

Abstract
Mutations in the MCOLN1 gene cause mucolipidosis type IV (MLIV), a severely debilitating, autosomal recessive, lysosomal storage disorder. Approximately 80% of patients with MLIV are of Ashkenazi Jewish (AJ) descent, and two mutations, IVS3−2A→G and 511del6434, account for >95% of the mutant alleles in this population. To determine the carrier frequencies of these two mutations, 2,029 anonymous, unrelated, unaffected AJ individuals from the greater New York metropolitan area were screened. A multiplex PCR method coupled with allele-specific oligonucleotide hybridization was developed, to enable large-scale screening. The frequencies of the IVS3−2A→G and 511del6434 mutations were 0.54% and 0.25%, respectively, for a combined carrier frequency of 0.79%, or 1 in 127 individuals (95% CI 0.40%–1.17%). The addition of both AJ mutations causing this neurodegenerative disorder should be considered for prenatal carrier screening in this population.
Mucolipidosis type IV (MLIV [MIM 252650]) is a neurodegenerative lysosomal storage disorder that is inherited as an autosomal recessive trait. Clinically, the disease, which occurs primarily among Ashkenazi Jewish (AJ) individuals, is characterized by growth and psychomotor retardation, as well as ophthalmologic abnormalities, which include corneal clouding, progressive retinal degeneration, and strabismus (Berman et al. 1974; Riedel et al. 1985). There is clinical variability, but most patients never develop the ability to speak or walk and remain at a developmental level of age 1–2 years. Although the disorder is associated with the accumulation of cytoplasmic storage bodies, normal levels of lysosomal hydrolases are present, with no specific identifiable storage compound (Amir et al. 1987). Electron photomicrographs of tissue biopsies from patients with MLIV have shown a widespread heterogeneous accretion of lipids and water-soluble compounds, indicating that the etiology of the disease is probably not related to the deficiency of a particular lysosomal enzyme but rather to a defect in endocytosis (Bargal and Bach 1997; Chen et al. 1998). It has been suggested that many cases may not be properly diagnosed, because of the variability in the observed phenotype and the lack of an indicative storage compound.
The MLIV locus was mapped to chromosome 19p13 in AJ families, and haplotype analysis of these families indicated that there were two founder chromosomes that accounted for 73% (major haplotype) and 23% (minor haplotype)—a total of 96%—of the carrier chromosomes in the AJ population (Slaugenhaupt et al. 1999; Sun et al. 2000). Recently, the gene that is mutated in patients with MLIV, MCOLN1 (MIM 605248), was isolated, and the two AJ founder mutations were identified. These mutations were (1) a splicing mutation, IVS3−2A→G, which resulted in skipping of exon 4, with the subsequent loss of the reading frame, and (2) a 6,434-bp deletion spanning genomic nucleotides 511–6,944, designated “511del6434,” which included the 5′ flanking region of the gene through exon 7 (Bargal et al. 2000; Bassi et al. 2000; Sun et al. 2000). Since both mutations resulted in the loss of a functional gene product, the severity of the phenotype was similar in patients who were homoallelic or heteroallelic for these mutations.
The MCOLN1 gene encodes the putative protein mucolipin-1, which is 580 amino acids long and is somewhat homologous to the polycystins, a family of cation channels (Somlo and Ehrlich 2001). In Caenorhibditis elegans, studies of cup-5, the mucolipin-1 homolog, have indicated that these proteins are vesicle membrane–associated regulators of the endocytic pathway, where loss of cup-5 function results in large lysosomal or late endosomal structures that exhibit both an increase in the rate of uptake of fluid phase markers and a decrease in the ability to degrade proteins (Fares and Greenwald 2001). The function of mucolipin-1 that is predicted on the basis of these studies is consistent with the lysosomal pathology observed in cultured cells derived from patients with MLIV.
To determine the frequency of carriers for MLIV in the AJ population, we screened for both the major IVS3−2A→G and the minor 511del6434 mutations in genomic DNAs isolated from blood samples obtained from 2,029 unrelated AJ individuals from the New York metropolitan area who were referred for prenatal carrier testing for other genetic disorders prevalent in the AJ population. DNA was extracted from peripheral blood obtained by venipuncture and archived with informed consent for use in research studies. All personal identifiers were removed, and the samples were tested anonymously. A multiplex PCR amplification was performed for 28 cycles with 100 ng of genomic DNA in a volume of 50 μl containing 10 pmol of each primer tagged with a 5′ universal primer sequence (UPS) (Shuber et al. 1995). The primer sequences designed to detect the major mutation were MLIV-1UPS (5′-GCGGTCCCAAAAGGGTCAGTATCTTCCATGCTGTGGACCA-3′) and MLIV-2UPS (5′-GCGGTCCCAAAAGGGTCAGTAACAGTGAAGCCTCGTCCTG-3′), and the primer sequences designed to detect the minor mutation were MLIV-3UPS (5′-GCGGTCCCAAAAGGGTCAGTGGCAGCTTTCTCAATGAAGG-3′ and MLIV-4UPS (5′-GCGGTCCCAAAAGGGTCAGTTCACCGTGCTGGAAGACACT-3′). In addition, 100 mM of each dNTP (Roche Molecular Biochemicals), 5 U of Taq DNA polymerase (Roche), 10 mM Tris-HCl, 50 mM KCl, 0.1% TritonX-100, and 1.5 mM MgCl2 were added to the PCRs. Dimethyl sulfoxide (Sigma) was added to a final concentration of 4%, because of the high (65%) GC content of the region amplified for analysis of the major mutation. The primers were designed from the genomic sequence of the MCOLN1 gene (Sun et al. 2000; GenBank accession number AF287270) to amplify a 410-bp PCR product encompassing the IVS3−2A→G mutation and a 396-bp PCR product when a 511del6434 mutation was present (fig. 1). Therefore, the multiplex PCR amplification of DNA from individuals with neither mutation and individuals who were heterozygous or homozygous for the IVS3−2A→G mutation would result in a single PCR product of 410 bp (fig. 1). Amplification of DNA from individuals who were carriers of the 511del6434 mutation or heteroallelic for both mutations would result in PCR products of 396 bp and 410 bp, respectively, whereas amplification of DNA from individuals who were homozygous for the 511del6434 mutation would result only in products of 396 bp (fig. 1). Control DNAs were obtained from the fibroblast cell lines GM02527, GM02529, and GM02525 (Coriell NIGMS Human Genetic Cell Repository), which were derived from patients with MLIV who were homozygous for the IVS3−2A→G mutation, compound heterozygous for the IVS3−2A→G/511del6434 mutations, and homozygous for the 511del6434 mutation, respectively (Bassi et al. 2000).
Figure 1.
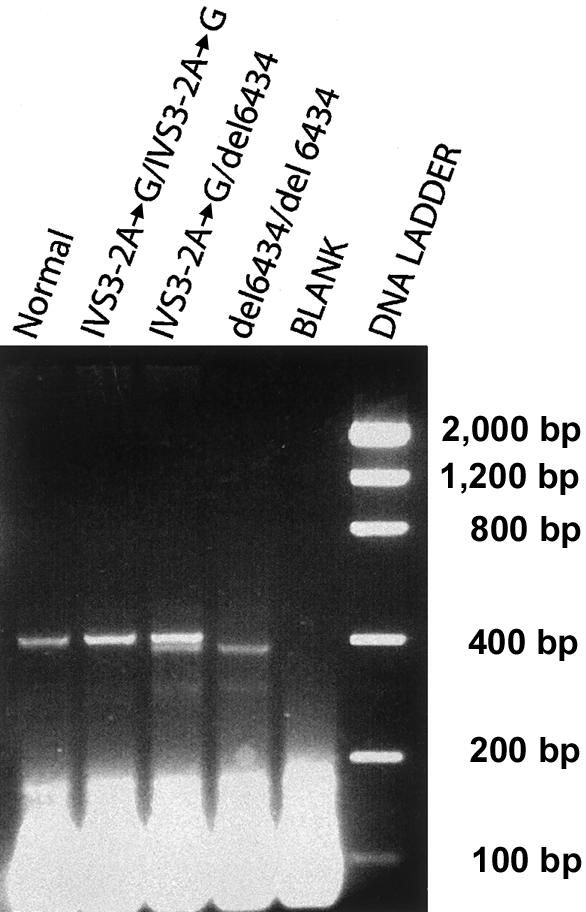
Multiplex PCR of the MCOLN1 gene; 2% agarose gel showing the products that result from the multiplex PCR amplification of the MCOLN1 gene with the IVS3−2A→G and 511del6434 primer pairs. Note that the 511del6434 primers amplify a product only when the deletion is present.
Aliquots (4 μl) of the PCR products were blotted in triplicate onto 8×12 cm Hybond −N+ membranes (Amersham Pharmacia Biotech) through use of a Biomek 2000 automated pipetting workstation (Beckman Coulter). Hybridization was performed with the following allele-specific oligonucleotides (ASOs): for the IVS3−2A→G mutation, 5′-TCTCTGCCCACAGTACCTG-3′ (normal) and 5′-TCTCTGCCCACGGTACCTG-3′ (mutant); and for the 511del6434 mutation, 5′-CCTGGGCTCAACAAAGCAC-3′. For radioactive detection, the membranes were hybridized, for a length of time ranging from 2 h to overnight, with ∼106 counts per minute of γ-[32P]ATP end-labeled probe/ml and 10-fold molar excess of the mutant or wild-type competitor oligonucleotide for the IVS3−2A→G mutation. The membranes were then washed sequentially in 5× saline sodium citrate (SSC) for 5 min at room temperature and in 5×SSC for 5 min at 45°C, followed by a wash in 0.1×SSC/0.1% SDS for 5 min at 45°C. Filters were then exposed to autoradiography by use of Biomax MR film (Kodak) (fig. 2).
Figure 2.

ASOs of the two AJ MCOLN1 mutations. Shown are autoradiographs of dot blots hybridized with the normal IVS3−2A→G ASO (left), the mutant IVS3−2A→G ASO (middle), and the 511del6434 ASO (right). PCR samples 1–95 were amplified from genomic DNAs; sample 96 is the ddH2O control. Sample 1 was amplified from the IVS3−2A→G homozygote (GM02527), sample 8 was amplified from the 511del6434 homozygote (GM02525), samples 18 and 67 were amplified from carriers of the IVS3−2A→G mutation, and sample 49 was amplified from a carrier of the 511del6434 mutation.
For the major IVS3−2A→G mutation, 2,029 AJ individuals were screened and 11 carriers were identified, corresponding to a frequency of 0.54%, or 1/184. Five carriers for the minor 511del6434 mutation were identified in a total of 2,029 AJ individuals tested, corresponding to a frequency of 0.25%, or 1/406. The combined frequency of the two mutations in the present study was, therefore, 0.79%, or 1/127 individuals (95% CI 0.40%–1.17%) (table 1). Because the haplotypes that correspond to these two mutations account for 96% of the carriers in the AJ population, the prevalences of the two mutations were 66% and 30% for the major and minor mutations, respectively. These percentages are in agreement with the results of Sun and colleagues (2000), who reported that the major and minor haplotypes are present on 73% and 23% of carrier chromosomes, respectively.
Table 1.
Carrier Frequency of the MCOLN1 Mutations Among Unrelated AJ Individuals
|
IVS3−2A→G |
511del6434 |
Combined |
|||||
| Study(Population) | No. of Individuals | No. ofCarriers | CarrierPercentage(Frequency) | No. ofCarriers | CarrierPercentage(Frequency) | No. ofCarriers | CarrierPercentage(Frequency) |
| Bargal et al. 2001 (Israel) | 2,000 | 17 | .85 (1/117) | 1 | .05 (1/2,000) | 19a | .95 (1/106)b |
| Wang et al. 2001 (New York) | 123 | 2 | 1.62 (1/62) | 0 | 0 | 2 | 1.62 (1/62) |
| Present study (New York) | 2,029 |
11 |
.54 (1/184) | 5 |
.25 (1/406) | 16 |
.79 (1/127)c |
| Combined | 4,152 | 30 | .72 (1/138) | 6 | .14 (1/692) | 37 | .89 (1/112) |
This number is based on an estimate of a 95% detection rate of MLIV carriers.
95% CI = .52%–1.39%.
95% CI = .40%–1.17%.
In a recent report (Wang et al. 2001), a small study of 123 AJ individuals was conducted, and a carrier frequency of 1/61 was reported for the major IVS3−2A→G mutation; however, no carriers for the minor 511del6434 mutation were identified (table 1). Therefore, the limited sample size prevented a reliable estimate of the frequency and distribution of MLIV carriers. Recently, Bargal et al. (2001) reported a carrier frequency of 1/100 for the two mutations in a sample of 2,000 AJ individuals from Israel (table 1). The distribution of the mutations was 94% for the major mutation and 6% for the minor mutation. This apparent disparity in the frequency of carriers between the AJ constituencies of the New York metropolitan area and Israel is not clear; however, the major mutation is more prevalent in the Israeli population and probably accounts for the observed higher frequency of total carriers of MLIV. These findings also may be indicative of different patterns of immigration of AJ individuals into the United States and Israel and/or may reflect distinct regional origins of the founders of the major and the minor mutation.
In a study of 17 Israeli AJ families with MLIV, familial origins were traced back to Poland or to neighboring Lithuania (Raas-Rothschild et al. 1999). The authors speculated that the underrepresentation of ultra-orthodox families among the 80 AJ families with MLIV worldwide might indicate a recent origin for the mutation, around the 18th and 19th centuries, in a founder that belonged to a secular family. These findings are likely to be specific for the major mutation, on the basis of its higher prevalence (94%) among the Israeli AJ population. Discordant carrier rates between Israeli and American Ashkenazi subpopulations have been reported recently for mutations in the Connexin 26 gene, which cause nonsyndromic sensorineural recessive deafness (Dong et al. 2001). Variation in the frequency of MLIV carriers between the United States and Israel thus provides further evidence that Ashkenazi subpopulations have not reached equilibrium.
A more recent origin for the two AJ MLIV founder mutations would explain why the frequency of carriers for MLIV is not as high as the frequencies of other mutations that are common in the AJ population, including some that originated in or were introduced into the European Jewish population >1,000 years ago (Diaz et al. 2000). The carrier frequencies of these disorders range from 1/18, for Gaucher disease (Beutler and Grabowski 2000), to 1/107, for Bloom syndrome (Li et al. 1998). The observed frequency of MLIV carriers within a large cohort of AJ individuals from the New York metropolitan area is 1/127, slightly lower than the frequency of disorders for which carrier screening is currently available. However, in light of the neurologic severity of the MLIV phenotype, for which there is no available treatment, and the fact that the two mutations account for >95% of carriers, carrier screening for the two mutations should be considered for this population.
Acknowledgments
The authors would like thank all the individuals who agreed to participate anonymously in this study. We also thank Asghar Bajwa and Jing Xu for their technical assistance. This work was supported, in part, by grants from the National Institutes of Health, including a Merit Award (grant 5 R37 DK34045), grant 5 M01 RR00071 to the Mount Sinai General Clinical Research Center, and grant 5 P30 HD28822 to the Mount Sinai Child Health Research Center.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Genbank, http://www.ncbi.nlm.nih.gov/Genbank/ (for Homo sapiens MCOLN1, complete coding sequence [accession number AF287270])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for MLIV [MIM 252650] and mucolipin-1 [MIM 605248])
References
- Amir N, Zlotogora J, Bach G (1987) Mucolipidosis type IV: clinical spectrum and natural history. Pediatrics 79:953–959 [PubMed] [Google Scholar]
- Bargal R, Avidan N, Ben-Asher E, Olender Z, Zeigler M, Frumkin A, Raas-Rothschild A, Glusman G, Lancet D, Bach G (2000) Identification of the gene causing mucolipidosis type IV. Nat Genet 26:118–123 [DOI] [PubMed] [Google Scholar]
- Bargal R, Avidan N, Olender T, Ben Asher E, Zeigler M, Raas-Rothschild A, Frumkin A, Ben-Yoseph O, Friedlender Y, Lancet D, Bach G (2001) Mucolipidosis type IV: novel MCOLN1 mutations in Jewish and non-Jewish patients and the frequency of the disease in the Ashkenazi Jewish population. Hum Mutat 17:397–402 [DOI] [PubMed] [Google Scholar]
- Bargal R, Bach G (1997) Mucolipidosis type IV: abnormal transport of lipids to lysosomes. J Inherit Metab Dis 20:625–632 [DOI] [PubMed] [Google Scholar]
- Bassi MT, Manzoni M, Monti E, Pizzo MT, Ballabio A, Borsani G (2000) Cloning of the gene encoding a novel integral membrane protein, mucolipin-1, and identification of the two founder mutations causing mucolipidosis type IV. Am J Hum Genet 67:1110–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman ER, Livni N, Shapira E, Merin S, Levij IS (1974) Congenital corneal clouding with abnormal systemic storage bodies: a new variant of mucolipidosis. J Pediatr 84:519–526 [DOI] [PubMed] [Google Scholar]
- Beutler E, Grabowski GA (2000) Gaucher’s disease. In: Beaudet A, Childs B, Kinzler K, Scriver CR, Sly WS, Valle D, Vogelstein B (eds) The metabolic and molecular bases of inherited disease, 8th ed. McGraw-Hill, New York, pp 3635–3668 [Google Scholar]
- Chen CS, Bach G, Pagano R E (1998) Abnormal transport along the lysosomal pathway in mucolipidosis type IV disease. Proc Nat Acad Sci USA 95:6373–6378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz GA, Gelb BD, Risch N, Nygaard TG, Frisch A, Cohen IJ, Miranda CS, Amaral O, Maire I, Poenaru L, Caillaud C, Weizberg M, Mistry P, Desnick RJ (2000) Gaucher disease: the origins of the Ashkenazi Jewish N370S and 84GG acid β-glucosidase mutations. Am J Hum Genet 66:1821–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Katz DR, Eng CM, Kornreich R, Desnick RJ (2001) Nonradioactive detection of the common Connexin 26 167delT and 35delG mutations and frequencies among Ashkenazi Jews. Mol Genet Metab 73:160–163 [DOI] [PubMed] [Google Scholar]
- Fares H, Greenwald I (2001)Regulation of endocytosis by CUP-5, the Caenorhabditis elegans mucolipin-1 homolog. Nat Genet 28:64–68 [DOI] [PubMed] [Google Scholar]
- Li L, Eng C, Desnick RJ, German J, Ellis NA (1998) Carrier frequency of the Bloom syndrome blmAsh mutation in the Ashkenazi Jewish population. Mol Genet Metab 64:286–290 [DOI] [PubMed] [Google Scholar]
- Raas-Rothschild A, Bargal R, DellaPergola S, Zeigler M, Bach G (1999) Mucolipidosis type IV: the origin of the disease in the Ashkenazi Jewish population. Eur J Hum Genet 7:496–498 [DOI] [PubMed] [Google Scholar]
- Riedel KG, Zwaan J, Kenyon KR, Kolodny EH, Hanninen L, Albert DM (1985) Ocular abnormalities in mucolipidosis IV. Am J Ophthalmol 99:125–136 [DOI] [PubMed] [Google Scholar]
- Shuber AP, Grondin VJ, Klinger KW (1995) A simplified procedure for developing multiplex PCRs. Genome Res 5:488–493 [DOI] [PubMed] [Google Scholar]
- Slaugenhaupt SA, Acierno JS Jr, Helbling LA, Bove C, Goldin E, Bach G, Schiffmann R, Gusella JF (1999) Mapping of the mucolipidosis type IV gene to chromosome 19p and definition of founder haplotypes. Am J Hum Genet 65:773–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlo S, Ehrlich B (2001) Human disease: calcium signaling in polycystic kidney disease. Curr Biol 11:R356–R360 [DOI] [PubMed] [Google Scholar]
- Sun M, Goldin E, Stahl S, Falardeau JL, Kennedy JC, Acierno JS Jr, Bove C, Kaneski CR, Nagle J, Bromley MC, Colman M, Schiffmann R, Slaugenhaupt SA (2000) Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum Mol Genet 9:2471–2478 [DOI] [PubMed] [Google Scholar]
- Wang ZH, Zeng B, Pastores GM, Raksadawan N, Ong E, Kolodny EH (2001) Rapid detection of the two common mutations in Ashkenazi Jewish patients with mucolipidosis type IV. Genet Test 5:87–92 [DOI] [PubMed] [Google Scholar]