

Abstract
Ammoxidized technical lignins are valuable soil-improving materials that share many similarities with native terrestrial humic substances. In contrast to lignins, the chemical fate of carbohydrates as typical minor constituents of technical lignins during the ammoxidation processes has not been thoroughly investigated. Recently, we reported the formation of N-heterocyclic, ecotoxic compounds (OECD test 201) from both monosaccharides (d-glucose, d-xylose) and polysaccharides (cellulose, xylan) under ammoxidation conditions and showed that monosaccharides are a source more critical than polysaccharides in this respect. GC/MS-derivatization analysis of the crude product mixtures revealed that ammoxidation of carbohydrates which resembles the conditions encountered in nonenzymatical browning of foodstuff affords also a multitude of nonheterocyclic nitrogenous compounds such as aminosugars, glycosylamines, ammonium salts of aldonic, deoxyaldonic, oxalic and carbaminic acids, urea, acetamide, α-hydroxyamides, and even minor amounts of α-amino acids. d-Glucose and d-xylose afforded largely similar product patterns which differed from each other only for those products that were formed under preservation of the chain integrity and stereoconfiguration of the respective monosaccharide. The kinetics and reaction pathways involved in the formation of the different classes of nitrogenous compounds under ammoxidation conditions are discussed.
Keywords: ammoxidation, ammonoxidation, glucose, xylose, humic substances, aminosugars, Maillard reaction
Introduction
Ammoxidation of lignin, that is, the oxidative conversion of lignin in aqueous ammonia, has been proposed as suitable approach to artificial humic substances, with their large-scale availability being able to greatly support the global efforts to combat soil erosion and desertification. The diversity of different nitrogenous moieties formed during ammoxidation is a key feature of the process. As the various nitrogenous moieties mineralize in soil at different rates, ammoxidized lignins can be used as organo–mineral soil improving materials that supply nitrogen to the plants over several vegetation periods.1−3 While older publications aimed at maximizing nitrogen incorporation by application of high concentrations of ammonia, high oxygen pressure, and temperature,4 it was later demonstrated that ammoxidation in aqueous medium under rather mild conditions (pO2 ≤ 0.2 MPa, T ≤ 100 °C, cNH3 ≤ 5%) affords products which resemble natural humic substances much better in terms of nitrogen contents (3–6%), C/N ratio, chemical structure and functional groups.2 The comparatively low technical expenditure and energy demand of near ambient pressure ammoxidation are strong pros with respect to economic viability and render this technology a promising approach for large-scale conversion of lignin to nitrogenous soil improving materials.
Next to (purified) technical lignins whose price and multipurpose utilization permanently increases, the huge quantities of globally generated lignocellulosic harvest residues (e.g., rice straw, cotton stalks, palm empty fruit bunches) are increasingly considered as cheap and suitable sources of organo–mineral soil improving materials. However, different from purified technical lignins lignocellulosic sources are associated with varying amounts of mono-, oligo-, and polysaccharides. The fate of saccharides under ammoxidative conditions, and in particular the formation of nonheteroyclic nitrogenous compounds has been therefore investigated in this study.
Recently we reported about the constitution of product mixtures obtained by ammoxidation of the monosaccharides glucose, xylose and the corresponding polysaccharides cellulose and xylan under different conditions.5 It was shown that the chemical integrity of cellulose and xylan is largely maintained at moderate temperature and oxygen pressure (70 °C, 0.2 MPa) which was evident from the low amounts of low-molecular degradation products, the only slightly reduced weight-average molecular weight (M̅w 153 vs 139 kg/mol) and the nearly constant amount of carbonyl groups, as shown for cellulose. At elevated temperature and oxygen pressure (140 °C, 1.0 MPa) cellulose was severely degraded (M̅w = 43/kg mol) and both alkaline peeling/stopping reactions and alkali-induced chain scission start playing a major role, resulting in an increased amount of monosaccharides and follow-up products.
N-Heterocyclic compounds such as 1H-imidazole-, pyridine-, and pyrazine derivatives were demonstrated to be formed from carbohydrates under ammoxidative conditions. While the amounts of N-heterocyclic compounds formed at 70 °C were extremely low for the polysaccharides (16–30 μg/g educt) and low for glucose (15.4 mg/g educt), they increased considerably at higher temperature (polysaccharides at 140 °C: 5.52–16.03 mg/g educt; monosaccharides at 100 °C: 122.4–160.5 mg/g educt). Extracts obtained from the crude ammoxidation products exhibited considerable ecotoxicity in terms of inhibiting the growth of the fresh water algae Pseudokirchneriella subcapitata (OECD test 201). 4-Methyl-1H-imidazole, 4-(hydroxymethyl)-1H-imidazole and 3-hydroxypyridine were those carbohydrate-derived ammoxidation products that had the highest ecotoxicity.
The reactions of carbohydrates under alkaline conditions in general, in aqueous ammonium hydroxide in particular, or in Maillard-type reactions with amino acids have been the subject of numerous studies.6−8 Lobry de Bruyn-van Ekenstein and Amadori rearrangements are the typical initial reactions that occur in alkaline medium and can convert one single aldose into a set of isomeric sugars and aminodeoxysugars, respectively. Direct condensation of ammonia with the hemiacetal functionality of the respective aldose leads to glycosylamines. Under basic conditions, reactive α-dicarbonyl compounds (osones) form from sugars. Aldonic and deoxyaldonic acids are follow-up products formed by degradation of sugars under alkaline conditions. Hence it was assumed that a broad spectrum of nitrogenous compounds besides the aforementioned N-heterocycles would also be present in the crude ammoxidation mixtures of mono- and polysaccharides.
The current study investigates the temperature-dependent and time-dependent formation of main classes of low-molecular, nitrogenous, water-soluble reaction products, with an emphasis on non-N-heterocyclic compounds (aminosugars, glycosylamines, ammonium salts of aldonic, deoxyaldonic and carbaminic acids, urea, α-hydroxyamides, and α-amino acids) under ammoxidative conditions, using d-glucose and d-xylose as model monosaccharides, which however are highly pertinent to carbohydrate residues in technical lignins. The kinetics of their formation and conversion to follow-up products are discussed in the context of proposed reaction mechanisms found in the literature.
Materials and Methods
All chemicals including d-glucose and d-xylose were purchased from Sigma Aldrich. Ethyl acetate was dried over anhydrous calcium chloride, distilled once with acetic anhydride and sulfuric acid and subsequently over potassium carbonate, in order to remove water, ethanol and acetic acid, respectively. Pyridine and ethyl acetate used for derivatization were stored over molecular sieves (4A), and filtered through a 0.45 μm syringe filter prior to use.
Ammoxidation
was accomplished in a 4566 C Series 100 mL laboratory-scale pressure vessel (Parr Instruments, Frankfurt, Germany) equipped with a glass liner and a septum valve. External heating was performed with an oil bath. Ten mL of 10% aqueous ammonia that contained 50 μL of 2-methyl quinoxaline and 20 μL of phosphoric acid (85%) as internal standards were placed into the above-described reactor. After flushing three times with oxygen the reactor was pressurized with O2 to 0.2 MPa and heated under continuous stirring to the respective temperature (70 °C, 100 or 140 °C). It was left in connection with the gas supply via a back-pressure valve in order to guarantee constant pressure throughout the reaction. The effective pressure including the respective vapor pressure of 5% aqueous ammonia was 0.27 MPa (70 °C), 0.38 MPa (100 °C), 0.68 MPa (140 °C). A solution of 1.0 g of the respective monosaccharide in 10 mL of water was added through a septum with a syringe, initiating the reaction (t = 0). Samples of approximately 80 μL were taken with a syringe after 1, 3, 5, 10, 15, 20, 30, 45 min, and 1, 1.5, 2, 3, 4, and 19 h. Each of those samples was split into three portions of 20.0 μL, which were immediately deep-frozen on dry ice, and subject to freeze-drying, silylation, and GC/MS analysis (n = 3).
Derivatization
Freeze-dried aliquots (−25 °C, 10–20 Pa, 24 h) of the crude reaction mixtures containing approximately 1 mg of dry matter were subject to GC/MS analysis after silylation which was performed by adding 200 μL of dry pyridine containing 1.5 g/L of the catalyst 4-(N,N-dimethylamino)-pyridine (DMAP), and 10 μL of a 10 g/L solution of the internal standard phenyl-α-glucopyranoside in pyridine. The mixture was vortexed for a minimum of 30 s, before 100 μL of the silylation mixture (BSTFA containing 10% trimethylchloro-silane) was added. Silylation was performed at 70 °C for 2 h. After cooling to room temperature, 900 μL of dry ethyl acetate was added, the mixture was vortexed again, and analyzed by GC/MS not later than 24 h after derivatization.
Synthesis of Di(Glucopyranosyl) Amine9
One gram of d-glucose (5.55 mmol) and 0.438 g of ammonium hydrogen carbonate (5.55 mmol) were dissolved in 27.75 mL of 26% aqueous ammonia, and stirred at 42 °C for 36 h. The solution was evaporated under reduced pressure to 10 mL and subsequently freeze-dried. The resulting pale-brown solid of glucopyranosylamine condenses at room temperature under emission of ammonia quantitatively to di(glucopyranosyl)amine within two weeks.
EI-MS (70 eV) of bis(2,3,4,6-tetra-O-(trimethylsilyl)-glucopyranosyl)amine: m/z 918 (M+; 0.03%), 903 (M+ −CH3; 1.4%), 815 (M+ −CH2OSiMe3, 2.3%), 581 (24.0%), 568 (58.0%), 450 (5.0%), 378 (3.8%), 361 (49.8%), 332 (15.3%), 319 (6.7%), 305 (3.4%), 271 (8.0%), 243 (8.3%), 232 (8.0%), 217 (71.9%), 204 (100%), 191 (9.6%), 169 (7.4%), 147 (36.9%), 129 (17.3%), 117 (11.5%), 103 (16.9%), 73 (82.4%).
GC-MS, data acquisition and processing, and tentative peak assignment were accomplished as described elsewhere.5
Data Presentation
Kinetic data following an exponential decay were fitted with functions 1 or 2:
| 1 |
| 2 |
Data that rise toward a limit were fitted with function 3
| 3 |
For concentrations running through a maximum, the Giddings peak function,10 eq 4, was used as implemented in the Origin 7.1 software package (OriginLab Corporation, Northampton, MA)
| 4 |
Results and Discussion
Per-trimethylsilylation and subsequent GC/MS analysis of the reaction mixtures obtained by ammoxidation of glucose and xylose revealed a far-reaching similarity of the low-molecular product pattern for the two monosaccharides within the studied temperature range (70–140 °C, Figure 1, Table 1). This is in good agreement with the results of a recent study.5 Differences between glucose and xylose were confirmed to be mainly related to those compounds formed under far-reaching preservation of the chain integrity and stereoconfiguration of the respective monosaccharide, as shown for d-glucose (Figure 2, 99). This is the case for > C3 sugar acids (e.g., hexonic (100), pentonic (101), and tetronic (102) acid and their 2- and 3-deoxy derivatives), amino sugars (e.g., glucosamine/mannosamine (103), fructosamine (107), di(glucopyranosyl)amine (104)), imidazoles (e.g., 4-(d-arabino-tetrahydroxybutyl)-1H-imidazole (105)) and pyrazines carrying oligohydroxyalkyl side chains that contain more than one carbon atom, such as, for example, fructosazine and deoxyfructosazine (106). The preservation of stereochemical information, however, is more pronounced with glucose than with xylose. The significantly larger number of very small peaks between 30 and 40 min retention time in the case of ammoxidized xylose is predominantly caused by isomeric oligohydroxyalkyl pyrazines. While for glucose, isomerization reactions in early stages of ammoxidation (Lobry de Bruijn-van Ekenstein reaction) are mostly restricted to the C1 and C2 stereocenters, they involve also other stereocenters (C3, possibly others) to a greater extent with xylose, which caused a larger variety of diastereomeric oligohydroxyalkyl pyrazines. However, apart from these differences, comparison of the product spectra of glucose and xylose upon ammoxidation show large similarities, and thus suggest that both cases share the same underlying reaction pathways. Therefore, d-glucose was further used as a representative monosaccharide to study the impact of the ammoxidation temperature on the product pattern.
Figure 1.

GC/MS spectra (after per-trimethylsilylation) of the low-molecular product fractions obtained from glucose and xylose, respectively, after ammoxidation (100 °C, 0.2 MPa O2, 3 h).
Table 1. Peak Numbers, Retention Times, and Peak Assignments for All Compounds Detected in the Silylated Ammoxidation Products of d-Glucose and d-Xylose.
| compound number | retention time (min) | compound name |
|---|---|---|
| Sugars | ||
| 58 | 31.08, 31.24, 31.33, 32.66 | fructose, pentakis(TMS) |
| 63 | 32.84, 34.67 | glucose, pentakis(TMS) |
| 64 | 32.94 | mannose, pentakis(TMS) |
| 88 | 30.06 | xylose, tetrakis(TMS) |
| Amino Sugars | ||
| 62 | 32.36 | glucosamine, tetrakis(TMS)a |
| 62 | 33.29 | glucosamine, hexakis(TMS) |
| 65 | 33.74 | fructosamine, hexakis(TMS)b |
| 69 | 37.98, 38.17, 38.58, 39.31 | aminohexopyranosid, hexakis(TMS)b |
| 71 | 41.15, 41.47, 41.76, 41.88, 42.01, 42.50, 46.26, 46.69, 47.10, 48.39, 49.57, 49.76, 49.94 | aminoglycosides of unknown constitutionb |
| 73 | 47.56 | di(glucopyranosyl)amine, octakis(TMS)b |
| Carboxylic Acids | ||
| 35 | 11.87 | lactic acid, bis(TMS)a |
| 36 | 12.30 | glycolic acid, bis(TMS)a |
| 37 | 14.01 | oxalic acid, bis(TMS) estera |
| 38 | 14.28 | 3-hydroxypropanoic acid, bis(TMS)a |
| 42 | 19.66 | glyceric acid, tris(TMS)d |
| 45 | 21.78 | 2,4-dihydroxybutyric acid, tris(TMS)a |
| 46 | 22.34 | 3,4-dihydroxybutyric acid, tris(TMS)a |
| 49 | 25.21 | erythronic acid, tetrakis(TMS)a |
| 50 | 25.62 | threonic acid, tetrakis(TMS)a |
| 53 | 27.79 | 2-deoxypentonic acid, tetrakis(TMS)d |
| 54 | 27.96 | 3-deoxypentonic acid, tetrakis(TMS)d |
| 56 | 30.26 | ribonic acid, pentakis(TMS)a |
| 57 | 30.61 | arabinoic acid, pentakis(TMS)a |
| 89 | 30.44 | xylonic acid, pentakis(TMS)a |
| 60 | 32.14 | 2-deoxyhexonic acid, pentakis(TMS)b |
| 61 | 32.25 | 3-deoxyhexonic acid, pentakis(TMS)d |
| 66 | 34.37, 35.06 | hexonic acid, hexakis(TMS)d |
| Carboxylic Acid Amides | ||
| 86 | 7.16 | acetic amidea |
| 6 | 15.03 | lactamide, bis(TMS)a |
| 7 | 15.63 | glycolamide, bis(TMS)a (coeluting with pyrazinylmethanol) |
| 10 | 17.25 | urea, N,N′-bis(TMS)a |
| 32 | 8.61, 11.00 | bis(TMS) formamide (two peaks)a |
| 33 | 8.95 | carbodiimide, N,N′-bis(TMS) (from urea)a |
| 34 | 11.00, 15.95 | carbamate, bis(TMS) and -tris(TMS)a |
| 39 | 17.51 | oxamic acid, bis(TMS)b |
| Amino Carboxylic Acids | ||
| 41 | 13.56, 18.83 | glycine, bis(TMS) and -tris(TMS)a |
| 47 | 23.20 | 2-aminomalonic acid, tris(TMS)d |
| 74 | 13.07 | alanine, bis(TMS)a |
| 75 | 17.50, 20.43 | serine, bisTMS and −trisTMSa |
| 52 | 21.59 | β-alanine, tris(TMS)d |
| 1H-Imidazoles | ||
| 2 | 12.87 | 1H-imidazole (TMS)a |
| 5 | 14.68 | 4-methyl-1H-imidazole (TMS)a |
| 43 | 20.90 | 4-hydroxymethyl-1H-imidazole, bis(TMS)a |
| 55 | 28.79 | 4-(1,2-dihydroxyethyl)-1H-imidazole, tris(TMS)b |
| 68 | 35.92 | 4-(d-arabino-tetrahydroxybutyl)-1H-imidazole, pentakis(TMS)a |
| 77 | 36.22 | 2-acetyl-4-(tetrahydroxybutyl)-1H-imidazole, pentakis(TMSb |
| 90 | 31.87 | 4(5)-(trihydroxypropyl)-1H-imidazole, tris-O-(TMS)b |
| Pyridines | ||
| 4 | 13.90 | 3-hydroxypyridine, TMSa |
| 20 | 24.60 | 2-(hydroxymethyl)-pyridin-5-ol, bis(TMS)b |
| Pyrazines | ||
| 1 | 11.85 | 2-pyrazinol, TMSa |
| 3 | 13.02 | 2-hydroxy-5-methylpyrazine, TMS or isomerb |
| 7 | 15.61 | 2-pyrazinylmethanol, TMSa |
| 8 | 15.83 | 2-hydroxy-3-methylpyrazine, TMS or isomerb |
| 12 | 17.94 | 2-hydroxymethyl-6-methylpyrazine, TMSa |
| 18 | 23.45 | 2-(dihydroxyethyl)pyrazine, bis(TMS)c |
| 21 | 24.77, 25.10, 25.36, 25.83, xylose only: 24.23 | 2-(dihydroxyethyl)-5-methyl-pyrazine, bis(TMS)b and isomers |
| 22 | 26.34, 26.57 | 2,5a- and 2,6b-bis(hydroxymethyl)pyrazine, bis(TMS) |
| 23 | 31.76 | 2-(dihydroxyethyl)-5(hydroxymethyl)pyrazine, tris(TMS)b |
| 24 | 33.07 | 2-(tetrahydroxybutyl)pyrazine, tetrakis(TMS)b |
| 25 | 33.45, 33.60, 33.75, 33.90 | 2-(tetrahydroxybutyl)-5-methyl-pyrazine, tetrakis(TMS)b and isomers |
| 28 | 28.76, 28.91 | 2-(trihydroxypropyl)pyrazine, tris(TMS)b, 2 isomers |
| 29 | 29.48, 29.58, 29.76, 29.86 | 2-(trihydroxybutyl)-5-methylpyrazine, tris(TMS) and isomersb |
| 30 | 34.90, 35.50 | 2-(trihydroxybutyl)-5-(hydroxymethyl)pyrazine, tetrakis(TMS) and isomersb |
| 78 | 36.10, 36.34 | 2-hydroxy-5 (and 6)-(tetrahydroxybutyl)pyrazine, pentakis(TMS)b |
| 79 | 37.96, 38.36 | 2-(hydroxymethyl)-5 (and 6)-(tetrahydroxybutyl)pyrazine, pentakis(TMS)b |
| 80 | 39.24, 39.79 | 2-(2-hydroxyethyl)-5 (and 6)-tetrahydroxybutyl pyrazine, pentakis(TMS)b |
| 81 | 41.23, 41.58 | 2-(dihydroxyethyl)-5 (and 6)-tetrahydroxybutyl pyrazine, hexakis(TMS)b |
| 82 | 42.92, 43.37 | 2-(2,3-dihydroxypropyl)-5 (and 6)-tetrahydroxybutyl pyrazine, hexakis(TMS)b |
| 83 | 43.92, 44.40 | 2-(trihydroxypropyl)-5 (and 6)-tetrahydroxybutyl pyrazine, hexakis(TMS)b |
| 84 | 46.03, 46.08 | 2,5- and 2,6-deoxyfructosazine, heptakis(TMS)b |
| 85 | 46.87, 7.11, 48.07,48.28 | 2,5- and 2,6-fructosazine and diastereomers, octakis(TMS)b |
| 87 | 29.51, 29.81 | 2-methyl-5 (and 6)-(trihydroxypropyl)pyrazine, tetrakis(TMS)b |
| 93 | 34.77, 34.83, 35.23, 35.44 | 2-hydroxymethyl-5(and 6)-(trihydroxypropyl) pyrazine, tetrakis (TMS)b |
| 94 | 35.96, 36.08, 36.54, 36.79, 36.95, 37.80 | 2-(hydroxyethyl)-5-(trihydroxypropyl)pyrazine, tetrakis(TMS) and isomersb |
| 95 | 38.15, 39.01 | 2-dihydroxyethyl-5 (and 6)-trihydroxybutyl pyrazine, pentakis TMSb |
| 96 | 39.88, 40.89 | 2-(dihydroxypropyl)-5 (and 6)-trihydroxypropyl pyrazine, hexakis(TMS)b |
| 97 | 40.94, 41.10, 41.55, 41.91 | 2,5- and 2,6-trihydroxypropyl pyrazine, hexakis(TMS)b |
| Others and Unknown | ||
| 11 | 17.75 | DMAP (catalyst)a |
| 13 | 18.66 | 2-methylquinoxaline (added)a |
| 31 | 8.44 | trifluoromethyl-bis-(trimethylsilyl)methyl ketone (silylation artifact)d |
| 40 | 18.09 | phosphate, tris(TMS) ester (internal standard)a |
| 51 | 27.05 | M+ = 272 |
| 59 | 32.00 | M+ = 525 |
| 70 | 40.50 | phenyl-α-glucopyranosid (internal standard)a |
| 9 | 16.69 | M+ = 196 |
| 14 | 19.40, 19.52 | M+ = 180 |
| 15 | 20.08–20.70 | mixture of low abundance compounds |
| 16 | 22.27 | M+ = 321 |
| 17 | 23.01 | M+ = 270 |
| 19 | 23.84 | M+ = 331 |
| 26, 27 | 21.18, 22.03 | M+ = 256 |
| 44 | 21.09 | M+ = 249 |
| 45 | 21.59 | M+ = 244 |
| 48 | 21.99 | M+ = 248 |
| 51 | 23.22 | M+ = 242 |
| 67 | 27.05 | M+ = 272 |
| 72 | 34.49 | M+ = 493 |
| 76 | 45.88 | M+ = 502 |
| 91 | 35.13 | M+ = 459 |
| 92 | 33.21, 33.70 | M+ = 400 |
| 98 | 31.59, 31.70 | M+ = 414 |
Figure 2.

Ammoxidation products of glucose with preserved glucose stereoconfiguration.
The gas chromatograms of the per-trimethylsilylated crude products obtained from glucose at 70 °C, 100 °C, and 140 °C ammoxidation temperature (0.2 MPa O2, 3 h) are shown in Figure 3. The pie diagrams give an overview of principal changes in the product pattern in terms of both overall concentration (size of pie chart) and the main classes of relevant compounds (parts of pie chart), that is, sugars, amino sugars (containing both Amadori products and glycosyl amines), organic acids including amides, and N-heterocyclic compounds. The latter were grouped into 1H-imidazole and pyrazine derivatives to better visualize the temperature-dependent contribution of the main reaction pathways to N-heterocyclic compounds. Pyridines, as another group of azabenzene derivatives formed upon ammoxidation, were integrated into the group “others” due to both their small amounts (highest value 0.7% at 140 °C) and the mechanism of their formation which substantially differs from that of the 1H-imidazole and pyrazine derivatives, respectively.5
Figure 3.

Chromatograms of the crude reaction mixtures obtained by ammoxidation of d-glucose at different temperature (70 °C, 100 °C, 140 °C; 0.2 MPa O2, 3 h) after freeze-drying and per-trimethylsilylation. Inserts: Pie diagrams show the semiquantitative constitution of the crude products in terms of main classes of organic compounds. The area of the pie diagrams corresponds to the total peak area of GC/MS-detectable compounds in the respective chromatogram.
It was evident that the product pattern varied largely with ammoxidation temperature. Sugars (18% of the total peak area), amino sugars (24%) and organic acids/amides (38%) constituted the largest product fraction at 70 °C, whereas only a very low quantity of N-heterocyclic compounds is formed at this temperature. At 100 °C, the overall yield of low molecular compounds decreased. Short-chain acids (C2 to C6), pyrazines and 1H-imidazoles accounted here for more than three-fourths of the total peak area, while sugars and amino sugars were already largely consumed by follow-up reactions, their fraction being reduced to about 10%. The amount of GC/MS-detectable compounds decreased further when raising the ammoxidation temperature to 140 °C. Organic acids/amides (47%) and pyrazines (42%) were the dominating products, while sugars and amino sugars were reduced to low levels (about 0.2% and 0.1%, respectively). Additional nonidentified low-abundance peaks were observed.
The significant reduction in the absolute amount of low-molecular compounds within the studied temperature range of 70–140 °C can be caused by both increasing oxidative degradation to gaseous compounds, such as CO2, and polymerization reactions of low-molecular intermediates. The occurrence of the latter was evident from the observed temperature-dependent discoloration of the reaction mixtures which was inversely correlated with the total GC/MS peak area. While an only faintly colored solution was obtained after 18 h of ammoxidation at 70 °C, the reaction mixture at 140 °C turned dark brown within the first five minutes. This observation is in accordance with literature, which stated that the color of Maillard-type reaction products originates almost exclusively from polymeric substances which can comprise up to 90% of the total product mixture.11−13
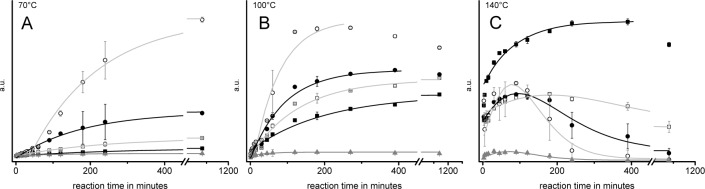
The time-dependent and temperature-dependent consumption of glucose and isomeric sugars, the formation of main classes of nitrogenous reaction products, and their involvement in onward reactions are summarized in Figure 4. The kinetics of the conversion of glucose and isomeric sugars confirm that the concentration of the monosaccharides in the reaction mixtures decreases exponentially with time, with the highest conversion rate at 140 °C. The shape of the respective curves, following the function f(x) = A1·(e-k1t+ e-k2t), furthermore indicates that glucose is consumed by at least two reaction pathways. In alkaline medium the Lobry - de Bruijn–van Ekenstein reaction leads to equilibria of glucose with isomeric sugars such as fructose and mannose. Simultaneously, the presence of ammonia leads to the formation of an equilibrium with glycosyl amine and diglycosyl amine. Until the respective equilibria are reached, glucose consumption is very fast. Then, the different compounds involved react in slower follow-up reactions, which is evident from the declining slope of the monosaccharide concentration curve.
Figure 4.

Change in concentration of different substance classes during ammoxidation (isobar 0.2 MPa O2) of glucose at 70 °C (A), 100 °C (B), and 140 °C (C). Legend: solid black squares, sugars; open white squares, aminosugars; solid black circles, acids and amides; open white circles, N-heterocyclic compounds; solid gray triangles, nonidentified compounds.
The concentration profiles of the amino sugars (Amadori products, glycosyl amines) were found to differ largely with ammoxidation temperature. While at 70 °C the concentration profile of amino sugars followed the typical kinetics of intermediate products, and the concentration proceeded through a maximum, their initial formation was so fast at 100 °C and 140 °C that the concentration increase could not be detected with the used analytical setup. After consumption of sugars and amino sugars, acids and amides formed the largest product fraction, followed by heterocyclic compounds.
Heterocycles, acids and amides can be considered end products of the reaction at 70 °C, as the respective maximum concentrations, once reached, did not further change within the studied time period. At 100 °C, the kinetics of the formation of acids and amides was quite similar to that observed at 70 °C, however, the decline in concentration at long reaction times (>240 min) suggests that some of the formed N-heterocyclic compounds were consumed in follow-up reactions. At 140 °C, not only the concentration of N-heterocycles started to decline even earlier (maximum at 30 min), but also the concentration of acids/amides was declining significantly at >120 min reaction time. The recurring increase of the acid/amide concentration as observed at 140 °C after about four hours of ammoxidation was owing to the formation of glycolic and oxalic acids.
As the above considerations on the kinetics of whole compound classes do not properly reflect the great differences in the product distribution within one class, a closer look into the reaction mechanisms leading to the formation of major representatives of the different compound classes seemed appropriate.
Glycosylamines, Aminodeoxy Sugars, and Isomerization Products of Glucose
Glycosyl amines, aminodeoxy sugars, and isomers of the nonaminated sugars are the very first reaction products of reducing sugars and ammonia. While glycosyl amines are simply formed by condensation of ammonia with the hemiacetal functionality of the respective aldose, isomeric sugars and aminodeoxysugars (i.e., 1-deoxy-1-amino ketoses and 2-deoxy-2-amino aldoses) are formed by Lobry de Bruyn-van Ekenstein and Amadori rearrangements, respectively. The 1,2-enediols and 1,2-enolamines, which are intermediates of these rearrangements, can be either oxidized or dehydrated to form osones, that is, α-dicarbonyl compounds, which in turn are key intermediates in many follow-up reactions. The multitude of these sugars, amino sugars, and dicarbonyl compounds forms a pool of compounds from which smaller fragments are formed by base-catalyzed retro-aldol14−16 and retro-claisen17 scissions, leading to shortened fragments, and small acids and aldehydes. Especially the latter are formed independently from the starting monosaccharide, and their generation is the main reason for the largely similar product spectra despite different starting carbohydrates (e.g., glucose versus xylose).
GC/MS analysis of the crude products after trimethylsilylation, however, did not show any carbohydrates of lower chain length than that of the educt, α-dicarbonyl compounds or small aldehydes. This is due to the high reactivity of such compounds which are usually immediately consumed by follow-up reactions affording products of higher stability. α-Dicarbonyl compounds under the respective ammoxidation conditions, for example, are so reactive that they can be only detected when trapped with a suitable reagent.18 Therefore, only the presence of fructose, mannose and glucosamine could be verified by comparison with commercially available compounds. Based on the fragmentation pattern of the prepared persilylated di(glucopyranosyl) amine (73), other smaller peaks in the reaction mixture of glucose at 70 °C were assigned, such as that of hexopyranosyl-pentosyl amine, or the major aminoglycoside of xylose ammoxidized at 70 °C, di(pentopyranosyl) amine.
The silylated crude ammoxidation product of glucose obtained at 70 °C consisted of 18 different monoglycosyl amines (retention time 36–40 min; Figure 1) and diglycosylamines (retention time 41–49 min), respectively, with largely similar mass spectra. Additional peaks in the same retention time ranges did show typical hexopyranosyl fragments (i.e., m/z 451, 361, 217, 204, 190), but could not be unambiguously assigned. The ratio of diglucosyl amines to monoglycosyl amines increased among otherwise identical samples with longer storage time, higher storage temperature and longer lyophilization duration. Just as for the sugars, also the glycosyl amines contributed a major fraction to the reaction mixture at 70 °C, they were found only in traces at 100 °C, and they were absent at 140 °C.
Aldonic Acids
A considerable fraction of the reaction products, in particular of those obtained at 70 °C, consisted of a homologous series of aldonic acids, i.e. aldoses with their terminal aldehyde/hemiacetal group having been converted into a carboxyl group (Table 2).
Table 2. Relative Amount of Aldonic Acids in the Crude Products of Glucose Ammoxidation at 70 °C, 100 °C, and 140 °C (0.2 MPa O2, 3 h).
| relative
percentagea |
|||||
|---|---|---|---|---|---|
| Rt (min) | aldonic acid | 70 °C | 100 °C | 140 °C | |
| C2 | 12.30 | glycolic acid (36) | 6.3 | 9.7 | 9.7 |
| C3 | 19.66 | glyceric acid (42) | 14.4 | 13.6 | 5.0 |
| C4 | 25.21 | erythronic acid (49) | 27.3 | 14.9 | 3.1 |
| C4 | 25.62 | threonic acid (50) | 2.4 | 2.7 | 1.5 |
| C5 | 30.26 | ribonic acid (56) | 6.1 | 5.2 | 2.4 |
| C5 | 30.61 | arabinonic acid (57) | 97.3 | 23.3 | 2.8 |
| C6 | 35.06 | mannonic acid (66) | 9.1 | 1.4 | 0.0 |
| C6 | 34.37 | gluconic acid (66) | 10.9 | 2.3 | 0.0 |
| sum | 173.8 | 73.1 | 24.5 | ||
Values were calculated as ratio of the relative peak areas of aldonic acids and the internal standard phenyl α-glucoside (200 μg).
The concentration of all detected aldonic acids covering the range from C2 (glycolic acid) to C6 acids (mannonic and gluconic acid) rose quickly as long as monosaccharides were present in the reaction mixture in significant amounts, with the reaction rate being proportional to the monosaccharide concentration (Figure 5). The highest total amounts of aldonic acids were obtained at 70 °C consisting mainly of C5/C6 acids. At 100 °C and above, the concentration of the longer acids (C5 and C6) declined in favor of those with shorter chains after consumption of monosaccharides. Glycolic and glyceric acid concentrations rose throughout the observed reaction time, while the tetronic acids had their maxima between 6 and 18 h reaction time. Upon ammoxidation at 140 °C, hexonic acids reached their maximum after 30 min and were completely consumed after 4 h, while glycolic acid concentration rose until 6.5 h reaction time, and only showed the beginning of a concentration decline at the last sampling time of 18 h. This is in contradiction to other authors,19 who claim that aldonic acids are stable end products of alkaline sugar degradation, albeit in their studies the effect of oxygen and ammonia beyond that of mere alkalinity was not considered.
Figure 5.
Change in concentration of different aldonic acids during ammoxidation (isobar 0.2 MPa O2) of glucose at 70 °C (A), 100 °C (B), and 140 °C (C). Legend: solid black squares, glycolic acid; open white squares, glyceric acid; solid black circles, tetronic acid; open white circles, pentonic acid; solid black triangles, hexonic acid. Values for pentonic and hexonic acids are sums of diastereomeric compounds.
The tetronic, pentonic, and hexonic acids were shown to afford two peaks, each of very similar mass fragment patterns due to the formation of diastereomers. The different diasteromers were identified by comparison with the retention times and mass spectra of silylated authentic samples. Of all the possible diastereomeric aldonic acids of respective chain lengths, only the corresponding C-2 epimers were found, which implies that the hexonic acids had the same configuration at C-3–C-5 as glucose (gluconic and mannonic acids, but not, for example, allonic acid). Similarly, in pentonic acids the configuration of C-4 and C-5 of glucose was preserved: arabinoic and ribonic acids were found, but not, for example, xylonic acid.
This observation allows for some important conclusions with regard to the mechanisms leading to the formation of aldonic acids. Base-catalyzed migration of the carbonyl group through the whole sugar backbone, which would lead to complete epimerization of the carbon centers, does not occur under the chosen conditions. Such carbonyl migrations, however, were observed for reactions in near-neutral, oxygen-free media (pH 7.4 phosphate buffer) and lower concentrations of amino acids20,21 and also for monosaccharides in N-methylmorpholine-N-oxide.22 Under the present, harsher conditions they are probably outrun by faster oxidation, rearrangement, condensation, and chain scission reactions. Also, build-up of pentoses and hexoses from smaller sugars by aldol-type or formose-type condensation can be ruled out as a major reaction pathway, as it would cause epimerized C3 or C4 centers of the hypothetic products, which however were not observed. Other higher carbohydrates formed by aldol-type condensation were also not detected in the ammoxidation products, such as various >C6 sugars and acids which were reported by De Bruijn et al.15 who treated glucose with 10 mM KOH at 78 °C for 7h.
Several possible formation mechanisms are discussed in literature for the formation of aldonic acids (Figure 6), most of which start from α-dicarbonyl compounds (109). Suggested pathways are oxidative cleavage (D);23 benzilic acid rearrangement (F); hydrolytic α-dicarbonyl cleavage via β–OH addition and subsequent elimination of a formyl anion (E);15,16,24,25 Isomerization to a β-dicarbonyl compound and subsequent retro-Claisen scission (G and H) lead to aldonic acids shortened by two carbon atoms. Oddly, direct oxidation of the aldehyde group without involvement of adjacent groups, for example, by Canizzarro reaction (I) or direct action of oxygen or reactive oxygen species (J), is usually not considered in the context of alkaline sugar degradation.
Figure 6.

Reaction mechanisms of the formation of aldonic acids and 2- and 3-deoxyaldonic acids. Pathway A: β-hydroxy elimination followed by keto–enol tautomerism.7B: keto-enediol tautomerism followed by enediol autoxidation. C: dehydration. D: oxidative α-dicarbonyl cleavage.23E: base-catalyzed α-dicarbonyl cleavage. F: benzilic acid rearrangement. G: carbonyl migration through keto-enediol tautomerism. H: retro-Claisen cleavage. I: Canizzarro reaction. J: aldehyde autoxidation.
Deoxy-Aldonic Acids
Besides aldonic acids, two series of deoxyaldonic acids were detected in the gas chromatograms: 2-deoxyaldonic acids and 3-deoxyaldonic acids. The homologous series of 3-deoxyaldonic acids comprised the chain lengths C3 (= lactic acid) to C6. Similar to the case of aldonic acids, we observed peak pairs of quite similar mass spectra for 3-deoxyaldonic acids of C5 and C6 chain lengths due to epimer formation, while only one peak was observed for the 2-deoxyaldonic acids, regardless of their chain lengths (Table 3).
Table 3. Relative Amount of Deoxyaldonic Acids in the Crude Products of Glucose Ammoxidation at 70 °C, 100 °C, and 140 °C (0.2 MPa O2, 3 h).
| relative
percentagea |
|||||
|---|---|---|---|---|---|
| Rt (min) | deoxyaldonic acids | 70 °C | 100 °C | 140 °C | |
| C3 | 11.87 | lactic acid (35) | 0.5 | 1.2 | 3.1 |
| C3 | 14.28 | 3-hydroxypropanoic acid (38) | 0.3 | 2.5 | 2.8 |
| C4 | 21.78 | 2,4-dihydroxybutyric acid (45) | 0.1 | 0.9 | 5.9 |
| C4 | 22.34 | 3,4-dihydroxybutyric acid (46) | 1.0 | 1.8 | 1.4 |
| C5 | 27.79 | 2-deoxypentonic acid (53) | 1.8 | 8.8 | 1.9 |
| C5 | 27.58 | 3-deoxypentonic acid, epimer I | 0.4 | 0.4 | 1.6 |
| C5 | 27.96 | 3-deoxypentonic acid, epimer II (54) | 0.8 | 0.8 | 1.5 |
| C6 | 32.14 | 3-deoxyhexonic acid, epimer I (60) | 0.7 | 0.5 | 1.4 |
| C6 | 32.25 | 3-deoxyhexonic acid, epimer II (61) | 1.3 | 5.4 | 24.7 |
| sum | 6.8 | 22.2 | 44.1 | ||
Values were calculated as ratio of the relative peak areas of aldonic acids and the internal standard phenyl α-glucoside (200 μg).
3-Deoxyaldonic acids are well-known products of alkaline sugar degradation (“metasaccharinic acids”). 2-Deoxyaldonic acids have also been reported,24,26,27 but less frequently. Under base catalysis, aldoses form 3-deoxy-1,2-dicarbonyl compounds (deoxyosones, Figure 6, pathway A). These undergo similar reactions as their nondeoxy counterparts: benzilic acid rearrangement (pathway F) affords 3-deoxyaldonic acids (110), while oxidative cleavage leads to 2-deoxyaldonic acids shortened by one carbon.7,15 An alternative literature pathway proceeding through a ketene intermediate (C)28 was deemed unlikely due to the high expected activation energy of such a process.
For the 3-deoxyhexonic acids, a preferential formation of one of the 2-epimers was observed, which has also been reported7,15,27 and explained27 in literature for similar reaction systems.7,15 As compared to the 3-deoxyhexonic acid epimers, the observed stereoselectivity in the formation of the 3-deoxypentonic acids was much smaller. This is likely due to the higher equilibrium concentration of the furanose form of the respective precursor molecule 3-deoxy-pentosone in comparison to 3-deoxyhexosone29,30 which leads to less stereochemical induction.
The amount of deoxyaldonic acids and of shorter-chain acids were shown to increase with ammoxidation temperature at the expense of longer-chain and nondeoxy acids. At 70 °C, shorter-chain (<C6) deoxyaldonic acids were found in relatively small amounts, and only the two products that can be directly formed from 3-deoxyglucosone, that is, 3-deoxygluconic acid and 2-deoxypentonic acid, were present in larger amounts. The formation of shorter-chain acids requires C–C bond cleavage by retro-aldol/retro-Claisen reactions, while 2-deoxypentonic acid is formed by oxidation reactions (enediol oxidation followed by oxidative cleavage, Figure 6), suggesting that the latter require less activation energy than C–C bond cleaving reactions.
All of the deoxyaldonic acids found originate from 3-deoxyosones as reactive intermediate, while no follow-up products of the frequently described 1-deoxyosones were found, such as parasaccharinic acids (i.e., 2-C-methylpentonic acids) or pyrazines with a 2-oligohydroxyalkyl-3-methyl substitution pattern, at least in glucose ammoxidation mixtures. (5) Formation of the 1-deoxyosone requires tautomeric migration of the aldose’s carbonyl group to form the respective 3-ketoaldose, followed by β-hydroxy carbonyl elimination of the 1-hydroxy group. This again indicates that, at least for glucose, tautomeric migration of the carbonyl group along the chain over more than one carbon does not occur to a measurable extent. However, parasaccharinic acids (C4–C5) were detected among the ammoxidation products of xylan (100 °C - 140 °C, 0.2 MPa O2, 3 h, data not shown). In xylan ammoxidation mixtures, low concentrations of free sugars were formed constantly by hydrolysis of the polysaccharide chain (data not shown). This demonstrates that the product spectrum and the prevalence of the different reaction pathways is also dependent on the sugar concentration: Higher sugar concentrations apparently facilitate condensation reactions leading to N-heterocycles and prevent extensive isomerization, while low sugar concentrations preferably promote base-catalyzed isomerizations and rearrangement reactions, thus affording a greater variety of carbonic acids, but less N-heterocyclic compounds.
The overall picture of the reaction kinetics of deoxyaldonic acids was similar to that of the aldonic acids: At 70 °C, all acids were following pseudo-first-order formation kinetics, while at 100 °C significant degradation of the longer-chain acids occurred as competitive consumption process, and at 140 °C, after a very short build-up period, most of the acids were progressively consumed (Figure 7). However, there are some differences: The stability of 3-deoxy acids was much higher as compared to the corresponding aldonic acids. While gluconic acid, for example, was consumed within 30 min at 140 °C (Figure 5), 3-deoxygluconic acid stayed the most abundant acid even after 3 h reaction time. This suggests that degradation and further consumption reactions of the onic acids involve the hydroxy groups in β- and γ-position to the carboxylic acid, for example, by a facilitated elimination of the γ-hydroxy group and formation of an enol in conjugation to the carboxyl group as the key step. At 140 °C however, even the deoxyaldonic acids are relatively rapidly degraded over time, with dehydration and fragmentation reactions in the polyhydroxyalkyl tail of the acids likely being responsible.
Figure 7.

Change in concentration of different deoxyaldonic acids during ammoxidation (isobar 0.2 MPa O2) of glucose at 70 °C (A), 100 °C (B) and 140 °C (C). Legend: solid black squares, lactic acid; open white squares, 3-hydroxypropanoic acid; solid black circles, 2,4-dihydroxybutyric acid; solid black triangles, 3,4-dihydroxybutyric acid; open white circles, 2-deoxypentonic acid; open white triangles, 3-deoxypentonic acid. Values for 3-deoxyhexonic acids are sums of peaks from epimers.
α-Amino Acids and α-Hydroxy Amides
Surprisingly, minor amounts of the α-amino acids glycine, alanine and serine were found in all crude products, together with larger amounts of the α-hydroxy amides glycol amide and lactamide.
The total amounts of both amino acids and hydroxy amides were shown to be a function of the ammoxidation temperature (Table 4). While the highest level of amino acids was reached at 100 °C, the total amount of α-hydroxy amides decreased from 70 to 140 °C. Among the amino acids, glycine contributed most and serine fewest at all temperature levels. Glycol amide as the major α-hydroxy amide was preferably formed at lower temperature (70 °C).
Table 4. Relative Amount of α-Amino Acids and α-Hydroxy Amides in the Crude Products of Glucose Ammoxidation at 70 °C, 100 °C, and 140 °C (0.2 MPa O2, 3 h).
| relative
percentage (‰)a |
||||
|---|---|---|---|---|
| Rt (min) | α-amino acids and α-hydroxy amides | 70 °C | 100 °C | 140 °C |
| 13.56b, 18.83c | glycine (41) | 1.8 | 3.3 | 0.2 |
| 13.07b | alanine (74) | 0.01 | 0.5 | 0.01 |
| 17.50b, 20.43c | serine (75) | 0.06 | 0.2 | 0 |
| 1.9 | 4.0 | 0.2 | ||
| 15.63 | glycolamide (7) | 10.0 | 6.1 | 1.1 |
| 15.03 | lactamide (6) | 0.04 | 1.8 | 1.2 |
| sum | 10.1 | 7.9 | 2.2 | |
Values were calculated as ratio of the relative peak areas of aldonic acids and the internal standard phenyl α-glucoside (200 μg).
Mono-N-TMS derivative.
Bis-N-TMS derivative.
The kinetics of α-hydroxy amide and α-amino acid formation and consumption at 70 °C, 100 °C, and 140 °C ammoxidation temperature is shown in Figure 8. As in the case of aldonic acids, α-amino acids and α-hydroxy amides followed pseudo-first-order kinetics at 70 °C. At 100 °C, the amino acids reached a plateau value, while both hydroxy amides start to undergo follow-up reactions within the studied time period, probably due to alkaline hydrolysis. This corroborates their formation via a rearrangement reaction rather than by direct conversion of the acid with hot aqueous ammonia. At 140 °C, both α-amino acids and α-hydroxy amides undergo degradation after a steep concentration increase in the first few minutes of the reaction. While for the amides, also a base-catalyzed hydrolysis can be assumed, the amino acids could take part in Maillard reaction cascades with their amino group. In that sense, amino acids may be regarded as important intermediary products in the reaction of ammonia with sugars.
Figure 8.

Change in concentration of the amino acids glycine, alanine and serine and of the α-hydroxy amides glycol amide and lactamide during ammoxidation (isobar 0.2 MPa O2) of glucose at 70 °C (A), 100 °C (B), and 140 °C (C). Legend: solid black squares, glycine; open white squares, glycol amide; solid black circles, alanine; open white circles, lactamide; solid black triangles, serine.
To our best knowledge, the formation of α-amino acids and α-hydroxy amides from saccharides and ammonia has not been reported so far. We suggest that the formation of α-amino acids and α-hydroxy amides follows a common mechanism, which is similar to the formation mechanism of aldonic acids from α-dicarbonyl compounds (see Discussion above).
First, α-carbonyl imines are formed (Figure 9). This can occur either by oxidation of amino sugars (pathway G), by condensation of a dicarbonyl compound with ammonia (pathway H) or by classic Maillard reaction from already formed amino acids and saccharides via Amadori rearrangement, Strecker degradation, oxidation, and dehydration (not depicted).8 The latter pathway allows the new formation and interconversion of amino acids in the Maillard reaction. The α-carbonyl imines are then attacked by a hydroxide ion (Figure 9, pathway K) either at the carbonyl group or at the imino group. Subsequent benzilic acid rearrangement affords either α-amino acids or α-hydroxy amides, depending on whether the rearrangement starts from the imino or the carbonyl group. This mechanism could apply to both α-keto aldimines and α-imino aldehydes (not depicted).
Figure 9.

Overview of main reactions of carbohydrates under ammoxidative conditions: pathway A: Lobry de Bruyn–van Ekenstein transformation. B: Retro-aldol scission. C: Amadori-rearrangement. D/F: β-Hydroxycarbonyl elimination. E/G: Enediol/enaminol oxidation. H: Imine condensation. I: Oxidative α-dicarbonyl cleavage. J/K: Benzilic acid rearrangements. L: Imidazole formation (Radziszewski reaction). M: Pyrazine formation. N: Pyrazinol formation. O: Cyclization/dehydration.
Direct conversion of the respective acids into amides in the presence of ammonia can be excluded, as this would require temperatures clearly above 100 °C and nonaqueous conditions.31
There is no satisfying explanation yet why only the above-mentioned three amino acids were formed, and not all products that are possible following pathway K (Figure 9), that are, all C2 to C6 2-deoxy-2-amino aldonic acids, 2,3-dideoxy-2-amino aldonic acids and the respective α-hydroxy amides. One explanation could be that the proposed mechanism occurs only in the more reactive open-chain C2 and C3 compounds, whereas longer-chain compounds are protected by hemiacetal formation. However, for most of these additional compounds no references are available so that the compounds may remain hidden in the crowded regions of the chromatograms and thus elude identification.
Other Nitrogenous Ammoxidation Products
Besides the already discussed compound classes, a few compounds not fitting into the other compound classes were also found. Urea and ammonium carbamate are formed from ammonia and carbon dioxide. Depending on the reaction temperature, either the carbamate (70 °C) or urea (higher temperatures) is formed.
Oxalic acid is formed continuously throughout the reaction at all temperatures, suggesting that it is the final oxidation product of C2 units that were split off in the various reaction pathways. Acetamide could form in a similar manner as acetic acid17,23 from respective aza-analogous intermediates; i.e. either by retro-Claisen scission of β-iminocarbonyl compounds or oxidative cleavage of α-iminocarbonyl compounds.
Overview of Reactions of Monosaccharides under Ammoxidative Conditions
The main pathways involved in the formation of the different classes of nitrogenous compounds from monosaccharides under ammoxidation conditions have been summarized in a simplified overall reaction scheme (Figure 9). First, sugars undergo base-catalyzed isomerizations and fragmentation (retro-aldol and retro-Claisen scissions) reactions, yielding a broad spectrum of sugars (and small aldehydes) of all chain lengths between C1 and C6 (in case of a hexose as starting material). Aldol condensations of shorter fragments back to larger carbohydrate derivatives were not observed under ammoxidation conditions. These different sugars undergo either condensation with ammonia, followed by Amadori rearrangement to afford amino sugars (Figure 9, pathway C); oxidation by oxygen to osones (pathway E), or base-catalyzed dehydration to deoxyosones (D). Oxidative cleavage (I) or benzilic acid rearrangement (J) of these compounds afford aldonic acids and their 2- and 3-deoxy derivatives. Oxidation (G) or dehydration (F) of amino sugars or condensation of osones with ammonia (H) affords α-carbonyl imines. These compounds, which also form through Strecker degradation in the reaction of α-amino acids and sugars,32 either undergo a benzilic acid rearrangement (K) to form an α-amino acid or an α-hydroxy amide or condense to form heterocyclic structures. Condensation with a reactive aldehyde and ammonia affords imidazoles (L), condensation with an α-amino carbonyl compound (i.e., amino sugar) affords pyrazines (M), and condensation with ammonia and an α-dicarbonyl compound affords pyrazinols (N).5 Cyclization and dehydration of amino sugars affords pyridinols (O).
For the sake of clarity, some known reaction pathways are omitted in Figure 9. For instance, the condensation of two amino-sugars forms dihydropyrazines, which are subsequently oxidized to pyrazines. Furthermore, amino acids can undergo subsequent Maillard reactions. While higher reaction temperatures favor fragmentation, dehydration, polymerization, and formation of pyrazines, lower temperatures promote product mixtures dominated by long-chain amino sugars, sugar acids, and imidazoles.
The results of the current study that investigated the fate of saccharides upon ammoxidation of lignocellulosic materials revealed that a large variety of partially phytotoxic5 nitrogenous compounds can be formed from this fraction, depending on the reaction conditions and amount of saccharides present in the parent material. However, ammoxidation using mild reaction conditions as realized by the near ambient pressure technology (pO2 ≤ 0.2 MPa, T ≈ 70 °C, cNH3 ≤ 5% in water) was confirmed to afford only low amounts of ecotoxic side-products. Their quantity can be further reduced by implementing a washing step prior to ammoxidation that is capable of reducing the monosaccharide content of the respective lignocellulosic source.
Supporting Information Available
Relative amount of other nitrogenous compounds in the crude products obtained by ammoxidation of glucose at 70 °C, 100 °C, and 140 °C (Table S1). EI-MS (70 eV) spectra of octakis-O-TMS di(glucopyranosyl) amine (Figure S1) and hexakis-O-TMS di(xylopyranosyl) amine (Figure S2). Proposed reaction mechanisms for the formation of α-amino acids and α-hydroxyamides (Figure S3). EI-MS (70 eV) fragmentation pattern of persilylated commercial N-heterocyclic compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The financial support by the Christian Doppler Research Society through the CD-laboratory for “Advanced Cellulose Chemistry and Analytics”, the Austrian Research Fund FWF (I154-N19), and the Austrian Research Promotion Agency FFG (834297 ENLIGMA) is gratefully acknowledged.
The authors declare no competing financial interest.
Supplementary Material
References
- Capanema E. A.; Balakshin M. Y.; Chen C. L.; Gratzl J. S. Oxidative ammonolysis of technical lignins. Part 4. Effects of the ammonium hydroxide concentration and pH. J. Wood Chem. Technol. 2006, 26, 95–109. [Google Scholar]
- Fischer K.; Schiene R., Nitrogenous fertilizers from lignins. In Chemical Modification, Properties and Usage of Lignin; Hu T., Ed.; Kluwer Academics/Plenum Publishers: New York, 2002; pp 167–198. [Google Scholar]
- Meier D.; Zuniga-Partida V.; Ramirez-Cano F.; Hahn N.-C.; Faix O. Conversion of technical lignins into slow-release nitrogenous fertilizers by ammoxidation in liquid phase. Bioresour. Technol. 1994, 49, 121–8. [Google Scholar]
- Flaig W.; Hingst G.; Wesselhoeft P.. Verfahren zur Herstellung von stickstoffreichen Ligninprodukten. Deutsches Patent 1 745 632, 1959.
- Klinger K. M.; Liebner F.; Fritz I.; Potthast A.; Rosenau T. Formation and ecotoxicity of N-hetercyclic compounds on ammoxidation of mono- and polysaccharides. J. Agric. Food Chem. 2013, 10.1021/jf4019596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kort M. J. Reactions of free sugars with aqueous ammonia. Adv. Carbohydr. Chem. 1970, 25, 311–349. [Google Scholar]
- Knill C. J.; Kennedy J. F. Degradation of cellulose under alkaline conditions. Carbohydr. Polym. 2002, 51, 281–300. [Google Scholar]
- Fay L. B.; Brevard H. Contribution of mass spectrometry to the study of the Maillard reaction in food. Mass Spectrom. Rev. 2005, 24, 487–507. [DOI] [PubMed] [Google Scholar]
- Lubineau A.; Auge J.; Drouillat B. Improved synthesis of glycosylamines and a straightforward preparation of N-acylglycosylamines as carbohydrate-based detergents. Carbohydr. Res. 1995, 266, 211–219. [DOI] [PubMed] [Google Scholar]
- Di M. V. B.; Bombi G. G. Mathematical functions for the representation of chromatographic peaks. J. Chromatogr., A 2001, 931, 1–30. [DOI] [PubMed] [Google Scholar]
- Dross A.; Baltes W. The fractionation of caramel ingredients according to their molecular weights. Z. Lebensm.-Unters. Forsch. 1989, 188, 540–4. [Google Scholar]
- Tressl R.; Wondrak G. T.; Garbe L.-A.; Krüger R.-P.; Rewicki D. Pentoses and hexoses as sources of new melanoidin-like Maillard polymers. J. Agric. Food Chem. 1998, 46, 1765–1776. [DOI] [PubMed] [Google Scholar]
- Rizzi G. P. chemical structure of colored Maillard reaction products. Food Rev. Int. 1997, 13, 1–28. [Google Scholar]
- Hodge J. E. Dehydrated foods, chemistry of browning reactions in model systems. J. Agric. Food Chem. 1953, 1, 928–943. [Google Scholar]
- De Bruijn J. M.; Kieboom A. P. G.; Van B. H. Alkaline degradation of monosaccharides. Part VII. A mechanistic picture. Starch/Staerke 1987, 39, 23–8. [Google Scholar]
- Tressl R.; Rewicki D. In Heat Generated Flavors and Precursors; Kluwer Academic/Plenum Publishers: 1999; pp 305–325. [Google Scholar]
- Davidek T.; Devaud S.; Robert F.; Blank I. Sugar fragmentation in the Maillard reaction cascade: isotope labeling studies on the formation of acetic acid by a hydrolytic β-dicarbonyl cleavage mechanism. J. Agric. Food Chem. 2006, 54, 6667–6676. [DOI] [PubMed] [Google Scholar]
- Liedke R.; Eichner K. Formation of α-dicarbonyl compounds in the reduction of amadori-rearrangement products. Lebensmittelchemie 2000, 54, 136–137. [Google Scholar]
- van der Poel P. W.; Schiweck H.; Schwartz T.. Zuckertechnologie; Verlag Dr. Albert Bartens KG: Berlin, 2000. [Google Scholar]
- Reihl O.; Rothenbacher T. M.; Lederer M. O.; Schwack W. Carbohydrate carbonyl mobility-the key process in the formation of [alpha]-dicarbonyl intermediates. Carbohydr. Res. 2004, 339, 1609–1618. [DOI] [PubMed] [Google Scholar]
- Biemel K. M.; Conrad J.; Lederer M. O. Unexpected carbonyl mobility in aminoketoses: The key to major Maillard crosslinks. Angew. Chem., Int. Ed. 2002, 41, 801–804. [DOI] [PubMed] [Google Scholar]
- Adorjan I.; Sjoberg J.; Rosenau T.; Hofinger A.; Kosma P. Kinetic and chemical studies on the isomerization of monosaccharides in N-methylmorpholine-N-oxide (NMMO) under Lyocell conditions. Carbohydr. Res. 2004, 339, 1899–1906. [DOI] [PubMed] [Google Scholar]
- Davidek T.; Robert F.; Devaud S.; Vera F. A.; Blank I. Sugar fragmentation in the Maillard reaction cascade: formation of short-chain carboxylic acids by a new oxidative α-dicarbonyl cleavage pathway. J. Agric. Food Chem. 2006, 54, 6677–6684. [DOI] [PubMed] [Google Scholar]
- Yang B. Y.; Montgomery R. Alkaline degradation of glucose: effect of initial concentration of reactants. Carbohydr. Res. 1996, 280, 27–45. [Google Scholar]
- Voigt M.; Smuda M.; Pfahler C.; Glomb M. A. Oxygen-dependent fragmentation reactions during the degradation of 1-deoxy-d-erythro-hexo-2,3-diulose. J. Agric. Food Chem. 2010, 58, 5685–5691. [DOI] [PubMed] [Google Scholar]
- Chetyrkin S. V.; Zhang W.; Hudson B. G.; Serianni A. S.; Voziyan P. A. Pyridoxamine protects proteins from functional damage by 3-deoxyglucosone: mechanism of action of pyridoxamine. Biochemistry 2008, 47, 997–1006. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Carmichael I.; Serianni A. S. Rearrangement of 3-deoxy-d-erythro-hexos-2-ulose in aqueous solution: NMR evidence of intramolecular 1,2-hydrogen transfer. J. Org. Chem. 2011, 76, 8151–8158. [DOI] [PubMed] [Google Scholar]
- Luijkx G. C. A.; van Rantwijk F.; van Bekkum H.; Antal M. J. The role of deoxyhexonic acids in the hydrothermal decarboxylation of carbohydrates. Carbohydr. Res. 1995, 272, 191–202. [DOI] [PubMed] [Google Scholar]
- Weenen H.; Van D. V. J. G. M.; Van D. L. L. M.; Van D. J.; Groenewegen A. C4, C5, and C6 3-deoxyglycosones: structures and reactivity. Spec. Publ. R. Soc. Chem. 1998, 223, 57–64. [Google Scholar]
- Köpper S.; Freimund S. The composition of keto aldoses in aqueous solution as determined by NMR spectroscopy. Helv. Chim. Acta 2003, 86, 827–843. [Google Scholar]
- Rosanoff M. A.; Gulick L.; Larkin H. K. Preparation of acetamide. J. Am. Chem. Soc. 1911, 33, 974–7. [Google Scholar]
- Hofmann T.; Schieberle P. Formation of aroma-active strecker-aldehydes by a direct oxidative degradation of amadori compounds. J. Agric. Food Chem. 2000, 48, 4301–4305. [DOI] [PubMed] [Google Scholar]
- Tsuchida H.; Morinaka K.; Fujii S.; Komoto M.; Mizuno S. Identification of novel non-volatile pyrazines in commercial caramel colors. Dev. Food Sci. 1986, 13, 85–94. [Google Scholar]
- Tsuchida H.; Kitamura K.; Komoto M.; Akomori N. Gas-liquid chromatography and mass spectrometry of trimethylsilyl ethers and butaneboronate-trimethyl-silyl derivatives of polyhydroxyalkylpyrazines. Carbohydr. Res. 1978, 67, 549–563. [Google Scholar]
- Patey A. L.; Startin J. R.; Rowbottom P. M.; Shearer G. Identification of substituted hydroxypyridines and hydroxypyrazines in caramel food colorings. Food Addit. Contam. 1987, 4, 9–15. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.