

Abstract
We report three distinct, peptide-based catalysts that enable site-selective phosphorylation of three distinct hydroxyl groups within the complex glycopeptide antibiotic teicoplanin A2-2. Two of the catalysts are based on a design that capitalizes on a catalyst-substrate interaction that mimics the biological mechanism of action for teicoplanin. These catalysts are based on a DXaa-DXaa peptide motif that is known to target the teicoplanin structure in a specific manner. The third was identified through evaluation of a set of catalysts that had been developed for historically distinct projects. Each catalyst contains additional functionality designed to dispose a catalytic moiety (a nucleophilic alkylimidazole) at a different region of the glycopeptide structure. A combination of mass spectrometry and 2D-NMR spectroscopy allowed structural assignment of the distinct phosphorylated teicoplanin derivatives. Mechanistic studies are also reported that support the hypotheses that led to the discovery of the catalysts. In this manner, small molecule catalysts have been achieved that allow rational, catalytic control over reactions at sites that are separated by 11.6 Å, 16.5 Å and nearly 17.7 Å, based on the X-ray crystal structure of teicoplanin A2-2. Finally, we report the biological activity of the new phosphorylated teicoplanin analogs, and compare the results to the natural product itself.
Keywords: teicoplanin, site-selective, phosphorylation, peptide-catalyzed, antibiotic
Introduction
Site-selective bond formation at redundantly occurring functional groups within complex molecules is a formidable challenge that, until recently, was largely viewed as the purview of enzymatic catalysis.1–5 As the sites become more remotely localized, and when the stereochemical relationship is not enantiotopic, particular challenges may exist. Catalysts that target large molecules may not exhibit high preferences for one sector of a structure over the other, which will increase the difficulty of function-alizing an intrinsically less reactive site in the presence of competing more reactive sites.6 Even so, advances have been recorded. Desymmetrization reactions provide inspirational examples of equivalent functional group differentiation, even when reaction sites are somewhat remotely localized.7–9 Site-selective reactions of increasingly complex molecules are also being developed.6,10–13 In our own laboratory, we observed that peptide-based catalysts could effect site-selective phosphorylation of the enantiotopic 1-and 3-positions of the inositol ring, with the reacting sites separated by 2.7 Å based on the X-ray crystal structure of myo-inositol14 (Figure 1a).15,16 Later, we developed catalysts that were effective for desymmetrization of a pharmaceutically relevant diarylmethane bis(phenol) where the competing enantiotopic sites were separated by nearly a nanometer (Figure 1b).17,18 More recently, we have endeavored to discover catalysts that enable rapid diversification of truly complex molecules, including the complex glycopeptide antibiotics vancomycin11,12 and teicoplanin.13 For example, we found two catalysts that can target the G6-hydroxyl group of a vancomycin derivative (1) with high selectivity, and another that functionalizes the Z6-position with high selectivity (Figure 1c). These findings, in the context of thiocarbonylation reactions, enabled the synthesis of the corresponding deoxyvancomycins. Distinct from vancomycin, teicoplanin A2-2 (2, Figure 1d) consists of three different sugar moieties located remotely from each other. Notably, the primary hydroxyl groups of each sugar subunits are separated by 11.6 Å, 16.5 Å, and 17.7 Å, based on the crystal structure of teicoplanin A2-2 (2, Figure 1d).19 This increased molecular complexity of teicoplanin A2-2 (2) necessitated the development of novel catalysts that can selectively functionalize the previously unexplored sugar units. In the present study, we report the identification of three distinct catalysts that exhibit high selectivity for phosphorylation of hydroxyl groups located at three different sugar moieties of teicoplanin A2-2 derivative 3.
Figure 1.

Representative examples of substrates for site-selective reactions with competing functional groups at a distance. (a) Enantioselective phosphorylation of inositol derivative. (b) Desymmetrizaton of bis(phenol). (c) Site-selective thiocarbonylation of vancomycin derivative 1. (d) Site selective functionalization of teicoplanin A2-2 derivative 3 and X-ray crystal structure of teicoplanin A2-2 (2, adapted from PDB 2XAD).19
Our interest in the glycopeptides stems from (a) the chemical challenge of developing effective site-selective catalysts that tolerate the dense arrays of chemical functionality, including the intrinsically varied reactivity of all the competing functional groups, and (b) the opportunity to contribute new analogs that might reveal aspects of the structure activity relationships for these important molecules. Vancomycin (4, Figure 2), teicoplanin20 and their relatives have emerged as important compounds in the campaign against antibiotic resistance.21–25 Studies of their chemical synthesis,26–32 semisynthesis,33–35 and biosynthesis36–40 continue to contribute mightily to our understanding of their modes of action, and also to promising new therapies in clinical settings. Teicoplanin in particular is an attractive compound for our interests not only because of its high degree of molecular complexity but also its higher degree of potency as an antibiotic.41 Yet, the onset of resistance to vancomycin (4), teicoplanin and their relatives motivates further the objective of developing new chemistry to manipulate the scaffold.42,43 Among the interesting glycopeptides in the clinic is the compound telavancin (5, Figure 2), a phosphonated vancomycin derivative.44 In part due to its phosphonated nature, we set out to learn if it might be possible to develop catalysts capable of catalytic phosphorylation of teicoplanin derivative in a site-selective manner.
Figure 2.

Structure of representative glycopletide drugs vancomycin (4) and telavancin (5).
Results and Discussion
Initial Design of the Catalysts
The mechanism of antibacterial activity for vancomycin or teicoplanin have been studied extensively.45 Among the conclusions is the canonical binding of the glycopeptides to the growing bacterial cell wall through a specific association between vancomycin/teicoplanin and the DAla-DAla moiety of the growing peptidoglycan. Five hydrogen bonds between the antibiotic and substrate play a key role in inhibiting the cell wall synthesis of the bacteria. We have been exploring the possibility of developing catalysts that specifically modify the glycopeptides by mimicking these biologically validated interactions. For example, we developed catalysts for selective thiocarbonylation12 and bromination11 of vancomycin, and bromination13 of teicoplanin on this basis.
Loll and coworkers reported the X-ray crystal structure of the complex of DAla-DAla peptide moieties and teicoplanin using a carrier protein strategy (Figure 3a).46 Based on this crystal structure data, we envisioned that a superposition of the terminal DAla1 with a D-π-methylhistidine (DPmh) would bring the nucleophilic alkylimidazole group in close proximity to the N-decanoylglucosamine unit (Figure 3b, N-decanoylglucosamine shown in red). Alkylimidazoles, such as that localized in Pmh, have been shown to be effective nucleophilic catalysts for the delivery of a variety of functional groups to hydroxyl sites.6,12,16,17,47,48 As noted above, these residues are quite well suited for catalytic phosphoryl group transfer, and may also be considered in some sense, mimics of kinases.49 Hence, we envisioned that a catalyst with Xaa-DAla-DPmh sequence would be optimal for the functionalization of the hydroxyl groups present in the “top/red” N-decanoylglucosamine moiety (Figure 3b). Indeed, this catalyst design was applied to the thiocarbonyl group transfer at G6 position of vancomycin derivative 1 (Figure 1c).12 For the selective functionalization of the mannose ring (“bottom/blue” sugar), we envisioned a catalyst where the DAla2 is substituted with DPmh (Figure 3c). The superposition of the DPmh residue in this locus suggested that the binding of Xaa-DPmh-DAla peptide to teicoplanin would position the nucleophilic alkylhistidine moiety proximal to the mannose sugar. The design of the peptide based catalyst for selective functionalization of the N-acetylglucosamine ring (“left/green” sugar) was most challenging due to the long distance from the DAla-DAla binding pocket to the sugar moiety (Figure 3d). The X-ray crystal structure and molecular model of teicoplanin A2-2 (2) bound to DAla-DAla based peptide show that the N-acetylglucosamine ring is positioned away from the peptide moiety due to the S-configuration of the Z6 carbon, which rendered the design of catalyst selective for this sugar unit uniquely challenging. We envisioned that an extended, well-defined peptide sequence, beyond a DAla-DAla initiator moiety, would be required for this goal.
Figure 3.

Design of peptide catalysts based on the X-ray crystal structure of DAla-DAla containing peptide bound to teicoplanin (a) X-ray crystal structure of Lys-DAla-DAla-O− (6, lines) bound to 6c–dechloroteicoplanin (7, sticks, adapted from PDB 3VFG).46 (b) Proposed model for delivery of diphenylphosphoryl group to the N-decanoylglucosamine ring by Z-Lys(Z)-DAla-DPmh-O−. (c) Proposed model for delivery of diphenylphosphoryl group to the mannose ring by Z-Ala-DPmh-DAla-O−. (d) Proposed model for delivery of diphenylphosphoryl group to the N-acetylglucosamine ring by Pmh-(Xaa)n-DAla-DAla-O−.
Development of the Catalyst for Selective Phosphorylation of the “Top/Red” N-Decanoylglucosamine
Our study commenced with the proper protection of the teicoplanin A2-2 (2). The primary amine present in teicoplanin A2-2 (2) was efficiently protected with N-(allyloxycarbonyloxy)succinimide in the presence of sodium bicarbonate. Next, the carboxylate and phenolic hydroxyl groups were allylated with allyl bromide and cesium carbonate.50–52 These functional groups had a dual role of masking the reactive amine and phenol functionalities and increasing solubility of the substrate to organic solvents.
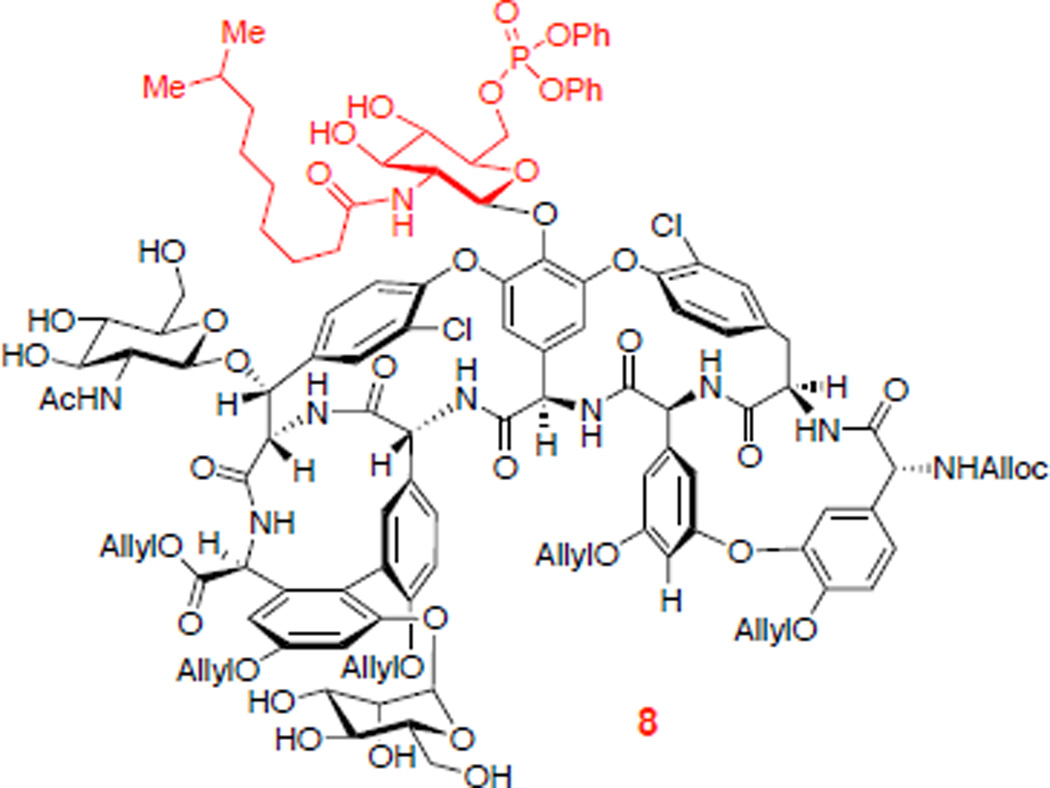
We then began to explore the phosphorylation of 3 (eq 1). When teicoplanin derivative 3 was treated with excess diphenyl phosphoryl chloride (DPCP, 6 equiv) and 1,2,2,6,6-pentamethylpiperidine (PEMP, 8 equiv) in the absence of nucleophilic catalyst, no reaction occurred (Table 1, entry 1). On the other hand, in the presence of N-methylimidazole (NMI, 1 equiv), DPCP (6 equiv), and PEMP (8 equiv) partial conversion to various mono- and bisphosphorylated products was observed (Table 1, entry 2). We could identify three different teicoplanin derivatives, each monophosphorylated at the primary hydroxyl groups of the N-decanoylglucosamine, N-acetylglucosamine, and mannose sugars (8 : 10 : 9 = 2.4 : 1 : 2.2), and a bisphosphorylated product 12. A description of the structural assignments of the products is presented below (vide infra).52 In terms of catalyst-dependent selectivity, when the reaction was run in the presence of designed catalyst Cbz-Lys(Cbz)-DAla-DPmh-ONBu4 (13; Table 1, entry 3), phosphorylation occurred selectively at the primary alcohol of the “top/red” N-decanoylglucosamine, in accord with our hypothesis (Figure 3b). The reaction could be run with 10 mol% of the peptide catalyst 13 to give 42% isolated yield of the “top/red” N-decanoylglucosamine phosphorylated product 8. This result is consistent with the selective G6 thiocarbonylation of vancomycin derivative 1 using the same catalyst 13.12 The portability of peptide-based catalyst from vancomycin thiocarbonylation to teicoplanin phosphorylation implies the potential application of this catalyst to broad arrays of glycopeptides for various transformations, enabling the generation of new antibiotic derivatives.21,23,25 It is interesting to note that the HPLC traces for these catalytic runs reflect reactions that are quite clean under the conditions we have developed. The isolated yield of 42% (as opposed to a higher yield) may reflect an intrinsic loss of material during the C18 HPLC purification process, as well as formation of undetermined minor byproducts.
Table 1.
Development of a “top/red” N-decanoylglucosamine-selective phosphorylation catalyst.
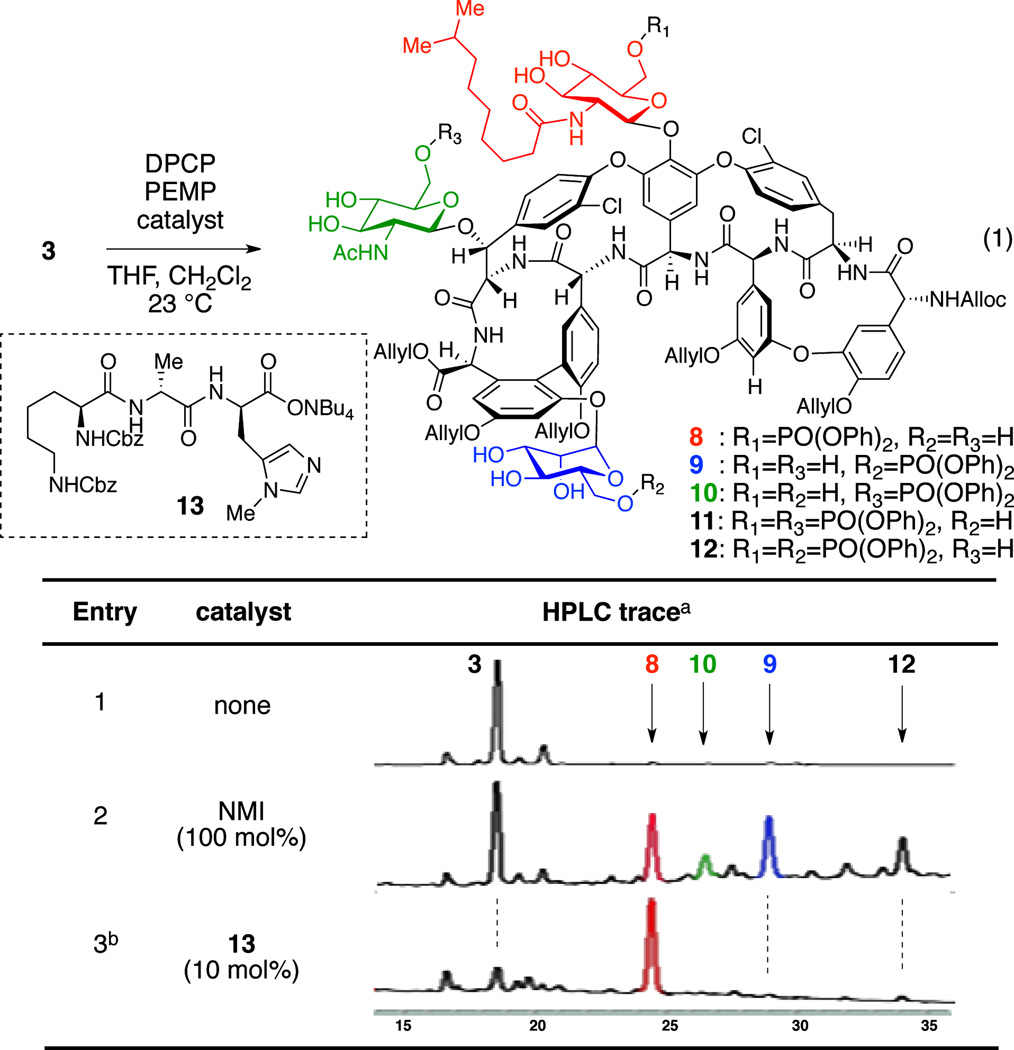 |
Reaction Conditions: DPCP (6 equiv), PEMP (8 equiv), catalyst, THF, CH2Cl2, 23 °C.
HPLC traces were recorded at 280 nm.
8 was isolated in 42% yield.
Development of the Catalyst for Selective Phosphorylation of the “Bottom/Blue” Mannose
We next sought to develop a catalyst for selective phosphorylation of the mannose ring of teicoplanin derivative 3 (eq 2). We first tested the catalyst 14 which contained DPmh in the second amino acid, therefore expected to functionalize the mannose unit of 3 (Figure 3c). Interestingly, when teicoplanin derivative 3 was treated with one equiv of peptide 14 along with excess DPCP and PEMP, mono- and bisphosphorylated products were obtained with a similar distribution ratio to that of the NMI control experiment (Table 2, entries 1 and 2). We reasoned that the DPmh caused a weaker binding between the peptide and the teicoplanin derivative 3, resulting in non-selective reaction. In fact, in 1971, Nieto and Perkins reported that the association constant of Ac-Lys(Ac)-DAla-DAla (5.9 × 105 l/mol) to structurally relevant ristocetin B is ten times larger than that of Ac-Lys(Ac)-DLeu-DAla (5.8 × 104) while it is similar to that of Ac-Lys(Ac)-DAla-DLeu (6.1 × 105).53 These historical results are consistent with the successful design of catalyst 13 (for the “top/red” sugar) and less successful catalyst 14 (designed for the “bottom/blue” sugar). Moreover, when teicoplanin derivative 3 was challenged with a mixture of catalysts 13 and 14 (13:14 = 1:1), DPCP, and PEMP only the “top/red” N-decanoylglucosamine phosphate 8 was observed (Table 2, entry 3), consistent with our rationalization.
Table 2.
Development of a “bottom/blue” mannose-selective phosphorylation catalyst.
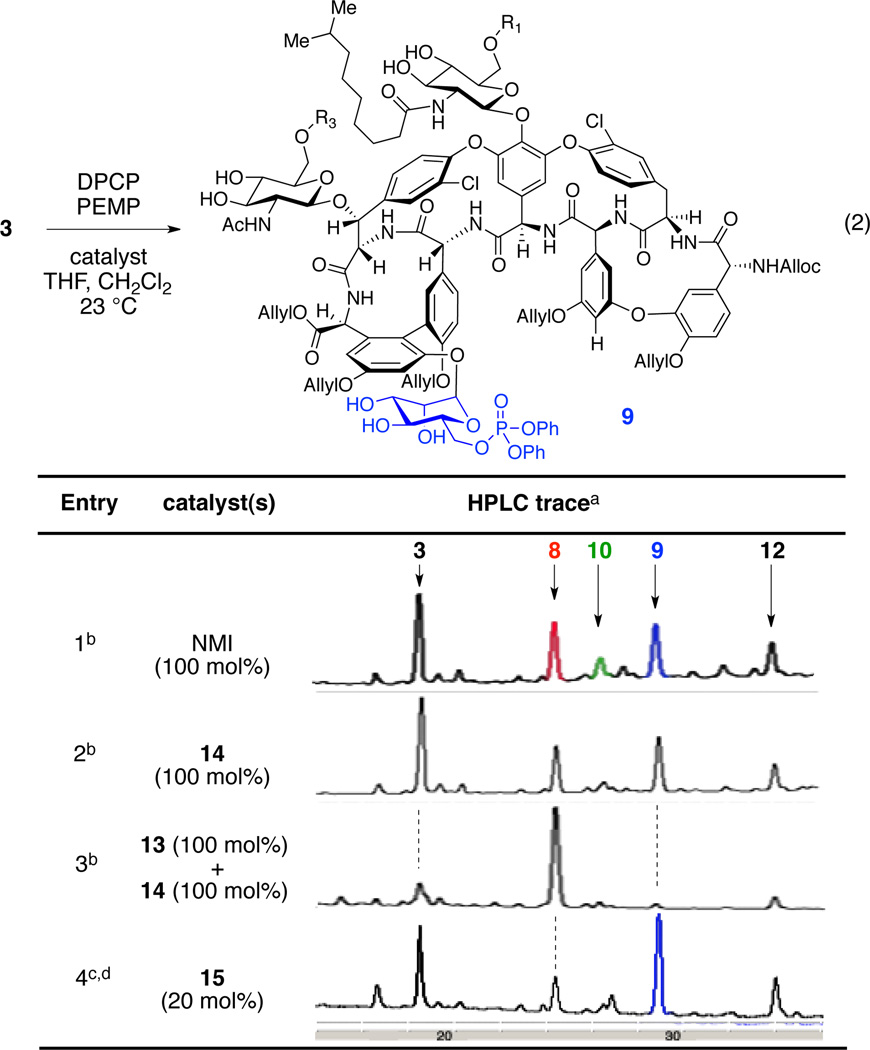 |
HPLC traces were recorded at 280 nm.
DPCP (6 equiv), PEMP (8 equiv), catalyst, THF, CH2Cl2, 23 °C.
DPCP (3 equiv), PEMP (4 equiv), catalyst, THF, CH2Cl2, 23 °C.
9 was isolated in 23% yield.
In light of these results, we performed a screen of fifteen additional catalysts available from previous projects in our group. Intriguingly, we found that peptide 15 (20 mol%) gave the desired mannose phosphorylated teicoplanin derivative 9 as a major product (23% isolated yield, Table 2, entry 4). Notably, only three equiv of DPCP were employed in this experiment, a function of the very high activity associated with catalyst 15.(Six equivalents of phosphorylating agent resulted in the formation of overphosphorylated teicoplanin derivatives.). The identification of catalyst 15 also presents philosophical questions. For example, catalyst 15 was initially reported in 2001, and later exploited for the desymmetrization of myoinositol derivatives.15,16,54–58 The fact that it emerges in the context of selective derivatization of teicoplainin analog 3 highlights its portability to a very different scaffold, suggesting that it may be “privileged” in some sense.59
Development of the Catalyst for Selective Phosphorylation of the “Left/Green” N-Acetylglucosamine
We next set our goal to develop a catalyst that can selectively phosphorylate the N-acetylglucosamine ring of teicoplanin derivative 3 (eq 3). Notably, the primary hydroxyl group of the “left/green” N-acetylglucosamine is the least reactive among those of three different sugar units present in 3 upon phosphorylation in the presence of NMI (Table 3, entry 1). The intrinsic low reactivity of the N-acetylglucosamine, coupled with its remote location to the DAla-DAla binding pocket (vide-supra), presented significant challenge regarding the development of a catalyst selective for this sugar unit. In fact, the localization of the “left/green” sugar is aptly described as on the opposite hemisphere of the teicoplanin core relative to the DAla-DAla binding site. Thus, a major alteration to our catalyst design strategy was necessary to target this site.
Table 3.
Development of a “left/green” acetylglucosamine-selective phosphorylation catalyst.
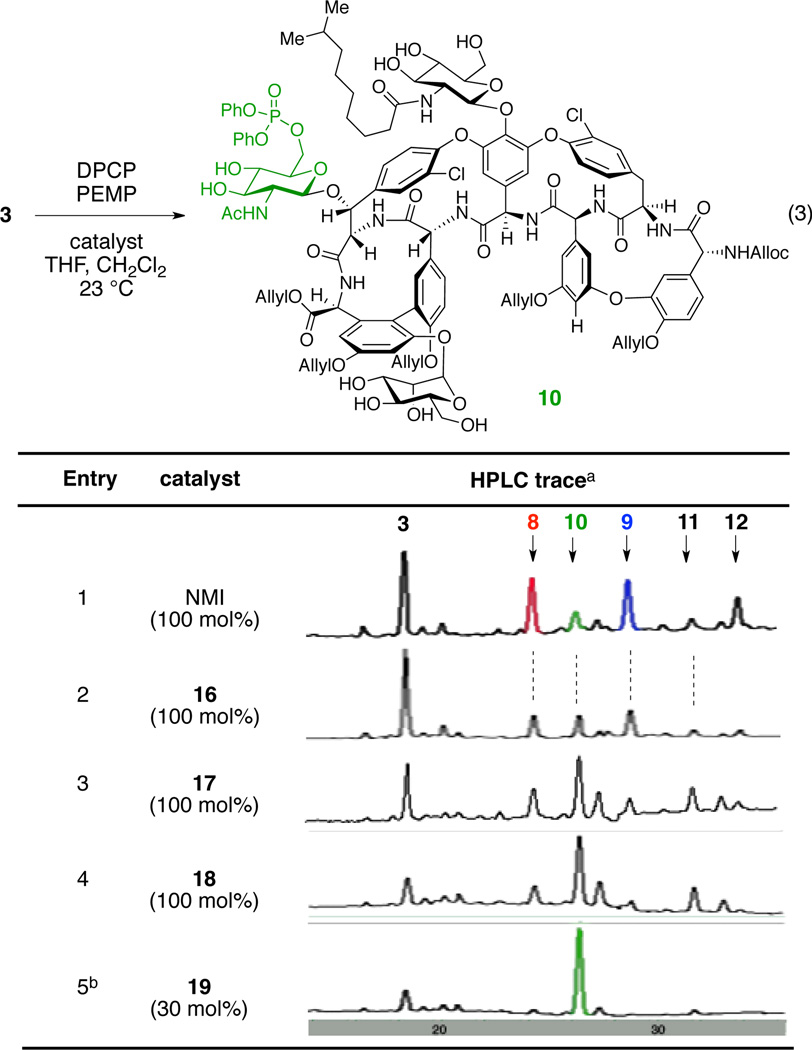 |
Reaction Conditions: DPCP (6 equiv), PEMP (8 equiv), catalyst, THF, CH2Cl2, 23 °C.
HPLC traces were recorded at 280 nm.
10 was isolated in 41% yield.
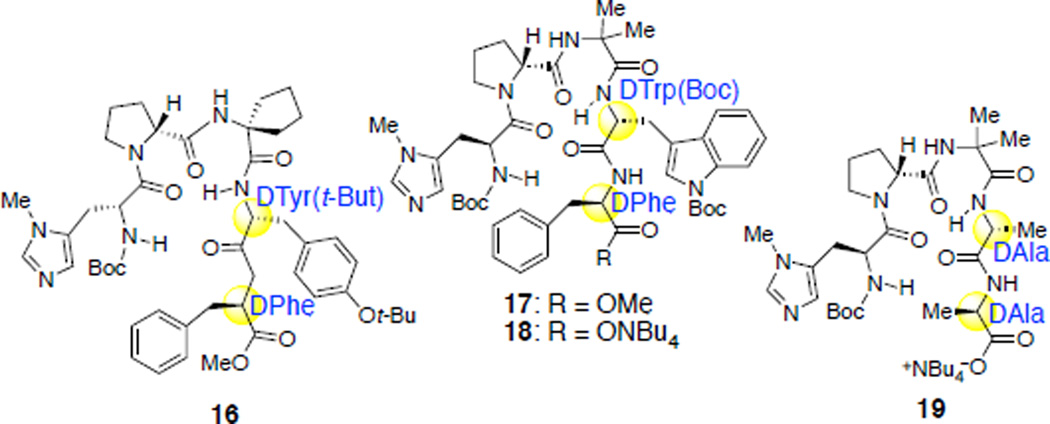
A clue emerged from our evaluation of the fifteen additional peptide catalysts noted above. In this screen, we identified catalysts 16 and 17, which showed modest increases in the extent of functionalization of the “left/green” sugar (leading to phosphorylated product 10) in comparison to NMI (Table 3, entries 1–3). In particular, peptide 16 showed moderate conversion to the top, left, and bottom phosphates 8, 10, and 9 (8:10:9 = 1:1:1.2); peptide 17 afforded N-acetylglucosamine phosphorylated teicoplanin derivative 10 as a major product, for the first time in our studies. Interestingly, both peptides contain β-turn-promoting motifs,60–63 possibly rendering structural rigidity to the catalysts via internal hydrogen bonding. Also striking, two D-amino acid residues were present at the C-terminal end of these hits (Table 3). We also noticed that peptide 18, synthetized via hydrolysis of the C-terminal methyl ester of peptide 17, showed increased selectivity and conversion to 10 (Table 3, entry 4). These observations, in conjunction with our initial peptide design plan for the left sugar (Figure 3d), stimulated us to merge these concepts in a new catalyst. These concepts culminated in the design of peptide 19, which merges both the DAla-DAla binding moiety and a β-turn-promoting motif within a unique catalyst.In accord with our design, when protected teicoplanin 3 was treated with DPCP and PEMP in the presence of 30 mol% of catalyst 19, the desired left phosphorylated teicoplanin derivative 10 was obtained with high selectivity and good conversion (41% isolated yield, Table 3, entry 5). Notably, in subsequent studies, we found that any stereochemical changes in the Pmh or DPro residues (Pmh-Pro, DPmh-DPro, or DPmh-Pro instead of Pmh-DPro) of the catalyst resulted in significantly lower reactivity and selectivity for this reaction.52 These observations reflect the importance of a stereochemical match of the catalyst to selectively phosphorylate the least reactive carbohydrate moiety, the “left/green” N-acetylglucosamine residue, within teicoplanin. In addition, they document a unique strategy for targeting hydroxyl sites that are substantially distal from the DAla-DAla binding of teicoplainin, with the capacity to reach a number of angstroms beyond the binding site itself. The merger of the DAla-DAla concept with the β-turn-promoting motif we have studied extensively previously64 thus constitutes a potentially new approach for accessing catalytic reactivity at quite remote sites.
In order to gain further insights about the importance of the DAla-DAla binding to the substrate for the catalysis, we envisioned experiments that might be conducted in the presence of competitive inhibitors for the catalysts. Thus, we ran the phosphorylation reaction of teicoplanin derivative 3 in the presence of one equivalent of Boc-Leu-DAla-DAla-OH peptide (20, eq 4), a putative competitor for the substrate-binding site. Under these conditions, when teicoplanin derivative 3 was treated with DPCP and PEMP in the presence of peptide catalyst 13, no reaction was observed (Table 4, entry 1).65 Similarly, in the presence of one equivalent of competitive Boc-Leu-DAla-DAla-OH (20) peptide, 50 mol% of left-sugar selective peptide 19 exhibited significantly lowered reactivity (Table 4, entry 2). On the other hand, catalyst 15, which does not contain a DAla-DAla based binding site, gave phosphorylated products 8 and 9, albeit in lower conversion and selectivity, perhaps due to the conformational change of the teicoplanin derivative upon binding to peptide 20 (Table 4, entry 3).66 These experiments are consistent with the targeted notions of teicoplanine/catalyst binding at the heart of our experimental designs.
Table 4.
Importance of the DAla-DAla binding for catalysts 13 and 19.
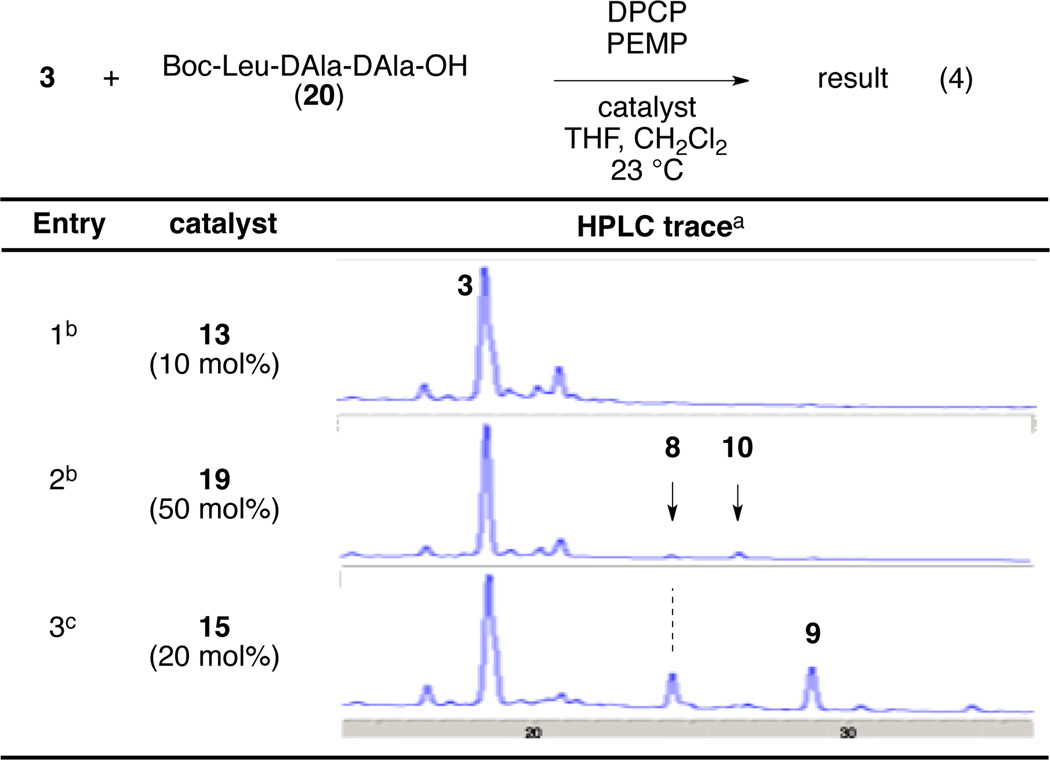 |
HPLC traces were recorded at 280 nm.
DPCP (6 equiv), PEMP (8 equiv), 20 (1equiv), catalyst, THF, CH2Cl2, 23 °C.
DPCP (4.0 equiv), PEMP (5.3 equiv), 20 (1 equiv), catalyst, THF, CH2Cl2, 23 °C.
Deprotection of the allyl/alloc Protecting Groups and Structural and Biological Characterization
With efficient synthetic access to three monophosphorylated teicoplanin derivatives 8, 9, and 10, deprotection conditions of the alloc and allyl groups were investigated. Palladium-mediated reduction using phenylsilane as reductant, conditions previously used in our group for the vancomycin derivatives deprotection,12 cleanly afforded the alloc and allyl deprotected products 21, 22, and 23. Straightforward reverse phase HPLC isolations resulted in yields of 53, 49 and 55%, respectively (Scheme 2). Structural assignments of phosphorylated teicoplanin derivatives were realized using mass spectrometry (MS) and NMR spectroscopy. ESI MS/MS data of 21, 22, and 23 provided distinct fragmentation patterns diagnostic of loss of each of the sugar moieties, which enabled the assessment of the site of phosphorylation (Figure 4). Furthermore, HSQC, HMBC, COSY, NOESY, and TOCSY data enabled unambiguous assignments of all proton and carbon chemical shifts of the sugar regions of 21, 22, and 23. Figure 4 shows the HSQC overlay of the sugar regions of each phosphorylated teicoplanin (grey) with teicoplanin A2-2 (2, colored) (Figure 4).52 In all cases, the biggest chemical shift difference was that of the methylene (Y6,6’, M6,6’, E6,6’) protons and carbons consistent with the phosphorylation at the primary hydroxyl groups.
Scheme 2.

Deprotection of the alloc and allyl protecting groups.
Figure 4.

Structural data for 21, 22, and 23. (a) ESI MS/MS data and overlay of HSQC spectrum (sugar region) of 21 (grey) with 2 (colored). (b) ESI MS/MS data and overlay of HSQC spectrum (sugar region) of 22 (grey) with 2 (colored). (c) ESI MS/MS data and overlay of HSQC spectrum (sugar region) of 23 (grey) with 2 (colored).
As a mode of biological characterization, we evaluated each of the new diphenylphosphoryl derivatives of teicoplanin A2-2 for their antibacterial activities (Table 4). The newly synthesized analogs 21, 22, and 23 showed attenuated activities in four of the standard strains that were examined (methicillin-susceptible Staphylococcus aureus (MSSA), methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-susceptible Enterococcus (VanS), and vancomycin-resistant Enterococcus VanB (VanB)) and somewhat improved activity against vancomycin-resistant Enterococcus VanA (VanA) in comparison to teicoplanin itself. It is conceivable that the site-selective functionalization capability we have established in this study may allow for design of new variants that will allow for a high precision SAR to be derived.
Conclusions
In summary, we identified three different peptide catalysts that selectively phosphorylate three distinct sugar moieties present in teicoplanin A2-2 (2). Rational design of catalysts based on the X-ray crystal structure and modeling enabled the identification of peptide 13, which selectively phosphorylated the “top/red” N-decanoylglucosamine moiety. However, our designs for the other sugar sites required editing based on experimental information, and new insights acquired through evaluation of fifteen additional, diverse catalysts. From this set, we identified peptide 15, which led to substantial selectivity for the “bottom/blue” phosphorylated teicoplanin 9. Finally through rational optimization of the initial hits from this screen, we conceived and synthesized peptide 13, an embodiment of both a well-ordered β-turn unit and DAla-DAla targeting moiety. This catalyst enabled the selective phosphorylation of the least reactive “left/green” sugar of teicoplanin A2-2 derivative 3. Notably, the new catalysts are considerably smaller than the substrates upon which they operate, an unusual situation for many classes of catalysts, including enzymes and many small molecule catalysts based on metal complexes and organo-catalysts. The chemical tools presented here expand our understanding of site-selective catalysis on complex chemical scaffolds. These results illustrate a possible step forward for the development of synthetic catalysts that operate distinctively and selectively over quite long dimensions. As noted, the hydroxyl groups we have been able to target in these studies are separated by as much as 17.7 Å in a teicoplanin crystal structure,46 which is a dimension characteristic of structural subunits in proteins.67
Supplementary Material
Scheme 1.

Synthesis of N-alloc-O-pentaallyl-teicoplanin (3). Reagents and conditions: a. N-(allyloxycarbonyloxy)succinimide, NaHCO3, H2O, 1,4-dioxane, 23 °C (89%). b. allylbromide, Cs2CO3, DMF, 50 °C (50%).
Table 5.
MIC data for new phosphorylated teicoplanin analogues 21–23.
| Compound |
S. aureus MSSAa |
S. aureus MSSAb |
E. faecalis VanSc |
E. faecalis VanSd |
E. faecalis VanSe |
|---|---|---|---|---|---|
| 21 | 4 | 4 | 1 | 4 | 32 |
| 22 | 8 | 8 | 1 | 8 | 32 |
| 23 | 4 | 4 | 0.5 | 4 | 62 |
| vancomycin | 1 | 1 | 2 | 16 | >64 |
| teicoplanin | 0.5 | 0.5 | 0.25 | 0.25 | >64 |
| Linexolid | 4 | 4 | 2 | 2 | 2 |
MIC values reported in µg/mL
MSSA: methicillin-susceptible S. aureus
MRSA: methicillin-resistant S. aureus
VanS: vancomycin-susceptoble
VanB: VanB resistance genotype/phenotype
VanA: VanA resistance genotype/phenotype
ACKNOWLEDGMENT
Funding Sources
We are grateful to the National Institutes of General Medical Sciences of the National Institute of Health (GM-068649) for support. We thank Mr. Seth Alexander and the Schepartz laboratory for assistance.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Description of the material included. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
REFERENCES
- 1.Riva S. Curr. Opin. Chem. Biol. 2001;5:106. doi: 10.1016/s1367-5931(00)00178-2. [DOI] [PubMed] [Google Scholar]
- 2.Gonzalez-Sabin J, Moran-Ramallal R, Rebolledo F. Chem. Soc. Rev. 2011;40:5321. doi: 10.1039/c1cs15081b. [DOI] [PubMed] [Google Scholar]
- 3.Chang S, Lee NH, Jacobsen EN. J. Org. Chem. 1993;58:6939. [Google Scholar]
- 4.Lee SH, Cheong CS. Tetrahedron. 2001;57:4801. [Google Scholar]
- 5.Bastian AA, Marcozzi A, Herrmann A. Nat. Chem. 2012;4:789. doi: 10.1038/nchem.1402. [DOI] [PubMed] [Google Scholar]
- 6.Lewis CA, Miller SJ. Angew. Chem. Int. Ed. 2006;45:5616. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]
- 7.Holland JM, Lewis M, Nelson A. Angew. Chem. Int. Ed. 2001;40:4082. [PubMed] [Google Scholar]
- 8.García-Urdiales E, Alfonso I, Gotor V. Chem. Rev. 2004;105:313. doi: 10.1021/cr040640a. [DOI] [PubMed] [Google Scholar]
- 9.Atodiresei I, Schiffers I, Bolm C. Chem. Rev. 2007;107:5683. doi: 10.1021/cr068369f. [DOI] [PubMed] [Google Scholar]
- 10.Lewis CA, Longcore KE, Miller SJ, Wender PA. J. Nat. Prod. 2009;72:1864. doi: 10.1021/np9004932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pathak TP, Miller SJ. J. Am. Chem. Soc. 2012;134:6120. doi: 10.1021/ja301566t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fowler BS, Laemmerhold KM, Miller SJ. J. Am. Chem. Soc. 2012;134:9755. doi: 10.1021/ja302692j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pathak TP, Miller SJ. J. Am. Chem. Soc. 2013;135:8415. doi: 10.1021/ja4038998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For an X-ray crystal structure of myo-inositol. Rabinovich IN, Kraut J. Act. Cryst. 1964;17:159. [Google Scholar]
- 15.Sculimbrene BR, Miller SJ. J. Am. Chem. Soc. 2001;123:10125. doi: 10.1021/ja016779+. [DOI] [PubMed] [Google Scholar]
- 16.Sculimbrene BR, Morgan AJ, Miller SJ. J. Am. Chem. Soc. 2002;124:11653. doi: 10.1021/ja027402m. [DOI] [PubMed] [Google Scholar]
- 17.Lewis CA, Chiu A, Kubryk M, Balsells J, Pollard D, Esser CK, Murry J, Reamer RA, Hansen KB, Miller SJ. J. Am. Chem. Soc. 2006;128:16454. doi: 10.1021/ja067840j. [DOI] [PubMed] [Google Scholar]
- 18.Lewis CA, Gustafson JL, Chiu A, Balsells J, Pollard D, Murry J, Reamer RA, Hansen KB, Miller SJ. J. Am. Chem. Soc. 2008;130:16358. doi: 10.1021/ja807120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan H-C, Huang Y-T, Lyu S-Y, Huang C-J, Li Y-S, Liu Y-C, Chou C-C, Tsai M-D, Li T-L. Mol. Biosys. 2011;7:1224. doi: 10.1039/c0mb00320d. [DOI] [PubMed] [Google Scholar]
- 20.Commercially available teicoplanin exists as a mixture of natural products with varying alkyl chain of the decanoylglucosamine ring. This study is conducted with purifyed teicoplanin A2-2 (2). See supporting information for details.
- 21.Bambeke F, Laethem Y, Courvalin P, Tulkens P. Drugs. 2004;64:913. doi: 10.2165/00003495-200464090-00001. [DOI] [PubMed] [Google Scholar]
- 22.Kahne D, Leimkuhler C, Lu W, Walsh C. Chem. Rev. 2005;105:425. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 23.Jeya M, Moon H-J, Lee K-M, Kim I-W, Lee J-K. Curr. Pharm. Biotechnol. 2011;12:1194. doi: 10.2174/138920111796117382. [DOI] [PubMed] [Google Scholar]
- 24.Li T-L, Liu Y-C, Lyu S-Y. Curr. Opin. Chem. Biol. 2012;16:170. doi: 10.1016/j.cbpa.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 25.Ashford P-A, Bew SP. Chem. Soc. Rev. 2012;41:957. doi: 10.1039/c1cs15125h. [DOI] [PubMed] [Google Scholar]
- 26.Evans DA, Wood MR, Trotter BW, Richardson TI, Barrow JC, Katz JL. Angew. Chem. Int. Ed. 1998;37:2700. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2700::AID-ANIE2700>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 27.Nicolaou KC, Mitchell HJ, Jain NF, Winssinger N, Hughes R, Bando T. Angew. Chem. Int. Ed. 1999;38:240. [Google Scholar]
- 28.Boger DL, Miyazaki S, Kim SH, Wu JH, Castle SL, Loiseleur O, Jin Q. J. Am. Chem. Soc. 1999;121:10004. [Google Scholar]
- 29.Boger DL, Kim SH, Miyazaki S, Strittmatter H, Weng J-H, Mori Y, Rogel O, Castle SL, McAtee JJ. J. Am. Chem. Soc. 2000;122:7416. [Google Scholar]
- 30.Boger DL, Kim SH, Mori Y, Weng J-H, Rogel O, Castle SL, McAtee JJ. J. Am. Chem. Soc. 2001;123:1862. doi: 10.1021/ja003835i. [DOI] [PubMed] [Google Scholar]
- 31.Evans DA, Katz JL, Peterson GS, Hintermann T. J Am Chem Soc. 2001;123:12411. doi: 10.1021/ja011943e. [DOI] [PubMed] [Google Scholar]
- 32.Xie J, Okano A, Pierce JG, James RC, Stamm S, Crane CM, Boger DL. J. Am. Chem. Soc. 2011;134:1284. doi: 10.1021/ja209937s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi Z, Griffin JH. J. Am. Chem. Soc. 1993;115:6482. [Google Scholar]
- 34.Nicolaou KC, Winssinger N, Hughes R, Smethurst C, Cho SY. Angew. Chem. Int. Ed. 2000;39:1084. doi: 10.1002/(sici)1521-3773(20000317)39:6<1084::aid-anie1084>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 35.Pace JL, Yang G. Biochem. Pharmacol. 2006;71:968. doi: 10.1016/j.bcp.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 36.Bischoff D, Pelzer S, Höltzel A, Nicholson GJ, Stockert S, Wohlleben W, Jung G, Süssmuth RD. Angew. Chem. Int. Ed. 2001;40:1693. doi: 10.1002/1521-3773(20010504)40:9<1693::aid-anie16930>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 37.Bischoff D, Pelzer S, Bister B, Nicholson GJ, Stockert S, Schirle M, Wohlleben W, Jung G, Sussmuth RD. Angew. Chem. Int. Ed. 2001;40:4688. doi: 10.1002/1521-3773(20011217)40:24<4688::aid-anie4688>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 38.Losey HC, Peczuh MW, Chen Z, Eggert US, Dong SD, Pelczer I, Kahne D, Walsh CT. Biochemistry. 2001;40:4745. doi: 10.1021/bi010050w. [DOI] [PubMed] [Google Scholar]
- 39.Losey HC, Jiang J, Biggins JB, Oberthür M, Ye X-Y, Dong SD, Kahne D, Thorson JS, Walsh CT. Chem. Biol. 2002;9:1305. doi: 10.1016/s1074-5521(02)00270-3. [DOI] [PubMed] [Google Scholar]
- 40.Bister B, Bischoff D, Nicholson GJ, Stockert S, Wink J, Brunati C, Donadio S, Pelzer S, Wohlleben W, Sussmuth RD. ChemBioChem. 2003;4:658. doi: 10.1002/cbic.200300619. [DOI] [PubMed] [Google Scholar]
- 41.Schmit JL. Clin. Infect. Dis. 1992;15:302. doi: 10.1093/clinids/15.2.302. [DOI] [PubMed] [Google Scholar]
- 42.Mainardi J-L, Shlaes DM, Goering RV, Acar JF, Goldstein FW. J. Infect. Dis. 1995;171:1646. doi: 10.1093/infdis/171.6.1646. [DOI] [PubMed] [Google Scholar]
- 43.Howden BP, Davies JK, Johnson PDR, Stinear TP, Grayson ML. Clin. Microbiol. Rev. 2010;23:99. doi: 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Higgins DL, Chang R, Debabov DV, Leung J, Wu T, Krause KM, Sandvik E, Hubbard JM, Kaniga K, Schmidt DE, Jr, Gao Q, Cass RT, Karr DE, Benton BM, Humphrey PP. Antimicrob. Agents Chemother. 2005;49:1127. doi: 10.1128/AAC.49.3.1127-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perkins HR. Pharmacol. Ther. 1982;16:181. doi: 10.1016/0163-7258(82)90053-5. [DOI] [PubMed] [Google Scholar]
- 46.Economou NJ, Zentner IJ, Lazo E, Jakoncic J, Stojanoff V, Weeks SD, Grasty KC, Cocklin S, Loll PJ. Act. Crys. 2013;D69:520. [Google Scholar]
- 47.Fierman MB, O’Leary DJ, Steinmetz WE, Miller SJ. J. Am. Chem. Soc. 2004;126:6967. doi: 10.1021/ja049661c. [DOI] [PubMed] [Google Scholar]
- 48.Evans JW, Fierman MB, Miller SJ, Ellman JA. J. Am. Chem. Soc. 2004;126:8134. doi: 10.1021/ja047845l. [DOI] [PubMed] [Google Scholar]
- 49.Endicott JA, Noble MEM, Johnson LN. Annu. Rev. Biochem. 2012;81:587. doi: 10.1146/annurev-biochem-052410-090317. [DOI] [PubMed] [Google Scholar]
- 50.Thompson C, Ge M, Kahne D. J. Am. Chem. Soc. 1999;121:1237. [Google Scholar]
- 51.Griffith BR, Krepel C, Fu X, Blanchard S, Ahmed A, Edmiston CE, Thorson JS. J. Am. Chem. Soc. 2007;129:8150. doi: 10.1021/ja068602r. [DOI] [PubMed] [Google Scholar]
- 52.See supporting information for details.
- 53.Nieto M, Perkins HR. Biochem. J. 1971;124:845. doi: 10.1042/bj1240845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sculimbrene BR, Morgan AJ, Miller SJ. Chemical Communications. 2003;0:1781. doi: 10.1039/b304015c. [DOI] [PubMed] [Google Scholar]
- 55.Sculimbrene BR, Xu Y, Miller SJ. J. Am. Chem. Soc. 2004;126:13182. doi: 10.1021/ja0466098. [DOI] [PubMed] [Google Scholar]
- 56.Morgan AJ, Wang YK, Roberts MF, Miller SJ. J. Am. Chem. Soc. 2004;126:15370. doi: 10.1021/ja047360x. [DOI] [PubMed] [Google Scholar]
- 57.Xu Y, Sculimbrene BR, Miller SJ. J. Org. Chem. 2006;71:4919. doi: 10.1021/jo060702s. [DOI] [PubMed] [Google Scholar]
- 58.Morgan AJ, Komiya S, Xu Y, Miller SJ. J. Org. Chem. 2006;71:6923. doi: 10.1021/jo0610816. [DOI] [PubMed] [Google Scholar]
- 59.Yoon TP, Jacobsen EN. Science. 2003;299:1691. doi: 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]
- 60.Awasthi SK, Raghothama S, Balaram P. Biochem. Biophys. Res. Comm. 1995;216:375. doi: 10.1006/bbrc.1995.2634. [DOI] [PubMed] [Google Scholar]
- 61.Haque TS, Little JC, Gellman SH. J. Am. Chem. Soc. 1996;118:6975. [Google Scholar]
- 62.Stanger HE, Gellman SH. J. Am. Chem. Soc. 1998;120:4236. [Google Scholar]
- 63.Fisk JD, Powell DR, Gellman SH. J. Am. Chem. Soc. 2000;122:5443. [Google Scholar]
- 64.Jarvo ER, Copeland GT, Papaioannou N, Bonitatebus PJ, Miller SJ. J. Am. Chem. Soc. 1999;121:11638. [Google Scholar]
- 65.For an example of the use of competitive inhibitor to test the specific binding. Wang H, Koshi Y, Minato D, Nonaka H, Kiyonaka S, Mori Y, Tsukiji S, Hamachi I. J. Am. Chem. Soc. 2011;133:12220. doi: 10.1021/ja204422r. [DOI] [PubMed] [Google Scholar]
- 66.Note the conformational difference between DAla-DAla peptide bound teicoplanin derivative (Figure 3a) and free teicoplanin (Figure 1d).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.