

Abstract
A study of the reactivity and diastereoselectivity of the Lewis acid-promoted cascade cyclizations of both acyclic and macrocyclic alkynones is described. In these reactions, a β-iodoallenolate intermediate is generated via conjugate addition of iodide to an alkynone, followed by an intramolecular aldol reaction with a tethered aldehyde to afford a cyclohexenyl alcohol. The Lewis acid magnesium iodide (MgI2) was found to promote irreversible ring closure, while cyclizations using BF3·OEt2 as promoter occurred reversibly. For both acyclic and macrocyclic ynones, high diastereoselectivity was observed in the intramolecular aldol reaction. The MgI2 protocol for cyclization was applied to the synthesis of advanced intermediates relevant to the synthesis of phomactin natural products, during which a novel transannular cation-olefin cyclization was observed. DFT calculations were conducted to analyze the mechanism of this unusual MgI2-promoted process.
Introduction
Significant progress has been made in the development of new cyclizations and carbon-carbon bond forming reactions initiated by the conjugate addition of halide nucleophiles to different unsaturated carbonyl systems.[1] The variant involving the addition of iodide to alkynone derivatives, which generates β-iodoallenolate intermediates, was first described by Kishi in 1986. 2 Since then, β-iodoallenolates have proven to be versatile nucleophilic intermediates in reactions with aldehydes,3 imines,4 oxiranes,5 and ketones.3a-d Asymmetric reactions have also been achieved using chiral Lewis acids6 or chiral auxiliaries.7 We have developed two related cascade cyclizations, promoted by two different Lewis acids, involving β-iodoallenolates II (Scheme 1).8
Scheme 1.
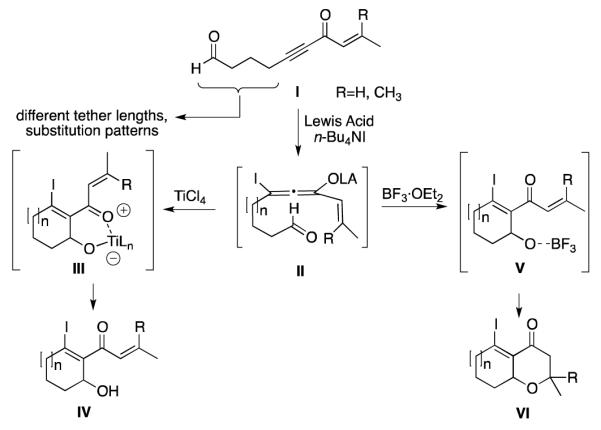
Lewis Acid-Initiated β-Iodoallenolate Cyclization.
We proposed that treatment of alkynones I with titanium tetrachloride (TiCl4) gave cyclohexenol products IV through chelated intermediates III, while treatment with boron trifluoride diethyl etherate (BF3·OEt2) led to intermediates of type V, which have rotational freedom to undergo oxa-Michael ring closure to produce oxacycles of type VI.
These cascades are some of the only examples of intramolecular reactions of β-iodoallenolates that have been reported,9 despite their potential value as a method for the synthesis of highly functionalized ring systems. To effectively apply this reaction chemistry to problems in natural product synthesis, it will be important to develop an understanding of the factors governing diastereoselectivity in β-iodoallenolate cyclizations. In this article, we assess diastereoselectivity and reversibility in the cyclizations of both acyclic and macrocyclic β-iodoallenolates using different Lewis acid promoters. We have also applied this method to the synthesis of the ABD core of phomactin A, and observed an unexpected transannular cyclization that we analyzed using DFT calculations.
Results and Discussion
Cascade Cyclization Strategy for the Synthesis of the ABD Core of Phomactin A
Over the past few years, we have sought to implement this cascade cyclization in the preparation of oxadecalin 1, which contains the ABD ring system of phomactin A and appropriate handles for further functionalization (Scheme 2).8,10,11 The idea was to generate a β-iodoallenolate intermediate from macrocyclic alkynone 2, which would undergo intramolecular aldol/oxa-Michael addition to deliver 1 via bicyclo[9.3.1]pentadecane 3.
Scheme 2.
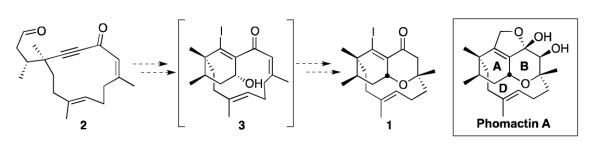
Strategy for the Synthesis of Phomactin A.
Macrocycle 2 was prepared as shown in Scheme 3.10
Scheme 3.
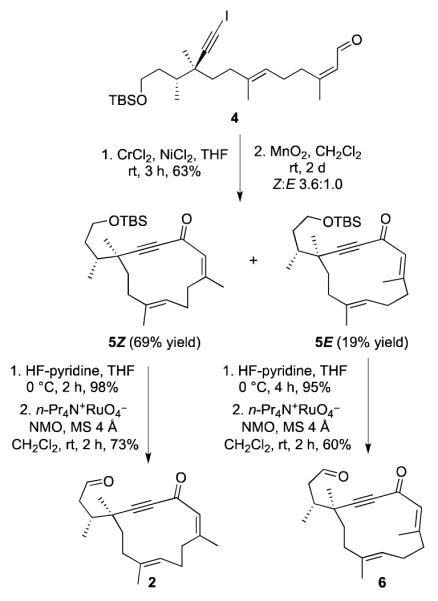
Synthesis of Macrocycles 2 and 6
Intramolecular Nozaki-Hiyama-Kishi Cr(II)/Ni(II)coupling[12] followed by MnO2 oxidation gave enone 5Z in two steps from iodoalkyne 4, along with isomeric enone 5E (3.6 : 1 ratio of Z and E isomers). After chromatographic separation of the E and Z-isomers, both could be desilylated using a HF-pyridine solution in tetrahydrofuran. Oxidation of each primary alcohol with the Ley-Griffith reagent13 afforded the alkynones 2 and 6, respectively (Scheme 3).
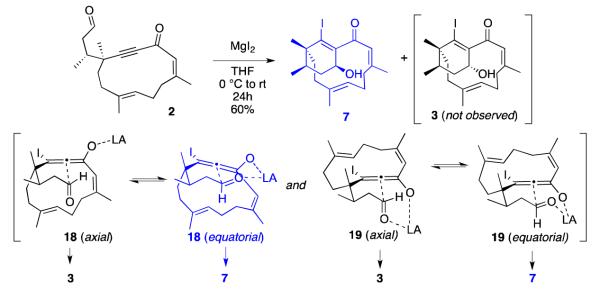
Attempts to cyclize macrocyclic alkynone 2 with the usual promoters (BF3·OEt2 and TiCl4) were unsuccessful, producing complex mixtures of products. Since magnesium iodide (MgI2) has been reported to promote β-iodoallenolate formation/ aldol reaction,3g,3i,9 we next tried cyclizing 2 using MgI2 (1.3 equiv) in dichloromethane. The reaction did not produce either cyclohexenyl alcohol 3 or oxadecalin 1; instead, a 1:1 mixture of products was generated: cyclohexenyl alcohol 7 (isolated as a single diastereomer) and tricycles 8a/8b (isolated as a 2.7:1 mixture of endo/ exo isomers; see Scheme 4, top).
Scheme 4.
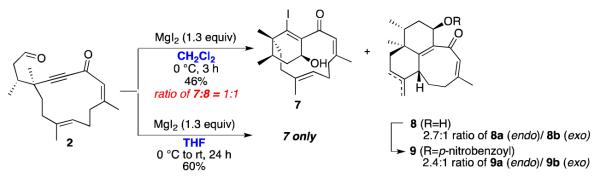
β-Iodoallenolate Cyclization with Alkynone 2.
We tried adding n-Bu4NI (1.3-5 equiv) to the reaction mixture,8 in an attempt to favor the formation of phomactin skeleton 7 over the tricyclic system 8, but the ratio of 7 to 8 did not change. However, we were able to avoid the formation of tricycles 8 by changing the solvent: if the reaction was run in tetrahydrofuran instead of dichloromethane, cyclohexenyl alcohol 7 was produced as the sole product in 60% yield and as a single diastereomer (Scheme 4, bottom).
We converted the mixture of tricycles 8a (endo) /8b (exo) into p-nitrobenzoyl esters 9a (endo) /9b (exo), which enabled us to obtain X-ray crystal structures of both the endo and exo isomers.14 The stereochemistry of the tricyclic system is shown in Scheme 4.
Since we needed cyclohexenyl 3 to assess the strategy outlined in Scheme 2, we performed a standard Mitsunobu inversion25 on cyclohexenyl alcohol 7, which furnished target 3 in 52% yield (Scheme 5). The oxa-Michael ring closure of 3 could be achieved under the standard conditions (BF3·OEt2 at low temperature)8 to afford target oxadecalin 1 in 20% yield.
Scheme 5.
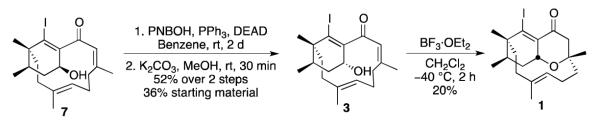
Synthesis of Oxadecalin 1 using BF3COEt2 as Promoter
To summarize, synthetic studies targeting phomactin revealed that macrocycles 2 and 3 exhibit unusual cyclization behavior. In particular: 1) MgI2 was identified as a mild alternative to BF3·OEt2 and TiCl4, and optimal for promoting the β-iodoallenolate cyclization of acid-sensitive alkynone 2; 2) the cyclization of 2 is highly diastereoselective; 3) tricycles 8 are produced unexpectedly from 2, through an unknown mechanism and 4) the BF3·OEt2-promoted oxa-Michael ring closure of cyclohexenyl alcohol 3 is inefficient. We conducted further cyclization studies on both acyclic and macrocyclic systems to improve our understanding of these four experimental observations.
MgI2-promoted cyclizations of acyclic and macrocyclic alkynones
Further experimentation with MgI2 as a promoter indicated that cyclization results were comparable to experiments employing TiCl4, producing cyclohexenols of type IV rather than oxadecalins of type VI (Scheme 1). Cyclization of 10 with TiCl4 gives cyclohexenyl alcohol 11 in 82% yield (Table 1, entry 1), while MgI2 produces 11 in 75% yield (entry 2). The analogous cyclization using BF3·OEt2 produces 12 (entry 3). The observed reactivity is readily explained by chelation: like TiCl4, MgI2 is able to bind both oxygens of the aldol product (cf. III, Scheme 1), which prevents oxa-Michael ring closure.8
Table 1.
β-Haloallenolate Cyclizations.[a]
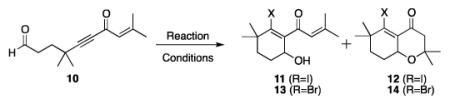
| Entry | Lewis Acid | Iodide | Conditions | product | Yield |
|---|---|---|---|---|---|
| 1 | TiCl4 | n-Bu4NI | −78 °C to 0 °C, 2 h | 11 | 82% |
| 2 | MgI2 | --- | 0 °C, 3 h | 11 | 75% |
| 3 | BF3·OEt2 | n-Bu4NI | −40 °C to 0 °C, 3 h | 12 | 77% |
| 4 | MgBr2 | --- | 0 °C to rt, 24 h | 13 | 52%[b] |
| 5 | BF3·OEt2 | n-Bu4NBr | −40 °C to rt, 7 h | 14 | 46% |
Reaction conditions: Alkynone (1.0 equiv), Lewis acid (1.3 equiv), and n-Bu4NX (1.3 equiv) in CH2Cl2 (0.10 M) for the indicated time at the indicated temperature.
65% conversion.
The two cyclization protocols were also successfully applied to the synthesis of β-bromocyclohexenyl alcohols. Cyclization of 10 using MgBr2 as promoter generated 13 in moderate yield (entry 4).[15] Treatment of 10 with BF3·OEt2/n-Bu4NBr promoted the cascade cyclization to produce oxadecalin 14 in 46% yield (entry 5). In general, these reactions required longer reaction times and warmer temperatures compared to the cyclizations carried out with iodide as the nucleophile (cf. entry 2 vs. 4 and entry 3 vs. 5).
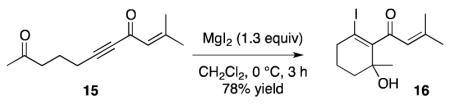
Finally, treatment of ketone 15 with MgI2 produced 16 in 78% yield (equation 1), whereas TiCl4 and BF3·OEt2 were not competent promoters.16 This result further demonstrates that MgI2 is a viable alternative to TiCl4 and BF3·OEt2 for acid sensitive substrates, and it is also convenient that the MgI2-promoted protocol does not require an external halide source (Table 1, entries 2 and 4, and equation 1).
 |
(1) |
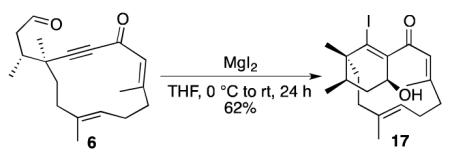
In additional experiments on the macrocyclic phomactin system, we found that E-enone 6 could be cyclized upon treatment with MgI2 in tetrahydrofuran, without isomerization of the α,β-unsaturated ketone. Cyclohexenyl alcohol 17 was obtained as a single diastereomer in 62% yield (equation 2).17 Importantly, compound 17 represents an alternative intermediate useful for synthesis of the phomactin skeleton, as it contains the relevant bicyclo[9.3.1]pentadecane core.
 |
(2) |
Diastereoselectivity of the β-Iodoallenolate Aldol Cyclization
To explain why the intramolecular aldol cyclization of alkynone 2 selectively produces diastereoisomer 7 rather than 3, it was helpful to perform a conformational analysis of β-iodoallenolate intermediates complexed with magnesium (Scheme 6). When macrocyclic alkynone 2 is exposed to MgI2, 1,4-addition of iodide is expected to produce two β-iodoallenolate diastereoisomers (18 and 19; Scheme 6). Cyclization of 18 via a Zimmerman-Traxler transition state is predicted to produce the major diastereoisomer 7.18,19 In contrast, β-iodoallenolate 18 (axial) is not aligned to form the magnesium chelate, while chelation of β-iodoallenolate 19 would produce two boat-like complexes, which may not form within the rigid macrocyclic system. Cyclization of 18 (equatorial) would deliver the observed cyclohexenol 7. To account for the high isolated yield of 7, it is reasonable to propose that β-iodoallenolate isomers 18 and 19 can equilibrate via reversible 1,4-addition of iodide,20 allowing selective cyclization via chelate 18 (equatorial).
Scheme 6.
Diastereoselectivity in the Intramolecular Aldol Reaction.
Cyclization studies on acyclic alkynone 20 provided further insight on the diastereoselectivity and reversibility of the intramolecular aldol reactions of β-iodoallenolate intermediates. Alkynone 20 was prepared as shown in Scheme 7. Selective mono-protection of the primary alcohol using tert-butyldimethylchlorosilane provided compound 22 in good yield. Then, oxidation of the neopentyl alcohol with Dess-Martin periodinane21 followed by a one-carbon homologation using the Ohira-Bestmann reagent 22 afforded desired alkyne 23. Then, addition of the lithium acetylide to aldehyde 2411g and oxidation of the resulting allylic alcohol gave the desired ketone 25 in 61% yield over two steps. Deprotection followed by oxidation of the resulting alcohol gave alkynone 20.
Scheme 7.
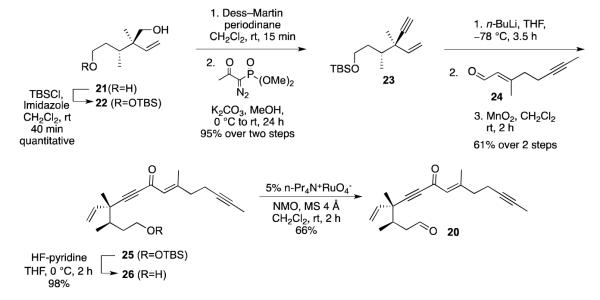
Synthesis of Alkynone 20
Cyclization of 20 with MgI2 in dichloromethane provided cyclohexenyl alcohols 27 and 28 in 77% yield as a 10:1 mixture of diastereomers (Scheme 8).23 This result is consistent with the model in Scheme 6, which predicts preferential formation of 27 through a magnesium chelate analogous to 18 (equatorial). The flexibility of the acyclic system must allow the intramolecular aldol reaction to occur through one of the axial conformations as well, resulting in formation of minor diastereomer 28.
Scheme 8.
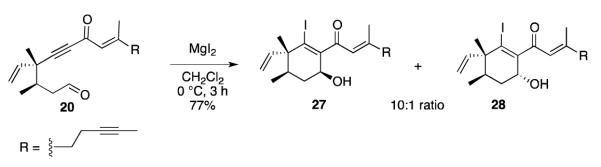
Intramolecular Aldol Reaction of Alkynone 20.
When pure samples of cyclohexenyl alcohol 27 and 2824,25 were treated with BF3·OEt2 to promote the oxa-Michael reaction,26 27 afforded oxadecalin 29 in 90% yield, but the reaction of 28 did not produce any of the corresponding oxadecalin 30. Instead, oxadecalin 29 was isolated in 30% yield (Scheme 9).
Scheme 9.
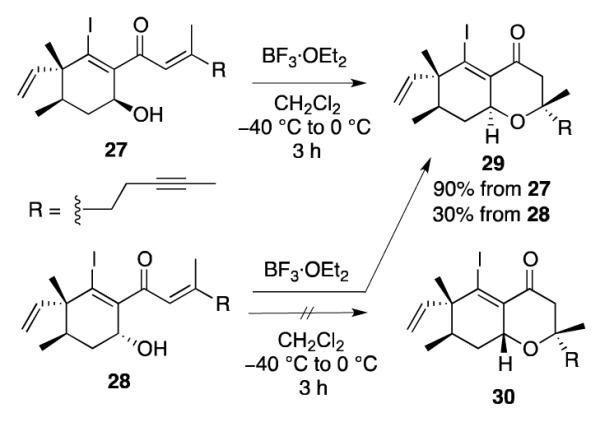
Oxa-Michael Reactions of 27 and 28
This result suggests that treatment of 28 with BF3·OEt2 can lead to the formation of 27 (with only moderate efficiency), via the corresponding β-iodoallenolate intermediates. The high-yielding, diastereoselective oxa-Michael ring closure of 27 then produces 29 (Scheme 10). The fact that only one oxadecalin isomer was obtained (29 and not 30) indicates that cyclohexenol 28 must undergo retro-aldol reaction more readily than oxa-Michael ring closure. In contrast, no reaction occurred upon treatment of either cyclohexenyl alcohol 28 or cyclohexenyl alcohol 3 (see Scheme 5) with MgI2. Taken together, these results suggest that intramolecular aldol reactions of β-iodoallenolate intermediates with BF3·OEt2 can occur reversibly, while the analogous MgI2-promoted cyclizations are irreversible.20 Thus, the inability to achieve efficient oxa-Michael ring closure in both 28 and the phomactin system 3 (see Scheme 5) using BF3·OEt2 may be attributed to a competing retro-aldol reaction. Fortunately, we were able to identify two other methods for inducing the oxa-Michael addition of 3. These results are described in the next section.
Scheme 10.
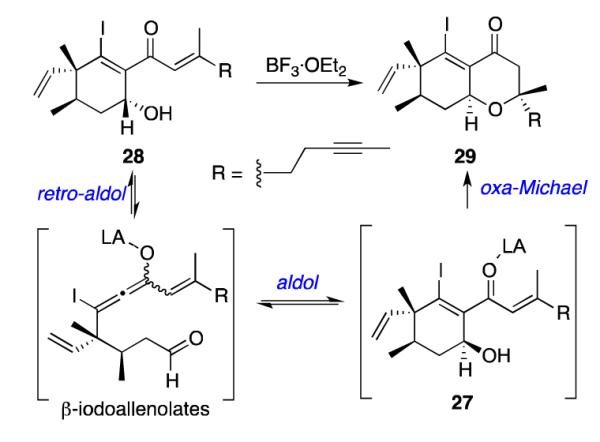
Cyclization of 28 via Retro-Aldol Reaction Pathway
Synthesis of the ABD Ring System of Phomactin
To advance the synthesis of phomactin A, we explored alternative strategies for obtaining oxadecalin 1 from cyclohexenyl alcohol 3. Treatment with 10 mol% of AuCl3 in dichloromethane at 0 °C effectively induced oxa-Michael addition, affording oxadecalin 1 in 50% yield,26 or alternatively, exposure to tert-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf) in the presence of 2,6-lutidine afforded silyl enol ether 31 in 54% yield (Scheme 11).10 This sequence is particularly advantageous because the oxa-Michael addition occurs with simultaneous protection of the ketone, providing a flexible intermediate that can be functionalized in different ways.
Scheme 11.
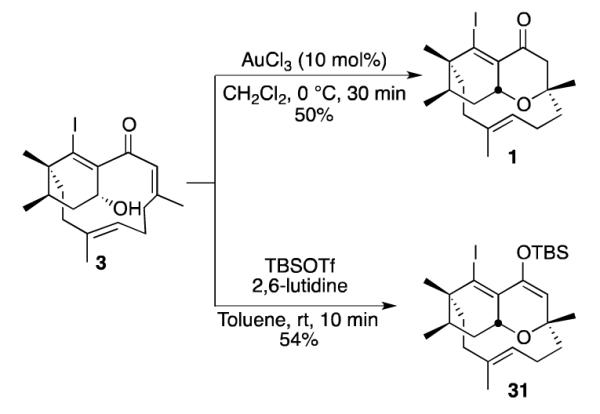
Alternative Methods for Inducing Oxa-Michael Cyclization in Cyclohexenyl Alcohol 3.
Mechanism of Formation of Tricycles 8a and 8b: DFT studies
The Lewis acid-promoted cyclization of 2 in dichloromethane resulted in significant production of tricycles 8 (see Scheme 4). Different reaction pathways can be proposed to rationalize this outcome. One possibility involves a cation-olefin cyclization cascade,27 with concerted formation of two new bonds to generate intermediate 32, followed by elimination to produce tricycles 8 (Scheme 12). Alternatively, stepwise mechanisms can be invoked, although these would require the formation of a high energy intermediate such as strained allene 33 or vinyl cation 34 (Scheme 12).
Scheme 12.

Proposed Mechanisms for the Formation of Tricycles 8.
We performed DFT calculations to assess the feasibility of these different reaction pathways.28,29 Although DFT is rarely used with magnesium,30 it is the only reasonable computational method that can be used with such a large system. Because MgI2 can decompose into several species in solution,31 we modeled multiple promoters: MgI2, MgI2·2THF, MgI+, MgI+·THF, and Mg2+. In each case, solvation corrections were obtained by the PCM method. We observed the systematic formation of a chelate as starting complex (see B, Table 2).
Table 2.
Computed Intermediates and Gibbs Free Energies After Solvation Correction (B3LYP/6-311G**[Mg,I]/6-31G*[other elements]//PCM; kcal/mol) Corresponding to the Formation of the Tricyclic Framework.
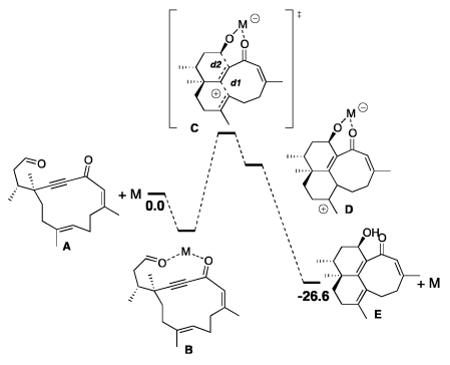
| Entry | M | ΔGAB | [ΔGAC]‡ | ΔGAD | d1 (Å) | d2 (Å) | |||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| DCM | THF | DCM | THF | DCM | THF | ||||
| 1 | MgI2 | −1.3 | −2.8 | 25.0 | 24.2 | 18.2 | 17.6 | 1.68 | 1.95 |
| 2 | MgI2·2THF | − | 10.0 | − | 39.2 | − | 3.0 | 1.68 | 1.97 |
| 3 | MgI+ | −1.3 | −1.3 | 26.8 | 25.0 | 26.0 | 23.6 | 1.85 | 2.64 |
| 4 | MgI+·THF | − | −15.3 | − | 8.9 | − | 0.2 | 1.66 | 2.12 |
| 5 | Mg2+ | −0.5 | −4.6 | 16.1 | 12.0 | 5.3 | 1.2 | 1.61 | 2.30 |
Its formation is weakly exothermic in CH2Cl2. In THF, it is moderately exothermic with MgI2, MgI+, or Mg2+, appreciably exothermic with MgI+·THF, but strongly endothermic with MgI2·2THF because of the steric strain.32 The cyclization of the chelate gave rise to the tricyclic core D in a concerted fashion (cf. intermediate 32, Scheme 11), via transition state C. The formation of the two rings is asynchronous, as shown by the very distinct values between d1 and d2 in C (Table 2), suggesting that C is more similar to 33 than to 34.33 Dissociation of the metallic fragment from D leads directly to the tricycle. In CH2Cl2, a reasonable free energy of activation was calculated with Mg2+. On the other hand, MgI+·THF gave rise to the lowest lying transition state in THF. All cyclizations were endothermic, but the decomplexation of the catalyst from D always proved exothermic. Overall, the cyclization of A into E liberates 26.6 kcal/mol of free energy. The subsequent isomerization of E into the observed product 8 presumably relieves strain in the tricyclic system.
Thus, DFT calculations support a Lewis acid-catalyzed cation-olefin cascade as the most reasonable reaction pathway for cyclization of 2 to 8. To the best of our knowledge, this is a unique example of a reaction in which activation of an aldehyde triggers a tandem cyclization involving an electron-deficient alkyne and an alkene.34 The transannular relationship of the alkyne and the alkene is probably an important factor. We did not make attempts to optimize the reaction to favor the formation of 8 over the desired target 7, but further experimentation is planned to further evaluate this interesting cyclization.
Conclusion
In summary, these studies provide new insight into the reactivity and diastereoselectivity of the Lewis acid-promoted cyclizations of both acyclic and macrocyclic alkynones. Our experiments indicate that the 1,4-addition of iodide to an alkynone is a reversible process using either MgI2 or BF3·OEt2, and generates a β-iodoallenolate intermediate. This intermediate can then undergo an intramolecular aldol reaction with a tethered aldehyde to afford a cyclohexenyl alcohol. We present evidence that MgI2 promotes irreversible ring closure, while the analogous BF3·OEt2-promoted cyclization occurs reversibly. For both acyclic and macrocyclic ynones, we found that the aldol reaction is highly diastereoselective. The MgI2 protocol was employed in the synthesis of a tricycle corresponding to the ABD ring system of phomactin A. Finally, we examined an interesting transannular cyclization generated under the Lewis acidic conditions, and gained insight into the process using DFT calculations.
Experimental Section
General
Reactions were carried out in oven-dried glassware under an argon atmosphere. Reagents were used as obtained from commercial suppliers without further purification. ACS grade hexanes and ethyl acetate were used for column chromatography. Thin layer chromatography (TLC) was performed on pre-coated silica gel 60 F254 glass-supported plates. Column chromatography was carried out on 60Å silica gel (230-400 mesh). Visualization on thin layer chromatography was done with a UV lamp followed by staining with either potassium permanganate/heat or p-anisaldehyde/heat. Infrared (IR) absorbance frequencies are given in cm−1 at the peak maximum. High resolution mass spectra were obtained a time-of-flight (TOF) mass spectrometer.
Spectroscopic Data
Structural assignment, including the identification of E/Z isomers and cis/trans isomers, was determined by either 1H and 13C NMR spectroscopy (at either 400 or 500 MHz and 100 or 125 MHz, respectively), and by nOe experiments and 2D COSY (when necessary), or an X-ray crystal structure. Chemical shifts are given in ppm, referenced to the residual proton resonance of the solvents (δ = 7.26 for CHCl3, δ = 7.16 for C6H6) or to the residual carbon resonance of the solvent (δ = 77.1 for CHCl3, δ = 128.0 for C6H6) Coupling constants (J) are given in Hertz (Hz). The terms m, s, d, and t refer to multiplet, singlet, doublet, and triplet. In all cases, unless otherwise noted, the major diastereomer is reported.
Experimental conditions and spectral data for the preparation of the following compounds have been reported previously: 10 and 15;8 4 and 21.10 Experimental details and spectral data for other compounds previously studied in our laboratories (1, 2, 3, 5Z, 5E, 7, 12, 14 and 31)8,10 are provided below.
General Procedure for β-Iodoallenolate Cyclizations Run with MgI2
Magnesium iodide (at the indicated equivalents) was added to a stirred solution of the alkynone (1.0 equiv) in dry CH2Cl2 (0.10 M) or THF (0.10 M) at 0 °C. The reaction was then carried out at the indicated temperature and time. After completion of the reaction, the mixture was diluted with ethyl acetate, quenched with saturated NaHCO3 solution, and extracted with ethyl acetate (3×). The combined organic layers were washed with saturated Na2S2O3 solution (2×), brine (1×), dried over MgSO4, and concentrated. The resulting residue was purified by flash chromatography on silica gel using different gradients of hexanes and ethyl acetate to afford the pure products.
General Procedure for β-Iodoallenolate Cyclizations Run with BF3·OEt2
Tetra-n-butylammonium iodide (at the indicated equivalents) was added to a stirred solution of the alkynone (1.0 equiv) in dry CH2Cl2 (0.10 M) at −40 °C. Boron trifluoride diethyl etherate (1.3 equiv) was then added dropwise. The reaction was carried out at the indicated temperature and time. After completion of the reaction, the mixture was diluted with ethyl acetate, quenched with saturated NaHCO3 solution, and extracted with ethyl acetate (3×). The combined organic layers were washed with saturated Na2S2O3 solution (2×), brine (1×), dried over MgSO4, and concentrated. The resulting residue was purified by flash chromatography on silica gel using different gradients of hexanes and ethyl acetate to afford the pure products.
(2Z,6E,10S,11R)-13-((tert-butyldimethylsilyl)oxy)-10-(iodoethynyl)-3,7,10,11-tetramethyltrideca-2,6-dienal (4):10
1H NMR (400 MHz, CDCl3): δ 9.94 (d, J = 8.2 Hz, 1H), 5.91 (d, J = 8.0 Hz, 1H), 5.18 (t, J = 6.9 Hz, 1H), 3.77–3.69 (m, 1H), 3.66–3.60 (m, 1H), 2.62 (t, J = 7.6 Hz, 2H), 2.32–2.24 (m, 2H), 2.16–2.05 (m, 2H), 2.02 (s, 3H), 1.96–1.93 (m, 1H), 1.69–1.55 (m, 2H), 1.64 (s, 3H), 1.48–1.40 (m, 1H), 1.32–1.23 (m, 1H), 1.12 (s, 3H), 0.93 (s, 12H), 0.09 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 190.5, 163.7, 137.4, 128.5, 122.0, 100.7, 61.8, 40.9, 37.5, 36.9, 35.5, 34.6, 32.5, 27.0, 25.9, 25.0, 22.5, 18.2, 16.2, 13.9, −5.3. (1 carbon is missing due to overlap). IR (neat) 2949, 2926, 2854, 1669, 1631.
(S,2Z,6E)-10-((R)-4-((tert-butyldimethylsilyl)oxy)butan-2-yl)-3,7,10-trimethylcyclododeca-2,6-dien-11-ynone (5Z) and (S,2E,6E)-10-((R)-4-((tert-butyldimethylsilyl)oxy)butan-2-yl)-3,7,10-trimethylcyclododeca-2,6-dien-11-ynone (5E)
As described previously,10 iodoalkyne 4 (295 mg, 0.556 mmol) was diluted in 6.3 mL of tetrahydrofuran, and slowly added, over 3 h, to a vigorously stirring solution of CrCl2 (509 mg, 4.1 mmol) and NiCl2 (0.07 mg, 0.055 mmol) in 44.8 mL of tetrahydrofuran. (NOTE: The tetrahydrofuran was thoroughly degassed (three times before each cyclization), and CrCl2 was dried for at least 3 h at 180 °C under vacuum. All operations were carried out in the glove box, the addition of the iodoalkyne was carried out in the atmosphere.) After approximately 3 h, the reaction mixture was quenched with 10 mL of saturated NH4Cl solution, extracted with diethyl ether (3 × 50 mL), washed with NaS2O3 (2 × 30 mL), H2O (2 × 30 mL), brine (2 × 30 mL), dried over MgSO4, and concentrated. The resulting residue was purified by flash chromatography on silica gel (hexanes:ethyl acetate 95:5) to give the macrocycle (142 mg, 63%) as an unidentified mixture of diastereomers with a complicated 1H-NMR spectrum, and was carried on to the next step without further purification.
The macrocycle (600 mg, 1.48 mmol), from above, was diluted in 15 mL of CH2Cl2 and MnO2 (2.50 g, 28.73 mmol) was added and rt. After 2 days, the reaction mixture was filtered over celite and concentrated. The resulting residue was purified by flash chromatography on silica gel (hexanes:ethyl acetate 99:1) to give the Z-ketone, (302 mg, 69%, traces of the E-isomer are present, only the Z-isomer is reported) as a yellow oil, and the E-ketone, (85 mg, 19%) as a yellow oil. The geometry of the Z-isomer was confirmed by nOe analysis (see supporting information). 5Z: 1H NMR (400 MHz, CDCl3): δ 5.84 (s, 1H), 5.47–5.39 (m, 1H), 3.74–3.66 (m, 1H), 3.65–3.58 (m, 1H), 2.61–2.46 (m, 2H), 2.27–2.15 (m, 4H), 2.00–1.91 (m, 1H), 1.88 (s, 3H), 1.86–1.75 (m, 2H), 1.66 (s, 3H), 1.51–1.43 (m, 1H), 1.30–1.20 (m, 1H), 1.18 (s, 3H), 0.92 (d, J = 6.8 Hz, 3H), 0.89 (s, 9H), 0.05 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 180.3, 152.4, 134.3, 128.2, 125.4, 103.0, 84.6, 61.5, 38.2, 36.7, 35.2, 35.0, 32.1, 31.8, 25.9, 25.4, 24.0, 22.6, 18.2, 15.6, 13.7, −5.3 (2C). IR (neat) 2947, 2928, 2366, 2335, 2193, 1654, 1633, 1604. HRMS (ESI-TOF) m/z calculated for C25H42O2Si [M+] 402.2954; found 402.2951.
5E
1H NMR (400 MHz, CDCl3) δ 6.11 (s, 1H), 5.11 (t, J = 7.4 Hz, 1H), 3.72-3.62 (m, 1H), 3.61–3.54 (m, 1H), 2.37–2.19 (m, 4H), 2.09 (t, J = 6.1 Hz, 2H), 1.91–1.88 (m, 2H), 1.84 (s, 3H), 1.75–1.68 (m, 1H), 1.58–1.47 (m, 1H), 1.52 (s, 3H), 1.23–1.16 (m, 1H), 1.10 (s, 3H), 0.87 (s, 12H), 0.02 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 178.7, 147.6, 137.2, 132.3, 123.5, 106.6, 84.5, 61.4, 38.2, 37.6, 36.4, 35.3, 34.3, 33.1, 27.2, 25.9, 22.0, 18.2, 18.0, 14.8, 13.5, −5.3, −5.4. IR (neat) 2928, 2858, 2187, 1666, 1631, 1462, 1435, 1384, 1253, 1207, 1091. HRMS (ESI-TOF) m/z calculated for C25H42O2Si [M+] 402.2954; found 402.2954.
(R)-3-((S,5Z,9E)-1,6,10-trimethyl-4-oxocyclododeca-5,9-dien-2-ynyl)butanal (2)
As described previously,10 in a plastic reaction vessel, 5Z (209 mg, 519 mmol) was dissolved in 4.2 mL of tetrahydrofuran and 0.42 mL of pyridine and cooled to 0 °C. Then, HF-pyridine (~70% HF in ~30% pyridine, 0.51 mL, 0.561 mmol) was slowly added. After 2 h, the reaction mixture was diluted in 2 mL of ethyl acetate and quenched with 10 mL of saturated solution of NaHCO3. The reaction mixture was extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with 50 mL of saturated solution of NaHCO3, 10 mL of saturated CuSO4 solution, 20 mL of brine, dried over MgSO4, and concentrated. The resulting residue was purified by flash chromatography on silica gel to afford the primary alcohol (146 mg, 98%, traces of the E-isomer are present, only the Z-isomer is reported) as a yellow oil.
1H NMR (400 MHz, CDCl3) δ 5.88 (s, 1H), 5.48 (t, J = 7.9 Hz, 1H), 3.86–3.74 (m, 1H), 3.70–3.59 (m, 1H), 2.69–2.60 (m, 1H), 2.57–2.48 (m, 1H), 2.33–2.22 (m, 4H), 2.08–1.98 (m, 2H), 1.92 (s, 3H), 1.90–1.80 (m, 2H), 1.69 (s, 3H), 1.57–1.48 (m, 1H), 1.43–1.32 (m, 1H), 1.21 (s, 3H), 0.97 (d, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 180.3, 153.3, 134.2, 128.1, 125.5, 103.2, 84.7, 61.2, 38.2, 37.4, 35.4, 35.1, 32.0, 31.9, 25.4, 24.1, 21.6, 15.5, 13.8. IR (neat) 3658–3090, 2973, 2939, 2874, 2195, 1627, 1600, 1442, 1377, 1348, 1280, 1249. HRMS (ESI-TOF) m/z calculated for C19H28O2 [M+] 288.2089; found 288.2092.
The primary alcohol (227 mg, 0.788 mmol), from above, was stirred with 4-methylmorpholine N-oxide (138 mg, 1.18 mmol) and 4Å molecular sieves in 7.6 mL of dry CH2Cl2. After 20 min at rt, tetra-n-propylammonium perruthenate (14 mg, 0.04 mmol) was added and the mixture was stirred at rt for 2 h. The reaction was quenched with 10 mL of saturated Na2SO3 solution, extracted with diethyl ether (3 × 20 mL), washed with brine, and saturated CuSO4 solution. The combined organic layers were dried over MgSO4, and filtered over celite. The resulting residue was concentrated to afford the aldehyde 2 (129 mg, 73%, traces of the E-isomer are present, only the Z-isomer is reported) as a yellow oil. The aldehyde was immediately used in the next reaction. (This compound was not stable to silica gel chromatography, but was sufficiently pure to use in the next step without further purification.)
1H NMR (500 MHz, CDCl3): δ 9.74 (s, 1H), 5.79 (s, 1H), 5.41 (t, J = 8.1 Hz, 1H), 2.84–2.75 (m, 1H), 2.54–2.44 (m, 1H), 2.34–2.24 (m, 2H), 2.24–2.14 (m, 4H), 1.84 (s, 3H), 1.79–1.69 (m, 1H), 1.61 (s, 3H), 1.59–1.44 (m, 2H), 1.14 (s, 3H), 0.91 (d, J = 6.1 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 201.6, 179.7, 153.2, 134.0, 128.1, 125.7, 101.0, 85.1, 47.4, 37.6, 35.0, 32.3, 32.0, 25.4, 24.1, 21.8, 15.5, 14.5 (1 carbon is missing due to overlap). IR (neat) 2966, 2935, 2877, 2854, 2198, 1724, 1627, 1600, 1466, 1377, 1280, 1249.
(R)-3-((S,5E,9E)-1,6,10-trimethyl-4-oxocyclododeca-5,9-dien-2-yn-1-yl)butanal (6)
In a plastic reaction vessel, ketone 5E (201 mg, 500 mmol) was dissolved in 4.0 mL of tetrahydrofuran and 0.40 mL of pyridine and cooled to 0 °C. Then, 0.10 mL of HF-pyridine (~70% HF in ~30% pyridine) was slowly added. After 1 h, an additional 0.10 mL of HF-pyridine was added, and this process was repeated until completion of the reaction as indicated by TLC. (NOTE: If HF-pyridine is added rapidly or in one portion isomerization of the double bond will occur.) The reaction mixture was diluted in 2 mL of ethyl acetate and quenched with 10 mL of saturated solution of NaHCO3. The reaction mixture was extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with saturated NaHCO3 solution, saturated CuSO4 solution, brine, dried over MgSO4, and concentrated. The resulting residue was purified by flash chromatography on silica gel (hexanes:ethyl acetate 80:20) to give the alcohol (137 mg, 95%) as a yellow oil.
1H NMR (400 MHz, CDCl3): δ 6.09 (s, 1H), 5.10 (t, J = 7.3 Hz, 1H), 3.72–3.66 (m, 1H), 3.58–3.52 (m, 1H), 2.58 (bs, 1H), 2.37–2.24 (m, 3H), 2.23–2.13 (m, 2H), 2.12–2.05 (m, 2H), 1.92–1.86 (m, 3H), 1.82 (s, 3H), 1.72–1.62 (m, 1H), 1.50 (s, 1H), 1.47–1.45 (m, 1H), 1.29–1.19 (m, 1H), 1.09 (s, 3H), 0.86 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 179.0, 148.2, 137.1, 132.1, 123.5, 106.6, 84.5, 61.1, 38.3, 37.6, 37.0, 35.3, 34.2, 33.1, 27.2, 21.5, 18.1, 14.8, 13.6. IR (neat): 3600–3045, 2974, 2931, 2858, 2719, 2191, 1730, 1675, 1637, 1450, 1343, 1275. HRMS (ESI-TOF) m/z calculated for C19H29O2 [M+H]+, 289.4244; found 289.4248.
The above alcohol (137 mg, 0.475 mmol) was stirred with 4-methylmorpholine N-oxide (83 mg, 1.18 mmol) and 4Å molecular sieves in 7.6 mL of dry CH2Cl2. After 20 min at rt, tetra-n-propylammonium perruthenate (8.0 mg, 0.04 mmol) was added and the mixture was stirred at rt for 2 h. The reaction was quenched with 10 mL of saturated Na2SO3 solution and extracted with diethyl ether (3 × 15 mL). The combined organic layers were washed with brine, saturated CuSO4 solution, dried over MgSO4, filtered over celite, and concentrated to give the aldehyde 6 (81 mg, 60%) as a yellow oil. (This compound was not stable to silica gel chromatography, but was sufficiently pure to use in the next step without further purification.)
1H NMR (400 MHz, CDCl3): δ 9.76 (s, 1H), 6.09 (s, 1H), 5.14 (t, J = 7.3 Hz, 1H), 2.77 (d, J = 13.8, 1H), 2.38–2.17 (m, 8H), 2.17–2.07 (m, 2H), 1.85 (s, 3H), 1.52 (s, 3H), 1.12 (s, 3H), 0.90 (d, J = 6.0 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 201.5, 178.4, 148.5, 136.1, 131.9, 123.8, 104.5, 85.1, 47.5, 37.8, 37.6, 34.6, 34.2, 33.5, 27.3, 21.4, 18.1, 14.8, 14.4. IR (neat): 2974, 2931, 2858, 2719, 2191, 1724, 1662, 1631, 1450, 1435, 1384, 1211. HRMS (ESI-TOF) m/z calculated for C19H26O2 [M+], 287.1933; found 287.1937.
(3Z,7E,11S,12R,14S)-14-hydroxy-15-iodo-4,8,11,12-tetra-methylbicyclo[9.3.1]pentadeca-1(15),3,7-trien-2-one (7)
As described previously,10 compound 7 was prepared from alkynone 2 using the general protocol for the MgI2-promoted cyclization. (Yield: 35 mg, 60%). (Eluent: hexanes:ethyl acetate, 90:10).
1H NMR (500 MHz, C6D6): δ 6.39 (s, 1H), 5.18 (dd, J = 11.3 Hz, 4.6 Hz, 1H), 4.37–4.32 (m, 1H), 2.65 (d, J = 8.2 Hz, 1H), 2.40–2.33 (m, 1H), 2.19–2.12 (m, 1H), 2.10 (d, J = 13.7 Hz, 1H), 2.02–1.97 (m, 2H), 1.97–1.88 (m, 1H), 1.86–1.81 (m, 1H), 1.70 (s, 3H), 1.67–1.66 (m, 1H), 1.63–1.59 (m, 1H), 1.55 (d, J = 1.2 Hz, 3H), 1.30–1.21 (m, 1H), 1.11–1.07 (m, 1H), 0.88 (s, 3H), 0.70 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 200.5, 147.9, 142.5, 133.2, 128.7, 128.3, 125.7, 122.4, 70.4, 46.8, 36.8, 34.5, 33.5, 28.7, 24.8, 24.6, 21.8, 18.0, 17.4. IR (neat) 3631–3108, 2966, 2935, 2854, 1689, 1602, 1556, 1446, 1381. HRMS (ESI-TOF) m/z calculated for C19H28IO2 [M+H]+, 415.1134; found 415.1146. [α]20D +82.9 (c 0.51, CHCl3).
(3Z,7E,11S,12R,14R)-14-hydroxy-15-iodo-4,8,11,12-tetra-methylbicyclo[9.3.1]pentadeca-1(15),3,7-trien-2-one (3)
As described previously,10 cyclohexenyl alcohol 7 (60 mg, 0.169 mmol) was dissolved in 1.34 mL of benzene and p-nitrobenzoic acid (442 mg, 1.69 mmol) and triphenylphosphine (282 mg, 1.69 mmol) were added. Then, diethyl azodicarboxylate (DEAD) (0.264 mL, 1.69 mmol) was slowly added to the reaction mixture at 0 °C. After completion of the reaction, as indicated by TLC, the reaction mixture was quenched with 4 mL of saturated solution of NaHCO3 and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were washed with H2O (1 × 4 mL), brine (1 × 4 mL), dried over MgSO4, filtered over silica gel, and concentrated. The resulting residue was dissolved in 2 mL of methanol and K2CO3 (46 mg, 0.338 mmol) was added at 0 °C. The reaction mixture was warmed to rt. After 30 min, the reaction mixture was quenched with 4 mL of 1N HCl and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were washed with H2O (1 × 4 mL), brine (1 × 4 mL), dried over MgSO4, and concentrated. The resulting residue was purified by flash chromatography on silica gel (hexanes:ethyl acetate 98:2 to hexanes:ethyl acetate 90:10 to hexanes:ethyl acetate 80:20) to afford the cyclohexenyl alcohol (31 mg, 52%) and the starting material (22 mg, 36%).
1H NMR (400 MHz, CDCl3): δ 6.14 (s, 1H), 5.34–5.24 (m, 1H), 4.76 (s, 1H), 2.91–2.81 (m, 1H), 2.80–2.75 (m, 1H), 2.41 (t, J = 13.5, 1H), 2.31–2.17 (m, 3H), 2.04–2.01 (m, 1H), 1.96–1.86 (m, 2H), 1.85 (s, 3H), 1.80–1.62 (m, 2H), 1.73 (s, 3H), 1.51 (dd, J = 15.2, 3.2, 1H), 1.06 (d, J = 6.8 Hz, 3H), 0.83 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 202.1, 145.8, 143.2, 136.4, 131.9, 128.8, 127.4, 67.2, 47.5, 36.4, 35.7, 34.6, 32.0, 27.8, 24.4, 23.5, 23.4, 18.0, 17.3. IR (neat) 3640–3189, 1639, 1592, 1450, 1380, 1298, 1249. HRMS (ESI-TOF) m/z calculated for C19H27IO2 [M+Na+], 437.0954; found 437.0954. [α]20D +298.7 (c 0.53, CHCl3).
(1R,3R,3aS,6aR,Z)-1-hydroxy-3,3a,6,9-tetramethyl-2,3,3a,4,7,8-hexahydro-1H-cycloocta[de]naphthalen-11(6aH)-one (8a) and (3aS,4R,6R,11aR,Z)-6-hydroxy-3a,4,9-trimethyl-1-methylene-2,3,3a,4,5,6,11,11a-octahydro-1H-cycloocta[de]naphthalen-7(10H)-one (8b)
Compounds 8a and 8b were prepared from alkynone 2 using the general protocol for the MgI2 promoted cyclization. Compounds 8a (endo) and 8b (exo) were obtained in a 2.7:1 ratio. (Yield: 10 mg, 23%). (Eluent: hexanes:ethyl acetate, 80:20). 1H NMR (400 MHz, CDCl3): δ 6.32 (s, 0.4H), 6.29 (s, 1H), 5.65 (d, J = 7.7 Hz, 1H), 4.98 (d, J = 7.4 Hz, 1H), 4.95 (d, J = 6.5 Hz, 0.4H), 4.89 (s, 0.4H), 4.80 (s, 0.4H), 3.46–3.39 (m, 0.4H), 3.28–3.18 (m, 1.4H), 3.17–3.10 (m, 1H), 2.68–2.46 (m, 1.1H), 2.38 (bs, 1H), 2.15–1.97 (m, 5.1H), 2.03 (s, 5.4H), 1.93–1.80 (m, 5.8H), 1.78 (s, 3.6H), 1.72–1.59 (m, 1.8 H), 0.96–0.92 (m, 5H), 0.88 (s, 1.2H), 0.08 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 196.5, 155.7, 155.4, 151.3, 150.9, 138.4, 137.7, 136.6, 132.8 (2C), 121.0, 110.0, 64.3, 44.0, 40.2, 40.0, 38.4, 36.5, 36.0, 35.1, 34.2, 33.7, 33.0, 32.3, 30.5, 30.1, 26.7, 26.3, 24.8, 21.6, 19.2, 17.4, 15.6, 15.4 (4 carbons are missing due to overlap). IR (neat): 3601–3202, 2950, 2923, 2872, 2815, 1730, 1636, 1603, 1554, 1484, 1452, 1435, 1376, 1298, 1258. HRMS (ESI-TOF) m/z calculated for C19H26O2 [M+], 286.1927; found 286.1928.
(1R,3R,3aS,6aR,Z)-3,3a,6,9-tetramethyl-11-oxo-2,3,3a,4,6a,7,8,11-octahydro-1H-cycloocta[de]naphthalen-1-yl 4-nitrobenzoate (9a) and (1R,3R,3aS,6aR,Z)-3,3a,9-trimethyl-6-methylene-11-oxo-2,3,3a,4,5,6,6a,7,8,11-decahydro-1H-cycloocta[de]naphthalen-1-yl 4-nitrobenzoate (9b)
The 8a/8b mixture from above (10 mg, 0.035 mmol) was stirred with p-nitrobenzoyl chloride (7 mg, 0.038 mmol) and pyridine (3.3 μL, 0.038 mmol) in 0.10 mL of CH2Cl2 at rt. After 1 h, the reaction mixture was quenched with 2 mL of 1 N HCl, extracted with diethyl ether (3 × 5 mL), washed with H2O, brine, dried over MgSO4, and concentrated. The reaction mixture was purified by flash chromatography on silica gel (hexanes:ethyl acetate, 90:10 to 70:30) and, then, recrystallized from hexane to give p-nitrobenzoates 9a and 9b (8 mg, 53%; obtained in 2.4:1 ratio.)
1H NMR (500 MHz, CDCl3): δ 8.21 (d, J = 8.4 Hz, 6H), 8.03 (d, J = 8.3 Hz, 6H), 6.24–6.16 (m, 6.2H), 5.61 (d, J = 7.5 Hz, 2.1H), 4.83 (d, J = 4.1 Hz, 2H), 3.46–3.39 (m, 1H), 3.35–3.28 (m, 2.2H), 3.27–3.20 (m, 1.1H), 3.15–3.09 (m, 2.2H), 2.63–2.56 (m, 1H), 2.55–2.46 (m, 1.1H), 2.24–2.15 (m, 2H), 2.15–2.09 (m, 3H), 2.09–2.01 (m, 4.3H), 1.99–1.96 (m, 9.1H), 1.92–1.80 (m, 12.1H), 1.74 (s, 1H), 0.92–0.86 (m, 12.2H), 0.80 (s, 6H). 13C NMR (125 MHz, CDCl3): δ (9a (endo) is reported) 194.3, 163.6, 155.3, 153.0, 150.3, 132.3, 130.5, 123.4, 121.1, 70.4, 40.2, 39.7, 34.4, 33.9, 33.6, 32.2, 30.3, 26.4, 21.6, 21.6, 20.0, 15.2 (2 carbons are missing due to overlap). IR (neat): 2924, 2854, 1720, 1653, 1608, 1527, 1450, 1342, 1265, 1099, 1014. HRMS (ESI-TOF) m/z calculated for C26H29NO5 [M+Na+], 458.1943; found 458.1933.
4,4,9-trimethyl-7-oxodec-8-en-5-ynal (10).8
1H NMR (500 MHz, CDCl3): δ 9.86 (s, 1H), 6.14 (s, 1H), 2.68 (t, J = 5 Hz, 2H), 2.23 (s, 3H), 1.96 (s, 3H), 1.84 (t, J = 5 Hz, 2H), 1.31 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 201.6, 176.7, 157.8, 126.2, 96.8, 83.6, 40.5, 34.4, 31.1, 28.3, 27.7, 21.1. IR (neat) 2970, 2924, 2854, 2206, 1724, 1651, 1608.
1-(6-hydroxy-2-iodo-3,3-dimethylcyclohex-1-en-1-yl)-3-methylbut-2-en-1-one (11)
Compound 11 was prepared from alkynone 10 using the general protocol for the MgI2-promoted cyclization. (Yield: 526 mg, 82%). (Eluent: hexanes:ethyl acetate, 80:20). All spectral data for 11 were in agreement with published data.8
1H NMR (500 MHz, CDCl3): δ 6.27 (s, 1H), 4.34 (m, 1H), 2.62 (s, 1H), 2.24 (s, 3H), 2.00 (s, 3H), 2.06-1.93 (m, 1H), 1.85-1.83 (m, 1H), 1.78-1.74 (m, 1H), 1.20 (s, 3H), 1.12 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 197.5, 158.4, 147.3, 123.7, 118.8, 68.0, 39.1, 32.6, 31.5, 29.0, 27.8, 21.3. IR (neat) 3623-3095, 2962, 2930, 2860, 1664, 1603. HRMS (ESI-TOF) m/z calculated for C13H19O2I1Na [M+Na+], 357.0321; found, 357.0322.
5-iodo-2,2,6,6-tetramethyl-6,7,8,8a-tetrahydro-2H-chromen-4(3H)-one (12)
As previously described,8 compound 12 was prepared from alkynone 10 using the general protocol for the BF3·OEt2-promoted cyclization. (Yield: 197 mg, 77%). (Eluent: hexanes:ethyl acetate, 90:10).
1H NMR (500 MHz, CDCl3): δ 4.48-4.45 (m, 1H), 2.63 (d, J = 15 Hz, 1H), 2.57 (d, J = 15 Hz, 1H), 2.08-2.02 (m, 1H), 1.95-1.91 (m, 1H), 1.87-1.83 (m, 1H), 1.72-1.66 (m, 1H), 1.34 (s, 3H), 1.31 (s, 3H), 1.25 (s, 3H), 1.21 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 199.7, 140.9, 121.6, 74.8, 71.7, 53.3, 41.3, 33.9, 30.6, 27.9, 26.5, 24.6. IR (neat) 2967, 2929, 2866, 1701, 1574. HRMS (ESI-TOF) m/z calculated for C13H19O2I1 [M+], 334.0424; found 334.0423.
1-(2-bromo-6-hydroxy-3,3-dimethylcyclohex-1-en-1-yl)-3-methylbut-2-en-1-one (13)
Prepared from alkynone 10 using the general protocol for the MgI2-promoted cyclization, except MgBr2 was used, and the reaction was warmed to rt for 24 h. (Yield: 41 mg, 52%). (Eluent: hexanes:ethyl acetate, 80:20).
1H NMR (400 MHz, C6D6): δ 6.30 (s, 1H), 4.33–4.30 (m, 1H), 2.69 (d, J = 4.1 Hz, 1H), 2.08 (s, 3H), 1.81–1.72 (m, 1H), 1.57–1.49 (m, 2H), 1.47 (s, 3H), 1.26–1.18 (m, 1H), 1.03 (s, 3H), 0.90 (s, 3H). 13C NMR (100 MHz, C6D6): δ 195.5, 156.3, 142.1, 135.8, 124.9, 68.2, 38.3, 34.0, 28.8, 27.6, 27.2, 26.9, 20.8. IR (neat) 3664–3140, 2966, 2935, 2866, 1666, 1608, 1442, 1381, 1238, 1168, 1067, 1041. HRMS (ESI-TOF) m/z calculated for C13H19BrO2 [M+Na+], 309.0466; found 309.0475.
5-bromo-2,2,6,6-tetramethyl-6,7,8,8a-tetrahydro-2H-chromen-4(3H)-one (14)
As described previously,8 compound 14 was prepared from alkynone 10 using the general protocol for the BF3·OEt2-promoted cyclization. (Yield: 35 mg, 46%). (Eluent: hexanes:ethyl acetate, 90:10).
1H NMR (500 MHz, CDCl3): δ 4.45-4.42 (m, 1H), 2.59 (d, J = 15 Hz, 1H), 2.53 (d, J = 15 Hz, 1H), 2.05-2.03 (m, 1H), 1.85-1.77 (m, 2H), 1.66-1.62 (m, 1H), 1.35 (s, 3H), 1.30 (s, 3H), 1.28 (s, 3H), 1.24 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 198.3, 139.0, 135.3, 74.7, 71.6, 53.9, 40.3, 35.1, 30.8, 30.5, 26.9, 26.3, 24.5. IR (neat) 2970, 2943, 2866, 1701, 1593. HRMS (ESI-TOF) m/z calculated for C13H19O2Br1 [M+], 286.0563; found, 286.0567.
10-methylundec-9-en-6-yne-2,8-dione (15):8
1H NMR (400 MHz, CDCl3): δ 6.12 (s, 1H), 2.58 (t, J = 8 Hz, 2H), 2.40 (t, J = 8 Hz, 2H), 2.19 (s, 3H), 2.15 (s, 3H), 1.86 (s, 3H), 1.85-1.80 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 207.7, 176.6, 157.8, 125.9, 91.1, 83.7, 41.9, 30.1, 27.8, 21.5, 21.1, 18.3. IR (neat) 2915, 2205, 1712, 1650, 1607.
1-(6-hydroxy-2-iodo-6-methylcyclohex-1-en-1-yl)-3-methylbut-2-en-1-one (16)
Prepared from alkynone 15 using the general protocol for the MgI2-promoted cyclization. (Yield 40 mg, 78%). (Eluent: hexanes:ethyl acetate, 90:10). All spectral data for 16 were in agreement with published data.8
1H NMR (500 MHz, CDCl3): δ 6.30 (s, 1H), 3.42 (bs, 1H), 2.79-2.75 (m, 1H), 2.70-2.63 (m, 1H), 2.26 (s, 3H), 2.01 (s, 3H), 1.92-1.91 (m, 1H), 1.89-1.88 (m, 1H), 1.69-1.62 (m, 2H), 1.32 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 198.1, 158.7, 149.6, 124.3, 100.1, 72.0, 41.5, 36.9, 28.5, 28.1, 21.7, 21.3. IR (neat) 3645, 3143, 2966, 2931, 2855, 1660, 1650, 1599. HRMS (ESI-TOF) m/z calculated for C12H17O2INa [M+Na+], 343.0165; found, 343.0166.
(3E,7E,11S,12R,14S)-14-hydroxy-15-iodo-4,8,11,12-tetramethylbicyclo[9.3.1]pentadeca-1(15),3,7-trien-2-one (17)
Prepared from alkynone 6 using the general protocol for the MgI2-promoted cyclization. (Yield: 49 mg, 62%). (Eluent: hexanes:ethyl acetate, 90:10). 1H NMR (400 MHz, C6D6): δ 6.11 (s, 1H), 4.89 (d, J = 9.7 Hz, 1H), 4.44–4.40 (m, 1H), 3.23 (bs, 1H), 2.34–2.18 (m, 1H), 2.07 (s, 3H), 1.99–1.86 (m, 4H), 1.83–1.70 (m, 4H), 1.66 (s, 3H), 1.47–1.38 (m, 1H), 1.20–1.09 (m, 1H), 0.86 (s, 3H), 0.75 (d, J = 6.9 Hz, 3H). 13C NMR (100 MHz, C6D6): δ 196.2, 151.7, 148.6, 133.9, 126.9, 125.6, 119.3, 70.0, 46.2, 38.7, 36.3, 34.2, 33.9, 30.3, 26.6, 23.6, 18.9, 17.6, 17.5. IR (neat) 3608–3084, 2966, 2928, 2877, 2858, 1689, 1627, 1435, 1381, 1225, 1053. HRMS (ESI-TOF) m/z calculated for C19H27IO2 [M+Na+], 437.0954; found 437.0953. [α]20D −7.8 (c 0.25, CHCl3).
(2S,3R)-2,3-dimethyl-2-vinylpentane-1,5-diol (21)
As described previously,10 an inseparable mixture of diastereomers (5.5:1) at the tertiary center was obtained. The mixture was carried on to the next step, and the ratio remained constant through all subsequent transformations. The major diastereomer is reported in all cases.
1H NMR (400 MHz, CDCl3): δ 5.85 (dd, J = 17.7, 10.9 Hz, 1H), 5.21–5.05 (m, 2H), 3.83–3.75 (m, 1H), 3.67–3.60 (m, 1H), 3.59–3.38 (m, 2H), 2.45 (bs, 2H), 1.86–1.71 (m, 2H), 1.27–1.16 (m, 1H), 0.94 (s, 3H), 0.89 (d, J = 6.5 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 143.7, 114.4, 68.5, 61.5, 44.8, 33.8, 33.3, 15.3, 14.7. IR (neat) 3721–3027, 2962, 2877, 1728, 1635, 1454, 1415, 1377. HRMS (ESI-TOF) m/z calculated for C9H19O2 [M+H]+, 159.1385; found 159.1387.
(2S,3R)-5-((tert-butyldimethylsilyl)oxy)-2,3-dimethyl-2-vinylpentan-1-ol (22)
Diol 21 (4.2 g, 26.7 mmol) was diluted in 104 mL of CH2Cl2, cooled to 0 °C, and imidazole (4.3 g, 66.7 mmol) was added. After 20 min, tert-butyldimethylchlorosilane (4.2 g, 26.9 mmol) was slowly added to the reaction mixture at 0 °C. After 20 min, the reaction mixture was quenched with 30 mL of saturated NH4Cl solution. The reaction mixture was then extracted with diethyl ether (3 × 50 mL), washed with H2O, brine, dried over MgSO4, and concentrated. The resulting residue was purified by flash chromatography on silica gel (hexanes:ethyl acetate, 90:10) to give the mono-protected alcohol (5.7 g, 80%, the major diastereomer is reported).
1H NMR (400 MHz, CDCl3): δ 5.81 (dd, J = 17.7, 10.9 Hz, 1H), 5.15 (d, J = 10.9 Hz, 1H), 5.03 (d, J = 17.7, 1H), 3.74–3.68 (m, 1H), 3.60–3.46 (m, 2H), 3.42–3.35 (m, 1H), 1.94 (dd, J = 9.4, 3.9 Hz, 1H), 1.77–1.72 (m, 1H), 1.70–1.62 (m, 1H), 1.15–1.04 (m, 1H), 0.90 (s, 12H), 0.83 (d, J = 6.9 Hz, 3H), 0.06 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 144.1, 114.0, 68.8, 62.1, 44.8, 34.1, 33.5, 25.9, 18.3, 15.0, 14.9, −5.3, −5.4. IR (neat): 3577–3130, 2954, 2927, 2882, 2856. HRMS (ESI-TOF) m/z calculated for C15H33O2Si [M+H]+, 273.2250; found 273.2253.
tert-butyl(((3R,4S)-4-ethynyl-3,4-dimethylhex-5-en-1-yl)oxy)dimethylsilane (23)
Alcohol 22 (2.1 g, 7.7 mmol) was dissolved in 228 mL of CH2Cl2 and Dess-Martin periodinane21 (4.2 g, 10.0 mmol) was added. After 15 min, the reaction mixture was diluted with 50 mL of CH2Cl2 and quenched with 80 mL of a saturated NaHCO3 solution and 80 mL of a saturated Na2S2O3 solution. After the solution became clear, the reaction mixture was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were washed with H2O, brine, dried over MgSO4, filtered over a short pad of silica gel, and concentrated. The resulting residue was used in the next step without further purification.
The aldehyde (1.9 g, 1.0 mmol), from above, was stirred with K2CO3 (1.9 g, 14.0 mmol) in 32 mL of methanol. The Ohira-Bestmann reagent22 (2.0 g, 10.5 mmol) was added at 0 °C. After 25 min, the reaction mixture was warmed to rt. After 24 h, the mixture was quenched with 10 mL of NaHCO3 and extracted with hexanes (3 × 50 mL). The combined organic layers were washed with brine, dried over MgSO4, and concentrated. The reaction mixture was purified by flash chromatography on silica gel (hexanes:ethyl acetate, 97:3) to give the alkyne (1.8 g, 95% over two steps, the major diastereomer is reported) as a yellow oil.
1H NMR (400 MHz, CDCl3): δ 5.65 (dd, J = 17.0, 10.2 Hz, 1H), 5.40 (dd, J = 17.0 Hz, 1.5 Hz, 1H), 5.10 (dd, J = 10.2 Hz, 1.5 Hz, 1H), 3.70–3.64 (m, 1H), 3.61–3.54 (m, 1H), 2.26 (s, 1H), 1.89–1.79 (m, 1H), 1.60–1.49 (m, 1H), 1.27 (s, 3H), 1.27–1.14 (m, 1H), 0.99 (d, J = 6.7 Hz, 3H), 0.89 (s, 9H), 0.04 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 142.4, 113.7, 86.9, 71.9, 61.6, 43.1, 38.1, 35.4, 25.9, 25.7, 18.2, 14.4, −5.3, −5.4. IR (neat): 3311, 2954, 2929, 2857, 2386. HRMS (ESI-TOF) m/z calculated for C16H30OSi [M+], 266.2066; 266.2061.
(E)-3-methyloct-2-en-6-ynal (24)
(E)-3-methyloct-2-en-6-yn-1-ol[Error! Bookmark not defined.] (1.0 g, 7.2 mmol) was diluted in 66 mL of CH2Cl2, MnO2 (12.6 g, 145.0 mmol) was added and the mixture was stirred at rt. After 2 d, the mixture was diluted with CH2Cl2, filtered over celite, and concentrated. The resulting residue was purified by flash chromatography on silica gel (hexane:ethyl acetate, 97:3) to give the aldehyde (800 mg, 80%) as a yellow oil.
1H NMR (400 MHz, CDCl3): δ 10.02 (d, J = 8.0 Hz, 1H), 5.92 (d, J = 8.0 Hz, 1H), 2.43–2.32 (m, 4H), 2.19 (s, 3H), 1.76 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 191.1, 161.7, 127.7, 77.0, 76.9, 39.4, 17.3, 16.8, 3.3. IR (neat): 2919, 2854, 1667, 1633, 1611. HRMS (ESI-TOF) m/z calculated for C9H12O [M+], 137.0602; found 137.0604.
(3R,4S,E)-1-((tert-butyldimethylsilyl)oxy)-3,4,9-trimethyl-4-vinyltetradeca-8-en-5,12-diyn-7-one (25)
Alkyne 23 (1.0 g, 3.7 mmol) was diluted in 11 mL of dry tetrahydrofuran and n-butyllithium (1.8 M in Hexanes 2.3 mL, 4.1 mmol) was slowly added at −78 °C. After 30 min, aldehyde 24 (6.13 mg, 4.5 mmol) was slowly added in 6.6 mL of tetrahydrofuran. After 3 h, the mixture was quenched with 10 mL of saturated NH4Cl solution, and extracted with diethyl ether (3 × 50 mL). The combined organic layers were washed with brine, dried over MgSO4, filtered over a short pad of silica gel, and concentrated. The resulting residue was used in the next step without further purification.
The alcohol, from above, was diluted in 22 mL of CH2Cl2, MnO2 (4.3 g, 49.7 mmol) was added and the mixture was stirred at rt. After 2 h, the mixture was diluted with CH2Cl2, filtered over celite, and concentrated. The resulting residue was purified by flash chromatography on silica gel (hexanes:ethyl acetate, 97:3) to give the ketone (924 mg, 61% over 2 steps, the major diastereomer is reported) as a yellow oil.
1H NMR (400 MHz, CDCl3): δ 6.16 (s, 1H), 5.76 (dd, J = 17.1, 10.3 Hz, 1H), 5.36 (dd, J = 17.1, 1.0 Hz, 1H), 5.13 (dd, J = 10.2 Hz, 1.0 Hz, 1H), 3.72–3.62 (m, 1H), 3.62–3.53 (m, 1H), 2.32 (s, 4H), 2.19 (s, 3H), 1.90–1.79 (m, 1H), 1.76 (s, 3H), 1.72–1.60 (m, 1H), 1.32 (s, 3H), 1.28–1.17 (m, 1H), 1.00 (d, J = 6.7 Hz, 3H), 0.88 (s, 9H), 0.03 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 176.7, 158.2, 141.2, 126.2, 114.4, 94.6, 86.8, 77.3, 76.7, 61.3, 43.5, 40.1, 38.3, 35.3, 25.8, 25.0, 19.4, 18.2, 17.2, 14.6, 3.3, −5.3, −5.4. IR (neat): 2951, 2927, 2855, 2206, 1654, 1651, 1608, 1384, 1360, 1255, 1220, 1125. HRMS (ESI-TOF) m/z calculated for C25H40O2Si [M+], 400.2797; found 400.2806.
(3R,4S,E)-1-hydroxy-3,4,9-trimethyl-4-vinyltetradeca-8-en-5,12-diyn-7-one (26)
In a plastic reaction vessel, ketone 25 (220 mg, 550 mmol) was dissolved in 4.5 mL of tetrahydrofuran and 0.45 mL of pyridine and cooled to 0 °C. Then, HF-pyridine (~70% HF in ~30% pyridine, 0.54 mL, 0.594 mmol) was slowly added. After 2 h, the reaction mixture was diluted in 2 mL of ethyl acetate and quenched with 10 mL of saturated NaHCO3 solution. The reaction mixture was extracted with ethyl acetate (3 ×20 20 mL). The combined organic layers were washed with a saturated NaHCO3 solution, a saturated CuSO4 solution, brine, dried over MgSO4, and concentrated. The resulting residue was purified by flash chromatography on silica gel to give the alcohol (154 mg, 98%, the major diastereomer is reported) as a yellow oil.
1H NMR (400 MHz, CDCl3): δ 6.21 (s, 1H), 5.72 (dd, J = 17.1, 10.2 Hz, 1H), 5.41 (d, J = 17.1 Hz, 1H), 5.19 (d, J = 10.3 Hz, 1H), 3.81–3.73 (m, 1H), 3.68–3.60 (m, 1H), 2.42–2.35 (m, 4H), 2.24 (s, 3H), 1.99–1.89 (m, 1H), 1.81–1.78 (m, 2H), 1.80 (s, 3H), 1.76–1.69 (m, 1H), 1.37 (s, 3H), 1.06 (d, J = 6.7 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 176.7, 158.6, 141.1, 126.1, 114.6, 94.5, 61.0, 43.5, 40.1, 38.3, 35.3, 24.6, 19.5, 17.1, 14.5, 3.3 (3 carbons are missing due to overlap). IR (neat): 3678–3126, 2974, 2920, 2877, 2206, 1651, 1604, 1438, 1381, 1334, 1280, 1226, 1130. HRMS (ESI-TOF) m/z calculated for C19H26O2 [M+Na+], 309.1831; found 309.1834.
(3R,4S,E)-3,4,9-trimethyl-7-oxo-4-vinyltetradeca-8-en-5,12-diynal (20)
Alcohol 26 (154 mg, 0.538 mmol) was stirred with 4-methylmorpholine N-oxide (94 mg, 0.807 mmol) and 4Å molecular sieves in 5.1 mL of dry CH2Cl2 at rt. After 20 min, tetra-n-propylammonium perruthenate (10 mg, 0.027 mmol) was added and the mixture was stirred for 2 h. The reaction was quenched with 10 mL of saturated Na2SO3 solution, extracted with diethyl ether (3 × 50 mL). The combined organic layers were washed with brine, a saturated CuSO4 solution, dried over MgSO4, filtered over celite, and concentrated. The resulting residue was purified by flash chromatography on silica gel (hexanes:ethyl acetate, 90:10) to give the aldehyde (101 mg, 66%, the major diastereomer is reported) as a yellow oil. (Note: Purification of alkynone 20 was rapid, and it was not allowed to remain on silica gel for extended periods of time.)
1H NMR (400 MHz, CDCl3): δ 9.71 (s, 1H), 6.15 (s, 1H), 5.67 (dd, J = 17.1, 10.2 Hz, 1H), 5.38 (d, J = 17.1, 1.0 Hz, 1H), 5.15 (d, J = 10.2 Hz, 1H), 2.67 (dd, J = 17.3, 2.9 Hz, 1H), 2.35–2.25 (m, 6H), 2.17 (s, 3H), 1.73 (s, 3H), 1.32 (s, 3H), 1.04 (d, J = 6.7 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 201.5, 176.3, 159.0, 140.8, 126.0, 115.4, 92.4, 87.5, 47.6, 43.2, 40.1, 36.1, 24.8, 19.5, 17.2, 15.5, 3.4 (2 carbons are missing due to overlap). IR (neat): 2974, 2920, 2854, 2723, 2210, 1774, 1724, 1651, 1604, 1442, 1441, 1381, 1338, 1280, 1222. HRMS (ESI-TOF) m/z calculated for C19H25O2 [M+H]+, 285.1855; found 285.1865.
(E)-1-((3S,4R,6S)-6-hydroxy-2-iodo-3,4-dimethyl-3-vinylcyclohex-1-en-1-yl)-3-methyloct-2-en-6-yn-1-one (27)
Prepared from alkynone 20 using the general protocol for the MgI2 promoted cyclization. (Yield: 23 mg, 77%). (Eluent: hexanes:ethyl acetate, 90:10).
1H NMR (400 MHz, CDCl3): δ 6.24 (s, 1H), 5.51 (dd, J = 17.3, 10.6 Hz, 1H), 5.24 (d, J = 10.7 Hz, 1H), 5.04 (d, J = 17.3 Hz, 1H), 4.64–4.57 (m, 1H), 2.36 (s, 5H), 2.20 (s, 3H), 2.06–1.98 (m, 1H), 1.89–1.80 (m, 1H), 1.76 (s, 3H), 1.67–1.55 (m, 1H), 1.14 (s, 3H), 0.96 (d, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 196.8, 159.0, 149.3, 145.8, 124.0, 114.9, 114.0, 77.6, 77.1, 70.2, 49.5, 40.4, 35.6, 35.4, 19.6, 18.0, 17.1, 16.6, 3.5. IR (neat) 3657–3126, 2966, 2920, 2874, 1666, 1608, 1446, 1411, 1377, 1320, 1176, 1141, 1060. HRMS (ESI-TOF) m/z calculated for C19H25IO2 [M+], 412.0899; found 412.0906.
(E)-1-((3S,4R,6R)-6-hydroxy-2-iodo-3,4-dimethyl-3-vinylcyclohex-1-en-1-yl)-3-methyloct-2-en-6-yn-1-one (28)
Cyclohexenyl alcohol 27 (30 mg, 0.072 mmol) was stirred with p-nitrobenzoic acid (60 mg, 0.363 mmol) and PPh3 (95 mg, 0.363 mmol) in 0.72 mL of dry benzene. Then, diethyl azodicarboxylate (DEAD) (56 μL, 0.363 mmol) was slowly added to the reaction mixture at 0 °C. After 3 h at rt, the reaction mixture was quenched with 4 mL of saturated NaHCO3 solution and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were washed with H2O, brine, dried over MgSO4, filtered over a short pad of silica gel, and concentrated. The resulting residue was dissolved in 2.2 mL of methanol and K2CO3 (19 mg, 0.144 mmol) was added at 0 °C. The reaction mixture was warmed to rt. After 30 min, the reaction mixture was quenched with 4 mL of saturated NaHCO3 solution and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were washed with H2O, brine, dried over MgSO4, and concentrated. The resulting residue was purified by flash chromatography on silica gel (hexanes:ethyl acetate 98:2 to hexanes:ethyl acetate 90:10 to hexanes:ethyl acetate 80:20) to give the inverted cyclohexenyl alcohol (23 mg, 76% over 2 steps, the major isomer is reported).
1H NMR (400 MHz, CDCl3): δ 6.33 (s, 1H), 5.63 (dd, J = 17.3, 10.6 Hz, 1H), 5.33 (d, J = 10.7 Hz, 1H), 5.12 (d, J = 17.3 Hz, 1H), 4.33 (s, 1H), 2.83–2.67 (m, 1H), 2.42 (s, 3H), 2.25 (s, 3H), 2.08 (d, J = 10.6 Hz, 1H), 1.90–1.69 (m, 6H), 1.10 (s, 3H), 1.01 (d, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 197.4, 159.8, 147.1, 145.6, 123.565, 118.1, 115.0, 77.5, 67.2, 49.7, 40.4, 34.2, 32.2, 19.7, 17.6, 17.0, 15.8, 3.5 (1 carbon is missing due to overlap). IR (neat) 3720–3098, 2924, 2874, 2854, 1735, 1666, 1604, 1446, 1373, 1238, 1165. HRMS (ESI-TOF) m/z calculated for C19H25IO2 [M+], 412.0899; found 412.0906.
(2S,6S,7R,8aS)-5-iodo-2,6,7-trimethyl-2-(pent-3-yn-1-yl)-6-vinyl-6,7,8,8a-tetrahydro-2H-chromen-4(3H)-one (29)
Prepared from cyclohexenyl alcohol 27 using the general protocol for the BF3·OEt2-promoted cyclization. (Yield: 10 mg, 90%). (Eluent: hexanes:ethyl acetate, 95:5).
1H NMR (500 MHz, CDCl3): δ 5.51 (dd, J = 17.3, 10.7 Hz, 1H), 5.29 (d, J = 12.9 Hz, 1H), 5.07 (d, J = 17.3 Hz, 1H), 4.51–4.46 (m, 1H), 2.60 (d, J = 2.8 Hz, 2H), 2.21–2.14 (m, 2H), 2.02–1.87 (m, 2H), 1.76 (s, 4H), 1.72–1.62 (m, 2H), 1.24 (s, 3H), 1.18 (s, 3H), 0.97 (d, J = 6.8 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ 198.6, 145.2, 140.9, 119.8, 115.3, 78.3, 76.0, 75.9, 71.6, 52.5, 51.9, 36.4, 34.7, 33.0, 26.7, 18.1, 16.8, 12.8, 3.4. IR (neat) 2974, 2931, 2870, 1747, 1701, 1582, 1449, 1379, 1327, 1230, 1165. HRMS (ESI-TOF) m/z calculated for C19H25IO2 [M+], 412.0899; found 412.0906.
Oxadecalin 1
Cyclohexenyl alcohol 3 (20 mg, 0.048 mmol) was diluted in 0.48 mL of dry CH2Cl2. AuCl3 (1.4 mg, 0.004 mmol) was then added at 0 °C, and warmed to rt. After completion of the reaction by TLC, the reaction mixture was purified by flash chromatography on silica gel (hexane:ethyl acetate, 99:1) to afford the oxadecalin (10 mg, 50%) as a pale yellow oil. All spectral data for 1 were in agreement with published data.10 1H NMR (400 MHz, CDCl3): δ 5.51–5.44 (m, 1H), 4.16–4.11 (m, 1H), 3.06-2.94 (m, 1H), 2.78 (d, J = 17.1 Hz, 1H), 2.56 (d, J = 17.1 Hz, 1H), 2.47–2.32 (m, 2H), 2.08–1.95 (m, 3H), 1.87–1.76 (m, 1H), 1.75–1.41 (m, 4H), 1.66 (s, 3H), 1.29 (s, 3H), 1.06 (d, J = 7.0 Hz, 3H), 0.94 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 197.1, 143.5, 136.9, 130.5, 130.0, 76.1, 68.7, 48.7, 47.1, 41.6, 35.7, 35.0, 31.8, 29.7, 29.6, 26.1, 23.6, 23.5, 17.3. IR (neat) 2966, 2924, 2854, 1693, 1570, 1450, 1373, 1292, 1207, 1140, 1053. HRMS (ESI-TOF) m/z calculated for C19H27IO2 [M+], 415.1134; found 415.1137. [α]20D +200.4 (c 0.15, CHCl3).
Tricycle 31
As described previously,10 compound 3 (20 mg, 0.048 mmol) was dissolved in 0.5 mL of toluene at rt. Then, 2,6-lutidine (0.11 mL, 0.48 mmol) and tert-butyldimethylsilyl trifluoromethanesulfonate (TBSOTf) (0.05 mL, 0.48 mmol) were rapidly added to the reaction mixture. After 10 min, the reaction mixture was diluted in 2 mL ethyl acetate, quenched with 0.5 mL of saturated NH4Cl solution, and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were washed with H2O (1 × 10 mL), brine (1 × 10 mL), dried of MgSO4, and concentrated. The resulting residue was purified by flash chromatography on silica gel (hexanes:ethyl acetate 98:2) to give the tricycle (15 mg, 54%) as a yellow oil.
1H NMR (400 MHz, CDCl3): δ 5.41 (d, J = 8.5 Hz, 1H), 4.56 (s, 1H), 3.86 (t, J = 2.8 Hz, 1H), 2.97–2.83 (m, 1H), 2.36–2.18 (m, 3H), 2.01–1.86 (m, 3H), 1.75 (s, 3H), 1.67–1.157 (m, 4H), 1.23 (s, 3H), 1.01 (d, J = 7.1 Hz, 3H), 0.99 (s, 9H), 0.89 (s, 3H), 0.22 (s, 3H), 0.19 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 150.2, 133.5, 131.3, 129.0, 120.4, 112.2, 77.9, 70.6, 46.2, 41.9, 35.8, 35.5, 32.3, 27.8, 26.4, 26.3, 24.8, 22.5, 19.5, 18.7, 17.5, −4.1, −4.3. IR (neat) 2958, 2928, 1635, 1462, 1323, 1253, 1200, 1085. HRMS (ESI-TOF) m/z calculated for C25H42IO2Si [M+H]+, 529.1999; found 529.1990. [α]20D +21.3 (c 0.37, CHCl3).
Supplementary Material
Acknowledgements
We thank the National Institute of Health (NIGMS R01 GM079364) for funding this work. We are also grateful to Dr. Alice Bergmann (University of Buffalo) and Dr. Furong Sun (University of Illinois, Urbana-Champaign) for carrying out high-resolution mass spectroscopy, and to Dr. William Brennessel (University of Rochester) for solving structures by X-ray crystallography. The authors thank Dr. Daniel P. Canterbury (University of Rochester) for helpful discussions. We used the computing facility of the CRIHAN (project 2006-013.)
Footnotes
Supporting Information: Spectra (1H and 13C) of all new compounds, DFT computational details, X-ray crystallographic data for 9a and 9b. This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- (1) a).Ressault B, Jaunet A, Geoffroy P, Goudedranche S, Miesch M. Org. Lett. 2012;14:366–369. doi: 10.1021/ol203118t. [DOI] [PubMed] [Google Scholar]; b) Yagi K, Turitani T, Shinokubo, Oshima K. Org. Lett. 2002;4:3111–3114. doi: 10.1021/ol026413h. [DOI] [PubMed] [Google Scholar]; c) Douelle F, Capes AS, Greaney MF. Org. Lett. 2007;9:1931–1934. doi: 10.1021/ol070482k. [DOI] [PubMed] [Google Scholar]; d) Koseki Y, Fujino K, Takeshita A, Sato H, Nagasaka T. Tetrahedron: Asymmetry. 2007;18:1533–1539. [Google Scholar]; e) Nazef N, Davies RDM, Greaney MF. Org. Lett. 2012;14:3720–3723. doi: 10.1021/ol301513h. [DOI] [PubMed] [Google Scholar]
- (2).Kishi Y, Taniguchi M, Hino T, Kobayashi S, Nakagawa M. Tetrahedron Lett. 1986;39:4767–4770. [Google Scholar]
- (3) a).Lee SI, Hwang GS, Ryu DH. Synlett. 2007:59–62. [Google Scholar]; b) Wei HX, Gao JJ, Li G, Paré PW. Tetrahedron Lett. 2002;43:5677–5680. [Google Scholar]; c) Ayed TB, Villìeras J, Amri H. Tetrahedron. 2000;56:805–809. [Google Scholar]; d) Zhang CM, Lu XY. Synthesis. 1996:586–588. [Google Scholar]; e) Yadav JS, Reddy BVS, Gupta MK, Eeshwaraiah B. Synthesis. 2005:57–60. [Google Scholar]; f) Deng G, Paré P, Hu H, Wei H. Helv. Chim. Acta. 2003;86:3510–3515. [Google Scholar]; g) Wei HX, Hu J, Jasoni RL, Li G, Paré PW. Helv. Chim. Acta. 2004;87:2359–2363. [Google Scholar]; h) Wei H, Hu J, Purkiss DW, Paré PW. Tetrahedron Lett. 2003;44:949–952. [Google Scholar]; i) Sharma V, McLaughlin ML. J. Comb. Chem. 2010;12:327–331. doi: 10.1021/cc100001e. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Lee SI, Hwang GS, Shin SC, Lee TG, Jo RH, Ryu DH. Org. Lett. 2007;9:5087–5089. doi: 10.1021/ol702134w. [DOI] [PubMed] [Google Scholar]
- (4) a).Li G, Banerjee S, Timmons S, Kattubonia A. Tetrahedron. 2006;62:7151–7154. S. [Google Scholar]; b) Li QJ, Shi M, Lyte JM, Li G. Tetrahedron Lett. 2006;47:7699–7702. [Google Scholar]
- (5).Kattuboina A, Kaur P, Timmons C, Li G. Org. Lett. 2006;13:2771–2774. doi: 10.1021/ol060828b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6) a).Chen D, Guo L, Kotti S, Li G. Tetrahedron: Asymmetry. 2005;16:1757–1762. [Google Scholar]; b) Senapati BHG, Lee S, Ryu DH. Angew. Chem. 2009;121:4462–4465. [Google Scholar]; Angew. Chem. Int. Ed. 2009;48:4398–4401. doi: 10.1002/anie.200900351. [DOI] [PubMed] [Google Scholar]; c) Li G, Wei H, Phelp B, Purkiss D, Kim S. Org. Lett. 2001;3:823–826. doi: 10.1021/ol000377+. [DOI] [PubMed] [Google Scholar]; d) Chen D, Timmons C, Liu J, Headley A, Li G. Eur. J. Org. Chem. 2004:3330–3335. [Google Scholar]
- (7) a).Wei HX, Chen DJ, Xu X, Li G, Paré PW. Tetrahedron: Asymmetry. 2003;14:971–974. [Google Scholar]; b) Xu X, Chen D, Wei H, Li G, Xiao TL, Armstrong DW. Chirality. 2003;15:139–142. doi: 10.1002/chir.10179. [DOI] [PubMed] [Google Scholar]
- (8).Ciesielski J, Canterbury DP, Frontier AJ. Org. Lett. 2009;11:4374–4377. doi: 10.1021/ol901721y. [DOI] [PubMed] [Google Scholar]
- (9).Sloman D, Bacon JW, Porco JA. J. Am. Chem. Soc. 2011;133:9952–9955. doi: 10.1021/ja203642n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ciesielski J, Cariou K, Frontier AJ. Org. Lett. 2012;14:4082–4085. doi: 10.1021/ol3017116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For recent reviews on total syntheses and approaches to the phomactins, see: Goldring WPD, Pattenden G. Acc. Chem. Res. 2006;39:354. doi: 10.1021/ar050186c. Cole KP, Hsung RP. ChemTracts. 2003;16:811. For total syntheses of phomactin A, see: Goldring WPD, Pattenden G. Chem. Commun. 2002:1736. doi: 10.1039/b206041h. Tang Y, Cole KP, Buchanan GS, Li G, Hsung RP. Org. Lett. 2009;11:1591. doi: 10.1021/ol900237e. Buchanan GS, Cole KP, Tang Y, Hsung RP. J. Org. Chem. 2011;76:7027. doi: 10.1021/jo200936r. Buchanan GS, Cole KP, Li G, Tang Y, You L, Hsung RP. Tetrahedron. 2011;67:10105. doi: 10.1016/j.tet.2011.09.111. Mohr PJ, Halcomb RL. J. Am. Chem. Soc. 2003;125:1712. doi: 10.1021/ja0296531. For approaches to phomactin A, see: Seth PP, Totah NI. Org. Lett. 2000;2:2507. doi: 10.1021/ol0061788. Teng D, Wang B, Augatis AJ, Totah NI. Tetrahedron Lett. 2007;48:4605. doi: 10.1016/j.tetlet.2007.04.122. Chemler SR, Iserloh U, Danishefsky SJ. Org. Lett. 2001;3:2949. doi: 10.1021/ol0161357. Mi B, Maleczka RE. Org. Lett. 2001;3:1491. doi: 10.1021/ol015807q. Shapland PDP, Thomas EJ. Tetrahedron. 2009;65:4201., and references cited therein. Schwartz KD, White JD. Org. Lett. 2011;13:248. doi: 10.1021/ol1026816. Huang S, Du G, Lee CJ. Org. Chem. 2011;76:6534. doi: 10.1021/jo200644t. You LF, Hsung RP, Bedermann AA, Kurdyumov AV, Tang Y, Buchanan GS, Cole KP. Adv. Synth. Catal. 2008;350:2885.
- (12) a).Elliott MR, Dhimane AL, Hamon L, Malacria M. Eur. J. Org. Chem. 2000:155. [Google Scholar]; b) Fürstner A. Chem. Rev. 1999;99:991. doi: 10.1021/cr9703360. For a comprehensive review of this reaction, see: [DOI] [PubMed] [Google Scholar]
- (13).Ley S, Norman J, Griffith W, Marsden S, Jung ME. Synthesis. 1994:639. [Google Scholar]
- (14).See supporting information.
- (15).For examples of β-bromoallenolates, see: Kataoka T, Kinoshita H, Kinoshita S, Iwamura T, Watanabe S. Angew. Chem. 2000;112:2448–2450. doi: 10.1002/1521-3773(20000703)39:13<2358::aid-anie2358>3.0.co;2-y. Angew. Chem. Int. Ed. 2000;39:2358–2360. doi: 10.1002/1521-3773(20000703)39:13<2358::aid-anie2358>3.0.co;2-y. Wei HX, Jasoni RL, Hu JL, Li G, Paré PW. Tetrahedron. 2004;60:10233–10237.
- (16).Cyclization of 15 to 16 could also be promoted using AlCl3 (6.5 equiv, 75% yield, see reference 8.)
- (17).The stereochemistry of the alcohol was assigned using nOe, see: Supporting Information.
- (18).Zimmerman HE, Traxler MD. J. Am. Chem. Soc. 1957;79:1920–1923. [Google Scholar]
- (19).For a related cyclization with a similar analysis of the stereochemical outcome, see reference 9.
- (20).For additional examples regarding the reversibility of reactions involving β-iodoallenolate intermediates, see Lee SI, Hwang GS, Shin SC, Lee TG, Jo RH, Ryu DH. Org. Lett. 2007;9:5087–5089. doi: 10.1021/ol702134w., and references cited therein.
- (21) a).Dess DB, Martin JC. J. Org. Chem. 1983;48:4155–4156. [Google Scholar]
- (22).The Ohira-Bestmann reagent was prepared according to a literature procedure: Pietruszba J, Witt A. Synthesis. 2006:4266–4268.
- (23).Similarly, cyclization of alkynone 20 with MgI2 in tetrahydrofuran gave cyclohexenyl alcohols 27 and 28 in 60% yield as a 7:1 mixture of diastereomers.
- (24).Cyclohexenyl alcohols 27 and 28 were easily separated via flash chromatography, or alternatively, 28 could be obtained in 76% yield from cyclohexenyl alcohol 27 using standard Mitsunobu conditions.
- (25).For a comprehensive review of the Mitsunobu reaction, see: Swamy KCK, Kumar NNB, Balaraman E, Kumar KVPP. Chem. Rev. 2009;109:2551–2651. doi: 10.1021/cr800278z.
- (26).For a detailed study of BF3·OEt2-promoted and AuCl3-catalyzed oxa-Michael addition reactions of related systems, see Ciesielski J, LeBœuf D, Stern HA, Frontier AJ. Adv. Synth. Catal. 2013;355 doi: 10.1002/adsc.201300265. (in press)
- (27).For examples of related cationic cyclizations involving alkynes, see: Johnson WS. Bioorg. Chem. 1976;5:51–98. Yoder RA, Johnston JN. Chem. Rev. 2005;105:4730–4756. doi: 10.1021/cr040623l. Gilmore K, Alabugin IV. Chem Rev. 2011;111:6513–6556. doi: 10.1021/cr200164y.
- (28).Computations were performed with the Gaussian ‘03 software package: Gaussian 03, Revision C.01, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian, Inc.; Wallingford CT: 2003.
- (29).Optimizations were carried out at the B3LYP/6-311G**[Mg,I]/6-31G*[other elements] level. Solvation corrections for dichloromethane and THF were carried out using the PCM method (UFF radii). Only the solvent corrected free energies are presented (kcal/mol).
- (30).Chen L, Zhang L, Lv J, Cheng J, Luo S. Chem. Eur. J. 2012;18:8891–8895. doi: 10.1002/chem.201201532. [DOI] [PubMed] [Google Scholar]
- (31) a).Ericson A, Persson IJ. Organomet. Chem. 1987;326:151–158. [Google Scholar]; b) Wellmar A, Persson I. J. Organomet. Chem. 1991;41:143–153. [Google Scholar]; c) Wehmschulte RJ, Twamley B, Khan MA. Inorg. Chem. 2001;40:6004–6008. doi: 10.1021/ic010513m. [DOI] [PubMed] [Google Scholar]
- (32).See Supporting Information for the geometries.
- (33).[D+MgI+] is the only intermediate with a structure closely related to 33. The distance between the central allene carbon and the aldehyde carbon is 2.54 Å, while the distance between the alkene carbon and the central allene carbon is 1.7 Å. However, dissociation of the metallic fragment directly leads to the cyclized product E.
- (34).In light of these findings, a concerted pathway must also be considered as a possible mechanism for the cyclization of type I alkynones into type IV β-iodocyclohexenyl alcohols (Scheme 1). By analogy, this cascade could be initiated by activation of the aldehyde with Lewis acid, which would suffer attack by the electron-deficient alkynone and intermolecular trapping with iodide. Studies are underway to assess the validity of this alternative mechanism.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.