

Abstract
Context and Objective:
Obesity in pregnancy is associated with increased risks of obesity in the offspring. We investigated the relationship between obesity in pregnancy and circulating maternal and fetal levels of adipose tissue-derived factors adipsin and acylation stimulating protein (ASP) in lean and obese mothers.
Design:
Paired peripheral and cord blood samples were taken. Paired fat and placenta tissue were taken for explant culture. Media were assayed for secreted adipsin and ASP. Clinical parameters assayed included fasting insulin, glucose, and adipsin.
Setting:
The study was conducted at a university hospital maternity unit.
Patients:
Patients included 35 lean [body mass index (BMI) 19–25 kg/m2, mean age 32 years and 39 obese (BMI) > 30 kg/m2, mean age 32.49 years] pregnant Caucasian women, delivered by cesarean section at term.
Main Outcome Measure:
Identification of placental macrophages [Hofbauer cells (HBCs)], as a source of adipsin and ASP was determined.
Results:
HBCs secreted both adipsin and ASP. Cord levels of adipsin (1663.78 ± 52.76 pg/mL) and ASP (354.48 ± 17.17 ng/mL) were significantly elevated in the offspring of obese mothers compared with their lean controls [1354.66 ± 33.87 pg/mL and 302.63 ± 14.98 ng/mL, respectively (P < .05 for both)]. Placentae from obese mothers released significantly more adipsin and ASP than placentae from lean mothers [546.0 ± 44 pg/mL · g vs 284.56 ± 43 pg/mL · g and 5485.75 ± 163.32 ng/mL · g vs 2399.16 ± 181.83 ng/mL · g, respectively (P < .05 for both)]. Circulating fetal adipsin and ASP positively correlated with maternal BMI (r = 0.611, P < .0001, and r = 0.391, P < .05, respectively). Fetal adipsin correlated positively with maternal (r = 0.482, P < .01) and fetal homeostasis model assessment of insulin resistance (r = 0.465, P < .01).
Conclusions:
We demonstrate novel secretion of adipsin and ASP by placental HBCs.
The prevalence of obesity is increasing and now affects more than 475 million individuals worldwide (1). The fact that obesity may be transmissible via nongenetic factors raises significant concerns for future generations. Obesity is proving a major clinical problem in women of reproductive age. It is currently estimated that one in four pregnant patients in the United Kingdom are classified as obese (2). Numerous studies have demonstrated that maternal obesity is related not only to increased risks for the mother but also the offspring (3). In addition to being associated with infertility, stillbirth, gestational diabetes and hypertension, preeclampsia and complication during delivery, maternal high body mass index (BMI) (>30 kg/m2) is positively correlated with neonatal and childhood obesity in the offspring (4), with negative consequences that include increased risk of metabolic syndrome (5) and type 2 diabetes later in life (6).
Although it is established that increased maternal adiposity directly affects the physiology of the fetus, the mechanisms by which these processes occur are incompletely understood (7, 8). More recently, animal studies have revealed mechanistic involvement of adipose tissue derived factors in maternal obesity as well as a crucial influence on metabolic pathways and programming in the fetus (9). However, comparatively limited work has been performed in humans examining the role of adipose tissue-derived factors in obese pregnancy.
Adipsin was originally identified as a 28-kDa adipocyte-secreted protease with close homology to human complement D. Adipsin was first described as an adipokine (10) before being also detected in muscle (11), lung, peripheral nerves (12), and, more recently, murine placenta (13). Adipsin is secreted in significant amounts from adipose tissue (12), and plasma adipsin levels are significantly higher in obesity and positively related to BMI (14). Adipsin is the rate-limiting enzyme in the formation of acylation stimulating protein (ASP) (15), a factor contributing to lipid storage in the adipose tissue (16–18). Adipsin catalyzes the breakdown of complement factor C3 into C3a (17), which is then converted into ASP. ASP is a 76-amino acid protein that enhances triacylglycerol clearance, resulting in reduced circulating triglyceride levels in the animal (19). Molecular mechanisms underlining ASP's metabolic function have been identified. ASP regulates lipid storage in the adipose tissue by increasing diacylglycerol O-acyltransferase 2 activity (20), enhancing glucose transport in adipocytes (17) and reducing hormone-sensitive lipase activity (21). Therefore, the combined increased in lipogenesis and decreased triglyceride breakdown results in ASP-driven enhancement of lipid accumulation in the adipose tissue and concomitantly a reduction of plasma triglyceride concentrations.
Moreover, ASP-induced triglyceride synthesis is not only more potent than that triggered by insulin but also appears to be independent but synergistic with insulin action (22). Plasma adipsin and ASP levels are increased in states of insulin resistance, and obese individuals display significantly increased ASP levels (23), potentially contributing to enhanced triglyceride storage in conditions of impaired insulin function (24). Interestingly, insulin resistance is a normal state developing in mothers during pregnancy and is associated with increased ASP concentrations in late gestation (25).
Despite the evidence that the adipsin-ASP pathway could contribute to increasing fetal fat mass (26), neither adipsin and ASP function nor the correlation between their levels in mothers and the developing fetus has been investigated in the context of obese pregnancy.
With this in mind, we undertook the following: 1) a systemic investigation of adipsin and ASP levels in obese and lean gestational age-matched women along with the analysis of adipsin and ASP levels in their corresponding cord blood, and 2) we examined paired adipose tissue and placental explants to identify a potential source of these molecules.
Materials and Methods
Subjects
All study participants were pregnant women scheduled for elective cesarean section delivering at 39–40 weeks of gestation. Thirty-five lean (BMI 19–25 kg/m2, mean age 32 y; range 18–44 y) and 39 obese (BMI > 30 kg/m2, mean age 32.49 y; range 22–44 y) Caucasian women were asked to fast overnight prior to undergoing elective cesarean section, which was performed at the same time of the day in all individuals to mitigate any possible diurnal influences on adipsin and ASP levels. The Coventry local research ethics committee approved the study, and all patients gave written informed consent (Research Ethics Committees 07/H1210/141). We defined the control group as lean pregnant women with prepregnancy BMI between 19 and 25 kg/m2 and the test group as obese pregnant women with pregravid BMI greater than 30.0 kg/m2. Oral glucose tolerance tests were performed at 26–28 weeks of gestation in all participants to characterize glucose metabolism and exclude diabetes. Women with multiple pregnancies as well as patients with cardiovascular disease, preeclampsia, or other relevant diseases were excluded. Paired maternal venous and cord blood samples were collected at the time of cesarean section and were spun (3000 × g; Beckman Coulter DS-9623C) immediately. The supernatant was stored immediately at −80°C until analysis. All chemicals and reagents were from Sigma-Aldrich unless otherwise stated.
Biochemical and hormone analysis
Venous blood samples were collected for the measurement of insulin, glucose, adipsin, ASP, nonesterified fatty acids (NEFAs), and leptin and for lipid profiling. Details are provided in Supplemental Methods, published on The Endocrine Society's Journals Online web site at http://jcem.endojournals.org.
Tissue explant culture
Paired explant experiments were performed using sc adipose tissue and placentae (27, 28) from obese and lean pregnant patients undergoing elective cesarean section at term (n = 4 for both). Details are provided in in supplemental Methods.
Immunohistochemistry
Eight-micrometer frozen placental and adipose tissue sections were obtained using a cryostat (Leica Microsystems) and fixed in 75% ethanol for 5 minutes. Immunohistochemistry analyses were performed on serial sections of placenta and adipose tissue as described previously (29). Immunostaining was performed using a Vector Polymer detection kit (Vector Laboratories), and adipsin positive cells were detected with mouse antihuman adipsin antibody (1:100; Santa Cruz Biotechnology). Assessment of Hofbauer cell (HBC) numbers was performed by staining with the Hofbauer Cell marker CD206 (R & D Systems), whereas the assessment of adipose tissue macrophages was performed with the pan-macrophage marker CD68 (Dako). Cell counting was performed using ImageJ software (National Institutes of Health) (30).
Western blotting
Proteins were harvested in radioimmunoprecipitation assay buffer with protease inhibitor cocktail added at 0.1% (vol/vol) (Cell Signaling). Further details are provided in Supplemental Methods.
Isolation of placental cytotrophoblast, fibroblast, and Hofbauer cells
Placental cells were isolated as previously described (31). Briefly, isolation of the different cell types comprising placenta was initiated through protocols previously used to obtain cytotrophoblasts (CTs) using trypsin/ deoxyribonuclease I digestion and discontinuous Percoll gradient fractionation. CTs (>95% purity) were generated after enzymatic digestion of placenta with trypsin, centrifugation on Percoll gradients, and negative immunoselection by simultaneous incubation with anti-CD45 and anti-CD9 antibodies. HBCs were isolated using collagenase digestion of trypsin-treated tissues, followed by centrifugation on Percoll gradients and negative immunoselection by sequential incubation with antiepithelial growth factor receptor and then with anti-CD10 antibodies. Cultures of fibroblasts (FIBs) (>95% purity) were obtained from cells attached to magnetic beads containing CD9 and CD45 antibodies from CT isolations and those attached to anti-CD10 beads from HBC preparations. HBCs are isolated with 98%–99% purity and a yield of 130–200 × 106 cells per 80–100 g of tissue assessed by flow cytometry as previously described (31).
Confocal microscopy
Placental sections were obtained as described in Immunohistochemistry. Details of confocal microscopy are provided in Supplemental Methods.
Statistical analyses
Data are expressed as mean ± SEM. Differences between two groups were compared using the Mann-Whitney U test, with significance set at a P < .05. Correlations between parameters were examined by bivariate analyses, using Pearson coefficients. Multiple linear regression analysis was performed using SPSS (IBM, version 19). Insulin resistance was estimated using the homeostasis model assessment of insulin resistance (HOMA-IR), calculated as [fasting insulin (microinternational units per milliliter) × fasting glucose (millimoles per liter)/22.5] (32).
Results
Patient characteristics and biochemical profiles
Patient clinical characteristics are presented in Table 1, whereas the maternal and fetal biochemical profiles are summarized in Table 2. There were no significant differences in glucose and high-density lipoprotein cholesterol levels between normal and obese women or their offspring (P > .05). However, statistically significant differences (P < .05) were found in total cholesterol, triglycerides, and low-density lipoprotein, which were all elevated in obese mothers as well as their corresponding fetal cord bloods when compared with the group with normal BMI. Similarly, serum insulin levels were significantly (P = .008) elevated in obese women compared with lean women. Insulin concentrations were not statistically different in the cord bloods of babies born to obese women compared with lean women. HOMA-IR was elevated in obese women (2.44 ± 0.21) and their offspring, as measured via cord blood (2.17 ± 0.29), compared with lean women (1.84 ± 0.28) and their babies (1.37 ± 0.08).
Table 1.
Clinical Characteristics of Control and Obese Patients
| Characteristics | Control (n = 35) | Obese (n = 39) | P Value |
|---|---|---|---|
| Maternal age, y | 32 (18–44) | 32.487 (22–47) | NS |
| Booking BMI, kg/m2 | 23.477 (20–24.9) | 33.723 (30–46) | <.01 |
| Gestational weight gain, kg | 12.6 ± 4.8 | 11.1 ± 5.6 | NS |
| Gestational age at delivery, wk | 39 ± 2 | 39 ± 2 | NS |
| OGTT 2 hours, mmol/L | 5.44 ± 0.31 | 5.60 ± 0.25 | NS |
| Birth weight, kg | 3.51 ± 0.04 | 3.61 ± 0.03 | <.05 |
| HOMA-IR mother | 1.84 ± 0.283 | 2.436 ± 0.21 | <.001 |
| HOMA-IR baby | 1.365 ± 0.08 | 2.171 ± 0.288 | <.001 |
Abbreviations: NS, not significant; OGTT, oral glucose tolerance test.
Table 2.
Biochemical Profiles of Maternal and Cord Blood From Control and Obese Patients
| Biochemical Parameters | Maternal |
Cord |
||||
|---|---|---|---|---|---|---|
| Normal BMI (n = 35) | High BMI (n = 39) | P Value | Normal BMI (n = 35) | High BMI (n = 39) | P Value | |
| Glucose, mmol/L | 4.288 ± 0.055 | 4.279 ± 0.098 | .938 | 3.732 ± 0.067 | 3.869 ± 0.072 | .168 |
| Triglycerides, mmol/L | 2.494 ± 0.12 | 2.887 ± 0.14 | .037a | 0.209 ± 0.013 | 0.269 ± 0.02 | .014a |
| Total cholesterol, mmol/L | 6.086 ± 0.235 | 6.743 ± 0.194 | .036a | 1.562 ± 0.059 | 1.734 ± 0.059 | .043a |
| HDL cholesterol, mmol/L | 1.744 ± 0.069 | 1.671 ± 0.07 | .459 | 0.813 ± 0.042 | 0.732 ± 0.039 | .163 |
| LDL cholesterol, mmol/L | 3.488 ± 0.175 | 4.011 ± 0.152 | .029a | 0.722 ± 0.028 | 0.879 ± 0.044 | .003a |
| Insulin, μIU/mL | 11.871 ± 1.267 | 17.815 ± 1.782 | .008a | 8.976 ± 0.763 | 11.4 ± 2.099 | .283 |
| Leptin, ng/mL | 23.933 ± 2.66 | 48.603 ± 2.801 | .001a | 2.674 ± 0.2 | 4.862 ± 0.54 | .05 |
Abbreviations: HDL, high density lipoprotein; LDL, low density lipoprotein; n, number of cases. Data are mean ± SEM.
Statistically significant.
Elevated plasma adipsin in obese pregnancy
Plasma adipsin levels (Figure 1A) were significantly elevated in obese pregnant women (843.42 ± 33.14 pg/mL) compared with pregnant women with normal BMI (720.63 ± 33.13 pg/mL). Cord blood samples revealed significantly higher levels of adipsin than maternal samples. Furthermore, babies born to obese mothers showed significantly higher levels of adipsin (1663.78 ± 52.76 pg/mL) in the cord bloods, as compared with babies of lean mothers (1354.37 ± 33.82 pg/mL) (P < .05).
Figure 1.

Measurements of adipsin, ASP, and NEFA concentrations. A, Adipsin concentration in maternal and cord plasma. B, ASP concentration in maternal and cord plasma. C, Measurement of secreted adipsin from paired placental (n = 5) and adipose tissue explants (n = 5) between normal and obese patients. D, Measurement of secreted ASP from paired placental (n = 5) and adipose tissues explants (n = 5) between normal and obese patients. E, NEFA concentration in maternal and cord plasma. Cont, control. *, P < .05
Elevated plasma ASP in obese pregnancy
Plasma ASP levels (Figure 1B) were significantly elevated in obese pregnant women (608.70 ± 67.15 ng/mL), compared with pregnant women with normal BMI (420.30 ± 39.98 ng/mL). In contrast to the observed increased levels of adipsin, cord blood sample levels for ASP were lower than maternal samples, but offspring of obese mothers had significantly (P < .05) higher levels of cord blood ASP (354.48 ± 12.12 ng/mL) than the offspring of lean mothers (302.63 ± 14.98 ng/mL).
Secretion of adipsin and ASP by paired human placental and adipose tissue explants
We identified adipsin secretion from placental explants, which was significantly greater in placentae from obese patients (446.0 ± 44.38 pg/mL · g) than from placentae from lean patients (384.56 ± 43 pg/mL · g; P < .05) (Figure 1C). Additionally and as expected, adipose tissue (AT) from obese patients produced significantly more adipsin (790.27 ± 64.60 pg/mL · g) than AT from lean mothers (504.74 ± 31.94 pg/mL · g; P < .05). To test whether human placenta was also producing ASP, similar analyses on placental explant media were performed and revealed that ASP secretion (Figure 1D) was markedly greater from placental tissues than from AT. ASP levels in the medium of placental tissues of obese women (5485.74 ± 163.32 ng/mL · g) were significantly higher than those of lean women (2399.16 ± 181.83 ng/mL · g). Similarly, although to a lesser extent, ASP secretion from adipose tissue of obese women (1508.40 ± 659.65 ng/mL · g) was significantly higher than ASP secretion from adipose tissue of lean women (807.21 ± 254.8 ng/mL · g).
Cord and maternal nonesterified fatty acid (NEFA) levels
Given the elevated levels of adipsin and ASP in our obese patient cohort, we examined circulating levels of NEFAs in the fetal and maternal compartments. Measurement of NEFAs showed that obese mothers had higher circulating NEFAs than their lean counterparts (1.35 ± 0.06 μmol/μL vs 1.09 ± 0.18 μmol/μL) (Figure 1E). The fetal compartment showed a similar pattern (0.19 ± 0.02 μmol/μL vs 0.12 ± 0.011 μmol/μL), with a 10-fold reduction in NEFA levels compared with the maternal circulation, which is consistent with previous findings (33). This was noted to be a similar order of magnitude to the reduction in triglyceride concentrations (Table 2) between the maternal and fetal compartments.
Fetal plasma adipsin and ASP correlations
A Pearson product-moment correlation coefficient was computed to assess the relationship between fetal adipsin and other clinical and biochemical parameters. Figure 2A reveals a significant relationship between fetal adipsin and maternal BMI (r = 0.611, P < .01). Correlations between adipsin and other parameters are presented in Figure 2C. The predictors of fetal adipsin levels were assessed by a multiple regression analysis. The model included maternal BMI, HOMA-IR, and leptin as independent variables. Maternal BMI explained 37.7% (P = .006) of the variation in fetal adipsin. Maternal HOMA-IR accounted for a further 8% (P = .036) of the variation in fetal adipsin.
Figure 2.

A, Relationship between fetal adipsin concentrations and maternal body mass index. B, Relationship between fetal ASP concentrations and maternal body mass index. C, Pearson correlation coefficients between fetal adipsin and fetal ASP with other covariates. P values are in brackets. NS, not significant; TG, triglycerides.
There was a significant correlation between fetal ASP and maternal BMI (r = 0.391, P < .05) (Figure 2B). The remaining correlations between ASP and other clinical/biochemical parameters are presented in Figure 2C. We also additionally observed that maternal adipsin levels correlated with maternal triglycerides (r = 0.329, P = .04) and fetal cholesterol (r = 0.280, P = .038), whereas maternal ASP correlated with both maternal triglycerides (r = 0.492, P = .004) as well as maternal cholesterol (r = 0.370, P = .04). Furthermore, fetal adipsin correlated with fetal triglycerides (r = 0.287, P < .01).
The predictors of fetal ASP levels were also estimated by multiple regression analysis. The model included maternal BMI, HOMA-IR, and leptin as independent variables. The model explained that only maternal BMI accounted for 26% of the variation in fetal ASP (P = .016).
Human placenta secretes adipsin
Immunohistochemistry experiments revealed adipsin positive areas in specific cells dispersed throughout the placenta (Figure 3A). Pronounced staining was particularly observed in the perivascular area, suggesting higher levels of adipsin in proximity to the fetal vasculature (Figure 3B). The distribution of these cells suggested that they were HBCs. Antibody specificity was confirmed by immunoblotting, with human adipose tissue as positive control (Figure 3C).
Figure 3.
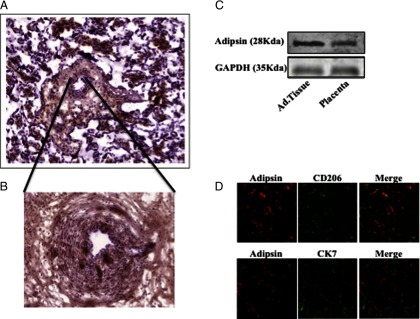
Immunohistochemistry, immunofluorescence, and Western blotting. A, Immunohistochemical detection of adipsin in placental tissues; image was taken at ×10. B, Perivascular localization image taken at ×20. C, Western blotting of adipsin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in adipose (Ad.Tissue) and placental tissues. D, Immunofluorescence images of adipsin and CD206 revealing colocalization (upper panels) and adipsin and CK7 revealing no colocalization (lower panels).
Confocal microscopy revealed that adipsin colocalized with CD206, a HBC marker (Figure 3D, upper panels), but adipsin did not colocalize with the trophoblast marker CK7 (Figure 3D, lower panels), therefore demonstrating that adipsin colocalized to the placental macrophages (HBCs).
HBCs secrete both adipsin and ASP
The previous observations strongly suggested that HBCs in the placenta produced adipsin and ASP. Therefore, to address this possibility and to exclude a contribution from other cell types within the placenta, negative magnetic selection of cell types from placenta was performed as previously described (31). FIBs, cytotrophoblasts (CTBs), and HBCs from 4 placentae were cultured and adipsin and ASP were quantified in their respective media. Although only minimal ASP or adipsin was found in either FIBs or CTBs, sustained secretion of both adipsin (49.75 pg/ml per 106 cells) and ASP (13.62 ng/mL per 106 cells) was detected from HBCs (Figure 4, A and B, respectively).
Figure 4.
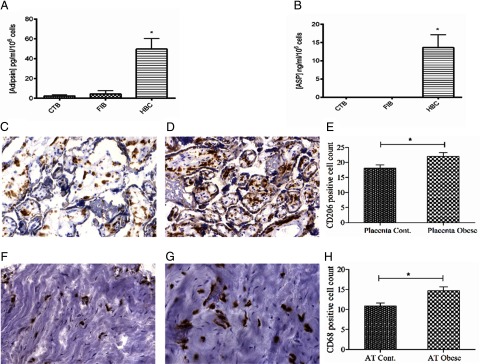
Measurement of adipsin (A) and ASP (B) secretions from isolated CTBs, FIBs, and HBCs. Significant secretion of adipsin and ASP was noted from HBCs. *, P < .001. HBC staining with CD206 in normal (C) and obese (D) placentae reveals increased numbers after counting using ImageJ (E). Macrophage staining with CD68 in normal (F) and obese (G) paired adipose tissue reveals increased numbers (H) after counting. Cont, control.
Quantification of macrophages and HBCs in obese vs normal placental and adipose tissue
ImageJ analysis of macrophage marker staining in adipose tissue (Figure 4, F and G) revealed increased macrophage numbers in obese adipose compared with normal [14.70 ± 0.92 vs 10.85 ± 0.78 (Figure 4H), P < .05] as well as significantly higher HBC counts in obese placentae (Figure 4, C and D) compared with normal placentae [22.05 ± 1.23 vs 18.09 ± 1.08, respectively (Figure 4E), P < .05] respectively, both results confirming the previous finding (34, 35).
Discussion
Adipsin and ASP play significant roles in triglyceride storage, and their levels rise both after high-fat meals and in obese states (36). Pregnancy is characterized by an insulin-resistant state with significant metabolic perturbations, which are exaggerated by maternal obesity. Although the adipsin/ASP pathway has been investigated in obesity (23) and plays a significant role in lipogenesis (37), limited information is available on the role of adipsin in pregnancy. ASP levels correlate with maternal hyperlipidemia in late gestation (25). Although our findings corroborate these observations, examining the role of ASP in hyperlipidemia in state of obesity was beyond the scope of this study. Instead, we report for the first time that circulating adipsin levels are increased in obese mothers compared with their lean controls. Most importantly, adipsin levels were significantly higher in the cord bloods than in the maternal circulation, with an even greater elevation in the cord bloods from obese mothers. In conjunction with this, elevated circulating ASP levels were noted in the obese group. Although ASP levels in the cord bloods were lower than those seen in the mothers, there were significantly higher levels in the cord bloods from obese mothers than cord bloods from lean mothers.
There were strong correlations between both fetal adipsin and fetal ASP with maternal BMI, suggesting potentially important roles for this pathway in pregnancy as well as correlations between maternal and fetal adipsin and ASP with maternal triglycerides, cholesterol, and fetal triglycerides and cholesterol. We found, in agreement with other studies (38), that our obese group did not gain more weight than our lean cohort. According to Institute of Medicine and Health Canada's gestational weight gain recommendations (39), a more moderate weight gain in obese mothers of 5–9 kg of gestational weight gain would be preferable, with the here observed mean weight gain of 11.1 kg being slightly higher. However, because absolute weight gain was comparable between lean and obese mothers, increased levels of adipsin and ASP in the obese mothers and their fetuses cannot be attributed to a higher increase of maternal weight in obese mothers in this cohort. We did note significant correlations between fetal adipsin and fetal birth weight, suggesting a possible contribution to fetal programming of obesity. Nevertheless, whether high adipsin and/or ASP levels in the fetus may be causally involved in increased birth weight remains to be investigated.
Given these data, the significantly elevated levels of adipsin observed in the cord blood suggest that the placenta might be a source of these molecules. We therefore undertook paired explant experiments using sc adipose tissue, which releases similar amounts of adipsin than visceral tissue (40), and placenta. We demonstrate that placental explants secreted both adipsin and ASP into the media. Adipsin secretion from placental explants was lower than that seen from the paired adipose tissue explants. However, ASP secretion had a strikingly opposite pattern, ie, ASP from placental explants was substantially greater than that seen from adipose tissue. Moreover, although the secretion of adipsin from the placental explants was translated into high levels of adipsin in cord blood, higher levels of ASP released by placental explants did not parallel levels of cord ASP. Given the normal levels of adipsin secretion from the explants, the elevated ASP seen in placental explants lends itself to the hypothesis that the placenta might use ASP locally.
Consistent with previous findings (33) was the observation that circulating triglyceride and NEFA levels were significantly lower in the fetal compartment. This is not unexpected when analyzing whole cord blood samples, given that whole cord blood represents both blood into fetal circulation via the umbilical vein and blood back to the placenta via the umbilical artery. Thus, returning blood would have reduced NEFAs because the fetus uses fatty acids. However, an additional physiological action of ASP at the placental barrier cannot be finally excluded. The finding that triglyceride and NEFA levels were lower in the fetal compartment does not obviate the finding that the offspring of obese mothers had higher levels of these lipid species than those of their lean counterparts. In healthy men and nonpregnant women, fasting ASP predicts postprandial triglyceride clearance (41). Moreover, fasting plasma ASP is increased in obesity, insulin resistance, coronary artery disease, and diabetes (42), whereas weight loss and exercise decrease ASP (43). It has therefore been suggested that increased levels of fasting ASP, in the presence of delayed triglyceride clearance, are suggestive of ASP resistance (44). Our data may indicate an enhanced adipsin/ASP response in obesity, which is insufficient to completely mitigate the elevated lipids in the fetus. Our data would support the hypothesis of ASP resistance in obese pregnancy, but further studies are needed to elucidate this.
Having demonstrated novel placental secretion of both adipsin and ASP, we aimed to identify the cell type(s) within the placenta that was responsible for the synthesis and/or secretion of these molecules. It has previously been demonstrated in adipose tissue that the stromal vascular compartment is responsible for the release of adipsin (40). The placenta is a complex organ, comprised of trophoblasts (cytotrophoblast and syncytiotrophoblast), endothelial cells, connective tissue, fibroblasts, and HBCs that are macrophages of mesenchymal origin and are present throughout pregnancy. Immunohistochemistry revealed perivascular staining of adipsin in a cell type that was neither endothelial nor trophoblast. Because positive staining was localized to areas with both fibroblasts and HBCs, we further aimed to identify the responsible cell type using confocal microscopy and a negative immunoselection technique that we have previously used to isolate cell types from the placenta (31). Confocal microscopy showed HBCs and adipsin colocalization. In addition, culturing separated cytotrophoblasts; fibroblasts, and HBCs revealed that both adipsin and ASP were released from HBCs, with negligible release from any of the other cell types seen. We also detected increased numbers of macrophages and HBCs in adipose tissues and placentae from obese mothers, as previously reported (34, 35), and these increased macrophage/HBC numbers might explain the circulating levels of these hormones.
This novel finding indicates that HBCs release molecules associated with the regulation of triglyceride metabolism. The presence of the adipogenic adipsin/ASP pathway in the HBCs of the placenta together with the elevated levels of adipsin in the fetal circulation and our correlation data suggest potential roles in regulation of triglyceride metabolism in the fetus.
Strengths of our study include the relatively large number of subjects investigated and the use of ex vivo explants, which supported our findings. Limitations include the use of estimates of insulin sensitivity rather than measuring this parameter using euglycemic hyperinsulinemic clamps and the lack of information about body fat distribution in the pregnant participants (although leptin was used as a surrogate marker). Finally, despite the clear evidence of placental secretion of adipsin and ASP via HBCs, the possibility that maternal adipsin/ASP might contribute to the fetal levels, perhaps by crossing the placental barrier, cannot completely be excluded, and further work beyond the scope of this manuscript is required to elucidate this.
In conclusion, we hypothesize that given the relative insulin resistance in pregnancy, and particularly in obese pregnancy, that adipsin and ASP might play a role in triglyceride metabolism in pregnancy. Our data place HBCs as a source of these metabolically active molecules.
Acknowledgments
M.V. acknowledges the Nuffield Department of Obstetrics and Gynaecology (University of Oxford, Oxford, United Kingdom). In addition, we acknowledge Ms C. S. Premananthan for assistance with clinical data collection.
This work was supported by the Nuffield Department of Obstetrics and Gynaecology (University of Oxford) and research and development funding from University Hospitals Coventry and Warwickshire National Health Service Trust. C.C.B. acknowledges support from the National Institutes of Health (DK81412).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ASP
- acylation stimulating protein
- AT
- adipose tissue
- BMI
- body mass index
- CT
- cytotrophoblast
- CTB
- cytotrophoblast
- FIB
- fibroblast
- HBC
- Hofbauer cell
- HOMA-IR
- homeostasis model assessment of insulin resistance
- NEFA
- nonesterified fatty acid.
References
- 1. Finkelstein EA, Khavjou OA, Thompson H, Trogdon JG, Pan L, Sherry B, Dietz W. Obesity and severe obesity forecasts through 2030. Am J Prev Med. 2012;42:563–570 [DOI] [PubMed] [Google Scholar]
- 2. Heslehurst N, Rankin J, Wilkinson JR, Summerbell CD. A nationally representative study of maternal obesity in England, UK: trends in incidence and demographic inequalities in 619 323 births, 1989–2007. Int J Obes (Lond). 2010;34:420–428 [DOI] [PubMed] [Google Scholar]
- 3. Sebire NJ, Jolly M, Harris JP, et al. Maternal obesity and pregnancy outcome: a study of 287,213 pregnancies in London. Int J Obes Relat Metab Disord. 2001;25:1175–1182 [DOI] [PubMed] [Google Scholar]
- 4. Dennedy MC, Avalos G, O'Reilly MW, O'Sullivan EP, Gaffney G, Dunne F. ATLANTIC-DIP: raised maternal body mass index (BMI) adversely affects maternal and fetal outcomes in glucose-tolerant women according to International Association of Diabetes and Pregnancy Study Groups (IADPSG) criteria. J Clin Endocrinol Metab. 2012;97:E608–E612 [DOI] [PubMed] [Google Scholar]
- 5. Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115:e290–e296 [DOI] [PubMed] [Google Scholar]
- 6. Dabelea D, Hanson RL, Lindsay RS, et al. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes. 2000;49:2208–2211 [DOI] [PubMed] [Google Scholar]
- 7. Akcakus M, Kurtoglu S, Koklu E, Kula M, Koklu S. The relationship between birth weight leptin and bone mineral status in newborn infants. Neonatology. 2007;91:101–106 [DOI] [PubMed] [Google Scholar]
- 8. Briana DD, Malamitsi-Puchner A. The role of adipocytokines in fetal growth. Ann NY Acad Sci. 2010;1205:82–87 [DOI] [PubMed] [Google Scholar]
- 9. Cripps RL, Martin-Gronert MS, Ozanne SE. Fetal and perinatal programming of appetite. Clin Sci (Lond). 2005;109:1–11 [DOI] [PubMed] [Google Scholar]
- 10. Cook KS, Min HY, Johnson D, et al. Adipsin: a circulating serine protease homolog secreted by adipose tissue and sciatic nerve. Science. 1987;237:402–405 [DOI] [PubMed] [Google Scholar]
- 11. Zhu L, Wigle D, Hinek A, et al. The endogenous vascular elastase that governs development and progression of monocrotaline-induced pulmonary hypertension in rats is a novel enzyme related to the serine proteinase adipsin. J Clin Invest. 1994;94:1163–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. White RT, Damm D, Hancock N, et al. Human adipsin is identical to complement factor D and is expressed at high levels in adipose tissue. J Biol Chem. 1992;267:9210–9213 [PubMed] [Google Scholar]
- 13. Takeshita A, Kondo T, Okada T, Kusakabe KT. Elevation of adipsin, a complement activating factor, in the mouse placenta during spontaneous abortion. J Reprod Dev. 2010;56:508–514 [DOI] [PubMed] [Google Scholar]
- 14. Maslowska M, Vu H, Phelis S, et al. Plasma acylation stimulating protein, adipsin and lipids in non-obese and obese populations. Eur J Clin Invest. 1999;29:679–686 [DOI] [PubMed] [Google Scholar]
- 15. Baldo A, Sniderman AD, St Luce S, et al. The adipsin-acylation stimulating protein system and regulation of intracellular triglyceride synthesis. J Clin Invest. 1993;92:1543–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cianflone K, Maslowska M, Sniderman A. The acylation stimulating protein-adipsin system. Int J Obes Relat Metab Disord. 1995;19(suppl 1):S34–S38 [PubMed] [Google Scholar]
- 17. Cianflone K, Roncari DA, Maslowska M, Baldo A, Forden J, Sniderman AD. Adipsin/acylation stimulating protein system in human adipocytes: regulation of triacylglycerol synthesis. Biochemistry. 1994;33:9489–9495 [DOI] [PubMed] [Google Scholar]
- 18. Millar CA, Meerloo T, Martin S, et al. Adipsin and the glucose transporter GLUT4 traffic to the cell surface via independent pathways in adipocytes. Traffic. 2000;1:141–151 [DOI] [PubMed] [Google Scholar]
- 19. Murray I, Sniderman AD, Cianflone K. Enhanced triglyceride clearance with intraperitoneal human acylation stimulating protein in C57BL/6 mice. Am J Physiol. 1999;277:E474–E480 [DOI] [PubMed] [Google Scholar]
- 20. Yasruel Z, Cianflone K, Sniderman AD, Rosenbloom M, Walsh M, Rodriguez MA. Effect of acylation stimulating protein on the triacylglycerol synthetic pathway of human adipose tissue. Lipids. 1991;26:495–499 [DOI] [PubMed] [Google Scholar]
- 21. Van Harmelen V, Reynisdottir S, Cianflone K, et al. Mechanisms involved in the regulation of free fatty acid release from isolated human fat cells by acylation-stimulating protein and insulin. J Biol Chem. 1999;274:18243–18251 [DOI] [PubMed] [Google Scholar]
- 22. Germinario R, Sniderman AD, Manuel S, Lefebvre SP, Baldo A, Cianflone K. Coordinate regulation of triacylglycerol synthesis and glucose transport by acylation-stimulating protein. Metabolism. 1993;42:574–580 [DOI] [PubMed] [Google Scholar]
- 23. Cianflone K, Xia Z, Chen LY. Critical review of acylation-stimulating protein physiology in humans and rodents. Biochim Biophys Acta. 2003;1609:127–143 [DOI] [PubMed] [Google Scholar]
- 24. Faraj M, Beauregard G, Tardif A, et al. Regulation of leptin, adiponectin and acylation-stimulating protein by hyperinsulinaemia and hyperglycaemia in vivo in healthy lean young men. Diabetes Metab. 2008;34:334–342 [DOI] [PubMed] [Google Scholar]
- 25. Saleh J, Cianflone K, Chaudhary T, Al-Riyami H, Al-Abri AR, Bayoumi R. Increased plasma acylation-stimulating protein correlates with hyperlipidemia at late gestation. Obesity (Silver Spring). 2007;15:646–652 [DOI] [PubMed] [Google Scholar]
- 26. Saleh J, Al-Riyami HD, Chaudhary TA, Cianflone K. Cord blood ASP is predicted by maternal lipids and correlates with fetal birth weight. Obesity (Silver Spring). 2008;16:1193–1198 [DOI] [PubMed] [Google Scholar]
- 27. Siman CM, Sibley CP, Jones CJ, Turner MA, Greenwood SL. The functional regeneration of syncytiotrophoblast in cultured explants of term placenta. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1116–R1122 [DOI] [PubMed] [Google Scholar]
- 28. Tan BK, Chen J, Lehnert H, Kennedy R, Randeva HS. Raised serum, adipocyte, and adipose tissue retinol-binding protein 4 in overweight women with polycystic ovary syndrome: effects of gonadal and adrenal steroids. J Clin Endocrinol Metab. 2007;92:2764–2772 [DOI] [PubMed] [Google Scholar]
- 29. Gould PS, Gu M, Liao J, et al. Upregulation of urotensin II receptor in preeclampsia causes in vitro placental release of soluble vascular endothelial growth factor receptor 1 in hypoxia. Hypertension. 2010;56:172–178 [DOI] [PubMed] [Google Scholar]
- 30. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tang Z, Tadesse S, Norwitz E, Mor G, Abrahams VM, Guller S. Isolation of Hofbauer cells from human term placentas with high yield and purity. Am J Reprod Immunol. 2011;66:336–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419 [DOI] [PubMed] [Google Scholar]
- 33. Hendrickse W, Stammers JP, Hull D. The transfer of free fatty acids across the human placenta. Br J Obstet Gynaecol. 1985;92:945–952 [DOI] [PubMed] [Google Scholar]
- 34. Challier JC, Basu S, Bintein T, et al. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta. 2008;29:274–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haghiac M, Vora NL, Basu S, et al. Increased death of adipose cells, a path to release cell-free DNA into systemic circulation of obese women. Obesity. 2012;20:2213–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xia Z, Cianflone K. Acylation-stimulating protein precursor proteins in adipose tissue in human obesity. Metabolism. 2003;52:1360–1366 [DOI] [PubMed] [Google Scholar]
- 37. Cianflone K, Maslowska M. Differentiation-induced production of ASP in human adipocytes. Eur J Clin Invest. 1995;25:817–825 [DOI] [PubMed] [Google Scholar]
- 38. Black MH, Sacks DA, Xiang AH, Lawrence JM. The relative contribution of prepregnancy overweight and obesity, gestational weight gain, and IADPSG-defined gestational diabetes mellitus to fetal overgrowth. Diabetes Care. 2013;36:56–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. In: Rasmussen KM, Yaktine AL, eds. Weight Gain During Pregnancy: Reexamining the Guidelines. Washington, DC, National Academies Press; 2009 [PubMed] [Google Scholar]
- 40. Fain JN, Nesbit AS, Sudlow FF, et al. Release in vitro of adipsin, vascular cell adhesion molecule 1, angiotensin 1-converting enzyme, and soluble tumor necrosis factor receptor 2 by human omental adipose tissue as well as by the nonfat cells and adipocytes. Metabolism. 2007;56:1583–1590 [DOI] [PubMed] [Google Scholar]
- 41. Cianflone K, Zakarian R, Couillard C, Delplanque B, Despres JP, Sniderman A. Fasting acylation-stimulating protein is predictive of postprandial triglyceride clearance. J Lipid Res. 2004;45:124–131 [DOI] [PubMed] [Google Scholar]
- 42. Yang Y, Lu HL, Zhang J, et al. Relationships among acylation stimulating protein, adiponectin and complement C3 in lean vs obese type 2 diabetes. Int J Obes (Lond). 2006;30:439–446 [DOI] [PubMed] [Google Scholar]
- 43. Schrauwen P, Hesselink MK, Jain M, Cianflone K. Acylation-stimulating protein: effect of acute exercise and endurance training. Int J Obes (Lond). 2005;29:632–638 [DOI] [PubMed] [Google Scholar]
- 44. Wen Y, Wang H, MacLaren R, Wu J, Lu H, Cianflone K. Palmitate and oleate induction of acylation stimulating protein resistance in 3T3-L1 adipocytes and preadipocytes. J Cell Biochem. 2008;104:391–401 [DOI] [PubMed] [Google Scholar]