

Abstract
The encapsulation of anticancer drugs in a liposome structure protects the drug during circulation and increases drug accumulation in the cancer tissue and antitumor activity while decreasing drug toxicity. This paper presents a new method of active drug loading based on a vitamin C pH/ion gradient. Formulations were characterized in terms of the following parameters: optimal external pH, time and drug-to-lipid ratio for the purpose of remote loading, and in vitro stability. In the case of the selected drug, epirubicin (EPI), its coencapsulation increases its anticancer activity through a possibly synergistic effect previously reported by other groups for a free nonencapsulated drug/vitamin C cocktail. The method also has another advantage over other remote-loading methods: it allows faster drug release through liposome destabilization at the tumor site, thanks to the very good solubility of the EPI vitamin C salt, as seen on cryogenic transmission electron microscopy images. This influences the drug-release process and increases the anticancer activity of the liposome formulation. The liposomes are characterized as stable, with very good pharmacokinetics (half-life 18.6 hours). The antitumor activity toward MCF-7 and 4T-1 breast cancer cells was higher in the case of EPI loaded via our gradient than via an ammonium sulfate gradient. Finally, the EPI liposomal formulation and the free drug were tested using the murine 4T-1 breast cancer model. The antitumor activity of the encapsulated drug was confirmed (tumor-growth inhibition over 40% from day 16 until the end of the experiment), and the free drug was shown to have no anticancer activity at the tested dose.
Keywords: liposomes, epirubicin, vitamin C, antitumor activity, remote loading, ascorbic acid
Introduction
Ascorbic acid (vitamin C) is a water-soluble vitamin with two ionizable groups (pKa: 4.2 and 11.6).1 It is a strong reducing agent that maintains iron in its ferrous state, making the catalysis of various biochemical processes by ferrous iron possible.2 This oxidizing ability of vitamin C, besides playing a role in many physiological functions, is probably responsible for its noteworthy property of selective toxicity against cancer cells.3–5 What should also be taken into account is that vitamin C not only shows cytotoxicity against cancer cells but also improves the antineoplastic activity of some anticancer drugs, in particular anthracyclines, because one of their cytotoxic activities is related to free radical formation. It was demonstrated that doxorubicin (DOX) and vitamin C exhibit synergistic activity against the breast cancer lines over a wide range of vitamin C concentrations.6 In a case study, Vollbracht et al7 showed that intravenous application of vitamin C improved the quality of life of breast cancer patients during radio- and chemotherapy. All these data might suggest that using a vitamin C gradient for remote loading of anthracyclines can be recommended, as it should improve the cytotoxic activity of the formulation compared to other liposomal formulations of anthracyclines.
In our research, we used epirubicin (EPI), which is an antineoplastic agent belonging to the group of second-generation anthracycline antibiotics. Like all cytostatic antibiotics, EPI has multiple mechanisms of action. EPI binds to DNA, bursts it, and stabilizes the fragmentation process of double-stranded DNA. In addition, EPI inhibits the processes of DNA and RNA synthesis by inhibiting topoisomerase II, disrupts the function of the cell membrane, and is responsible for the formation of free radicals.8 The latter property is also responsible for the cardiotoxic effects of EPI.9 It is used in the treatment of breast, ovarian, bladder, lung, bronchus, pancreatic, stomach, esophagus, and liver cancers, soft-tissue sarcoma, and cancers of the lymphatic system.10 As with any use of anthracyclines, side effects may occur when using EPI. These include damage to the myocardium (heart failure, arrhythmia) and bone marrow, gastrointestinal damage (vomiting, diarrhea, and nausea), hepatitis, blood vessel inflammation at the site of injection, mouth inflammation, fever, or hair loss. EPI is given as an intravenous infusion once every 3 weeks at a dose of 60–90 mg/m2 of body surface area. EPI is mainly excreted in bile by the liver and in smaller quantities by the kidneys in urine as unaltered drug and as epirubicinol, which is the oxidized form. EPI has a three-stage biological half-life (t½) as follows: 5 minutes, 1 hour, and 20–40 hours.11
Liposomes are one of the leading intravenous drug-delivery systems. As found for DOX (Doxil) and daunorubicin (DaunoXome), using a liposomal formulation of EPI is expected to improve its efficacy further.12–14 However, only a few liposomal formulations of EPI have been proposed.10,15,16
In this study, we decided to use a novel method applying a vitamin C gradient for active EPI loading. This gives the opportunity to transfer both the drug and vitamin C at the molar concentration required for its activity; it was documented in the case of other anticancer substances that they required specific molar combinations for synergistic activity to come into effect.17,18 As we mentioned before, vitamin C exhibits certain cytotoxic activity and improves the cytotoxicity of DOX, so by using a vitamin C gradient, we intended to improve the cytotoxicity of the liposomal formulation of EPI and possibly discover evidence to show that the method could also be used for other anthracyclines.
Using the ammonium salt of vitamin C for pH/ion-gradient formation resulted in stable and efficient EPI encapsulation in a Doxil-like, long-circulating liposomal formulation. Compared to idarubicin, EPI is much more water-soluble, so there is no need to ensure that EPI forms a complex of very low solubility in the liposomes for the purposes of drug stability.19 The cryogenic transmission electron microscopy (cryo-TEM) images of the resulting liposomal formulation showed that the drug is precipitated in the form of diffused fibrous structures and not the dense bundles observed in the case of other anthracycline liposomal formulations obtained using the ammonium sulfate or citrate methods.16,20 The vitamin C EPI salt solubility at pH 4 is very high: more than 10 mg/mL (0.23 mg/mL for EPI sulfate). This suggests that an essential part of the drug will be entrapped in a soluble, nonprecipitated form, which is our ultimate goal in the case of EPI or DOX formulations. This means a rather fast drug release at the tumor site and a further increase in drug activity due to hypothetical vitamin C synergism. Additionally, to maintain the drug’s anticancer activity, only its monomeric form is active. Barenholz reports that for DOX sulfate, drug polymers are formed for salt concentrations as low as 1 μM.21 This means a low free drug-molecule population at the cancer site during liposome destabilization. In the case of EPI vitamin C salt, a much higher concentration seems to be needed for drug polymerization, so an increased amount of free drug molecules can reach the cancer cell nucleus. Since our pharmacokinetic studies confirmed very good blood drug circulation of the vitamin C-loaded EPI formulation, at least in this case, the partially soluble drug state in the liposomes does not affect its rapid release into the circulation.22 We also showed that a liposomal formulation of EPI based on a vitamin C gradient improved the drug’s in vitro anticancer activity against the MCF-7 and 4T-1 breast cancer cell lines. Furthermore, it showed activity against the murine 4T-1 EPI-resistant tumor model in vivo, whereas the free drug was not active at the used dose.
Materials and methods
Materials
Hydrogenated soy phosphatidylcholine (HSPC), 1,2-dis tearoyl-sn-glycero-phosphoethanolamine-N-(poly[ethylen e glycol]2000) (DSPE-PEG 2000) and cholesterol (Chol) were purchased from Northern Lipids (Vancouver, BC, Canada). Sodium dihydrogen phosphate, disodium hydrogen phosphate, sodium chloride, ascorbic acid, and ammonium hydroxide were purchased from POCH (Gliwice, Poland). Sephadex G-50 fine and Sepharose CL-4B were obtained from Sigma-Aldrich (Poznan, Poland). EPI hydrochloride was donated by the Pharmaceutical Research Institute (Warsaw, Poland).
Preparation of liposomes
HSPC/Chol/DSPE-PEG 2000 (5.5:4:0.5 molar ratio) liposomes were prepared using the extrusion method. Briefly, 30 mg of lipids dissolved in cyclohexane were mixed together and frozen in liquid nitrogen. The sample was then freeze-dried overnight at low pressure using a Savant Modulyo apparatus (Thermo Savant, Inc., Holbrook, NY, USA). Multilamellar vesicles were formed by hydrating the lipid film with 1 mL of 300 mM ascorbic acid (pH 2.4) or 2 mL of 300 mM ammonium ascorbate (pH 4.0) at 64°C, followed by seven freeze–thaw cycles. Additionally, for the in vitro plasma-stability test and in vitro cytotoxicity experiments, liposomes containing an internal solution of 300 mM ammonium sulfate (pH 5.5) and citric buffer (pH 4.0) were prepared. Large unilamellar vesicles were prepared by extrusion through Nucleopore polycarbonate filters (Whatman, Maidstone, UK) with a pore size of 100 nm (ten times) on a Thermobarrel Extruder (PPHU Marker, Wrocław, Poland). The extruder was heated to a temperature of 64°C prior to liposome extrusion.
Preparation of the ion/pH gradient for drug encapsulation
The ion and/or pH gradient was subsequently generated by exchanging the extravesicular liposomal solution on Sephadex G-50 (1 × 20 cm) columns equilibrated with phosphate-buffered saline (PBS; 20 mM sodium phosphate, 150 mM NaCl) of various pH. Buffers of pH 5.5, 6.5, 7.5, and 8.5 were used in the experiments to determine the influence of the external pH on EPI encapsulation. For other experiments, PBS of pH 7.5 was used. In the collected liposomal fractions, the lipid concentration was then determined. EPI hydrochloride solution in 150 mM NaCl (6 mg/mL) was added to the large-unilamellar-vesicle suspension at drug-to-lipid ratios (w/w) of 1:2, 1:3, 1:4, and 1:5 to determine the encapsulation efficiency (EE) at different ratios. In all other experiments, a 1:5 drug-to-lipid ratio was used. The loading process was carried out at 60°C for 15 minutes. To determine the kinetics of drug encapsulation, 50 μL liposomal samples were taken after loading for 2.5, 5, 15, 30, or 60 minutes.
Liposome characterization
The mean diameter, polydispersity index, and zeta potential of the prepared liposomes was measured using a Zetasizer Nano-ZS (Malvern Instruments, Malvern, UK). The lipid concentration was measured using the ammonium ferrothiocyanate assay23 on a UV-2401 PC spectrophotometer (Shimadzu, Kyoto, Japan). The EPI concentration was measured with a high-performance liquid chromatography (HPLC) Waters (Milford MA, USA) 660 pump, an XTerra RP18 (250 × 4.6 mm, 5 μm; Waters) column, and a water/acetonitrile/tetrahydrofuran/H3PO4/triethylamine (312:165:20:1:2, v/v) mobile phase with a flow rate of 1 mL/minute. The detection was done using a Waters 474 Scanning Fluorescence Detector at λ ex 482 nm and λ em 542 nm. A Waters Millennium Version 3.20 processing module was used to record and process the chromatograms. The relative amount of EPI was calculated from the appropriate calibration curve in the range of 0.25–20 μg/mL. The physicochemical parameters of the liposomes used for the study are summarized in Table 1.
Table 1.
Physicochemical parameters of the liposomes used for the study, initially and after 360 days of storage at 4°C
| Liposome composition | Salt gradient | Size before drug loading (nm) | Size after drug loading (nm) | PDI | Size after 360 days of storage (nm) | PDI after 360 days of storage |
|---|---|---|---|---|---|---|
| HSPC/Chol/DSPE-PEG 2000 5.5:4:0.5 |
Ammonium ascorbate | 112 ± 3 | 115 ± 3 | 0.097 ± 0.011 | 117 ± 12 | 0.112 ± 0.023 |
| HSPC/Chol/DSPE-PEG 2000 5.5:4:0.5 |
Ascorbic acid | 110 ± 4 | 118 ± 5 | 0.066 ± 0.023 | 122 ± 14 | 0.144 ± 0.034 |
Abbreviations: HSPC/Chol/DSPE-PEG 2000, hydrogenated soy phosphatidylcholine/cholesterol/1,2-distearoyl-sn-glycero-phosphoethanolamine-N-(poly[ethylene glycol]2000); PDI, polydispersity index.
Determination of encapsulation efficiency
The nonencapsulated drug was removed from the EPI-containing liposomes using size-exclusion chromatography on a Sephadex G-50 minicolumn (5.5 × 70 mm) equilibrated with PBS. The 50 μL liposome samples were placed on a column, and free EPI was separated from EPI-containing liposomes. Then, the liposome fraction was collected, and the concentration of lipid and EPI was determined to calculate the EE.
Liposomal release of EPI in the presence of human plasma in vitro
EPI was encapsulated in HSPC/Chol/DSPE-PEG 2000 (mol/mol 5.5:4:0.5) liposomes at a 1:5 drug-to-lipid ratio (w/w) using the following gradients: 300 mM ammonium sulfate (pH 5.5), citrate buffer (pH 4.0), 300 mM ascorbic acid (pH 2.4), and 300 mM ammonium ascorbate (pH 4.0). The liposomal samples were diluted to obtain a total lipid concentration of 4 mM. Two milliliters of each liposomal suspension was then mixed with 2 mL of fresh human plasma to a final lipid concentration of 2 mM. The liposomes were incubated at 37°C for 24 hours. At 0, 1, 3, 6, and 24 hours, 100 μL samples were placed on Sepharose CL 4B minicolumns to separate the released drug from liposomes. Then, the drug and lipid contents were measured in the collected liposomal fractions and compared with the initial values.
Long-term EPI retention in liposomes
HSPC/Chol/DSPE-PEG 2000 5.5:4:0.5 (mol/mol) liposomes containing EPI (drug-to-lipid ratio 1:5 w/w) encapsulated using ascorbic acid and ammonium ascorbate gradients were used to measure long-term EPI retention. Nonencapsulated drug was subsequently removed on Sephadex G-50 minicolumns (5.5 × 70 mm) equilibrated with PBS containing 0.1% NaN3. Liposomes at a final lipid concentration of 4 mM were stored for 30 days at 37°C, 180 days at 25°C, and 360 days at 4°C. At selected time intervals, 50 μL liposomal samples were placed on Sephadex G-50 minicolumns (5.5 ×70 mm) equilibrated with PBS. The liposomal samples were collected and the drug and lipid contents were measured in the collected liposomal fractions and compared with the initial values.
Cryo-transmission electron microscopy
For the cryo-TEM experiments, EPI-loaded liposomes consisting of HSPC/Chol/DSPE-PEG 2000 (mol/mol 5.5:4:0.5) with a 1:5 drug-to-lipid ratio (w/w) were prepared. The liposomal formulations were prepared to a total lipid concentration of 7–9 mM. Copper grids (S7/2 Cu 400 mesh, holey carbon films; Quantifoil Micro Tools, Jena, Germany) were prepared according to a standard procedure. After placing a drop of the sample on the grid most of the liquid was removed with filter paper, leaving a thin film stretched over the holes. The samples were immediately shock-frozen by plunging into liquid ethane, stored at 90K in liquid nitrogen and loaded into a cryogenic sample holder (D262; Gatan, Pleasanton, CA, USA). The samples were transferred to the microscope (Leo 912 Omega; Carl Zeiss, Oberkochen, Germany) and then examined at 100K. Digital images were recorded with a slow-scan charge-coupled-device camera system (Proscan HSV 2; Oxford Instruments, Abingdon, UK) under low-dose conditions.
Breast cancer cell culture
Human breast cancer cells from the MCF-7 line (Institute of Immunology and Experimental Therapy, Wrocław, Poland) were cultured in Eagle’s minimal essential medium (Institute of Immunology and Experimental Therapy) supplemented with 10% fetal bovine serum and 2 mM glutamine (Sigma-Aldrich), 100 U/mL penicillin, 100 μg/mL streptomycin and 25 μg/mL amphotericin B (Polfa Tarchomin, Warsaw, Poland). Murine mammary cancer cell line 4T-1 (Institute of Immunology and Experimental Therapy) were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Lonza, Basel, Switzerland) supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, and 25 μg/mL amphotericin B. The cells were cultured at 37°C in a humid atmosphere saturated with 5% CO2.
Cytotoxicity measurements
Cell-growth inhibition was assessed using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) test.24 Briefly, MCF-7 and 4T-1 cells were seeded into 96-well culture plates at a density of 104 cells/well and incubated at 37°C in a humid atmosphere saturated with 5% CO2. After 24 hours, the medium was changed to serum-free medium with serial concentrations of different EPI liposomal formulations or free EPI. The concentration of EPI ranged from 0.05 to 50 μM. Empty liposomes were added to the wells as a control. At 48 and 72 hours after the medium change, the MTT test was performed. For this purpose, the medium was carefully removed, and a 50 μL solution of MTT was added to each well at a concentration of 0.5 mg/mL in culture medium without serum. Incubation was carried out for 4 hours at 37°C in an atmosphere containing 5% CO2. Then, the medium with EPI or liposomes was removed, and 50 μL of dimethyl sulfoxide was added to each well. The plate was shaken for about 10 minutes to dissolve the formazan crystals resulting from the MTT test. Then, 6.25 μL of Sørensen buffer was added to the wells. The absorbance was detected using an enzyme-linked immunosorbent assay microplate reader at a wavelength of 570 nm. This series of experiments was carried out in triplicate.
Plasma elimination of liposomal EPI
HSPC/Chol/DSPE-PEG 2000 5.5:4:0.5 (mol/mol) liposomes containing EPI (drug-to-lipid ratio 1:5 w/w) encapsulated using the ammonium ascorbate-gradient method were injected into mice (BALB/c, male, five per group, 18–22 g) via the lateral tail vein at a dose of 25.87 μmol/kg (15 mg/kg) body weight. At selected intervals (15 minutes, 2 hours, 6 hours, and 24 hours), the mice were anesthetized with isoflurane (Forane), and blood was collected from the retro-orbital sinus. The blood samples were immediately centrifuged (2,000 g, 10 minutes, 4°C) to separate the plasma and erythrocytes. For HPLC determination, 100 μL of plasma samples were frozen for 12 hours and then deproteinized with 100 μL of acetonitrile. The samples were mixed vortexed for 2 minutes, and then centrifuged (25,000 g, 5 minutes, 25°C). The supernatant was injected onto the column. The concentration of EPI was determined using HPLC analysis. Ten microliters of each sample was injected on the chromatographic column and separated at 25°C. The method was validated for liposomal EPI mixed with the human plasma and applied as described earlier with a recovery efficiency of 92%–104%. Values for the pharmacokinetic parameters of all of the formulations, including the observed parameters (area under the curve [AUC], t½, area under the first moment curve [AUMC], mean residence time, and biliary clearance [Clb]), were calculated using standard noncompartmental methods.25
Animal experiments
EPI and liposome formulations were prepared in PBS as described previously and administered to mice at a dose of 6 mg/kg of body weight. The 4T-1 murine mammary cancer cell line (Cell Culture Collection, Institute of Immunology and Experimental Therapy) was cultured in vitro in RPMI 1640 medium (Life Technologies, Carlsbad CA, USA) supplemented with 4.5 g/L glucose, 1.0 mM sodium pyruvate, and 10% fetal bovine serum (Sigma-Aldrich), and 100 U/mL penicillin and 100 μg/mL streptomycin (Polfa Tarchomin, Warsaw, Poland). The cells were cultured at 37°C in a humid atmosphere saturated with 5% CO2. The 7- to 8-week-old female BALB/c mice (weighing 18–23 g) were obtained from the Center for Experimental Medicine, Medical University of Bialystok and maintained under the standard specific pathogen free (SPF). All of the experiments were approved by the First Local Committee for Experiments with the Use of Laboratory Animals, Wrocław, Poland. Mice were implanted orthotopically (right mammary gland) with 3 × 105 4T-1 cells suspended in 50 mL of liquid Hanks medium (Institute of Immunology and Experimental Therapy). When the tumor volume reached 100 mm3, the animals were divided into four homogeneous groups with eight mice in each. Each mouse was injected intravenously once with free EPI or liposome formulations with EPI. The EPI and liposomes with EPI were administered to the mice in a single dose of 6 mg/kg body mass. The control group received saline.
The increase in tumor volume was monitored over the course of the experiment, with measurements performed every second day after the injection. The tumor volume was calculated using the formula (a2 × b)/2, where a = small tumor diameter in millimeters and b = large tumor diameter in millimeters. The inhibition of tumor growth was calculated from the following formula: tumor growth inhibition (TGI; %) = WT/WC × 100 − 100%, where WT is the median tumor volume found for the treated mice and WC is that of the untreated control animals. The average body-weight change (BWC) in all of the groups was calculated using the formula BWC = (ABWn/ABW1) × 100 − 100%, where ABWn is the average body weight on the nth day of the experiment (during treatment), and ABW1 is the average body weight on the first day of treatment.
Statistical evaluation
Statistical analysis was performed with Statistica version 7.1 (StatSoft, Tulsa, OK, USA). For the tumor growth-inhibition analysis, the Kruskal-Wallis analysis of variance multiple-comparison P-value (two-tailed) test was used. Differences were considered statistically significant for P-values lower than 0.05.
Results
Influence of external pH on EPI encapsulation
Liposomes with an ion and pH gradient of 300 mM ascorbic acid (pH 2.4) or 300 mM ammonium ascorbate (pH 4.0) showed the ability to encapsulate EPI. To observe the effect of external pH on drug encapsulation, several liposomal samples were prepared in 300 mM ascorbic acid (pH 2.4) or 300 mM ammonium ascorbate (pH 4.0). The external solution was then exchanged for PBS with various pH values (5.5, 6.5, 7.5, and 8.5). The following parameters were chosen for drug encapsulation: 1:5 (w/w) drug-to-lipid ratio, 60°C and 5 minutes’ incubation. A gradual increase in EPI encapsulation with increasing external pH and ΔpH was observed. There was no significant difference in EE between liposomes containing 300 mM ascorbic acid (pH 2.4) and 300 mM ammonium ascorbate (pH 4.0) at higher pH. At lower pH, EPI was encapsulated with slightly better efficiency. A summary of the results of these experiments is shown in Figure 1.
Figure 1.
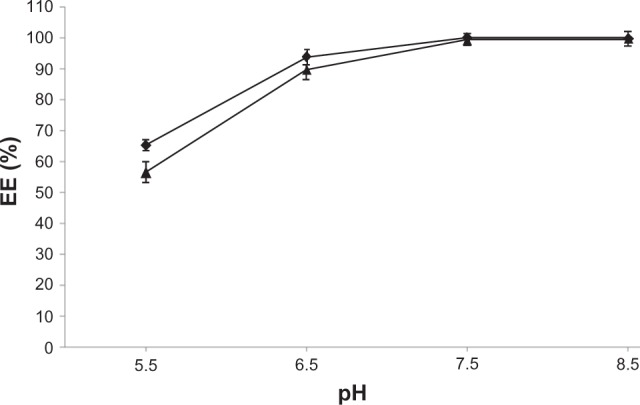
The influence of the external buffer on the encapsulation efficiency (EE) of EPI in HSPC/Chol/DSPE-PEG 2000 LUVs containing 300 mM ascorbic acid (♦) or 300 mM ammonium ascorbate (▲) after 5 minutes of incubation.
Abbreviations: EPI, epirubicin; HSPC/Chol/DSPE-PEG 2000, hydrogenated soy phosphatidylcholine/cholesterol/1,2-distearoyl-sn-glycero-phosphoethanolamine-N-(poly[ethylene glycol]2000); LUVs, large unilamellar vesicles.
Determination of encapsulation efficiency at different drug-to-lipid ratios
For all drug-to-lipid ratios, both formulations (300 mM ascorbic acid and 300 mM ammonium ascorbate) exhibited very high EE, and more than 90% EPI encapsulation could be achieved. The EE decreased with increasing drug-to-lipid ratio. Liposomes with 300 mM ascorbic acid (pH 2.4) exhibited slightly higher EE than liposomes with 300 mM ammonium ascorbate (pH 4.0). The results of these experiments are shown in Figure 2.
Figure 2.
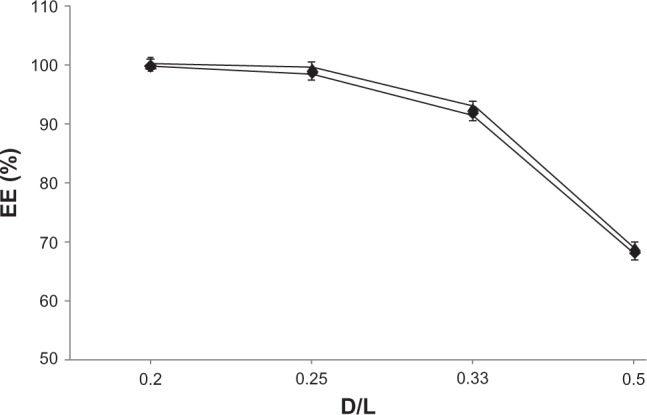
Encapsulation efficiency (EE) for EPI in liposomes at different drug-to-lipid ratios (w/w). Internal buffer containing 300 mM ammonium ascorbate (♦) or 300 mM ascorbic acid (▲).
Abbreviations: EPI, epirubicin; D/L, drug to lipid.
Kinetics of EPI encapsulation in HSPC/Chol/DSPE-PEG 2000 (5.5:4:0.5) liposomes
The EE of EPI loaded using a 300 mM ascorbic acid (pH 2.4) or 300 mM ammonium ascorbate (pH 4.0) gradient was measured at different time intervals to determine the time required for complete drug encapsulation in liposomes. A 1:5 (w/w) drug-to-lipid ratio and a temperature of 60°C were chosen for drug encapsulation. As shown in Figure 3, the kinetics of the process is very rapid, and after 2.5 minutes almost all of the drug had been encapsulated (>90% EE). The accumulation of EPI is little bit faster in liposomes containing 300 mM ascorbic acid (pH 2.4). After 1 hour, marginal drug leakage was observed in both formulations. All the data are summarized in Figure 3.
Figure 3.
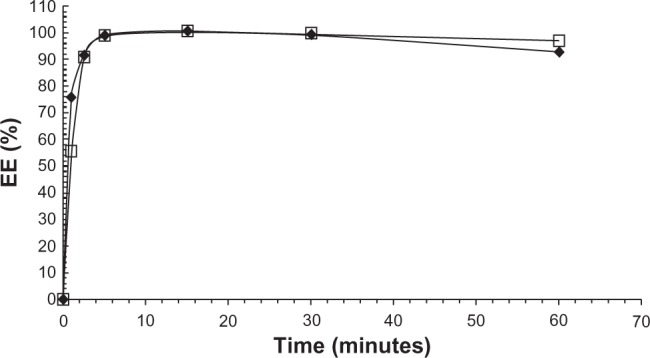
The effect of incubation time on EE of EPI in 110 nm HSPC/Chol/DSPE-PEG 2000 LUVs containing 300 mM ascorbic acid (♦) or 300 mM ammonium ascorbate (□). The loading process was performed at 60°C, and the drug-to-lipid ratio was 1:5 (w/w). Error bars (<5%) are not shown for clarity reasons.
Abbreviations: EPI, epirubicin; HSPC/Chol/DSPE-PEG 2000, hydrogenated soy phosphatidylcholine/cholesterol/1,2-distearoyl-sn-glycero-phosphoethanolamine-N-(poly[ethylene glycol]2000); LUVs, large unilamellar vesicles; EE, encapsulation efficiency.
EPI long-term stability experiment
Figures 4 and 5 show the long-term stability plot for the liposomes (in terms of drug retention) loaded using a 300 mM ascorbic acid (pH 2.4) or 300 mM ammonium ascorbate (pH 4.0) gradient at a drug-to-lipid ratio of 1:5 w/w. The liposomes were stored at 4°C, 24°C, or 37°C. Both formulations were stable at 4°C for at least 1 year, and there was only marginal drug leakage observed at 24°C over a 6-month storage period (data not shown). Only at 37°C did EPI leak from the liposomes to an appreciable degree. In all cases, EPI loaded using a 300 mM ammonium ascorbate (pH 4.0) gradient is slightly more stable in terms of retention. Also, there was not any hydrolysis of lipids observed during storage (monitored by thin layer chromatography).26
Figure 4.
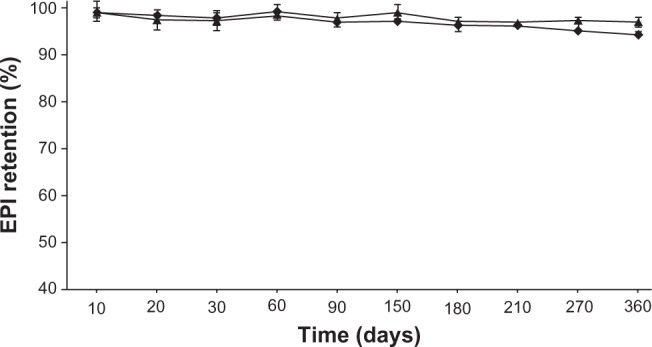
The long-term stability of HSPC/Chol/DSPE-PEG 2000 (5.5:4:0.5 mol/mol) liposomes loaded with EPI using a 300 mM ascorbic acid (♦) or 300 mM ammonium ascorbate (▲) gradient and stored at 4°C for 12 months in phosphate-buffered saline at a lipid concentration of 2 mM.
Abbreviations: EPI, epirubicin; HSPC/Chol/DSPE-PEG 2000, hydrogenated soy phosphatidylcholine/cholesterol/1,2-distearoyl-sn-glycero-phosphoethanolamine-N-(poly[ethylene glycol]2000).
Figure 5.
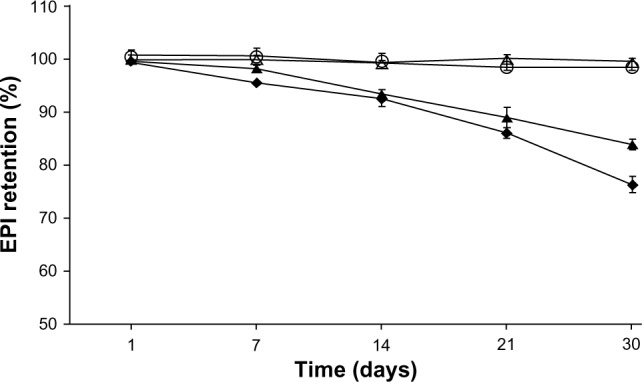
The 30-day stability of HSPC/Chol/DSPE-PEG 2000 (5.5:4:0.5 mol/mol) liposomes loaded with EPI using a 300 mM ascorbic acid (♦) or 300 mM ammonium ascorbate (▲) gradient and stored at 37°C in phosphate-buffered saline at a lipid concentration of 2 mM and that of liposomes loaded with EPI using a 300 mM ascorbic acid (⋄) or 300 mM ammonium ascorbate (O) gradient and stored at 24°C in phosphate-buffered saline at a lipid concentration of 2 mM.
Abbreviations: EPI, epirubicin; HSPC/Chol/DSPE-PEG 2000, hydrogenated soy phosphatidylcholine/cholesterol/1,2-distearoyl-sn-glycero-phosphoethanolamine-N-(poly[ethylene glycol]2000).
Determination of liposomal release of EPI in the presence of human plasma in vitro
Four different liposomal formulations were prepared to determine and compare the stability of liposomes containing EPI encapsulated using a transmembrane gradient of ascorbic acid. HSPC/Chol/DSPE-PEG 2000 EPI-loaded liposomes were prepared as described earlier. The release of EPI in blood-like conditions is summarized in Figure 6. EPI release in both cases (EPI loaded using a 300 mM ascorbic acid or 300 mM ammonium ascorbate gradient) was negligible and slightly smaller than for EPI loaded using an ammonium sulfate gradient. More rapid drug leakage was observed in the case of liposomes where EPI was loaded using a citrate gradient.
Figure 6.
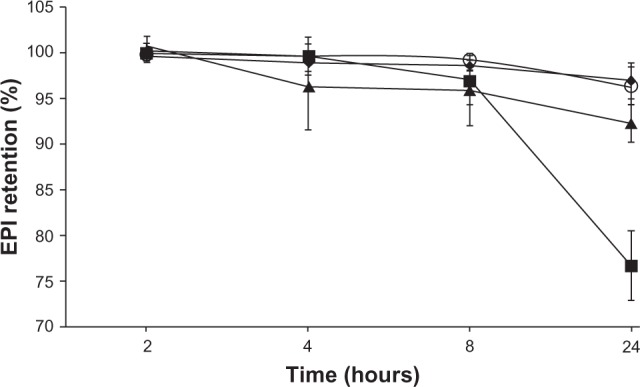
Comparison of the release of EPI in 50% human plasma from the HSPC/Chol/DSPE-PEG 2000 (5.5:4:0.5 mol/mol) liposomes into which it had been encapsulated using different ion and/or pH gradients: 300 mM ammonium ascorbate at pH 4.0 (♦), 300 mM ascorbic acid at pH 2.4 (O), 300 mM ammonium sulfate at ph 5.5 (▲), and 300 mM citrate buffer at pH 4.0 (■).
Abbreviations: EPI, epirubicin; HSPC/Chol/DSPE-PEG 2000, hydrogenated soy phosphatidylcholine/cholesterol/1,2-distearoyl-sn-glycero-phosphoethanolamine-N-(poly[ethylene glycol]2000).
Cryo-TEM
The cryo-TEM technique was used to determine and visualize the physical state of the drug inside the liposomes. This state can directly influence the dissolution rate of the drug from the liposomes, and thus its antineoplastic activity. As shown in Figure 7A, EPI did not precipitate inside liposomes loaded using an ascorbate acid gradient (pH 2.4). Only in liposomes containing EPI loaded using ammonium ascorbate (pH 4.0) was a precipitate observed (Figure 7B). However, even in those liposomes, the precipitate appears in several uncondensed fibers, unlike anthracyclines loaded using ammonium sulfate or citrate gradients, where the precipitate takes the form of elongated bundles (“coffee-bean” structure).20,27 This suggests that EPI-fiber formation occurs in ammonium ascorbate liposomes only because of the excess of its aqueous solubility under these conditions, which may lead to a better dissolution rate and better antineoplastic activity.
Figure 7.
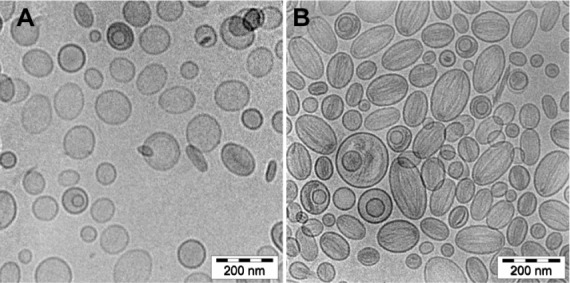
(A and B) Cryogenic transmission electron micrographs of HSPC/Chol/DSPE-PEG 2000 (5.5:4:0.5 mol/mol) liposomes loaded with EPI using an ammonium ascorbate pH gradient at a 1:5 (w/w) drug-to-lipid ratio. The internal buffer was 300 mM ammonium ascorbate, pH 2.4 (A) or 300 mM ammonium ascorbate, pH 4.0 (B). The external buffer was 150 mM phosphate-buffered saline, pH 7.4. At an internal pH of 2.4, no drug precipitates are seen in most of the liposomes. Occasionally, a thin rod-like structure can be found. The circular liposomes shape remains unchanged. At pH 4.0, the drug forms thin diffused filaments, filling the whole liposome interior, and the liposomes can be characterized as melon-shaped. The entire bar represents 200 nm.
Abbreviations: EPI, epirubicin; HSPC/Chol/DSPE-PEG 2000, hydrogenated soy phosphatidylcholine/cholesterol/1,2-distearoyl-sn-glycero-phosphoethanolamine-N-(poly[ethylene glycol]2000).
Cytotoxicity
Figure 8A and B illustrate cell viability after 48 and 72 hours’ incubation of MCF-7 cells. Figure 8C and D illustrate cell viability after 48 and 72 hours’ incubation of 4T-1 cells with different EPI liposomal formulations and free EPI. The experiment used EPI encapsulated in HSPC/Chol/DSPE-PEG 2000 (5.5:4:0.5 mol/mol) liposomes using 300 mM ascorbic acid (pH 2.4), 300 mM ammonium ascorbate (pH 4.0), or 300 mM ammonium sulfate (pH 5.5) gradients. Liposomal formulations containing EPI loaded using the ascorbic acid gradients showed higher cytotoxic activity against all of the tested cell lines. The novel method of EPI formulation based on an ascorbic acid gradient shows superior cytotoxicity, especially when the drug is loaded to liposomes using an ascorbic acid gradient at pH 2.4. EPI loaded using an ammonium ascorbate gradient shows much higher cytotoxicity than EPI loaded via an ammonium sulfate gradient. Empty liposomes containing ascorbic acid, ammonium ascorbate, and ammonium sulfate did not show any cytotoxicity (data not shown).
Figure 8.


(A–D) Comparison of cytotoxicity of EPI encapsulated in HSPC/Chol/DSPE-PEG 2000 (5.5:4:0.5 mol/mol) liposomes using an ascorbic acid gradient (EPI lip ascorbic acid pH 2.4), ammonium ascorbate gradient (EPI lip ascorbic acid pH 4.0) or ammonium sulfate gradient (EPI am sulfate) and that of free EPI (EPI free). The cytotoxicity was measured on MCF-7 cells after 48 (A) and 72 hours (B), and on 4T-1 cells after 48 (C) and 72 hours (D).
Abbreviations: EPI, epirubicin; HSPC/Chol/DSPE-PEG 2000, hydrogenated soy phosphatidylcholine/cholesterol/1,2-distearoyl-sn-glycero-phosphoethanolamine-N-(poly[ethylene glycol]2000).
In vivo antitumor activity
Long-circulating liposomes are known to accumulate inside tumors through the so-called enhanced-permeability-and-retention (EPR) effect, which is caused by the increased leakage of the fenestrated blood vessels in inflamed tissues.28 There are a lot of factors, such as liposome composition, drug–bilayer interactions, and encapsulation method, which have a direct influence on drug leakage from liposomes and indirect influence on the antitumor activity of the drug. To estimate the anticancer potential of the newly designed long-circulating liposomal formulation of EPI, we used mice inoculated with the 4T-1 murine mammary cancer cell line. On day 9, free and liposomal EPI encapsulated using ascorbic acid and ammonium ascorbate gradients were injected into the mice at a dose of 6 mg/kg body weight.
The applied concentration of free EPI (6 mg/kg body weight) did not inhibit 4T-1 tumor growth (Figure 9). Both tested liposomal formulations of EPI given via intravenous injection had a similar effect on 4T-1 tumor-growth kinetics, significantly inhibiting tumor growth compared to the effect of the treatment with free EPI. EPI in liposomal formulations encapsulated using an ammonium ascorbate gradient showed 40% tumor-growth inhibition on day 14. The highest activity was observed on day 18 (TGI = 59.99%), and the activity of this formulation remained at 40%–60% until day 30 of the experiment (Figure 10). EPI in the liposomal formulation encapsulated using ascorbic acid showed its highest activity on day 22 of the experiment (TGI = 51.9%), and the activity of the preparation remained high (40%–55%) from day 16 to 24. Both liposomal formulations of EPI exhibited enhanced antitumor activity compared to free EPI and similar antitumor activity when compared to each other. The experiment also showed no toxicity for free EPI or the two EPI liposomal formulations at the intended doses of 6 mg/kg body weight.
Figure 9.
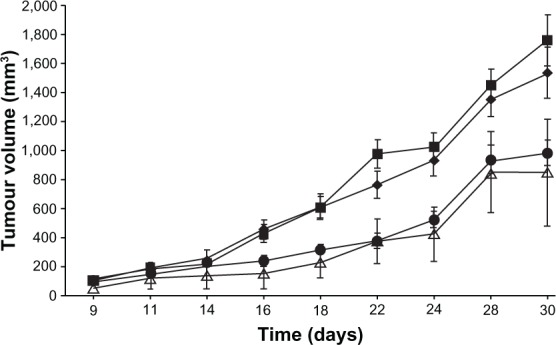
Effects of EPI (free or in liposomal formulation) given at a dose of 6 mg/kg in the 4T-1 murine mammary cancer model. Control, saline-injected mice (■), free EPI (♦), EPI encapsulated in liposomes using ascorbic acid gradient (•), and EPI encapsulated in liposomes using ammonium ascorbate gradient (Δ).
Abbreviation: EPI, epirubicin.
Figure 10.

Inhibition of tumor growth determined for EPI encapsulated in HSPC/Chol/DSPE-PEG 2000 (5.5:4:0.5 mol/mol) liposomes using ascorbic acid gradient (EPI lip ascorbic acid pH 2.4) or ammonium ascorbate gradient (EPI lip ascorbic acid 4.0) and for free EPI (EPI free).
Abbreviations: EPI, epirubicin; HSPC/Chol/DSPE-PEG 2000, hydrogenated soy phosphatidylcholine/cholesterol/1,2-distearoyl-sn-glycero-phosphoethanolamine-N-(poly[ethylene glycol]2000); TGI, tumor-growth inhibition.
Plasma elimination of liposomal EPI
As seen in Figure 11, the plasma elimination of free EPI is very rapid. After 15 minutes, only 1% of the drug remains in the plasma. In the case of the drug encapsulated in liposomes using an ammonium ascorbate gradient, the plasma elimination of EPI is much slower, as expected. AUC0–24 and t½ in the case of the liposomal EPI increased significantly, whereas Clb (clearance) decreased. After 24 hours, about 40% of the injected drug is still present in the plasma. All of the data are summarized in Figure 11 and Table 2.
Figure 11.
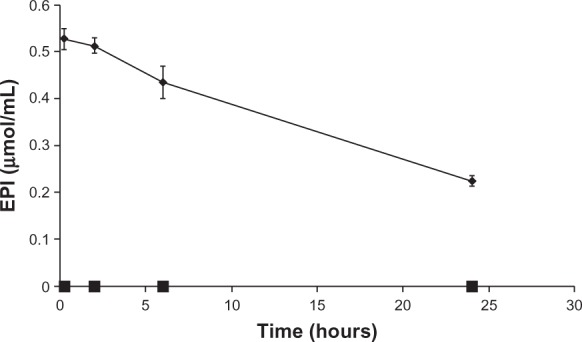
The plasma concentrations of EPI in mice over time after the injection of the HSPC/Chol/DSPE-PEG 2000 (5.5:4:0.5 mol/mol) ammonium ascorbate ion gradient-loaded EPI liposomes (♦) and free EPI (■) at a dose of 25.87 μmol/kg body weight (15 mg/kg); n = 5.
Abbreviations: EPI, epirubicin; HSPC/Chol/DSPE-PEG 2000, hydrogenated soy phosphatidylcholine/cholesterol/1,2-distearoyl-sn-glycero-phosphoethanolamine-N-(poly[ethylene glycol]2000).
Table 2.
Summary of the pharmacokinetic parameters of the free and liposomal EPI
| Sample | AUC0–24 (μmol/hour/mL) | AUMC (μmol/hour/mL) | MRT (hours) | t½ (hours) | Clb (mL/kg/hour) |
|---|---|---|---|---|---|
| Free EPI | 0.009745 | 0.085920 | 8.816941 | 6.111438 | 3540.322526 |
| HSPC/Chol/DSPE-PEG 2000 5.5:4:0.5 | 14.1791 | 395.871 | 27.9193 | 18.6165 | 2.510540 |
Note: For the clarity of the presented data, standard deviations (<3%) are not shown.
Abbreviations: EPI, epirubicin; AUC, area under the curve; AUMC, area under the first moment curve; MRT, mean residence time; t½, half-life; Clb, biliary clearance; HSPC/Chol/DSPE-PEG 2000, hydrogenated soy phosphatidylcholine/cholesterol/1,2-distearoyl-sn-glycero-phosphoethanolamine-N-(poly[ethylene glycol]2000).
Discussion
In this study, we assessed a novel method of liposomal encapsulation for anthracyclines and other weak bases. Our main goal was to obtain a stable liposomal formulation of EPI with a fast dissolution/release rate for the drug in vivo to yield strong antitumor activity. It is generally acknowledged that the main tumor-targeting principles for liposomes are based on the EPR effect and increasing the blood-circulation time to enhance drug delivery to the tumor site. Novel liposomal formulations are typically characterized using EE, long-term stability, in vitro stability in the presence of human serum, in vitro cytotoxicity, and in vivo antitumor activity. We used these parameters here.
For the drug encapsulation, we used a novel method based on ascorbic acid with a very low internal pH (2.4) and a low internal pH (4.0). The method uses an ascorbic acid ion gradient as a driving force, enabling efficient drug accumulation inside the lipid vesicles, and yields high EE.
As shown, the liposomes with an internal pH of 2.4 had a slightly better EE than liposomes with an internal pH of 4.0 when the external pH was 5.5. However, when the external pH was 6.5 or higher, there was no significant difference in the EE between these two formulations (Figure 1). Surprisingly, EPI is very soluble in the presence of ascorbic acid at both pH values (2.4 and 4.0). This is not a disadvantage, because with EPI, which is more hydrophilic than idarubicin, there is no need to form a highly insoluble salt to prevent drug leakage. Inside the liposomes, it is probable that part of the EPI is in an aggregated state and another part is in the form of a soluble complex with ascorbic acid, similar to the situation seen in liposomes containing mitoxantrone29 encapsulated using a copper ion gradient. However, this hypothesis requires further investigation.
This novel method of encapsulation gives a very high EE, ie, when the drug-to-lipid ratio was 1:3, the EE was >90%, and when the drug-to-lipid ratio was 1:2, the EE was >65% (but in the latter case, the formulation was not stable). The formulation is very stable in suspension: when stored at 4°C for 1 year, no significant drug leakage was observed (Figure 4). The formulation was also very stable (Figure 5) when stored at 24°C, with only marginal drug release over a 6-month period. Neither liposome aggregation nor an increase in the polydispersity index were observed during liposome storage (data not shown). Only when liposomes were stored at 37°C was any significant drug release observed (Figure 5). The internal initial pH of 2.4 only marginally affected drug leakage (Figures 4 and 5). Only when liposomes with an internal pH of 2.4 were stored at 37°C was their stability significantly lower than that of liposomes with an internal pH of 4.0. The novel liposomal formulation of EPI is very stable in the presence of 50% human plasma (Figure 6). Under these conditions, liposomes containing EPI encapsulated using an ascorbic acid or ammonium sulfate gradient had very similar stability (>90%), and only EPI encapsulated using a citrate gradient was released to a significant degree.
In vitro cytotoxicity measurements show that liposomes containing EPI encapsulated using an ascorbic acid gradient exhibit superior activity when compared to liposomes containing EPI encapsulated using an ammonium sulfate gradient. The superior activity of those liposomes may be due to several factors. The most important factor is the enhanced activity of anthracyclines in the presence of ascorbic acid. Kurbacher et al6 showed that in high doses, ascorbic acid itself presented cytotoxic activity and also enhanced the antineoplastic activity of several anticancer drugs. This effect was particularly clear with DOX, as ascorbic acid and DOX exhibited synergism. In our research, empty liposomes containing ascorbic acid did not show any cytotoxicity (data not shown), but as shown in Figure 8, liposomes containing EPI loaded using an ascorbic acid gradient exhibited superior activity when compared to liposomes containing EPI loaded using an ammonium sulfate gradient.
Another important feature that increases the cytotoxic activity of the liposomes made using this novel method of encapsulation is the physical state of the drug inside the vesicles. As mentioned, EPI does not form a low-soluble precipitate with ascorbic acid like it does in case of ammonium sulfate. Cryo-TEM photography of EPI loaded using an ascorbic acid gradient reveals that drug inside the vesicles did not form elongated bundles. As shown by Li et al,20 the physical state of the drug inside the liposomes directly affects its stability. The poorly water-soluble complex of EPI with sulfate ion might be released from liposomes very slowly inside tumor cells when compared to EPI encapsulated in liposomes with an ascorbic acid gradient. In the latter case, a faster release under certain conditions, such as in lysosomes, may be observed, but this hypothesis requires further investigation. The better cytotoxic activity of the liposomal formulation containing EPI loaded using ascorbic acid at low pH seems to confirm that where the drug is in its soluble form, its dissolution rate can be faster, which seems to translate to better antineoplastic activity.
It is also noteworthy that although the drug inside liposomes did not form a low-soluble precipitate, this novel method of encapsulation provides very good pharmacokinetic parameters. After 24 hours in the blood, about 40% of the drug is still there, which is comparable with EPI loaded using ammonium sulfate.30
The in vivo activity of the novel liposomal formulation was investigated on mice with 4T-1 murine mammary cancer, which is a physiological mouse model that closely resembles breast cancer in humans.31,32 The novel tested liposomal formulations improved EPI antitumor activity compared to that of the free drug at a dose of 6 mg/kg body weight. The free EPI antitumor activity at this dose was statistically nonsignificant when compared to that of the liposomal formulation of EPI. There was a small difference between the liposomal formulation of EPI loaded using an ascorbic acid gradient at pH 2.4 and EPI loaded using ammonium ascorbate at pH 4.0 in terms of in vivo activity. The observed results indicate that the formulation of EPI loaded using an ammonium ascorbate gradient exhibits slightly higher antitumor activity compared to EPI loaded using an ascorbic acid gradient (TGI for ammonium ascorbate gradient remained above 40% from day 16 to the end of the experiment and for ascorbic acid gradient only between day 16 and 24 of the experiment).
The increased antitumor effect observed for EPI encapsulated using an ammonium ascorbate gradient may be associated with prolonged circulation of the drug in the body. As mentioned in the Results section, no body-weight loss was observed for free EPI or the two EPI liposomal formulations at the intended dose of 6 mg/kg body weight. Increased antitumor activity of liposomal formulations is the result of the EPR effect, which leads to the accumulation of lipid vesicles at the tumor site. In the case of the EPR effect, the most important feature is the size of particles, as shown by Yuan et al,33 who found that particles with a size of around 100 nm or less accumulate within the tumor effectively and do not diffuse back to the bloodstream even after a week. They recommended that liposomes should be around 100 nm in diameter. However, Popović et al34 claimed that particles smaller than 100 nm showed better accumulation within the tumor. This novel method of encapsulation deposited liposomes filled with EPI and ascorbic acid near the tumor, and increased the antitumor activity of EPI through the (putative) synergistic activity of anthracyclines together with ascorbic acid and/or the increased drug-leakage rate in the cancer tissue.
This novel method of encapsulation enables remote loading of weak bases to liposomes, and is exceptionally useful for anthracycline loading because it improves their antineoplastic activity. We applied this method in our laboratory to encapsulate other anthracyclines, such as mitoxantrone, and other drugs, such as ciprofloxacin. The EE for all of the tested anthracyclines loaded using an ascorbic acid gradient led us to the general conclusion that this method is suitable for the preparation of commercial liposomal formulations of anthracyclines. In order to increase the antitumor activity of these formulations, we are currently carrying out investigations on active targeting by attaching specific ligands to the liposomes.
Conclusion
In this paper, we discussed a new method of active anthracycline loading based on an ascorbic acid gradient. The method ensures rapid and efficient EPI encapsulation (in less than 5 minutes, the EE was around 100% at a 0.33 drug-to-lipid ratio) within lipid vesicles. Enhanced EPI antitumor activity was a result of the synergistic antineoplastic activity of anthracyclines together with ascorbic acid. Another factor contributing to high anticancer activity of our formulation was good solubility of EPI in the ascorbic acid gradient, as seen on the cryo-TEM images. It may result in better dissolution rate of the drug from liposomes and higher bioavailability. In vitro results showed the superior activity of this method compared with ammonium sulfate method used generally for anthracycline encapsulation. The method resulted in stable formulation both in vitro (negligible release at 4°C during 360 days’ storage) and in vivo (around 40% of the injected dose was still present in circulation after 24 hours). The in vivo study showed high antitumor activity of this liposomal EPI encapsulated with an ascorbic acid gradient against 4T-1 murine mammary cancer.
Acknowledgments
This study was supported by the Wrocław Research Centre EIT+ under the Biotechnologies and Advanced Medical Technologies project – BioMed (POIG.01.01.02-02-003/08), financed by the European Regional Development Fund (Operational Programme Innovative Economy, 1.1.2).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Svirbely JL, Szent-Györgyi A. The chemical nature of vitamin C. Biochem J. 1933;27(1):279–285. [PMC free article] [PubMed] [Google Scholar]
- 2.Englard S, Seifter S. The biochemical functions of ascorbic acid. Annu Rev Nutr. 1986;6:365–406. doi: 10.1146/annurev.nu.06.070186.002053. [DOI] [PubMed] [Google Scholar]
- 3.Chen Q, Espey MG, Krishna MC, et al. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A. 2005;102(38):13604–13609. doi: 10.1073/pnas.0506390102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Q, Espey MG, Sun AY, et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc Natl Acad Sci U S A. 2007;104(21):8749–8754. doi: 10.1073/pnas.0702854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du J, Martin SM, Levine M, et al. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin Cancer Res. 2010;16(2):509–520. doi: 10.1158/1078-0432.CCR-09-1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kurbacher CM, Wagner U, Kolster B, Andreotti PE, Krebs D, Bruckner HW. Ascorbic acid (vitamin C) improves the antineoplastic activity of doxorubicin, cisplatin, and paclitaxel in human breast carcinoma cells in vitro. Cancer Lett. 1996;103(2):183–189. doi: 10.1016/0304-3835(96)04212-7. [DOI] [PubMed] [Google Scholar]
- 7.Vollbracht C, Schneider B, Leendert V, Weiss G, Auerbach L, Beuth J. Intravenous vitamin C administration improves quality of life in breast cancer patients during chemo-/radiotherapy and aftercare: results of a retrospective, multicentre, epidemiological cohort study in Germany. In Vivo. 2011;25(6):983–990. [PubMed] [Google Scholar]
- 8.Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57(7):727–741. doi: 10.1016/s0006-2952(98)00307-4. [DOI] [PubMed] [Google Scholar]
- 9.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56(2):185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 10.Tian W, Ying X, Du J, et al. Enhanced efficacy of functionalized epirubicin liposomes in treating brain glioma-bearing rats. Eur J Pharm Sci. 2010;41(2):232–243. doi: 10.1016/j.ejps.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Robert J. Epirubicin. Clinical pharmacology and dose-effect relationship. Drugs. 1993;45(Suppl 2):20–30. doi: 10.2165/00003495-199300452-00005. [DOI] [PubMed] [Google Scholar]
- 12.O’Shaughnessy J. Liposomal anthracyclines for breast cancer: overview. Oncologist. 2003;8(Suppl 2):1–2. doi: 10.1634/theoncologist.8-suppl_2-1. [DOI] [PubMed] [Google Scholar]
- 13.Safra T, Muggia F, Jeffers S, et al. Pegylated liposomal doxorubicin (Doxil): reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann Oncol. 2000;11(8):1029–1033. doi: 10.1023/a:1008365716693. [DOI] [PubMed] [Google Scholar]
- 14.Sakakibara T, Chen FA, Kida H, et al. Doxorubicin encapsulated in sterically stabilized liposomes is superior to free drug or drug-containing conventional liposomes at suppressing growth and metastases of human lung tumor xenografts. Cancer Res. 1996;56(16):3743–3746. [PubMed] [Google Scholar]
- 15.Todorov DK, Ninjo SS, Zeller WJ. Antiproliferative activity of liposomal epirubicin on experimental human gliomas in vitro and in vivo after intratumoral/interstitial application. J Cancer Res Clin Oncol. 1995;121(3):164–168. doi: 10.1007/BF01198098. [DOI] [PubMed] [Google Scholar]
- 16.Mayhew EG, Lasic D, Babbar S, Martin FJ. Pharmacokinetics and antitumor activity of epirubicin encapsulated in long-circulating liposomes incorporating a polyethylene glycol-derivatized phospholipid. Int J Cancer. 1992;51(2):302–309. doi: 10.1002/ijc.2910510221. [DOI] [PubMed] [Google Scholar]
- 17.Mayer LD, Harasym TO, Tardi PG, et al. Ratiometric dosing of anticancer drug combinations: controlling drug ratios after systemic administration regulates therapeutic activity in tumor-bearing mice. Mol Cancer Ther. 2006;5(7):1854–1863. doi: 10.1158/1535-7163.MCT-06-0118. [DOI] [PubMed] [Google Scholar]
- 18.Tardi PG, Dos Santos N, Harasym TO, et al. Drug ratio-dependent antitumor activity of irinotecan and cisplatin combinations in vitro and in vivo. Mol Cancer Ther. 2009;8(8):2266–2275. doi: 10.1158/1535-7163.MCT-09-0243. [DOI] [PubMed] [Google Scholar]
- 19.Gubernator J, Chwastek G, Korycinska M, et al. The encapsulation of idarubicin within liposomes using the novel EDTA ion gradient method ensures improved drug retention in vitro and in vivo. J Control Release. 2010;146(1):68–75. doi: 10.1016/j.jconrel.2010.05.021. [DOI] [PubMed] [Google Scholar]
- 20.Li X, Hirsh DJ, Cabral-Lilly D, et al. Doxorubicin physical state in solution and inside liposomes loaded via a pH gradient. Biochim Biophys Acta. 1998;1415(1):23–40. doi: 10.1016/s0005-2736(98)00175-8. [DOI] [PubMed] [Google Scholar]
- 21.Barenholz Y. Doxil – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 22.Maurer N, Wong KF, Hope MJ, Cullis PR. Anomalous solubility behavior of the antibiotic ciprofloxacin encapsulated in liposomes: a 1H-NMR study. Biochim Biophys Acta. 1998;1374(1–2):9–20. doi: 10.1016/s0005-2736(98)00125-4. [DOI] [PubMed] [Google Scholar]
- 23.Stewart JC. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal Biochem. 1980;104(1):10–14. doi: 10.1016/0003-2697(80)90269-9. [DOI] [PubMed] [Google Scholar]
- 24.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 25.Riviere JE. Pharmacokinetics of nanomaterials: an overview of carbon nanotubes, fullerenes and quantum dots. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1(1):26–34. doi: 10.1002/wnan.24. [DOI] [PubMed] [Google Scholar]
- 26.Hossann M, Wiggenhorn M, Schwerdt A, et al. In vitro stability and content release properties of phosphatidylglyceroglycerol containing thermosensitive liposomes. Biochim Biophys Acta. 2007;1768(10):2491–2499. doi: 10.1016/j.bbamem.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 27.Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151(2):201–215. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 28.Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153(3):198–205. doi: 10.1016/j.jconrel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li C, Cui J, Li Y, et al. Copper ion-mediated liposomal encapsulation of mitoxantrone: the role of anions in drug loading, retention and release. Eur J Pharm Sci. 2008;34(4–5):333–344. doi: 10.1016/j.ejps.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 30.Sha X, Guo J, Chen Y, Fang X. Effect of phospholipid composition on pharmacokinetics and biodistribution of epirubicin liposomes. J Liposome Res. 2012;22(1):80–88. doi: 10.3109/08982104.2011.627513. [DOI] [PubMed] [Google Scholar]
- 31.Tao K, Fang M, Alroy J, Sahagian GG. Imagable 4T1 model for the study of late stage breast cancer. BMC Cancer. 2008;8:228. doi: 10.1186/1471-2407-8-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52(6):1399–1405. [PubMed] [Google Scholar]
- 33.Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK. Microvascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res. 1994;54(13):3352–3356. [PubMed] [Google Scholar]
- 34.Popović Z, Liu W, Chauhan VP, et al. A nanoparticle size series for in vivo fluorescence imaging. Angew Chem Int Ed Engl. 2010;49(46):8649–8652. doi: 10.1002/anie.201003142. [DOI] [PMC free article] [PubMed] [Google Scholar]