

Abstract
(−)-Deguelin is a rotenoid natural product that possesses significant potential as a chemopreventive and chemotherapeutic agent. While several racemic syntheses of deguelin have been reported, a formal evaluation of the anticancer activity of both the natural and unnatural enantiomers remains lacking. We describe herein the successful application of a flexible and selective thiourea-catalyzed cyclization strategy toward the enantioselective total synthesis of deguelin, which allows access to either stereoisomer for biological studies. The synthesis was completed in six steps (longest linear) with no protecting groups. The evaluation of both enantiomers of the natural product demonstrated potent inhibition of several cancer cell lines by these compounds, but interestingly showed that the unnatural (+)-deguelin preferentially inhibited the growth of MCF-7 breast cancer and HepG2 liver carcinoma cells when compared to the natural product.
Introduction
(−)-Deguelin (1) is a compact natural product isolated from the flowering plants Lonchocarpus utilis and urucu1 that is related to the pesticide rotenone (2) (Fig. 1). This rotenoid has demonstrated significant anticancer activity in a number of in vitro and in vivo studies and is a compelling target for further therapeutic lead optimization and development. Deguelin has been shown to induce apoptosis in premalignant and malignant human bronchial epithelial (HBE) cells without affecting the growth of normal HBE cells, indicating that it could act as a specific chemopreventive agent.2 In addition, deguelin has demonstrated chemopreventive activity in skin and mammary carninogenesis models,3 and also inhibits the metastatic spread of lung cancer cells in a murine model.4 Like rotenone, deguelin derives its cytotoxicity from inhibition of ornithine decarboxylase, an enzyme associated with tumor progression,5,6 and NADH/ubiquinone oxidoreductase, the first protein in the electron-transport chain (ETC) for oxidative phosphorylation.7 This interaction was further confirmed by a recent report by Winssinger, which demonstrated that fluorescently-labeled deguelin conjugates bind selectively to mitochondria, the site of the ETC in human cells.8 These in vitro and in vivo results suggest that deguelin has significant potential as a chemotherapeutic agent, since it is able to inhibit relevant anticancer cellular targets, thereby reducing tumor growth and metastasis in several models.
Fig. 1.

The natural product (−)-deguelin (1) and related compound rotenone (2).
Depsite this promising biological activity, there remains a need for the development of a concise and enantioselective synthesis of (−)-deguelin and related structures. While racemic syntheses of deguelin and a formal synthesis from rotenone have been reported,9-13 there is only one asymmetric synthesis of this compound reported by Winssinger, who relied on a chiral epoxide opening/Mitsunobu sequence to set the key C13a stereocenter.14 Furthermore, much like the abyssinone family of natural products previously studied by our group,15 the effect of the absolute stereochemistry on the anticancer activity of deguelin has not been formally studied/reported to the best of our knowledge. Given the power of enantioselective catalysis to address this type of question with asymmetric synthesis, we saw the opporutnity to apply a thiourea-catalyzed asymmetric cyclization strategy to the construction of (−)-degeulin based on our program in asymmetric pyran methodology.15-19 Furthermore, since pseudoenantiomeric thiourea catalysts15,16,20 could be utilized in the key cyclization step to set the C13a stereocenter, this approach would facilitate efficient access either enantiomer of the natural product and analogs for biological evaluation. Herein we report the use of this synthetic strategy to obtain the first catalytic, enantioselective total synthesis of the natural product (−)-deguelin, and the biological evaluation of this important anticancer natural product and its unnatural enantiomer in relevant cytotoxicity assays.
Results and discussion
To construct the natural product (−)-deguelin, we wanted to capitalize on our chiral thiourea-catalyzed intramolecular cyclization strategy to control the stereochemistry of the center at C13a.21 We envisioned a late-stage oxidative alpha-arylation22 to generate (−)-deguelin, which would be obtained from the unsubstituted chromanone precursor 3 (Scheme 1). The enantioenriched chromanone material 3 could be synthesized through the thiourea-catalyzed intramolecular cyclization of alkylidene 4, similar to the strategy that was used for the construction of the abyssinone natural products by our group.15 Using both Hiemstra's and “epi”-Hiemstra's thiourea catalysts,16,20 both the (R) and (S) configurations at the C13a stereocenter could be obtained, which would allow for the generation of the natural and unnatural enantiomers of deguelin for biological testing. Similar to our approach with the abyssinones, the alkylidene precursor 4 would be obtained through a Knoevenagel condensation between the β-ketoester 5 and aldehyde 6. These starting materials would be easily obtained from 2,4-dihydroxyacetophenone and 3,4-dimethoxyphenol, two achiral and readily available starting materials.
Scheme 1.

Retrosynthetic analysis of (−)-deguelin, featuring a late-stage oxidative alpha-arylation and a thiourea-catalyzed intramolecular cyclization.
The synthesis of the natural product deguelin commenced with the construction of the requisite β-ketoester starting material, which followed a similar route to that which was used to construct the β-ketoester substrate for the abyssinones.15 Using the same copper-catalyzed etherification strategy, we alkylated 2,4-dihydroxyacetophenone (7) selectively at the C4 phenol to yield alkyne 8 in 80% yield on multi-gram scale (Scheme 2).23 This alkyne 8 was then subjected to microwave conditions at 180 °C to yield the cyclized material 9. Although there have been reports supporting the regiochemical outcome of this type of thermal cyclization reaction,24-28 it does appear to go against conventionally held steric arguments which would dictate that the cyclization occur in the opposite direction. Therefore, the structure of 9 was confirmed by x-ray diffraction before moving onto the next step of the synthesis, with the results supporting the formation of the desired product (Scheme 2).
Scheme 2.

Synthesis of the β-ketoester starting materials 10 and 13. Reagents and conditions: (a) 3-chloro-3-methylbutyne (1.8 equiv), K2CO3 (2 equiv), KI (1.7 equiv), CuI (5 mol %), DMF, 65 °C, 15 h, 80%; (b) μwave, PhMe, 180 °C, 30 min, >99%; (c) hexamethyldisilazane (4 equiv), nBuLi (4 equiv), THF, −78 °C, then 9 (1 equiv), −20 °C, 2 h, then diallyl carbonate (4 equiv), −78 °C → 23 °C, 16 h, 72%; (d) μwave, PhMe, 180 °C, 30 min, >99%; (e) hexamethyldisilazane (6 equiv), nBuLi (6 equiv), THF, −78 °C, then tert-butyl acetate (3.5 equiv), −78 °C, 2 h, then 12 (1 equiv), −78 °C → 23 °C, 15 h, 70%
With the correct crystal structure confirmed, the substituted acetophenone 9 was then deprotonated with excess LiHMDS to form the dianion, which was trapped with diallyl carbonate to give the desired allyl β-ketoester 10 in 72% yield. The allyl β-ketoester was chosen for this synthesis since it had worked well in the synthesis of the abyssinones, allowing for the deprotection and decarboxylation of the ester in one pot using previously reported Pd(0) conditions.29 However, since cyclizations with thiourea catalysts in our laboratory have typically given higher enantioselectivity when the ester substituent is bulky,30,31 the t-butyl β-ketoester 13 was also synthesized. This t-butyl β-ketoester was synthesized through a similar method to the allyl β-ketoester 10,32 except 2,4-dihydroxymethyl benzoate was used as the initial substrate. The lithium anion of t-butyl acetate was then used in the last step to displace the methyl ester of intermediate 12 to generate the required β-ketoester in 70% yield.16
Once the desired β-ketoester substrates were in hand, it was necessary to synthesize the requisite aldehyde coupling partner 6 for the desired Knoevenagel cyclization (Scheme 3). Several approaches toward this aldehyde were attempted, including reduction of the ethyl ester33 and Weinreb amide34,35 substrates with DIBAL-H,36,37 cleavage of the allyl ether with OsO4/NaIO4,38 and oxidation of the alcohol precursor with TPAP•NMO or Dess-Martin periodinane.17 However, the desired product could not be obtained using any of these routes. Ultimately, we were able to obtain the desired material in 88% yield through the mildly acidic deprotection of dimethyl acetal 15 with Amberlyst-15 resin,39,40 although the material had to be stored at −20 °C at all times to prevent decomposition.
Scheme 3.

Synthesis of the aldehyde 6. Reagents and conditions: (a) bromoacetaldehyde dimethyl acetal (1.4 equiv), K2CO3 (6 equiv), DMF, 150 °C, 16 h, 97%; (b) Amberlyst-15 resin (10 mol %), MeCN:water (5:1), 85 °C, 20 h, 88%.
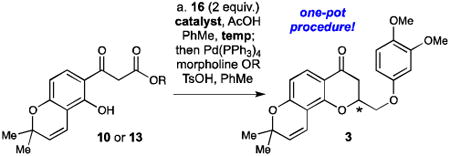
After developing the routes to construct the desired β-ketoester and aldehyde starting materials, our attention turned to the asymmetric synthesis of the chromanone precursor 3. Initially, we attempted to directly couple the allyl β-ketoester 10 with the aldehyde 6 under standard Knoevenagel conditions (piperidine, acetic acid, benzene, Dean-Stark)16 to yield the desired alkylidene 17, but the reaction gave extremely poor conversion and resulted in the generation of the racemic cyclized material. Other conditions utilizing the dimethyl acetal 15 as the coupling partner were also attempted, but none of the reactions yielded the desired material. Finally, switching to the bis-morpholine aminal substrate 16 (actually a 3:1:1 mixture of the two possible enamine isomers) led to the generation of the desired alkylidene substrate 17, although it was only isolated in a 26% yield due to spontaneous cyclization when the material was exposed to silica gel (Scheme 4). While the majority of the product that was obtained from this reaction was the racemic cyclized material, the alkylidene product was isolated and tested in cyclization reactions with pseudoenantiomeric thiourea catalysts 18 and 19. As expected, the cyclization with the thiourea catalysts proceeded smoothly under standard conditions, and the desired enriched chromanones 3 were obtained in 85:15 er (with 18) and 84:16 er (with 19) after deprotection/decarboxylation with Pd(0). A second repeat of each experiment confirmed the results of this first attempt, giving the cyclized material 3 with the same levels of stereoinduction.
Scheme 4.

Initial synthesis of enantioenriched chromanone 3. Reagents and conditions: (a) morpholine (2 equiv), benzene, Dean-Stark, 110 °C, 1 h, 93%; (b) AcOH (4 equiv), 16 (2 equiv), PhMe, 23 °C, 2 h, 26%; (c) 10 mol % 18 or 19, PhMe, −25 °C, 2 days; (d) Pd(PPh3)4 (5 mol %), morpholine (25 equiv), THF, 23 °C, 1 h, 82% over two steps (with 18) and 80% (with 19).
Although we were encouraged by this initial success, there was a need to devise another strategy that avoided purification of the alkylidene precursor 17. This intermediate was exquisitely sensitive to silica gel and would readily cyclize upon purification to give the racemic cyclized products. In our 2007 report on the thiourea-catalyzed synthesis of flavanones,16 a similar problem had been encountered with the synthesis of the chromanone natural product flindersiachromanone, which also has an aliphatic substituent at C2. Since both the Knoevenagel reaction to form the alkylidene and the thiourea-catalyzed cyclization were run in toluene, these two steps could be combined successfully in a one-pot procedure, thereby avoiding purification of the sensitive alkylidene intermediate. Hence, combining allyl β-ketoester 10 and bis-morpholine aminal 16 with 20 mol % thiourea catalyst 18 under these one-pot conditions led to the formation of the desired chromanone product 3 in 34% yield (over 3 steps) and 79:21 er (Table 1, entry 1) after deprotection/decarboxylation. While this one-pot yield was a slight improvement when compared to the original two-step procedure described in Scheme 4, the enantioselectivity was still unacceptably low for this process. Several attempts at optimization of this reaction were made by decreasing the temperature to −30 °C (Table 1, entry 2) or changing the catalyst loading (Table 1, entries 3 and 4). However, none of these optimization attempts led to the formation of the desired chromanone 3 in acceptable yield and enantioselectivity.
Table 1.
Optimization of One-Pot Synthesis of Chromanone 3.
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | R | catalyst | mol % | temp (°C) | % yieldb | er |
| 1 | allyl | 18 | 20 | 23 | 34 | 79:21 |
| 2 | allyl | 18 | 20 | −30 | NRc | NR |
| 3 | allyl | 18 | 5 | 23 | 25 | 60:40 |
| 4 | allyl | 18 | 50 | 23 | 42 | 78:22 |
| 5 | t-Bu | 18 | 50 | 23 | 67 | 90:10 |
| 6 | t-Bu | 18 | 20 | 23 | 46 | 87:13 |
| 7 | t-Bu | 19 | 20 | 23 | 33 | 84:16 |
Reagents and conditions:
10 or 13 (1 equiv), 16 (2 equiv), catalyst 18 or 19, AcOH (4 equiv), 0.1 M in PhMe, 23 °C, 3 days; then Pd(PPh3)4 (5 mol %), morpholine (25 equiv), THF, 23 °C, 1 h (with 10) or TsOH (50 mol%), 0.1 M in PhMe, 80 °C, 16 h (with 13);
yield over three steps (Knoevenagel, conjugate addition, and decarboxylation;
NR = no reaction.
At this point, other modifications that might increase the enantioselectivity of the one-pot process were considered. As mentioned earlier, it has been shown that bulkier β-ketoesters are more reactive and provide high enantioselectivity in this type of intramolecular cyclization reaction.30,31 Therefore, we attempted the one-pot process using the t-butyl β-ketoester 13 instead of the allyl β-ketoester 10. With this modification, both the yield and enantioselectivity of the one-pot reaction increased significantly, to 67% and 90:10 er, respectively (Table 1, entry 5). These results demonstrated that we could efficiently access the desired chromanone material 3 in very high enantioselectivity by utilizing this one-pot procedure. Furthermore, reducing the catalyst loading to 20 mol % (Table 1, entry 6) provided the desired material 3 with minimal loss of yield (46%) and enantioselectivity (87:13), and allowed us to advance material through the end of the synthesis with minimal use of catalyst. Therefore, these conditions with 20 mol % of the Hiemstra and “epi”-Hiemstra catalysts 18 and 19 were utilized to complete the synthesis of both enantiomers of the chromanone precursor 3 (Table 1, entries 6 and 7).
With the enantioenriched chromanone product 3 constructed via the one-pot sequence described above, the last steps of the synthesis were accomplished through an oxidative arylation based upon a report by Snider.22 The requisite TBS silyl enol ether 20 was first synthesized through treatment of enantioenriched chromanone 3 with triethylamine, TBSCl and NaI.41 The resulting TBS ether (20) was then exposed to Cu(OTf)2/Cu2O in the presence of 2,6-di-t-butylpyridine (DTBP). These oxidative conditions directly yielded the natural product (−)-deguelin in 25% yield, with the remainder of the mass balance identified as over-oxidized decomposition products. Interestingly, there was no evidence of the transfused product in this reaction, despite the fact that the report by Snider demonstrated formation of both cis- and trans-fused [5,6] systems.22 However, calculations of energy differences between the cis- and trans-fused systems in this reaction demonstrated that the natural configuration was energetically preferred, which likely explains the lack of trans-fused product. Therefore, the efficiency of this particular bond disconnection made it an attractive route to access the natural product in an efficient and convergent manner.
Although a number of in vitro cell-based toxicity studies have been performed on deguelin, there are no investigations into the effect of stereochemistry on its anticancer potential. Since the successful thiourea-catalyzed cyclization strategy described above provides both enantiomers of deguelin, we were able to evaluate each pure stereoisomer42 in cytotoxicity assays against a panel of cancer cell lines. Selected IC50 data from this preliminary biological screen is shown in Fig. 2. As shown in the figure, both enantiomers of deguelin inhibited the growth of PC-3 (prostate), MCF-7 (breast), HepG2 (liver) and Jurkat (leukemia) cells at low micromolar IC50 values, confirming that this natural product does possess promising anticancer potential. However, the most interesting finding was that the unnatural enantiomer (+)-deguelin inhibited the growth of both the MCF-7 and HepG2 cell lines more effectively than the natural enantiomer (−)-deguelin (p=0.006 and p=0.04, respectively, by unpaired t-test). In fact, (+)-deguelin was 2-fold more effective than the natural product against the MCF-7 breast cancer line, and demonstrated nanomolar activity against the HepG2 cell line. Similar to our studies on the abyssinone natural products15 which also demonstrated variable biological activity depending on the configuration of the C2 stereocenter, these initial results confirm the importance of stereochemical control during the synthesis of deguelin. Furthermore, since (+)-deguelin has never been previously tested in biological assays, these preliminary results demonstrate the therapeutic potential of this unnatural antipode and reiterate the importance of a modular synthetic approach which can be used to access both enantiomers of this chemotherapeutic target.
Fig. 2.

IC50 studies for (−)-deguelin and (+)-deguelin against a panel of cancer cell lines. The average IC50 values for each enantiomer are shown in the graph as well as the table, along with the unpaired t-test values for comparison between enantiomers.
Conclusions
(−)-Deguelin is a rotenoid natural product that has demonstrated significant chemotherapeutic and chemopreventive potential in a number of in vitro and in vivo cancer models. Motivated by this potential, we have established a stereoselective synthesis of (−)-deguelin empolying a thiourea-catalyzed intramolecular cyclization to successfully access the unnantural and natural enantiomers of deguelin. When coupling this asymmetric approach with a copper-promoted arylation, (−)-deguelin was accessed the shortest asymmetric synthesis to date (six linear steps with no protecting groups) from 2,4-dihydroxymethyl benzoate with good levels of enantioselectivity. Additionally, our ability to access either enantiomer of this rotenoid with our tandem cyclization/arylation strategy allowed us to demonstrate for the first time that (+)-deguelin and (−)-deguelin have differential cytotoxicity profiles when evaluated against a panel of relevant cancer cell lines. These results not only confirm the importance of a general catalytic, enantioselective approach, but also present new possibilities for the therapeutic development of deguelin and related rotenoids.
Supplementary Material
Scheme 5.

Completion of the synthesis of the anticancer natural product (−)-deguelin (1). Reagents and conditions: (a) NEt3 (2 equiv), TBSCl (2 equiv), NaI (2 equiv), MeCN, 23 °C, 16 h, 58%; (b) Cu(OTf)2 (2 equiv), Cu2O (3.5 equiv), DTBP (4 equiv), MeCN, −30 °C, 5 min, 25%.
Acknowledgments
The authors thank Dr. Andrew Mazar, Dr. Irawati Kandela, and the Developmental Therapeutics Core of the Chemistry of Life Processes Institute for their help with the biological screening assays. This work has been supported by the American Cancer Society (Research Scholar award 09-016-01 CDD). R. L. F. thanks the NIH for a predoctoral fellowship (F31CA138097), as well as the Malkin Scholars Program for a cancer research grant. We thank John Roberts and Michael Wang (NU) for assistance with X-ray cyrstallography. Purification support was provided by the Center for Molecular Innovation and Drug Discovery ChemCore (supported in part by an NIH instrumentation grant, S10RR025690) and the Chicago Biomedical Consortium with support from The Searle Funds at The Chicago Community Trust (Lever Award). Funding for the NU Integrated Molecular Structure and Education Research Center (IMSERC) has been furnished in part by the NSF (CHE-9871268).
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures and spectroscopic data for all new compounds. See DOI: 10.1039/b000000x/
Notes and references
- 1.Fang N, Casida JE. J Agric Food Chem. 1999;47:2130–2136. doi: 10.1021/jf981188x. [DOI] [PubMed] [Google Scholar]
- 2.Chun KH, Kosmeder JW, Sun S, Pezzuto JM, Lotan R, Hong WK, Lee HY. J Natl Cancer Inst. 2003;95:291–302. doi: 10.1093/jnci/95.4.291. [DOI] [PubMed] [Google Scholar]
- 3.Udeani GO, Gerhauser C, Thomas CF, Moon RC, Kosmeder JW, Kinghorn AD, Moriarty RM, Pezzuto JM. Cancer Res. 1997;57:3424–3428. [PubMed] [Google Scholar]
- 4.Hu J, Ye HY, Fu A, Chen XA, Wang YS, Chen XC, Ye X, Xiao WJ, Duan XM, Wei YQ, Chen LJ. Int J Cancer. 2010;127:2455–2466. doi: 10.1002/ijc.25253. [DOI] [PubMed] [Google Scholar]
- 5.Gerhauser C, Lee SK, Kosmeder JW, Moriarty RM, Hamel E, Mehta RG, Moon RC, Pezzuto JM. Cancer Res. 1997;57:3429–3435. [PubMed] [Google Scholar]
- 6.Gerhauser C, Mar W, Lee SK, Suh N, Luo Y, Kosmeder J, Luyengi L, Fong HH, Kinghorn AD, Moriarty RM, et al. Nat Med. 1995;1:260–266. doi: 10.1038/nm0395-260. [DOI] [PubMed] [Google Scholar]
- 7.Fang N, Casida JE. Proc Nat Acad Sci U S A. 1998;95:3380–3384. doi: 10.1073/pnas.95.7.3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia J, Barluenga S, Gorska K, Sasse F, Winssinger N. Bioorg Med Chem. 2012;20:672–680. doi: 10.1016/j.bmc.2011.09.064. [DOI] [PubMed] [Google Scholar]
- 9.Anzeveno PB. J Org Chem. 1979;44:2578–2580. [Google Scholar]
- 10.Fukami H, Oda J, Nakajima M, Sakata G. Agric Biol Chem. 1961;25:252–253. [Google Scholar]
- 11.Fukami H, Oda J, Sakata G, Nakajima M. Bull Agric Chem Soc Jpn. 1960;24:327–328. [Google Scholar]
- 12.Omokawa H, Yamashita K. Agric Biol Chem. 1974;38:1731–1734. [Google Scholar]
- 13.Pastine SJ, Sames D. Org Lett. 2003;5:4053–4055. doi: 10.1021/ol035419j. [DOI] [PubMed] [Google Scholar]
- 14.Garcia J, Barluenga S, Beebe K, Neckers L, Winssinger N. Chem Eur J. 2010;16:9767–9771. doi: 10.1002/chem.201001080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farmer RL, Biddle MM, Nibbs AE, Huang XK, Bergan RC, Scheidt KA. ACS Med Chem Lett. 2010;1:400–405. doi: 10.1021/ml100110x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biddle MM, Lin M, Scheidt KA. J Am Chem Soc. 2007;129:3830–3831. doi: 10.1021/ja070394v. [DOI] [PubMed] [Google Scholar]
- 17.Crane EA, Zabawa TP, Farmer RL, Scheidt KA. Angew Chem Int Ed. 2011;50:9112–9115. doi: 10.1002/anie.201102790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Custar DW, Zabawa TP, Scheidt KA. J Am Chem Soc. 2008;130:804–805. doi: 10.1021/ja710080q. [DOI] [PubMed] [Google Scholar]
- 19.Morris WJ, Custar DW, Scheidt KA. Org Lett. 2005;7:1113–1116. doi: 10.1021/ol050093v. [DOI] [PubMed] [Google Scholar]
- 20.Marcelli T, van der Haas RNS, van Maarseveen JH, Hiemstra H. Angew Chem Int Ed. 2006;45:929–931. doi: 10.1002/anie.200503724. [DOI] [PubMed] [Google Scholar]
- 21.Luyengi L, Lee IS, Mar W, Fong HHS, Pezzuto JM, Kinghorn AD. Phytochemistry. 1994;36:1523–1526. [Google Scholar]
- 22.Snider BB, Kwon T. J Org Chem. 1992;57:2399–2410. [Google Scholar]
- 23.Bell D, Davies MR, Geen GR, Mann IS. Synthesis-Stuttgart. 1995:707–712. [Google Scholar]
- 24.Joshi SC, Trivedi KN. Tetrahedron. 1992;48:563–570. [Google Scholar]
- 25.Quillinan AJ, Scheinmann FJ. J Chem Soc Perkins Trans 1. 1972:1382–1387. [Google Scholar]
- 26.Subramanian RS, Balasubramanian KK. Tetrahedron Lett. 1988;29:6797–6800. [Google Scholar]
- 27.Thomas P, Whiting DA. Tetrahedron Lett. 1984;25:1099–1102. [Google Scholar]
- 28.Yamaguchi S, Ishibashi M, Akasaka K, Yokoyama H, Miyazawa M, Hirai Y. Tetrahedron Lett. 2001;42:1091–1093. [Google Scholar]
- 29.Tsuji J, Shimizu I, Minami I, Ohashi Y, Sugiura T, Takahashi K. J Org Chem. 1985;50:1523–1529. [Google Scholar]
- 30.Connon SJ. Chem Commun. 2008:2499–2510. doi: 10.1039/b719249e. [DOI] [PubMed] [Google Scholar]
- 31.Connon SJ. Synlett. 2009:354–376. [Google Scholar]
- 32.Donnelly DMX, Fitzpatrick BM, Finet JP. J Chem Soc Perkins Trans 1. 1994:1791–1795. [Google Scholar]
- 33.WO2007119984. Korea Pat. 2007
- 34.Shen ZM, Dornan PK, Khan HA, Woo TK, Dong VM. J Am Chem Soc. 2009;131:1077–1091. doi: 10.1021/ja806758m. [DOI] [PubMed] [Google Scholar]
- 35.Takikawa H, Suzuki K. Org Lett. 2007;9:2713–2716. doi: 10.1021/ol070929p. [DOI] [PubMed] [Google Scholar]
- 36.Ahmed A, Hoegenauer EK, Enev VAS, Hanbauer M, Kaehlig H, Ohler E, Mulzer J. J Org Chem. 2003;68:3026–3042. doi: 10.1021/jo026743f. [DOI] [PubMed] [Google Scholar]
- 37.WO2009141399. US Pat. 2009
- 38.Arndt HC, Carroll SA. Synthesis-Stuttgart. 1979:202–204. [Google Scholar]
- 39.Phillips EM, Wadamoto M, Roth HS, Ott AW, Scheidt KA. Org Lett. 2009;11:105–108. doi: 10.1021/ol802448c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Speranza G, Mueller B, Orlandi M, Morelli CF, Manitto P, Schink B. Helv Chim Acta. 2003;86:2629–2636. [Google Scholar]
- 41.Dratch S, Charnikhova T, Saraber FCE, Jansen BJM, de Groot A. Tetrahedron. 2003;59:4287–4295. [Google Scholar]
- 42.The samples of (−)-deguelin and (+)-deguelin used in the cytotoxicity assay were first purified by preparatory-scale chiral-phase HPLC to ensure optically pure material for biological evaluation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.