70.2.264. [DOI] [PubMed] [Google Scholar]
- 44.Vink H, Duling BR. Identification of distinct luminal domains for macromolecules, erythrocytes, and leukocytes within mammalian capillaries. Circ Res. 1996;79:581–89. doi: 10.1161/01.res.79.3.581. [DOI] [PubMed] [Google Scholar]
- 45.Van Haaren PM, VanBavel E, Vink H, Spaan JA. Localization of the permeability barrier to solutes in isolated arteries by confocal microscopy. Am J Physiol Heart Circ. 2003;285:H2848–56. doi: 10.1152/ajpheart.00117.2003. [DOI] [PubMed] [Google Scholar]
- 46.Adamson RH. Permeability of frog mesenteric capillaries after partial pronase digestion of the endothelial glycocalyx. J Physiol. 1990;428:1–13. doi: 10.1113/jphysiol.1990.sp018197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ueda A, Shimomura M, Ikeda M, Yamaguchi R, Tanishita K. Effect of glycocalyx on sheardependent albumin uptake in endothelial cells. Am J Physiol Heart Circ Physiol. 2004;287:H2287–94. doi: 10.1152/ajpheart.00808.2003. [DOI] [PubMed] [Google Scholar]
- 48.Grichnik KP, D’Amico T. Acute lung injury and acute respiratory distress syndrome after pulmonary resection. Semin Cardiothorac Vasc Anesth. 2004;8(4):317–34. doi: 10.1177/108925320400800405. [DOI] [PubMed] [Google Scholar]
- 49.Maniatis NA, Orfanos SE. The endothelium in acute lung injury/acute respiratory distress syndrome. Curr Opin Crit Care. 2008;14:22–30. doi: 10.1097/MCC.0b013e3282f269b9. [DOI] [PubMed] [Google Scholar]
- 50.Kuebler WM, Ying X, Bhattacharya J. Pressure-induced endothelial Ca2+ oscillations in lung capillaries. Am J Physiol Lung Cell Mol Physiol. 2002;282(5):L917–23. doi: 10.1152/ajplung.00275.2001. [DOI] [PubMed] [Google Scholar]
- 51.Dull RO, Jo H, Sill H, Hollis TM, Tarbell JM. The effect of varying albumin concentration and hydrostatic pressure on hydraulic conductivity and albumin permeability of cultured endothelial monolayers. Microvasc Res. 1991;41(3):390–407. doi: 10.1016/0026-2862(91)90037-c. [DOI] [PubMed] [Google Scholar]
- 52.Dull RO, Dinavahi R, Schwartz L, Humphries DE, Berry D, Sasisekharan R, Garcia JG. Lung endothelial heparin sulfates mediate cationic peptide-induced barrier dysfunction: a new role for the glycocalyx. Am J Physiol Lung Cell Mol Physiol. 2003;285:L986–95. doi: 10.1152/ajplung.00022.2003. [DOI] [PubMed] [Google Scholar]
- 53.Kuebler WM, Uhlig U, Goldmann T, Schael G, Kerem A, Exner K, Martin C, Vollmer E, Uhlig S. Stretch activates nitric oxide production in pulmonary vascular endothelial cells in situ. Am J Respir Crit Care Med. 2003;168:1391–98. doi: 10.1164/rccm.200304-562OC. [DOI] [PubMed] [Google Scholar]
- 54.Florian JA, Kosky JR, Ainslie K, Pang Z, Dull RO, Tarbell J. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ Res. 2003;93:e136–42. doi: 10.1161/01.RES.0000101744.47866.D5. [DOI] [PubMed] [Google Scholar]
- 55.Zharikov SI, Sigova AA, Chen S, Bubb MR, Block ER. Cytoskeletal regulation of the L-arginine/NO pathway in pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;280:L465–73. doi: 10.1152/ajplung.2001.280.3.L465. [DOI] [PubMed] [Google Scholar]
- 56.Bucci M, Roviezzo F, Posadas I, Yu J, Parente L, Sessa WC, Ignarro LJ, Cirino G. Endothelial nitric oxide synthase activation is critical for vascular leakage during acute inflammation in vivo. PNAS. 2005;102(3):904–08. doi: 10.1073/pnas.0408906102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giantsos KM, Kopeckova P, Dull RO. The use of an endothelium-targeted cationic copolymer to enhance the barrier function of lung capillary endothelial monolayers. Biomaterials. 2009;30:5885–91. doi: 10.1016/j.biomaterials.2009.06.048. [DOI] [PubMed] [Google Scholar]
- 58.Thi MM, Tarbell JM, Weinbaum S, Spray DC. The role of the glycocalyx in reorganization of the actin cytoskeleton under fluid shear stress: a “bumper-car” model. Proc Natl Acad Sci USA. 2004;101:16483–88. doi: 10.1073/pnas.0407474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Henry CB, Duling BR. TNF-alpha increases entry of macromolecules into luminal endothelial cell glycocalyx. Am J Physiol Heart Circ Physiol. 2000;279:H2815–23. doi: 10.1152/ajpheart.2000.279.6.H2815. [DOI] [PubMed] [Google Scholar]
- 60.Vink H, Constantinescu AA, Spaan JA. Oxidized lipoproteins degrade the endothelial surface layer: implications for platelet-endothelial cell adhesion. Circulation. 2000;101:1500–02. doi: 10.1161/01.cir.101.13.1500. [DOI] [PubMed] [Google Scholar]
- 61.Huxley VH, Williams DA. Role of a glycocalyx on coronary arteriole permeability to proteins: evidence from enzyme treatments. Am J Physiol Heart Circ Physiol. 2000;278:H1177–85. doi: 10.1152/ajpheart.2000.278.4.H1177. [DOI] [PubMed] [Google Scholar]
- 62.Vanteeffelen JW, Dekker S, Fokkema DS, Siebes M, Vink H, Spaan JA. Hyaluronidase treatment of coronary glycocalyx increases reactive hyperemia but not adenosine hyperemia in dog hearts. Am J Physiol Heart Circ Physiol. 2005;289:H2508–13. doi: 10.1152/ajpheart.00446.2005. [DOI] [PubMed] [Google Scholar]
- 63.van den Berg B, Vink H. Glycocalyx perturbation: cause or consequence of damage to the vasculature? Am J Physiol Heart Circ Physiol. 2006;290:H2174–75. doi: 10.1152/ajpheart.00197.2006. [DOI] [PubMed] [Google Scholar]
- 64.Becker BF, Chappell D, Bruegger D, Annecke T, Jacob M. Therapeutic strategies targeting the endothelial glycocalyx: acute deficits, but great potential. Cardiovasc Res. 2010;87:300–10. doi: 10.1093/cvr/cvq137. [DOI] [PubMed] [Google Scholar]
- 65.Park BJ. Respiratory failure following pulmonary resection. Semin Thorac Cardiovasc Surg. 2007;19(4):374–9. doi: 10.1053/j.semtcvs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 66.Jordan S, Mitchell JA, Quinlan GJ, Goldstraw P, Evans TW. The pathogenesis of lung injury following pulmonary resection. Eur Respir J. 2000;15:790–99. doi: 10.1034/j.1399-3003.2000.15d26.x. [DOI] [PubMed] [Google Scholar]
- 67.Turner WS, Lunn JJ. Postpneumonectomy pulmonary edema. A retrospective analysis of associated variables. Chest. 1993;103:1646–50. doi: 10.1378/chest.103.6.1646. [DOI] [PubMed] [Google Scholar]
- 68.Marthru M, Blakeman B, Dries DJ, Kleinman B, Kumar P. Permeability pulmonary edema following lung resection. Chest. 1990;98:1216–18. doi: 10.1378/chest.98.5.1216. [DOI] [PubMed] [Google Scholar]
- 69.Turnage WS, Lunn JL. Postpneumonectomy pulmonary edema: a retrospective analysis of associated variables. Chest. 1993;103:1646–50. doi: 10.1378/chest.103.6.1646. [DOI] [PubMed] [Google Scholar]
- 70.van der Werff YD, van der Houwen HK, Heilmans PJ, Duurkens VA, Leusink HA, van Heesewijk HP, de Boer A. Postpneumonectomy pulmonary edema: a retrospective analysis of incidence and possible risk factors. Chest. 1997;111:1278–80. doi: 10.1378/chest.111.5.1278. [DOI] [PubMed] [Google Scholar]
- 71.Licker M, De Perrot M, Spiliopoulos A, Robert J, Diaper J, Chevalley C, Tschopp JM. Risk factors for acute lung injury after thoracic surgery for lung cancer. Anesth Analg. 2003;97:1558–65. doi: 10.1213/01.ANE.0000087799.85495.8A. [DOI] [PubMed] [Google Scholar]
- 72.Schilling T, Kozain A, Huth C, Buhling F, Kretzschmar M, Welte T, Hachenb T. The pulmonary immune effects of mechanical ventilation in patients undergoing thoracic surgery. Anesth Analg. 2005;101:957–65. doi: 10.1213/01.ane.0000172112.02902.77. [DOI] [PubMed] [Google Scholar]
- 73.Fernandez E, Keegan M, Brown DR. Intraoperative tidal volume as a risk factor for respiratory failure after pneumonectomy. Chest. 2005;128:129s. doi: 10.1097/00000542-200607000-00007. abstract. [DOI] [PubMed] [Google Scholar]
- 74.Wrigge H, Uhlig U, Zinserling J, Behrends-Callsen E, Ottersbach G, Fischer M, Uhlig S, Putensen C. The effects of different ventilatory settings on pulmonary and systemic inflammatory responses during major surgery. Anesth Analg. 2004;98(3):775–81. doi: 10.1213/01.ane.0000100663.11852.bf. [DOI] [PubMed] [Google Scholar]
- 75.Parquin F, Marchal M, Mehiri S, Hervé P, Lescot B. Post-pneumonectomy pulmonary edema: analysis and risk factors. Eur J Cardiothoracic Surg. 1996;10:929–33. doi: 10.1016/s1010-7940(96)80392-7. [DOI] [PubMed] [Google Scholar]
- 76.Waller DA, Keavey P, Woodfine L, Dark JH. Pulmonary endothelial permeability changes after major lung resection. Ann Thorac Surg. 1996;61:1435–40. doi: 10.1016/0003-4975(96)00103-8. [DOI] [PubMed] [Google Scholar]
- 77.Filaire M, Fadel E, Decante B, Seccatore F, Mazmanian GM, Hervé P. Inhaled nitric oxide does not prevent postpneumonectomy pulmonary edema in pigs. J Thorac Cardiovasc Surg. 2007;133:770–4. doi: 10.1016/j.jtcvs.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 78.Nettelbladt O, Hallgren R. Hyaluronan (hyaluronic acid) in bronchoalveolar lavage fluid during the development of bleomycin-induced alveolitis in the rat. Am Rev Respir Dis. 1989;140:1028–32. doi: 10.1164/ajrccm/140.4.1028. [DOI] [PubMed] [Google Scholar]
- 79.Liu YY, Lee CH, Dedaj R, Zhao H, Mrabat H, Sheidlin A, Syrkina O, Huang PM, Garg HG, Hales CA, Quinn DA. High-molecular-weight hyaluronan – a possible new treatment for sepsis-induced lung injury: a preclinical study in mechanically ventilated rats. Crit Care. 2008;12:R102. doi: 10.1186/cc6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Singleton PA, Mirzapoiazova T, Guo Y, Sammani S, Mambetsariev N, Lennon FE, Moreno-Vinasco L, Garcia JG. High-molecular-weight hyaluronan is a novel inhibitor of pulmonary vascular leakiness. Am J Physiol Lung Cell Mol Physiol. 2010;299:L639–51. doi: 10.1152/ajplung.00405.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, Zemans RL, Bowman JC, Koyanagi DE, Yunt ZX, Smith LP, Cheng SS, Overdier KH, Thompson KR, Geraci MW, Douglas IS, Pearse DB, Tuder RM. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nature Medicine. 2012;18(8):1217–23. doi: 10.1038/nm.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zeldin RA, Normandin D, Landtwing D, Peters RM. Postpneumonectomy pulmonary edema. J Thorac Cardiovasc Surg. 1984;87:359–65. [PubMed] [Google Scholar]
- 83.Crouch JD, Lucas CL, Keagy BA, Wilcox BR, Ha B. The acute effects of pneumonectomy on pulmonary vascular impedance in the dog. Ann Thorac Surg. 1987;43:613–616. doi: 10.1016/s0003-4975(10)60231-7. [DOI] [PubMed] [Google Scholar]
- 84.Slinger P. Postpneumonectomy pulmonary edema: good news, bad news. Anesthesiology. 2006;105:2–5. doi: 10.1097/00000542-200607000-00003. [DOI] [PubMed] [Google Scholar]
- 85.Williams EA, Quinlan GJ, Goldstraw P, Gothard JW, Evans TW. Postoperative lung injury and oxidative damage in patients undergoing pulmonary resection. Eur Respir J. 1998;11:1028–34. doi: 10.1183/09031936.98.11051028. [DOI] [PubMed] [Google Scholar]
- 86.Broccard AF, Hotchkiss JR, Kuwayama N, Olson DA, Jamal S, Wangensteen DO, Marini JJ. Consequences of vascular flow on lung injury induced by mechanical ventilation. Am J Respir Crit Care Med. 1998;157(6 Pt 1):1935–42. doi: 10.1164/ajrccm.157.6.9612006. [DOI] [PubMed] [Google Scholar]
- 87.Briot R, Bayat S, Anglade D, Martiel JL, Grimbert F. Increased cardiac index due to terbutaline treatment aggravates capillary-alveolar macromolecular leakage in oleic acid lung injury in dogs. Crit Care. 2009;13(5):R166. doi: 10.1186/cc8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Broccard AF, Vannay C, Feihl F, Schaller MD. Impact of low pulmonary vascular pressure on ventilator- induced lung injury. Crit Care Med. 2002;30(10):2183–90. doi: 10.1097/00003246-200210000-00002. [DOI] [PubMed] [Google Scholar]
- 89.Hotchkiss JR, Jr, Blanch L, Murias G, Adams AB, Olson DA, Wangensteen OD, Leo PH, Marini JJ. Effects of decreased respiratory frequency on ventilator-induced lung injury. Am J Respir Crit Care Med. 2000;161(2 Pt 1):463–68. doi: 10.1164/ajrccm.161.2.9811008. [DOI] [PubMed] [Google Scholar]
- 90.Hotchkiss JR, Jr, Blanch L, Naveira A, Adams AB, Carter C, Olson DA, Leo PH, Marini JJ. Relative roles of vascular and airspace pressures in ventilator-induced lung injury. Crit Care Med. 2001;29(8):1593–98. doi: 10.1097/00003246-200108000-00016. [DOI] [PubMed] [Google Scholar]
- 91.Anglade D, Corboz M, Menaouar A, Parker JC, Sanou S, Bayat S, Benchetrit G, Grimbert FA. Blood flow vs. venous pressure effects on filtration coefficient in oleic acid-injured lung. J Appl Physiol. 1998;84(3):1011–23. doi: 10.1152/jappl.1998.84.3.1011. [DOI] [PubMed] [Google Scholar]
- 92.Ohkuda K, Nakahara K, Weidner WJ, Binder A, Staub NC. Lung fluid exchange after uneven pulmonary artery obstruction in sheep. Circ Res. 1978;43(2):152–61. doi: 10.1161/01.res.43.2.152. [DOI] [PubMed] [Google Scholar]
- 93.Landolt CC, Matthay MA, Albertine KH, Roos PJ, Wiener-Kronish JP, Staub NC. Overperfusion, hypoxia, and increased pressure cause only hydrostatic pulmonary edema in anesthetized sheep. Circ Res. 1983;52(3):335–41. doi: 10.1161/01.res.52.3.335. [DOI] [PubMed] [Google Scholar]
- 94.Garcia-Delgado M, Colmenero-Ruiz M, Fernandez-Sacristan MA, Rus-Mansilla C, Fernandez-Mondejar E. Effect of a catecholamine-induced increase in cardiac output on extravascular lung water. Crit Care Med. 2001;29(5):931–35. doi: 10.1097/00003246-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 95.Townsley MI, Parker JC, Korthuis RJ, Taylor AE. Alterations in hemodynamics and Kf,c during lung mass resection. J Appl Physiol. 1987;63(6):2460–66. doi: 10.1152/jappl.1987.63.6.2460. [DOI] [PubMed] [Google Scholar]
- 96.Licker M, de Perrot M, Spiliopoulos A, Robert J, Diaper J, Chevalley C, Tschopp JM. Risk factors for acute lung injury after thoracic surgery for lung cancer. Anesth Analg. 2003;97(6):1558–65. doi: 10.1213/01.ANE.0000087799.85495.8A. [DOI] [PubMed] [Google Scholar]
- 97.Schreiber T, Hueter L, Gaser E, Schmidt B, Schwarzkopf K, Rek H, Karzai W. PEEP has beneficial effects on inflammation in the injured and no deleterious effects on the noninjured lung after unilateral lung acid instillation. Intensive Care Med. 2006;32(5):740–49. doi: 10.1007/s00134-006-0117-6. [DOI] [PubMed] [Google Scholar]
- 98.Motosugi H, Quinlan WM, Bree M, Doerschuk CM. Role of CD11b in focal acid-induced pneumonia and contralateral lung injury in rats. Am J Respir Crit Care Med. 1998;157(1):192–198. doi: 10.1164/ajrccm.157.1.9602095. [DOI] [PubMed] [Google Scholar]
- 99.Schreiber T, Hueter L, Gaser E, Schmidt B, Schwarzkopf K, Karzai W. Effects of a catecholamine-induced increase in cardiac output on lung injury after experimental unilateral pulmonary acid instillation. Crit Care Med. 2007;35(7):1741–48. doi: 10.1097/01.CCM.0000269374.85160.BF. [DOI] [PubMed] [Google Scholar]
- 100.Lopez-Quintero SV, Amaya R, Pahakis M, Tarbell JM. The endothelial glycocalyx mediates shear-induced changes in hydraulic conductivity. Am J Physiol Heart Circ Physiol. 2009;296:H1451–56. doi: 10.1152/ajpheart.00894.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Czarnowska E, Karwatowska-Prokopczuk E. Role of oxygen free radicals in cardiocyte injury in the reperfused rat heart. Folia Histochem Cytobiol. 1996;34(Suppl 1):25–26. [PubMed] [Google Scholar]
- 102.Nogae C, Makino N, Hata T, Nogae I, Takahashi S, Suzuki K, Taniquchi N, Yanaga T. Interleukin 1 alpha -induced expression of manganous superoxide dismutase reduces myocardial reperfusion injury in the rat. J Mol Cell Cardiol. 1995;27(10):2091–99. doi: 10.1016/s0022-2828(95)91155-3. [DOI] [PubMed] [Google Scholar]
- 103.Chen EP, Bittner HB, Davis RD, Folz RJ, Van Trigt P. Extracellular superoxide dismutase transgene overexpression preserves postischemic myocardial function in isolated murine hearts. Circulation. 1996;94(9 Suppl):II412–17. [PubMed] [Google Scholar]
- 104.Rubio-Gayosso I, Platts SH, Duling BR. Reactive oxygen species mediate modification of glycocalyx during ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2006;290(6):H2247–56. doi: 10.1152/ajpheart.00796.2005. [DOI] [PubMed] [Google Scholar]
- 105.Kurzelewski M, Czarnowska E, Beresewicz A. Superoxide- and nitric oxide-derived species mediate endothelial dysfunction, endothelial glycocalyx disruption, and enhanced neutrophil adhesion in the post- ischemic guinea-pig heart. J Physiol Pharmacol. 2005;56(2):163–78. [PubMed] [Google Scholar]
- 106.Bruegger D, Rehm M, Jacob M, Chappell D, Stoeckelhuber M, Welsch U, Conzen P, Becker BF. Exogenous nitric oxide requires an endothelial glycocalyx to prevent post-ischemic coronary vascular leak in guinea pig hearts. Crit Care. 2008;12(3):R73. doi: 10.1186/cc6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rehm M, Bruegger D, Christ F, Conzen P, Thiel M, Jacob M, Chappell D, Stoeckelhuber M, Welsch U, Reichart B, Peter K, Becker BF. Shedding of the endothelial glycocalyx in patients undergoing major vascular surgery with global and regional ischemia. Circulation. 2007;116(17):1896–1906. doi: 10.1161/CIRCULATIONAHA.106.684852. [DOI] [PubMed] [Google Scholar]
- 108.Bruegger D, Rehm M, Abicht J, Paul JO, Stoeckelhuber M, Pfirrmann M, Reichart B, Becker BF, Christ F. Shedding of the endothelial glycocalyx during cardiac surgery: onpump versus off-pump coronary bypass graft surgrey. J Thorac Cardiovasc Surg. 2009;138(6):1445–47. doi: 10.1016/j.jtcvs.2008.07.063. [DOI] [PubMed] [Google Scholar]
- 109.Chappell D, Jacob M, Hofmann-Kiefer K, Rehm M, Welsch U, Conzen P, Becker BF. Antithrombin reduces shedding of the endothelial glycocalyx following ischaemia/reperfusion. Cardiovasc Res. 2009;83(2):388–96. doi: 10.1093/cvr/cvp097. [DOI] [PubMed] [Google Scholar]
- 110.Nieuwdorp M, Meuwese MC, Mooij HL, Ince C, Broekhuizen LN, Kastelein JJ, Stroes ES, Vink H. Measuring endothelial glycocalyx dimensions in humans: a potential novel tool to monitor vascular permeability. J Appl Physiol. 2008;104:845–52. doi: 10.1152/japplphysiol.00440.2007. [DOI] [PubMed] [Google Scholar]
- 111.The ARDS Definition Task Force. Acute Respiratory Distress Syndrome: The Berlin Definition. JAMA. 2012;307(23):2526–33. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 112.Williams EA, Quinlan GJ, Anning PB, Goldstraw P, Evans TW. Lung injury following pulmonary resection in the isolated, blood-perfused rat lung. Eur Respir J. 1999;14:745–50. doi: 10.1034/j.1399-3003.1999.14d04.x. [DOI] [PubMed] [Google Scholar]
- 113.Ley K. The role of selectins in inflammation and disease. Trends Mol Med. 2003;9:263–68. doi: 10.1016/s1471-4914(03)00071-6. [DOI] [PubMed] [Google Scholar]
- 114.Kaslovsky RA, Horgan MJ, Lum H, McCandless BK, Gilboa N, Wright D, Malik AB. Pulmonary edema induced by phagocytosing neutrophils. Protective effect of monoclonal antibody against phagocyte CD18 integrin. Circ Res. 1990;67:795–802. doi: 10.1161/01.res.67.4.795. [DOI] [PubMed] [Google Scholar]
- 115.Chappell D, Hofmann-Kiefer K, Jacob M, Rehm M, Briegel J, Welsch U, Conzen P, Becker BF. TNF-alpha induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res Cardiol. 2009;104:78–89. doi: 10.1007/s00395-008-0749-5. [DOI] [PubMed] [Google Scholar]
- 116.Giantsos-Adams KK, Lopez-Quintero VV, Kopeckova PP, Kopecek JJ, Tarbell JM, Dull RO. Study of the therapeutic benefit of cationic copolymer administration to vascular endothelium under mechanical stress. Biomaterials. 2011;32(1):288–94. doi: 10.1016/j.biomaterials.2010.08.092. [DOI] [PMC free article] [PubMed] [Google Scholar]