

Abstract
A biophysical model of the glycogen molecule is developed, which takes into account the points of attack of synthase and phosphorylase at the level of the individual glucose chain. Under the sole assumption of steric effects governing enzyme accessibility to glucosyl residues, the model reproduces the known equilibrium structure of cellular glycogen at steady state. In particular, experimental data are reproduced assuming that synthase (1) operates preferentially on inner chains of the molecule and (2) exhibits a faster mobility than phosphorylase in translocating from an attacked chain to another. The model is then used to examine the turnover of outer versus inner tiers during the labeling process of isotopic enrichment (IE) experiments. Simulated data are fitted to in vivo 13C nuclear magnetic resonance spectroscopy measurements obtained in the human brain under resting conditions. Within this experimental set-up, analysis of simulated label incorporation and retention shows that 7% to 35% of labeled glucose is lost from the rapidly turning-over surface of the glycogen molecule when stimulation onset is delayed by 7 to 11.5 hours after the end of [1-13C]glucose infusion as done in actual procedures. The substantial label washout before stimulation suggests that much of the subsequent activation-induced glycogenolysis could remain undetected. Overall, these results show that the molecular structure significantly affects the patterns of synthesis and degradation of glycogen, which is relevant for appropriate design of labeling experiments aiming at investigating the functional roles of this glucose reserve.
Keywords: glycogen β-particle, human brain, mathematical modeling
Introduction
Virtually all organisms, from unicellular to humans, store D-glucose residues in the form of the highly branched polysaccharide glycogen.1 The structural features of the mature glycogen molecule (called β-particle, or macroglycogen) have been optimized in the course of evolution to achieve an efficient storage and fast release of glucose.2, 3 In mammals, the major deposits of glycogen are in skeletal muscle and liver, but significant glycogen levels are found in other organs including the heart and the brain. The liver is unique in that it stores glycogen as molecular aggregates of β-particles called α-particles (or α-rosettes), while most cell types including cerebral astrocytes accumulate and metabolize glycogen in the form of β-particles.4
In the human brain, glycogen is the sole energy store and it is present at concentrations of 3 to 4 μmol/g in terms of glucose equivalents.5 This corresponds to 12 to 16 μmol/g in astrocytes (assuming a tissue volume fraction of 25%), which is significantly higher than tissue glucose level at euglycemia. Although the involvement of cerebral glycogen in the metabolic response to the glycemic and hormonal state of the tissue is well established, controversy exists about its function during physiologic brain activation (reviewed by ref. 6). In particular, the early indications of substantial stimulation-induced glycogen utilization measured in rodent brain and cell cultures (see ref. 7 and references therein) have failed to be confirmed in the human brain using 13C nuclear magnetic resonance (NMR) spectroscopy.5 This finding motivated a subsequent study, which supported the notion of cerebral glycogen as a small emergency depot in case of energy failure.8 Previous theoretical work9 suggested that the inability in detecting glycogen mobilization could be inherent to the kinetic model used to fit experimental data, which assumed that the probability of detaching a glucosyl residue from glycogen is homogeneous and proportional to the total tissue glycogen content.5, 10 However, the effective concentration of glycogen has been found to change according to the actual tier being mobilized.4 Therefore, the organization and size of the glycogen particles is potentially of considerable importance to their metabolism, and knowledge of the patterns of glycogen metabolism is necessary for the assessment of changes in the rates of glycogen deposition and utilization.11 For example, very different kinetics can be produced by a ‘last on-first off' pattern compared with a random synthesis and degradation pattern.12 The potential erroneous interpretation of studies involving incorporation of labeled glucose into glycogen was clearly showed in cell cultures, where it was found that release of 3H- or 14C-labeled glucose from glycogen was opposite to the order of their isotopic enrichment (IE) during glycogen synthesis.13 This is consistent with the fact that the accessibility of glycogen degrading and synthesizing enzymes to glucosyl residues within the glycogen particles results in a highly heterogeneous turnover of the molecule, with external regions turning over much more rapidly than internal ones.14
The outermost tier of the glycogen molecule contains about half of the total glucose,15 of which only ∼36% is directly accessible to phosphorylase before any debranching occurs.2 Accordingly, purified phosphorylase (i.e., in the absence of debranching enzyme) was found to degrade glycogen to the extent of ∼40% only, giving rise to a so-called limit dextrin (LD), which cannot be further metabolized due to the inability of the enzyme to bypass a branching point.16 Thus, the glycogen molecule is not metabolically homogeneous and the probability of replacement of a glucosyl residue depends on its location, whether peripheral or central. Accordingly, labeling experiments after 14C-glucose injection have shown that the outermost tier is always more radioactive than the residual LD, indicating that the turnover of the glycogen molecule primarily involves the nonreducing peripheral surface of the polysaccharide.17 An LD is obtained for each tier of the glycogen molecule, and different LD can be produced in vitro by repetitive alternating treatment with phosphorylase and debranching enzyme. Assays for 14C within these experimental procedures have indicated that, for muscle glycogen, the specific activity in subsequent LD (i.e., LD-1 to LD-3) is greater than the polysaccharide from which it is derived.18 As an illustration, turnover time of the first LD (LD-1) in the brain was found to be twice that of total glycogen.19
To address the impact of the molecular structure on the patterns of glycogen turnover, a biophysical model of the glycogen β-particle is developed taking into account the points of attack of glycogen synthase (GS) and glycogen phosphorylase (GP) enzymes at the level of the individual glucosyl chain. The model is then used to examine the IE process of brain glycogen, as measured by 13C-NMR spectroscopy during continuous [1-13C]glucose infusion.5 On the basis of modeling outcomes, the potential utilization of the stored sugar that can be inferred by label kinetics upon brain stimulation is discussed. To this end, some quantitative comparison will be made with previously published as well as some unpublished 13C-NMR experimental data obtained by Dr Gulin Oz and coworkers in the human brain.5
Materials and methods
Glycogen is synthesized from glycogenin, a self-glucosylating protein that acts as a primer for glycogen growth. Polymerization of glucose proceeds via α-1,4-glycosydic linkages, with branch points between individual glycogen chains introduced by the occurrence of α-1,6-glycosydic linkages. It has been proposed that glycogen synthesis occurs in discrete steps, leading first to the formation of proglycogen, a low molecular weight polysaccharide (∼400 kDa compared with 107 Da of mature glycogen) possibly with its own enzymology.20, 21 However, the existence of a well-defined proglycogen molecular species is presently questioned and there is strong evidence for a homogeneous polydispersity in vivo.4, 22 Thus, in the present model the possibility of different isoforms for the glycogen synthesizing and glycogen degrading enzymes is neglected.
The synthesis of glycogen is performed by the coordinated action of GS and glycogen branching enzyme (GBE). Glycogen synthase builds the polymer by adding glucose residues from the activated monomer UDP glucose, while GBE cuts a stretch from a long chain and attaches it to another, thereby forming a new branch. The GBE is in high excess over synthase, as indicated by the 200-fold higher activity of the former compared with the latter.3 The degradation of glycogen requires two enzymes as well, namely GP and glycogen debranching enzyme (GDE). Glycogen phosphorylase catalyzes the removal of glucose residues from nonreducing ends (i.e., A-chains) with tails longer than four residues, while GDE transfers those branches maximally trimmed by phosphorylase to adjacent glucosyl chains. Noteworthy, glycogen degradation has been shown to be independent of GDE, as A-chains constantly provide a substrate for phosphorylase.23 In agreement with previous modeling analyses,14 branching/debranching activity is incorporated in the action of GS and GP enzymes, which is compatible with the fact that both GBE and GDE are not generally considered to be rate limiting for glycogen accumulation and breakdown.4
The basic assumption of the present model is that the basal activities (i.e., in the absence of external stimulation) of synthase and phosphorylase are governed solely by steric factors. This means that any chemical effectors other than the availability of glucose in the medium and glucose content within the glycogen molecule are ignored. To formalize this assumption, it is to be noted that the ‘surface' of the molecule is where the enzymes actually work, which is consistent with its homogeneous growth (i.e., glycogen remains approximately spherical while new residues are attached). Indeed, only the more external tiers are usually involved in the regular metabolic turnover of glycogen synthesis and degradation,14 while the few percent of glycogen stored in the inner tiers has essentially the role of molecule skeleton.21 Although the cascade processes that regulate the activity of enzymes involved in glycogen synthesis and breakdown are well known, the process controlling cessation of biosynthesis remains an unanswered issue.24 The current hypothesis is that the self-limiting size of glycogen is the results of the high density of glucose residues in the outer tier, which impedes further activity of the synthase.25 In particular, the size of the enzymes participating in glycogen synthesis is thought to be optimized for growth stopping before the degeneration of chain length (i.e., incomplete chains) hinders the homogeneity of the molecule.14
Given the complete branched structure of glycogen, the total number of glucose residues GT in the whole molecule can be calculated as:
 |
where gc is the average number of glucose molecule per chain, r is the degree of branching, and t is the relevant tier. Inverting the latter equation for t gives:
 |
Equation (2) expresses the tier number as a function of the glucose content of the glycogen particle (note that t is rational not integer). To determine the enzymatic reaction rates, the free volume for enzyme activity is calculated by taking into account the space occupied by glucose in each tier. The effective length per tier has been estimated2 to be:
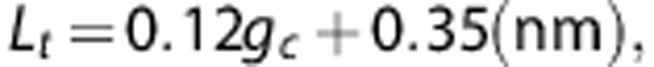 |
which gives a volume Vt of the tier t of:
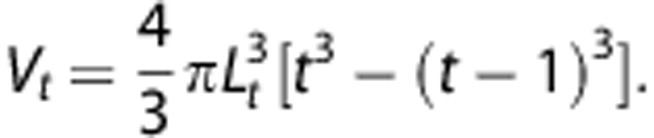 |
Therefore, the quantity Ltt with t(GT) gives the actual radius of the glycogen particle. The volume occupied by glucose in the tier t is:
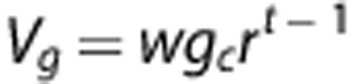 |
where w=0.113 nm3 is the Van der Waals volume of an individual glucose molecule. Thus, the accessible volume for the action of enzymes is the difference Vt−Vg, which is non zero, for a given tier t, only when applied to the incomplete tier. The rate equations for synthase and phosphorylase activity can be written as:
 |
where Gm is the number of glucose molecules in the medium (kept fixed to, e.g., 1 mmol/g=0.0006 molecules/nm3) and GPT=(gc−4)r(t−1) is the total amount of glucose residues available to phosphorylase within A-chains.2 With the methodology described above, a balance equation depending (through the tier number t) only on the total glucose content GT (equation (1)) of the glycogen molecule can be formulated as
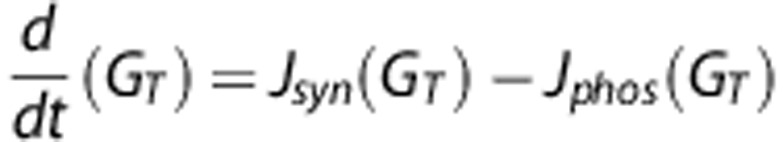 |
with parametric dependence of both Jsyn and Jphos on several known properties of the glycogen particle. Table 1 shows the properties of cellular glycogen β-particle according to the generally accepted model of Whelan.26, 27
Table 1. Structural parameters of cellular glycogen β-particle.
| Description | Symbol | Value |
|---|---|---|
| Branching degree | r | 2 |
| Chain length (glucosyl residues) | gc | 13 |
| Tiers | t0 | 12 |
| Tier length (nm) | Lt | 1.9 |
| Total glucosyl residues | GT,0 | 53,000 |
Glycogen β-particles have a spherical shape and a molecular weight of ∼107 Da. The mature glycogen molecule is spherical, with a total radius of ∼23 nm. It is organized in 12 concentric tiers, all having the same length of 1.9 nm. The inner tiers contain branched type B-chains, whereas nonbranched type A-chains are in the outermost tier. An individual chain has a mean length of 13 glucose residues. The branching of B-chains is uniformly distributed and every B-chain has two branches.
In the rate equations (3) and (4), the constants Ksyn=7.5/minute and Kphos=2.65 × 10−7/(nm3 min) are obtained by imposing Jsyn=Jphos and adjusted to match the experimental turnover rate at steady state (i.e., when t=t0=12) measured by 13C-NMR spectroscopy in the human brain in vivo.5 This study suggests a glycogen turnover of 2.67 nmol/g per minute and a total cerebral glycogen content of 3.5 to 4 μmol/g. By assuming homogeneous glycogen pool of particles containing GT,0=53,000 glucose residues, a turnover rate of 40 residues per minute is obtained. It should be noted that total glycogen content might be underestimated when using the measured 13C-glycogen data, because the internal tiers may never become enriched. For example, if turnover is restricted to only the last three tiers, which contain 87.5% of the glucose residues, then the total glycogen content can be as high as 4.5 μmol/g.
Specific interaction times between glycogen and synthase or phosphorylase are taken into account as well. By defining the enzyme interaction time as the duration before occurrence of random displacement from the attacked chain, the relevant interaction times for the two enzymes are adjusted to reproduce the experimental time course5 as well as the tier-specific pattern17 of 13C-label incorporation in brain glycogen (see ‘Results' section). It is assumed that GS and GP cannot attack the same glucosyl residue (i.e., the same A-chain) simultaneously. To the author's knowledge, a difference in synthase versus phosphorylase interaction times with the glycogen molecule was not previously reported nor hypothesized. Therefore, in the absence of direct experimental proof this type of enzyme action should be interpreted as purely phenomenological under the computational point of view.
It is possible that different glycogen deposits might be in different states of synthesis and degradation both spatially and temporally, even sharing the same kinetic patterns. Nevertheless, while this is certainly true for the short time scale, it is unlikely that significant inhomogeneities at pool level persist for periods as long as 50 hours. In cultured rat myotubes, the largest departure from complete coherence of glycogen molecules breakdown occurred in the range of 0.5 to 3.5 hours, and the contribution of the simultaneous component substantially increased already at 7 hours.13 These numbers could be even smaller for the human brain, as the metabolic rate of rat myotubes in vitro is lower than that of human astrocytes. Therefore, in the analysis of IE experiments a homogeneous pool of glycogen is considered, in which each particle behaves in the same way on average under steady-state conditions.
Simulations were performed under SWI Prolog programming language Version 6.2 (University of Amsterdam; http://www.swi-prolog.org/). In particular, the code determines the activity of GS and GP at each time step according to rate equations derived above (see also Figure 1). The two enzymes act in parallel on the glycogen molecule by randomly attacking a nonreducing end within an A-chain. One enzyme is displaced from the attacked chain when the corresponding interaction time vanishes, and then another A-chain is randomly attacked for a new cycle. New branches are initiated by GS when the growing A-chain reaches the fifth and tenth residues (mimicking the action of GBE). Completely trimmed A-chains (four residues long) are automatically attached to another nonreducing end (mimicking the action of GDE). The medium can contain variable concentrations of unlabeled and/or labeled glucose, which are used to determine the relative substrate probability for the synthase, while the exact localization of each glucosyl residue that is incorporated into the glycogen molecule is recorded. The full Prolog source code is available on request.
Figure 1.

Model reaction rates of glycogen synthase and phosphorylase as a function of molecule size. Theoretical dependency between the size of the molecule, here represented either as tier number t (A) or as total glucose GT (B), and synthase and phosphorylase reaction rates (expressed as residues per minute), Jsyn and Jphos, respectively. The point of intersection between the curves identifies the steady-state position of the mature glycogen particle corresponding to 12 tiers and a content of ∼53,000 glucosyl units. Specifically, the rate of synthase is higher than that of phosphorylase when the glucose content is smaller than 53,299 in our simulations, and vice versa. Note that tier number is calculated from total glycogen according to equation (2), thus the two plots are equivalent.
Results
First, steady-state analysis is performed to match model outcomes to the known properties of cellular glycogen. To examine in detail how the high structural homogeneity of the polysaccharide is realized, several assumptions are tested about the affinity of enzymes for inner versus outer chains within the glycogen molecule. It is found that synthase but not phosphorylase is required to operate in internal zones of the glycogen particle to obtain structural homogeneity, which is otherwise compromised by the excessive growth of outer chains (Figure 2). Previous theoretical analyses have indicated that glycogen synthesis must operate under constraints promoting homogeneous growth, thus leaving no internal chains unfinished.28 Accordingly, it is assumed that GS preferentially works on the internal chains to avoid the excessive growth of the external chains when those in the inner tiers are not yet finished, as previously suggested.14 In addition, because GP functions during the biosynthesis process, corrections of any mistake of the synthase are ‘spontaneously' achieved by removing those chains that are too external. As the high structural homogeneity of the glycogen molecule is thought to form the basis of its metabolic function as a quick fuel supplier,28 theoretical investigation of how such uniform organization can be realized is all important. The minimal set of assumptions required to reproduce the known equilibrium structure of cellular glycogen, which is identified by defining the spatial constraints on the activity of synthesizing and degrading enzymes together with the preference of GS for inner chains, proved sufficient to produce a saturable growth of the glycogen molecule (Figure 3).
Figure 2.

Effect of synthase affinity for inner chains on the homogeneity of the glycogen molecule. (A) In the absence of constraints on the preferential sites of enzyme work, the molecule of glycogen results highly inhomogeneous. In particular, the molecule shows two types of structural irregularities. First, there is excessive branching up to tier ∼30 and hence many incomplete chains. Second, the branching is asymmetric because left branch is created before right branch (at fifth and tenth residues, respectively). (B) When synthase is assumed to operate on inner chains, the glycogen molecule displays the homogeneity reported in the literature for cellular glycogen (see Table 2). The affinity of synthase for inner chains represents the minimal assumption to fulfill almost complete homogeneity. The phosphorylase is not required to exhibit affinity for inner or outer chains, although the homogeneity of glycogen slightly increases if phosphorylase is assumed to have preference for outer chains (not shown). Nonetheless, in the simulations there are no imposed constraints on phosphorylase. It should be realized that both the inhomogeneous (A) and the homogeneous (B) molecules contain the same number of glucose residues and that the time courses of total glucose content during the growth process for the two molecules are nearly identical (not shown). Note that the images are bidimensional representations (not projections of the tridimensional structure) of the actual glycogen structure, and the visualization parameters (e.g., the variation of the angle between branches as a function of the distance from the center) have been chosen only for illustration purposes. The filled circle in the middle of the molecule represents glycogenin.
Figure 3.

Simulated growth process of an individual glycogen molecule. (A) Simulated synthase and phosphorylase reaction rates (Jsyn and Jphos, respectively) during the growth process. The molecule is not complete before about 120 hours. (B) Time course of total glucose content GT. Note that the amount of A-chains (i.e., non reducing ends) in the whole glycogen molecule fluctuates during turnover even when the steady state is established (inset in B). Note also the apparent peaks in the number of A-chains that occur during the transition from a tier to the next (i.e., creation of two new branches per chain at fifth and tenth residues). The subsequent plateau is established when the ‘mother' chains become full, or 13 residues long.
Next, to study the incorporation of labeled glucose in the different tiers of the glycogen molecule, the experimental data for IE in glycogen LD17 are reproduced by estimating the relative interaction time of synthase and phosphorylase. In particular, determination of radioactivity in rabbit muscle glycogen after intravenous injection of 14C-glucose have shown that total radioactivity was distributed in tiers 9, 10, 11, and 12 in a proportion of roughly 1%, 7%, 20%, and 72%, respectively (estimated from Table 1 of ref. 17). To reproduce this pattern of IE in simulations, it is found that, once a chain is attacked, phosphorylase has to interact with glycogen with greater mobility than synthase, the latter exploring the ‘surface' of the polysaccharide more rapidly. In other words, it is found that GS associates to and dissociates from the glycogen molecule several times during a single association/dissociation step of GP. This requirement can possibly be explained by the difference between the glycogen-storage site through which the enzymes are thought to bind to glycogen granules in vivo.29, 30 The enzyme mobility could depend on the relative number of GS and GP acting simultaneously on an individual glycogen molecule as well. The exact value for enzyme–glycogen interaction time needs to be also adjusted to reproduce the experimental IE time course (i.e., exponential growth) and eventually to correct for tissue/species differences in the rate of glycogen turnover. The best fit with experimental data of absolute amount label incorporation as well IE time course is obtained assuming a geometric distribution (i.e., discrete exponential distribution) with mean value approximately equal to the length of a glucosyl chain (Figure 4). Note that the geometric distribution has no upper bound, so occasionally the phosphorylase continues degrading the glycogen molecule up to inner tiers. A faster release of GS relative to GP from glycogen is perfectly consistent with the role of GS in maintaining the homogeneity of the whole glycogen particle, as the resulting greater mobility would allow the enzyme to rapidly ‘find-and-fill' the incomplete chains left behind from GP.
Figure 4.

Determination of enzyme interaction time and relative probability distribution. (A) Exponential growth time course for label incorporation into glycogen, and (B) enrichment of last 3 to 4 tiers (i.e., limit dextrin 1 to 3). These requirements cannot be met at the same time by fixed or uniformly distributed interaction times. Specifically, correct labeling of inner tiers is obtained when phosphorylase does not detach before ∼200 residues (required to expose tier 9), but this gives a wrong time course for glycogen IE. Conversely, a correct time course for IE is obtained with a relatively low interaction time (e.g., uniformly distributed in the range of 1 to 25, i.e., mean of 13 residues), but this hinders the incorporation of the label in inner tiers (simulations not shown). To obtain an exponential growth time course for IE and an LD-specific label incorporation in agreement with experimental results, it is thus necessary to include constraints on the probability distribution of enzyme interaction time. Specifically, if the detachment of phosphorylase is described as a memorylessness change of state occurring with probability p, then the appropriate probability distribution is geometric. This means that the probability for phosphorylase to attack k residues is given by (1−p)k−1p for k=1,2,3,... (note that the mean value of the distribution is 1/p, as shown in the figure). The value of p can be estimated by calculating the integral of the probability distribution in the interval given by the number of residues contained in a specific limit dextrin, knowing its percent enrichment. The best result of both time course and tier labeling pattern is obtained assuming a mean value (1/p) nearly equal to the length of a glucosyl chain, i.e., 13 residues, which is the value used for all subsequent simulations.
The structural properties of the simulated mature glycogen β-particle are found to be in good agreement with the known molecular structure of the polysaccharide (Table 2). Some small deviations are found within the external tiers of the molecule compared with the Whelan model,2 which predicts that the twelfth tier is the last one and that it is fully complete. Simulations show instead that the twelfth tier is nearly 90% complete, and the thirteenth tier is slightly (6%) populated. This is due to the stochastic character of the turnover process in the present model, and means that the homogeneity of the molecule is not complete near its surface. Interestingly, this conclusion stems from the different mobility of GS and GP. Indeed, almost complete homogeneity of the glycogen molecule (i.e., fully populated twelfth tier) is obtained in simulations where the two enzymes are assumed to displace at the same time from an attacked chain (data not shown). However, this assumption of equal interaction times of GS and GP could not be accepted due to the failure of the label incorporation process.
Table 2. Theoretical and simulated glucosyl residue content in each tier of the glycogen molecule.
|
Glucosyl residues |
|||||
|---|---|---|---|---|---|
| Tier | Theoretical maximum | Whelan model | Simulation | Simulated populationa (%) | Simulated populationb (%) |
| 1 | 13 | 13 | 13 | 100 | 0.024 |
| 2 | 26 | 26 | 26 | 100 | 0.049 |
| 3 | 52 | 52 | 52 | 100 | 0.098 |
| 4 | 104 | 104 | 104 | 100 | 0.19 |
| 5 | 208 | 208 | 208 | 100 | 0.39 |
| 6 | 416 | 416 | 416 | 100 | 0.78 |
| 7 | 832 | 832 | 832 | 100 | 1.56 |
| 8 | 1,664 | 1,664 | 1,664 | 100 | 3.13 |
| 9 | 3,328 | 3,328 | 3,328 | 100 | 6.25 |
| 10 | 6,656 | 6,656 | 6,656 | 100 | 12.5 |
| 11 | 13,312 | 13,312 | 13,304 | >99.9 | 25.0 |
| 12 | 26,624 | 26,624 | 23,286 | 87.5 | 43.7 |
| 13 | 53,248 | 0 | 3,340 | 6.3 | 6.3 |
The simulated values are in good agreement with the prediction of the Whelan model for β-glycogen. The surface of the molecule (i.e., the part undergoing turnover) is characterized by a lower homogeneity compared with the prediction of the Whelan model, which is identified in the outermost ∼3,300 glucosyl residues belonging to the thirteenth tier.
As compared with the theoretical maximum.
Relative to the total glucose content in the mature glycogen molecule given by the simulations (53,299 glucosyl residues).
Validation of the model is obtained by comparing the simulated IE with the experimental time course of [1-13C]glucose incorporation into human brain glycogen.5 The fit between simulated and experimental labeling process for different infusion durations is shown in Figure 5. The assumptions made during model design are found to be compatible with the kinetics of the labeling process, as evidenced by the qualitative and quantitative agreement between simulated and experimental IE time courses. The simulated curves are nearly indistinguishable from exponential grow and decay trends, which were previously used to fit experimental results (compare Figure 5 with Figure 3 in ref. 5). However, only the present model provides additional information behind the fit of total label content. These include localization of the label within the glycogen molecule, which is examined next.
Figure 5.

Simulated and experimental time course of label incorporation in glycogen for different infusion durations. Fitting between simulated and nuclear magnetic resonance (NMR) experimental data of 13C labeled glucose content within the glycogen molecule corresponding to total infusion time of (A) 6 hours, (B) 11 hours, (C) 22 hours, (D) 40 hours, (E) 48 hours, and (F) 50 hours. The simulated curves are obtained by rescaling the label content of a single glycogen molecule to a pool of glycogen molecules and assuming a total glycogen concentration of 3.5 μmol/g. The experimental data points (open squares) represent the 13C-NMR measurements obtained in human brain by Oz et al.5 The error bar on experimental data is assumed to be fixed at 0.4 μmol/g reflecting 20% sensitivity on individual measurements (Dr Gulin Oz, personal communication). For details about the [1-13C]glucose infusion protocol and subject management, see ref. 5.
The detectability of net glycogen utilization is examined by studying the simulated label localization underlying the changes from immediately after the end of the labeled glucose infusion to the stimulation onset several hours later. Figure 6 shows the label loss relative to the entire glycogen molecule as well as that relative to the last tier as a function of the delay between end of infusion and stimulation onset. Notably, most if not all of the label loss during the above-mentioned delay occurs in the external tier of the molecule, which is expected as this tier displays the fastest turnover rate. The disappearance of labeled glucose strongly depends on the delay before stimulation onset, while it is less dependent on the total infusion duration. The fraction of label loss spans values from 7% to 35% in correspondence to delays in the range of 7 to 11.5 hours, which identify the time intervals adopted in the 13C-NMR spectroscopy experiment (Table 3). Thus, the distribution of the label inside the glycogen molecule is substantially and inhomogeneously altered after cessation of infusion, with label retention restricted to the more internal tiers. The requirement for the above-mentioned delay was originally rationalized by the need for returning of plasma 13C-glucose to natural abundance.5 This was a reasonable attempt to maximize the detectability of potential stimulus-induced glycogen utilization, according to the idea that glucosyl units removed from glycogen during stimulation would have been replaced by unlabeled glucose. The simulations shown in Figure 6 indicate that this approach has the drawback of unlabeling the more active regions of the glycogen molecule. This suggests that the utilization of glycogen can be better estimated by measuring the change in the rate of label incorporation during the stimulation without stopping the infusion, or alternatively the change in the rate of label washout during the stimulation immediately after stopping the infusion.
Figure 6.

Simulated label localization at cessation of the infusion and at stimulation onset. Localization of labeled glucosyl residues as a function of particle radius corresponding to (A) 11 hours infusion without and with 8 hours delay, (B) 22 hours infusion without and with 11.5 hours delay, (C) 22 hours infusion without and with 7 hours delay, (D) 40 hours infusion without and with 8.4 hours delay, and (E) 50 hours infusion without and with 7.4 hours delay. These simulations identify the actual data sets obtained from experimental measurements (see Table 3). Glucosyl residue localization for the steady-state condition (i.e., all unlabeled) is showed for comparison in each plot. (F) Percent label loss for the different experiments. Physiologic brain stimulation initiated from 7 to 11.5 hours after cessation of glucose infusion, which was necessary for return of plasma [1-13C]glucose to natural abundance. The interval between the cessation of the infusion and the presentation of the stimulation substantially alters the distribution of the label within the glycogen molecule. In particular, the last tier is the most significantly affected with a reduction in labeled glucose ranging from 7% to 35%. On the contrary, the label incorporated in the inner tiers is almost totally retained in the molecule (maximum decrease <1%).
Table 3. Experimentally determined changes of 13C-labeled glycogen in stimulated human visual cortex.
| Infusion duration (hours) | Delay (hours) | Labeled glycogen content change (%) |
|---|---|---|
| 11 | 8 | +22 |
| 22 | 11.5 | +1 |
| 22 | 7 | −2 |
| 40 | 8.4 | −4 |
| 50 | 7.4 | −5 |
Percent changes of 13C label retention in human brain glycogen after 20 minutes of visual stimulation are those measured in individual experiments (Dr Gulin Oz, personal communication). Each row identifies a single subject.
Delays represent the temporal interval between cessation of infusion and onset of visual stimulation, which were necessary to allow complete washout of plasma [1-13C]glucose (see ref. 5). Only fractional enrichment measured before and after the stimulation is available.
Note the increase in apparent glycogen mobilization, which can be inferred by changes in 13C-labeled glycogen, for higher infusion times and lower delays.
Discussion
Glycogen is designed to maximize the capacity for storing a large amount of glucose in the least possible volume.2 At the molecular level, the polymeric structure of glycogen imposes a number of geometrical constraints on the enzymes involved in the incorporation and release of glucose, which is thought to be part of the optimization of its metabolic role in the availability of large amount of glucose in short time31 as well as fast recovery,28 which is attainable only if the outer tier is complete. By formalizing this principle, a biophysical model of the glycogen particle is developed, which reproduced the known high structural homogeneity of the molecule and the pattern of IE under the following two conditions: (1) synthase must preferentially work on internal chains (of course, acting on nonreducing ends) and (2) displacement of synthase from the attacked site is much more rapid than that of phosphorylase.
The model is validated with in vivo 13C-NMR data of labeled glucose incorporation obtained in human brain, and subsequently used to examine the change in glycogen content that could be inferred by measurements of 13C-glycogen level before and after stimulation.5 It is found that the turnover of glycogen is highly heterogeneous and it is restricted to a great extent within the outer tiers of the molecule. Thus, the isotopic incorporation of glucose into a complex macromolecule such as glycogen may result in erroneous conclusions if the structure of the molecule is not taken into account.15 The delays between cessation of [1-13C]glucose infusion and brain stimulation (7 to 11.5 hours in the 13C-NMR spectroscopy experiment) are found to disperse 7% to 35% of the label in the outer tiers. This means that any subsequent upregulation of glycogen breakdown might be underestimated because the glucose residues that are eventually mobilized are those with the fastest turnover at the periphery of the molecule, thus nonlabeled and hence undetected. Interestingly, there is a positive correlation between the observed signal changes and the total infusion time as well as the delay between end of infusion and stimulation (see Table 3). This makes particularly sense, as a long infusion time would result in full labeling of the outer tier, and a brief delay would avoid significant washout of the label in the outer more active tier, both effects concurring to increase the contrast and sensitivity of the experimental determination of glycogenolytic rate.
A limitation of the present study is that concentrations of plasma and brain glucose as well as tissue glycogen are assumed to be constant during the whole duration of the experiments. A closer examination of the simulations shown in Figure 5 indicates that simulated data match experimental measurements more consistently during the infusion than after (although the difference is very small). This might originate from changes in tissue glucose level, or alternatively, from a modest de novo glycogen synthesis probably due to the mild hyperglycemic conditions during infusion.10 These possibilities are distinct, as changes in brain glucose concentration would result in corresponding changes of glycogen turnover not glycogen content. In any case, it is possible to obtain a better fit of experimental data after the infusion (i.e., slow-down of label washout) if one assumes that during this time interval either (1) glycogen turnover decreases (due to decreased glucose level) or (2) total glycogen have increased (due to hyperglycemia), or a combination of both.
Unfortunately, without further assumptions the model cannot be used to directly analyze the stimulation-induced difference between the amount of glycogen that can be revealed by IE measurements versus that actually mobilized. Indeed, to simulate the activation of glycogenolysis, the reaction rate of phosphorylase relative to synthase has to be upregulated during stimulation compared with basal conditions. This imbalance is difficult to estimate in absolute terms: first, the reaction rates underlying net glycogen breakdown cannot be obtained without the prior knowledge of how much glycogen is mobilized, and second, there are multiple ways of upregulating net glycogen breakdown, as also the absolute rate of synthase is likely to change along with that of phosphorylase. Furthermore, it is presently unknown whether the mechanisms of action of synthase and phosphorylase change during stimulation because of, e.g., conformational changes of activated/phosphorylated enzymes. For example, a higher reaction rate of the phosphorylase might parallel a higher mobility of the enzyme, thus resulting in a degradation pattern confined to the surface of the glycogen molecule. Conversely, a low mobility of the phosphorylase results in transients ‘holes' in the molecule that are subsequently refilled by synthase, thus leading to a greater degradation of internal residues. Finally, as the velocity of glycogenolysis is likely limited to few minutes after the onset of stimulation,32 it cannot be established to what extent glycogen resynthesis hides the mobilization of the label. Indeed, synthase and phosphorylase may cooperate during the glycogen breakdown induced by stimulation by remaining close to each other.
The latter point is also relevant to the choice of stimulus duration during measurements of changes in glycogen content in different species. For example, cerebral metabolic rate in rodents is nearly threefold higher than in humans. The different carbohydrate demand may affect the rate and/or the amount of glycogen utilization. It has been suggested that the stimulus duration to be used in human experiments should be correspondingly made longer than that used in rodent experiments to obtain the same decrease in glycogen content.5 However, if glycogenolysis stops few minutes after stimulus onset, then any extra increase in stimulus duration would translate in net rise, not fall, of glycogen content (see discussion in ref. 9). Further research is required to evaluate the optimal, eventually stimulus specific, duration that minimizes the resynthesis-induced loss of sensitivity for the determination of glycogen utilization. It is suggested that retention of the polysaccharide is needed to preserve the available glycogenolytic response in the long term, thus net utilization before resynthesis can be as small as few percent.33 It has been previously showed by kinetic modeling that glycogen breakdown as small as 10% would be sufficient to modulate the cell glucose uptake to a significant extent.9 Specifically, by altering the intracellular level of hexose phosphates, glycogen metabolism can control the channeling of endogenous glucose both within an individual cell and between different cells.6 Accordingly, it has been suggested that the enzyme regulation which triggers the rapid phosphorolysis of glycogen is an efficient mechanism to control the glycolytic flux by balancing the availability of phosphorylated glucose.2 It is clear that a relatively modest reduction in glycogen content after stimulation makes glycogenolytic rate determination experimentally difficult, as few percent changes can be under the detection limit of most experimental techniques.
In conclusion, in the present paper a model of the glycogen molecule is developed and validated with experimental data of glycogen turnover in the human brain. The results indicate that 13C-MRS-based assessment of glycogenolysis during physiologic brain stimulation is model dependent. The model further provides several insights about the dependence of enzyme action on the structural properties of the glycogen molecule, which can be investigated experimentally. As a general principle, the determination of glycogen use by labeling methods will benefit from the analysis that is described here using biophysical modeling. Additionally, the present theoretical account can be used to estimate glycogen growth, turnover, and utilization in a variety of conditions. The study of the effects exerted by molecular structure on the patterns of glycogen mobilization appears to be prerequisite for design of labeling experiment and proper evaluation of IE data.
Acknowledgments
The author gracefully thanks Dr Gulin Oz for the 13C-MRS data and for her constructive suggestions on the paper. Dr Silvia Mangia and Dr Federico Giove are cordially acknowledged for their useful comments on this work.
The author declares no conflict of interest.
References
- Preiss J, Walsh DA.The comparative biochemistry of glycogen and starch metabolism and their regulationIn: Ginsburg V, (eds). Biology of Complex Carbohydrates John Wiley and Sons: New York; 1981199–314. [Google Scholar]
- Melendez-Hevia E, Waddell TG, Shelton ED. Optimization of molecular design in the evolution of metabolism: the glycogen molecule. Biochem J. 1993;295:477–483. doi: 10.1042/bj2950477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez R, Melendez-Hevia E, Cascante M. How did glycogen structure evolve to satisfy the requirement for rapid mobilization of glucose? A problem of physical constraints in structure building. J Mol Evol. 1997;45:446–455. doi: 10.1007/pl00006249. [DOI] [PubMed] [Google Scholar]
- Roach PJ. Glycogen and its metabolism. Curr Mol Med. 2002;2:101–120. doi: 10.2174/1566524024605761. [DOI] [PubMed] [Google Scholar]
- Oz G, Seaquist ER, Kumar A, Criego AB, Benedict LE, Rao JP, et al. Human brain glycogen content and metabolism: implications on its role in brain energy metabolism. Am J Physiol Endocrinol Metab. 2007;292:E946–E951. doi: 10.1152/ajpendo.00424.2006. [DOI] [PubMed] [Google Scholar]
- DiNuzzo M, Maraviglia B, Giove F. Why does the brain (not) have glycogen. Bioessays. 2011;33:319–326. doi: 10.1002/bies.201000151. [DOI] [PubMed] [Google Scholar]
- Dienel GA, Cruz NF. Astrocyte activation in working brain: energy supplied by minor substrates. Neurochem Int. 2006;48:586–595. doi: 10.1016/j.neuint.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Oz G, Kumar A, Rao JP, Kodl CT, Chow L, Eberly LE, et al. Human brain glycogen metabolism during and after hypoglycemia. Diabetes. 2009;58:1978–1985. doi: 10.2337/db09-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNuzzo M, Mangia S, Maraviglia B, Giove F. Glycogenolysis in astrocytes supports blood-borne glucose channeling not glycogen-derived lactate shuttling to neurons: evidence from mathematical modeling. J Cereb Blood Flow Metab. 2010;30:1895–1904. doi: 10.1038/jcbfm.2010.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz G, Tesfaye N, Kumar A, Deelchand DK, Eberly LE, Seaquist ER. Brain glycogen content and metabolism in subjects with type 1 diabetes and hypoglycemia unawareness. J Cereb Blood Flow Metab. 2012;32:256–263. doi: 10.1038/jcbfm.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Yu AC, Chan PH, Sharp FR. Glutamate increases glycogen content and reduces glucose utilization in primary astrocyte culture. J Neurochem. 1990;54:490–496. doi: 10.1111/j.1471-4159.1990.tb01898.x. [DOI] [PubMed] [Google Scholar]
- Youn JH, Bergman RN. Patterns of glycogen turnover in liver characterized by computer modeling. Am J Physiol. 1987;253:E360–E369. doi: 10.1152/ajpendo.1987.253.4.E360. [DOI] [PubMed] [Google Scholar]
- Elsner P, Quistorff B, Hansen GH, Grunnet N. Partly ordered synthesis and degradation of glycogen in cultured rat myotubes. J Biol Chem. 2002;277:4831–4838. doi: 10.1074/jbc.M108226200. [DOI] [PubMed] [Google Scholar]
- Melendez R, Melendez-Hevia E, Mas F, Mach J, Cascante M. Physical constraints in the synthesis of glycogen that influence its structural homogeneity: a two-dimensional approach. Biophys J. 1998;75:106–114. doi: 10.1016/S0006-3495(98)77498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahorski SR, Rogers KJ. The incorporation of glucose into brain glycogen and the activities of cerebral glycogen phosphorylase and synthetase: some effects of amphetamine. J Neurochem. 1974;23:579–587. doi: 10.1111/j.1471-4159.1974.tb06062.x. [DOI] [PubMed] [Google Scholar]
- Stetten D, Jr., Stetten MR. Glycogen metabolism. Physiol Rev. 1960;40:505–537. doi: 10.1152/physrev.1960.40.3.505. [DOI] [PubMed] [Google Scholar]
- Stetten MR, Stetten D., Jr. A study of the nature of glycogen regeneration in the intact animal. J Biol Chem. 1954;207:331–340. [PubMed] [Google Scholar]
- Stetten MR, Stetten D., Jr. Glycogen regeneration in vivo. J Biol Chem. 1955;213:723–732. [PubMed] [Google Scholar]
- Watanabe H, Passonneau JV. Factors affecting the turnover of cerebral glycogen and limit dextrin in vivo. J Neurochem. 1973;20:1543–1554. doi: 10.1111/j.1471-4159.1973.tb00272.x. [DOI] [PubMed] [Google Scholar]
- Lomako J, Lomako WM, Whelan WJ, Dombro RS, Neary JT, Norenberg MD. Glycogen synthesis in the astrocyte: from glycogenin to proglycogen to glycogen. FASEB J. 1993;7:1386–1393. doi: 10.1096/fasebj.7.14.8224611. [DOI] [PubMed] [Google Scholar]
- Lomako J, Lomako WM, Whelan WJ. Proglycogen: a low-molecular-weight form of muscle glycogen. FEBS Lett. 1991;279:223–228. doi: 10.1016/0014-5793(91)80154-u. [DOI] [PubMed] [Google Scholar]
- Katz A. Glycogenin, proglycogen, and glycogen biogenesis: what's the story. Am J Physiol Endocrinol Metab. 2006;290:E757–E758. doi: 10.1152/ajpendo.00532.2005. [DOI] [PubMed] [Google Scholar]
- Nelson TE, White RC, Watts TE. The action of the glycogen debranching enzyme system in a muscle protein particle. Biochem Biophys Res Commun. 1972;47:254–259. doi: 10.1016/s0006-291x(72)80036-6. [DOI] [PubMed] [Google Scholar]
- Skurat AV, Roach PJ.Regulation of Glycogen SynthesisIn: LeRoith D, Taylor SI, Olefsky JM, (eds). Diabetes Mellitus: A Fundamental and Clinical Text Lippincott Williams & Wilkins: Philadelphia; 2004317–333. [Google Scholar]
- Madsen NB, Cori CF. The binding of glycogen and phosphorylase. J Biol Chem. 1958;233:1251–1256. [PubMed] [Google Scholar]
- Gunja-Smith Z, Marshall JJ, Mercier C, Smith EE, Whelan WJ. A revision of the Meyer-Bernfeld model of glycogen and amylopectin. FEBS Lett. 1970;12:101–104. doi: 10.1016/0014-5793(70)80573-7. [DOI] [PubMed] [Google Scholar]
- Goldsmith E, Sprang S, Fletterick R. Structure of maltoheptaose by difference Fourier methods and a model for glycogen. J Mol Biol. 1982;156:411–427. doi: 10.1016/0022-2836(82)90336-9. [DOI] [PubMed] [Google Scholar]
- Melendez R, Melendez-Hevia E, Canela EI. The fractal structure of glycogen: A clever solution to optimize cell metabolism. Biophys J. 1999;77:1327–1332. doi: 10.1016/S0006-3495(99)76982-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LN, Acharya KR, Jordan MD, McLaughlin PJ. Refined crystal structure of the phosphorylase-heptulose 2-phosphate-oligosaccharide-AMP complex. J Mol Biol. 1990;211:645–661. doi: 10.1016/0022-2836(90)90271-M. [DOI] [PubMed] [Google Scholar]
- Buschiazzo A, Ugalde JE, Guerin ME, Shepard W, Ugalde RA, Alzari PM. Crystal structure of glycogen synthase: homologous enzymes catalyze glycogen synthesis and degradation. EMBO J. 2004;23:3196–3205. doi: 10.1038/sj.emboj.7600324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LN. Glycogen phosphorylase: control by phosphorylation and allosteric effectors. FASEB J. 1992;6:2274–2782. doi: 10.1096/fasebj.6.6.1544539. [DOI] [PubMed] [Google Scholar]
- DiNuzzo M, Mangia S, Maraviglia B, Giove F. The Role of Astrocytic Glycogen in Supporting the Energetics of Neuronal Activity. Neurochem Res. 2012;37:2432–2438. doi: 10.1007/s11064-012-0802-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman RG, Hyder F, Rothman DL. Cerebral energetics and the glycogen shunt: neurochemical basis of functional imaging. Proc Natl Acad Sci USA. 2001;98:6417–6422. doi: 10.1073/pnas.101129298. [DOI] [PMC free article] [PubMed] [Google Scholar]