

Abstract
Pancreatic cancer is the fifth leading cause of cancer death in the United States. Nearly every person diagnosed with pancreatic cancer will die from it, usually in <6 mo. Familial clustering of pancreatic cancers is commonly recognized, with an autosomal dominant inheritance pattern in ∼10% of all cases. However, the late age at disease onset and rapid demise of affected individuals markedly hamper collection of biological samples. We report a genetic linkage scan of family X with an autosomal dominant pancreatic cancer with early onset and high penetrance. For the study of this family, we have developed an endoscopic surveillance program that allows the early detection of cancer and its precursor, before family members have died of the disease. In a genomewide screening of 373 microsatellite markers, we found significant linkage (maximum LOD score 4.56 in two-point analysis and 5.36 in three-point analysis) on chromosome 4q32-34, providing evidence for a major locus for pancreatic cancer.
Adenocarcinoma of the pancreas typifies the most dreaded of all cancers, because it is difficult to detect, early to metastasize, and resistant to treatment (Schnall and Macdonald 1996). Of the estimated 29,200 new cases currently diagnosed in the United States each year, 28,900 will die of the disease, usually <4–6 mo after diagnosis. Although pancreatic cancer is relatively common, very few dominant genetic or environmental risk factors have been identified (Kernan et al. 1999). There is, however, growing epidemiological evidence for important genetic susceptibility factors (Silverman et al. 1999; Tersmette et al. 2001). In addition, familial and inherited pancreatic cancer syndromes may provide clues to pancreatic cancer susceptibility (Lynch et al. 1990; Banke et al. 2000; Efthimiou et al. 2001; Rulyak and Brentnall 2001; Tersmette et al. 2001; Whitcomb et al. 2001).
Several familial cancer syndromes increase the risk of pancreatic cancer. The best-characterized include the hereditary nonpolyposis-colon-cancer syndrome (HNPCC [MIM 114500]) associated with mutations mainly on chromosomes 2 and 3, hereditary breast-ovarian cancer syndrome (BRCA2 [MIM 600185]) associated with the BRCA2 gene mutations on chromosome 13q, Peutz-Jeghers polyposis with mutations in the serine/threonine protein kinase 11 (STK11 [MIM 602216]) on chromosome 19p, and familial atypical multiple-mole-melanoma syndrome (FAMMM [MIM 600160]) with mutations on chromosome 9p (Efthimiou et al. 2001; Rulyak and Brentnall 2001; Whitcomb et al. 2001). In general, these syndromes increase the risk of pancreatic cancer ∼10-fold, although the risk of pancreatic cancer in FAMMM may be higher. Another hereditary predisposition to pancreatic cancer results from the gain-of-function protease serine-1 (PRSS1 [MIM 276000]) gene mutations associated with hereditary pancreatitis (Whitcomb et al. 1996; Whitcomb 1999, 2000). Individuals affected with pancreatitis have a lifetime pancreatic-cancer risk ratio of 57 and cumulative incidence, to age 70 years, of 40% (Lowenfels et al. 1997). However, the critical PRSS1-gene mutations are rare, and PRSS1 is a digestive enzyme causing childhood-onset pancreatitis, with cancer developing decades later, probably because of the environment of chronic pancreatic inflammation (Whitcomb 2000; Ponder 2001).
Many familial-pancreatic-cancer kindreds have been identified, most notably the family of the former President of the United States, Jimmy Carter (Lynch et al. 1990; Brentnall et al. 1999; Banke et al. 2000; Hruban et al. 2001). These families inherit pancreatic cancer in the absence of other types of cancer (colon, breast, melanoma, etc.) and in the absence of chronic pancreatitis. However, the late age at onset (60–70 years), unknown penetrance, absence of recognized premalignant phenotype, and short survival (average 6 mo) after the diagnosis of pancreatic cancer have made genetic linkage analyses nearly impossible.
Families with pancreatic cancer are relatively uncommon, and some groupings of pancreatic cancers within a family are likely due to chance. However, recent segregation analysis suggests that ⩾10% of patients with pancreatic cancer inherit the risk of pancreatic cancer in an autosomal dominant pattern (Banke et al. 2000). The most striking example is “family X” (fig. 1), which we have described elsewhere (Brentnall et al. 1999; Meckler et al. 2001). The primary characteristics include early age at onset (median age 43 years) and high penetrance (i.e., >80%) of pancreatic cancer. Moreover, some, but not all, family members can develop pancreatic insufficiency prior to the onset of cancer. We have been able to develop a surveillance program in this and other kindreds with familial pancreatic cancer, using endoscopic ultrasound and endoscopic retrograde pancreatography to help identify patients with precancerous changes. All of the precancerous changes (dysplasia) are verified histologically. The early identification of cancer/precancer through surveillance, the large family size, and the high level of family-member participation in family X enabled the first successful pancreatic-cancer genetic linkage study to be performed.
Figure 1.

Pedigree of family X. Blackened symbols denote affected family members. The “X” marks where the pedigree was split for GENEHUNTER analysis.
The study was performed according to approved Institutional Review Board guidelines at the University of Washington. The cancer in family X can be associated with a prodrome of pancreatic endocrine and/or exocrine insufficiency, possibly due to the increasing metaplastic and dysplastic changes in the pancreas. The prodrome of pancreatic insufficiency has also been described in some patients with sporadic pancreatic cancer (Silverman et al. 1999). No other types of cancer or distinctive features are notable in family X. Family members were considered affected if they had histologic evidence of either cancer or dysplasia (precancer) or if there were records (medical or death certificate) documenting pancreatic cancer. Family members who had the prodrome of pancreatic insufficiency but for whom further histologic confirmation was lacking were considered to be affected also. There are 20 cases, in four generations (fig. 1), of either dysplasia/cancer (n=18) or, in lieu of histologic diagnosis (n=2), the phenotypic prodrome equivalent. All of the affected family members from generations II and III had either pancreatic cancer or histologic evidence of pancreatic dysplasia; in generation IV, two members had evidence of pancreatic insufficiency (phenotypic prodrome), and two others had histologic evidence of pancreatic dysplasia. Blood from all living individuals was obtained, and DNA was extracted. Genetic material from pathology specimens was obtained from three deceased individuals, and the DNA was recovered by standard deparaffinization techniques.
The study subjects were initially genotyped at 373 marker loci, by the ABI PRISM Linkage Mapping Set Version 2 (Applied Biosystems). Up to 12 PCR products from single DNA samples were pooled together, heat denatured, and loaded into ABI 377 DNA Sequencers (Applied Biosystems), for GENESCAN data collection. The data were analyzed and the alleles were assigned by either TrueAllele software (Cybergenetics) or Genotyper 2.5 (Applied Biosystems). The genetic spacing between the markers averaged 9.4 cM, according to comprehensive genetic maps of microsatellite loci (Broman et al. 1998). The data were checked for Mendelian inconsistencies, by the PedCheck program, which uses the individual's genotype information to check for parent-child inconsistencies and too many alleles in a sibship and then uses genotype elimination to find more-subtle errors (O’Connell and Weeks 1998). Two-point and three-point analyses were performed by FASTLINK (Schaffer et al. 1994). Multipoint analysis and haplotype analyses were performed by GENEHUNTER version 2.1 (Markianos et al. 2001). Because of the size of this pedigree, the family was split into two subpedigrees, for calculation of LOD scores by GENEHUNTER.
We performed a genome scan of family X, with 373 microsatellite markers (average spacing 9.4 cM). Of the 373 markers, 6 showed LOD scores >1 in two-point linkage analysis. These six markers were located on chromosome 3 (two markers), chromosome 4 (two markers), chromosome 7 (one marker), and chromosome 14 (one marker). Markers D3S1278 and D3S1267 were adjacent to each other and were separated by 9.3 cM, and markers D4S1597 and D4S1539 were adjacent to each other and were separated by 12 cM. Two additional markers flanking the positive markers on chromosome 4 also gave positive LOD scores. The four chromosome 4 markers were the only contiguous set of four markers with positive LOD scores in the scan. All genomic regions where two consecutive markers gave positive LOD scores were followed up with three-point analysis. Each of these analyses resulted in LOD scores <1.5, except on chromosome 4, where consecutive pairs D4S413, D4S1597, D4S1539, and D4S415 gave LOD scores >2.1 and where markers D4S1597 and D4S1539, and the interval between them, showed LOD scores of ∼3.0.
On the basis of these analyses, we determined that the D4S413–D4S415 interval was the most likely location of the disease gene. We genotyped 17 additional markers within this interval and included additional family members in the analysis. Two-point analyses of these additional markers provided further support for localization of the gene to this region (table 1). The highest LOD score was 4.56 at a recombination fraction (θ) of 0, for D4S3030. In three-point analysis, the maximum LOD score increased to 5.36, between markers D4S1595 and D4S2991.
Table 1.
Two-Point LOD Scores for Various θ Values
|
LOD Score at θ = |
|||||||||
| Marker | .00 | .01 | .05 | .10 | .20 | .30 | .40 |
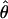 a a
|
Maximum LOD Score |
| D4S413 | 1.18 | 1.15 | 1.02 | .86 | .54 | .24 | .05 | .00 | 1.18 |
| D4S393 | 4.02 | 3.95 | 3.66 | 3.30 | 2.53 | 1.67 | .75 | .00 | 4.02 |
| D4S1603 | 2.30 | 2.25 | 2.06 | 1.81 | 1.31 | .81 | .35 | .00 | 2.30 |
| D4S2952 | 3.62 | 3.55 | 3.24 | 2.85 | 2.01 | 1.15 | .37 | .00 | 3.62 |
| D4S1566 | .98 | .95 | .84 | .71 | .46 | .24 | .07 | .00 | .98 |
| D4S620 | .97 | .95 | .88 | .78 | .57 | .37 | .18 | .00 | .97 |
| D4S1596 | 1.13 | 1.09 | .97 | .81 | .51 | .23 | .05 | .00 | 1.13 |
| D4S2979 | −∞ | −4.95 | −1.75 | −.62 | .10 | .17 | .06 | .27 | .18 |
| D4S1597 | 3.72 | 3.64 | 3.31 | 2.88 | 1.99 | 1.11 | .36 | .00 | 3.72 |
| D4S2910 | 1.15 | 1.11 | .98 | .83 | .54 | .28 | .08 | .00 | 1.15 |
| D4S1545 | −∞ | .38 | .91 | .98 | .82 | .52 | .20 | .10 | .98 |
| D4S1617 | 2.92 | 2.87 | 2.66 | 2.36 | 1.72 | 1.06 | .45 | .00 | 2.92 |
| D4S621 | −∞ | 1.40 | 1.85 | 1.82 | 1.42 | .88 | .36 | .07 | 1.87 |
| D4S1595 | 4.16 | 4.08 | 3.74 | 3.30 | 2.39 | 1.46 | .60 | .00 | 4.16 |
| D4S2991 | 2.33 | 2.27 | 2.07 | 1.82 | 1.31 | .80 | .33 | .00 | 2.33 |
| D4S2977 | −∞ | 2.43 | 2.80 | 2.67 | 2.08 | 1.32 | .51 | .05 | 2.80 |
| D4S1539 | −∞ | .17 | .67 | .73 | .54 | .27 | .08 | .09 | .73 |
| D4S1555 | −∞ | .25 | .79 | .88 | .74 | .46 | .17 | .10 | .88 |
| D4S3028 | −∞ | 2.68 | 3.04 | 2.89 | 2.25 | 1.44 | .60 | .05 | 3.04 |
| D4S3030 | 4.56 | 4.47 | 4.12 | 3.67 | 2.70 | 1.68 | .67 | .00 | 4.56 |
| D4S415 | −∞ | −.73 | −.14 | .02 | .07 | .05 | .03 | .19 | .07 |
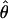 is the θ at which the maximum LOD score is obtained.
is the θ at which the maximum LOD score is obtained.
In multipoint analysis, the maximum LOD score was 4.9, at D4S2952/D4S1566, and the 2-LOD support interval extended from D4S413 to D4S1596 (fig. 2). Haplotype analysis showed that a single haplotype is, in general, present in all of the affected individuals and is absent in the unaffected individuals. Crossovers indicate that the gene lies between markers D4S413 and D4S2910, consistent with the multipoint analysis.
Figure 2.

LOD scores for multipoint analysis
The new pancreatic cancer–susceptibility locus identified in the present study appears to encompass the first pancreatic cancer–specific genetic defect. All of the known pancreatic cancer–associated genes and cancer syndromes have been excluded by localization of the susceptibility gene to chromosome 4q32-34. Furthermore, the autosomal dominant inheritance pattern argues against the hypothesis that this syndrome is polygenic. Other inherited-cancer syndromes usually involve alteration of a rate-limiting event needed for the development of cancer (e.g., the Rb gene in retinoblastoma), defective DNA repair, or genomic instability (Knudson 1971; Cavenee et al. 1983; Ponder 2001). Thus, the chromosome 4q32-34 region appears to represent a unique pancreatic cancer–specific locus.
Affected family members develop metastatic pancreatic adenocarcinoma that is histologically identical to sporadic pancreatic cancer (Brentnall et al. 1999; Meckler et al. 2001). Histologic review reveals that dysplasia (precancer) involves the small- and medium-size ducts. The dysplasia can be widespread—but it is not uniform, although the underlying fibrocystic change is. These histologic findings raise the possibility that the gene may involve a pathway associated with the desmoplastic reaction common in pancreatic cancer. Some late-stage–affected members of family X have developed multiple types of pancreatic cancer within a single organ. Ductal adenocarcinoma is always present, but, occasionally, small-cell undifferentiated carcinoma, anaplastic giant carcinoma, and/or cystadenocarcinoma may accompany it. Such tumor heterogeneity suggests the possibility of unregulated growth of either pancreatic stem cells or a mutator phenotype. The precursor changes of widespread fibrosis and multifocal dysplasia, together with the very-late-stage tumor heterogeneity, may reflect a common genetic pathway associated with this pancreatic cancer–susceptibility gene on chromosome 4q.
We have studied 28 families that inherit pancreatic cancer (authors' unpublished data). Although family X is the largest of the kindreds, it is not unique. There are other families that have high penetrance, inheriting pancreatic cancer in an autosomal fashion—these families are, in general, smaller than family X. There appears to be some variety in the phenotypic presentation in families with familial pancreatic cancer: some kindreds have pancreatic insufficiency prior to cancer onset, whereas others do not; and some families, like family X, have anticipation (earlier onset of cancer in each succeeding generation), whereas other families do not. Whether the current loci are associated with susceptibility to pancreatic cancer in all families with familial pancreatic cancer remains to be determined. Indeed, familial pancreatic cancer may be a heterogeneous disorder, with separate genes causing similar, but not identical, phenotypes—just as is the case in hereditary breast cancer syndromes BRCA1 (MIM 113705 and BRCA2, respectively).
The availability of BACs and ESTs that map to the 4q32-34 region will greatly facilitate the localization of the gene. With the use of these tools, we will be able to identify regions of 4q32-34 that show allelic loss or gain, as well as changes in gene expression during tumor development. We have constructed an array of all of the ESTs that map to this region, we are currently analyzing the expression pattern, and we are screening patient samples, for allelic loss, with the BAC clones that map to the area. This information will aid in the identification of the mutant gene in this family.
Progress in understanding and treating pancreatic cancer has been slow (Kern et al. 2001). The identification of a susceptibility gene on chromosome 4q should shed new light on the mysteries that surround pancreatic cancer. By understanding the identity of the gene, it is possible to fully understand the underlying mechanism and the possibilities for intervention. The discovery of a gene for familial pancreatic cancer would also allow identification of affected families—and thus help tailor surveillance methods to those who are at highest risk (e.g., gene carriers). As with the familial adenomatous-polyposis paradigm of colon cancer, it is possible that the gene causing the familial form pancreatic cancer plays an important role in the sporadic form of the disease. Finally, understanding the function of the gene may be the cornerstone for chemoprevention efforts.
Acknowledgments
This work was supported by grants DK54709 (to D.C.W.) and MH59520 (to L.K.); by a grant from the National Pancreas Foundation (to D.C.W.); and by the Lustgarten Foundation (support to D.C.W.), the Center for Genomic Sciences, University of Pittsburgh, and the Chiron Corporation. L.K. is a James S. McDonnell Centennial Fellow. L.K. dedicates his work on this project to the memory of Herb Sobel, and D.C.W. dedicates this work to Doug Edmiston.
Electronic-Database Information
Accession numbers and the URL for data in this article are as follows:
- Online Mendelian Inheritance in Man (OMIM) http://www.ncbi.nlm.nih.gov/Omim/ (for HNPCC [MIM 114500], BRCA2 [MIM 600185], STK11 [MIM 602216], FAMMM [MIM 600160], PRSS1 [MIM 276000], and BRCA1 [MIM 113705])
References
- Banke MG, Mulvihill JJ, Aston CE (2000) Inheritance of pancreatic cancer in pancreatic cancer-prone families. Med Clin North Am 84:677–690 [DOI] [PubMed] [Google Scholar]
- Brentnall TA, Bronner MP, Byrd DR, Haggitt RC, Kimmey MB (1999) Early diagnosis and treatment of pancreatic dysplasia in patients with a family history of pancreatic cancer. Ann Intern Med 131:247–255 [DOI] [PubMed] [Google Scholar]
- Broman, KW, Murray JC, Sheffield VC, White RL, Weber JL (1998) Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am J Hum Genet 63:861–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavenee WK, Dryja TP, Phillips RA, Benedict WF, Godbout R, Gallie BL, Murphree AL, Strong LC, White RL (1983) Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature 305:779–781 [DOI] [PubMed] [Google Scholar]
- Efthimiou E, Crnogorac-Jurcevic T, Lemoine NR, Brentnall TA (2001) Inherited predisposition to pancreatic cancer. Gut 48:143–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruban RH, Canto MI, Yeo CJ (2001) Prevention of pancreatic cancer and strategies for management of familial pancreatic cancer. Dig Dis 19:76–84 [DOI] [PubMed] [Google Scholar]
- Kern, S, Hruban R, Hollingsworth MA, Brand R, Adrian TE, Jaffee E, Tempero MA (2001) A white paper: the product of a pancreas cancer think tank. Cancer Res 61:4923–4932 [PubMed] [Google Scholar]
- Kernan GJ, Ji BT, Dosemeci M, Silverman DT, Balbus J, Zahm SH (1999) Occupational risk factors for pancreatic cancer: a case-control study based on death certificates from 24 U.S. states. Am J Ind Med 36:260–270 [DOI] [PubMed] [Google Scholar]
- Knudson AG (1971) Mutations and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 68:820–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenfels A, Maisonneuve P, DiMagno EP, Elitsur Y, Gates LK Jr, Perrault J, Whitcomb DC (1997) Hereditary pancreatitis and the risk of pancreatic cancer. J Natl Cancer Inst 89:442–446 [DOI] [PubMed] [Google Scholar]
- Lynch, HT, Fitzsimmons ML, Smyrk TC, Lanspa SJ, Watson P, McClellan J, Lynch JF (1990) Familial pancreatic cancer: clinicopathologic study of 18 nuclear families. Am J Gastroenterol 85:54–60 [PubMed] [Google Scholar]
- Markianos K, Daly MJ, Kruglyak L (2001) Efficient multipoint linkage analysis through reduction of inheritance space. Am J Hum Genet 68:963–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meckler, KA, Brentnall TA, Haggitt RC, Crispin D, Byrd DR, Kimmey MB, Bronner MP (2001) Familial fibrocystic pancreatic atrophy with endocrine cell hyperplasia and pancreatic carcinoma. Am J Surg Pathol 25:1047–1053 [DOI] [PubMed] [Google Scholar]
- O'Connell JR, Weeks DE (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponder BAJ (2001) Cancer genetics. Nature 411:336–341 [DOI] [PubMed] [Google Scholar]
- Rulyak, SJ, Brentnall, TA (2001) Inherited pancreatic cancer: surveillance and treatment strategies for affected families. Pancreatology 1:477–485 [DOI] [PubMed] [Google Scholar]
- Schaffer AA, Gupta SK, Shriram K, Cottingham RW Jr (1994) Avoiding recomputation in linkage analysis. Hum Hered 44:225–237 [DOI] [PubMed] [Google Scholar]
- Schnall, SF, Macdonald JS (1996) Chemotherapy of adenocarcinoma of the pancreas. Semin Oncol 23:220–228 [PubMed] [Google Scholar]
- Silverman DT, Schiffman M, Everhart J, Goldstein A, Lillemoe KD, Swanson GM, Schwartz AG, Brown LM, Greenberg RS, Schoenberg JB, Pottern LM, Hoover RN, Fraumeni JF Jr (1999) Diabetes mellitus, other medical conditions and familial history of cancer as risk factors for pancreatic cancer. Br J Cancer 80:1830–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tersmette AC, Petersen GM, Offerhaus GJ, Falatko FC, Brune KA, Goggins M, Rozenblum E, Wilentz RE, Yeo CJ, Cameron JL, Kern SE, Hruban RH (2001) Increased risk of incident pancreatic cancer among first-degree relatives of patients with familial pancreatic cancer. Clin Cancer Res 7:738–744 [PubMed] [Google Scholar]
- Whitcomb DC (1999) Hereditary pancreatitis: new insights into acute and chronic pancreatitis. Gut 45:317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (2000) Genetic predispositions to acute and chronic pancreatitis. Med Clin North Am 84:531–547 [DOI] [PubMed] [Google Scholar]
- Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD, Martin SP, Gates LK Jr, Amann ST, Toskes PP, Liddle R, McGrath K, Uomo G, Post JC, Ehrlich GD (1996) Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 14:141–145 [DOI] [PubMed] [Google Scholar]
- Whitcomb DC, Ulrich CD, Lerch MM, Durie P, Neoptolemos JP, Maisonneuve P, Lowenfels AB (2001) Third International Symposium on Inherited Disease of the Pancreas. Pancreatology 1:423–431 [DOI] [PubMed] [Google Scholar]