

Abstract
The transcription factor STAT3 has been implicated in axon regeneration. Here we investigate a role for STAT3 in sympathetic nerve sprouting after myocardial infarction (MI) - a common injury in humans. We show that NGF stimulates serine phosphorylation (S727) of STAT3 in sympathetic neurons via ERK1/2, in contrast to cytokine phosphorylation of Y705. Maximal sympathetic axon regeneration in vitro requires phosphorylation of both S727 and Y705. Furthermore, cytokine signaling is necessary for NGF-induced sympathetic nerve sprouting in the heart after MI. Transfection studies in neurons lacking STAT3 suggest two independent pools of STAT3, phosphorylated on either S727 or Y705, that regulate sympathetic regeneration via both transcriptional and non-transcriptional means. Additional data identify STAT3-microtubule interactions that may complement the well-characterized role of STAT3 stimulating regeneration associated genes. These data show that STAT3 is critical for sympathetic axon regeneration in vitro and in vivo, and identify a novel non-transcriptional mode of action.
Keywords: ciliary neurotrophic factor, myocardial infarction, nerve growth factor, signal transducer and activator of transcription 3, sympathetic axon regeneration
Introduction
Sympathetic neurons, like other peripheral neurons, can regenerate following injury although the mechanisms are not completely understood. Nerve growth factor (NGF) is required for sympathetic neuron survival and enhances sympathetic axon outgrowth during development (Glebova & Ginty, 2005), but its role in adult nerve regeneration is less clear. Mature sympathetic neurons no longer require NGF for their survival (Ruit et al, 1990; Sofroniew et al, 2001), and regeneration of adult sympathetic axons to the skin after nerve transection does not require NGF (Gloster & Diamond, 1995). However, collateral sprouting of sympathetic axons into injured skin (Gloster & Diamond, 1992; Gloster & Diamond, 1995), arthritic joints (Ghilardi et al, 2012), or damaged myocardium (Hasan et al, 2006; Wernli et al, 2009) is stimulated by NGF in the target tissue and can be blocked by NGF antibodies.
The milieu produced by tissue damage is complex and includes many factors in addition to NGF, including the inflammatory cytokines CNTF (Ciliary Neurotrophic Factor) and LIF (Leukemia Inhibitory Factor) (Adler, 1993; Rao et al, 1993). These cytokines act via the gp130 receptor (Ip et al, 1992; Taga & Kishimoto, 1997) to promote axon regeneration in the central and peripheral nervous systems (Cafferty et al, 2001; Ekstrom et al, 2000; Homs et al, 2011; Leibinger et al, 2009). In sympathetic neurons cytokines are involved in the “conditioning lesion” response whereby prior injury enhances the subsequent regeneration (Hyatt Sachs et al, 2010; McQuarrie & Grafstein, 1973; Navarro & Kennedy, 1990; Shoemaker et al, 2005). Cytokines are thought to enhance nerve regeneration after injury through stimulating transcription of regeneration associated genes via tyrosine phosphorylation of Signal Transducer and Activator of Transcription 3 (STAT3) (Ben-Yaakov et al, 2012; Habecker et al, 2009; Lee et al, 2004; Liu & Snider, 2001; O’Brien & Nathanson, 2007; Qiu et al, 2005; Smith & Skene, 1997). A second subcellular locus of STAT3 action was identified recently in embryonic motor neurons, where tyrosine-phosphorylated STAT3 enhanced microtubule stability in motor axons from pmn (progression motor neuronopathy) mice, which contain unstable microtubules (Selvaraj et al, 2012).
NGF acting through the TrkA receptor can also stimulate phosphorylation of STAT3, but NGF triggers phosphorylation of STAT3 on serine (Ng et al, 2006b; Zhou & Too, 2011) rather than tyrosine. Likewise, activation of TrkB by Brain Derived Neurotrophic Factor (BDNF) leads to serine 727 (S727) phosphorylation of STAT3 in hippocampal (Ng et al, 2006b) and cortical (Zhou & Too, 2011) neurons. Thus, STAT3 serves as a downstream mediator for neurotrophin signaling as well as cytokine signaling. Interestingly, serine phosphorylation of STAT3 is required for neurotrophin-stimulated neurite extension in PC12 cells and axon regeneration in hippocampal and cortical neurons (Ng et al, 2006b; Zhou & Too, 2011). NGF stimulates transcription of regeneration associated genes in PC12 cells via STAT3 (Ng et al, 2006b), but serine phosphorylated STAT3 is also found in axons and growth cones (Ng et al, 2006b; Zhou & Too, 2011), suggesting that it may play a non-transcriptional role.
We have investigated the role of NGF and gp130 cytokines on sympathetic nerve sprouting after myocardial infarction (MI), which is a common source of nerve damage in humans. Over 1 million people in the U.S. suffer an MI each year (Roger et al, 2012), resulting in the loss of sympathetic nerve terminals in undamaged peri-infarct myocardium (Barber et al, 1983; Inoue & Zipes, 1988; Vaseghi et al, 2012). NGF is elevated in the heart following MI (Abe et al, 1997; Hiltunen et al, 2001; Meloni et al, 2010; Zhou et al, 2004), and blocking NGF with antibodies or preventing infiltration of NGF-producing immune cells into the heart inhibits post-infarct sympathetic nerve sprouting (Hasan et al, 2006; Wernli et al, 2009). However, LIF and several other gp130 cytokines are also elevated in the heart following myocardial infarction (Aoyama et al, 2000; Fischer & Hilfiker-Kleiner, 2007; Frangogiannis et al, 2002; Gwechenberger et al, 1999; Hilfiker-Kleiner et al, 2010), and their role in sympathetic regeneration has not been examined in vivo. In this study, we identify STAT3 as an integrator of neurotrophin and inflammatory cytokine signaling. We show that gp130 cytokine signaling is necessary for the NGF-induced sympathetic nerve sprouting in the heart after MI, and provide evidence that STAT3 plays a non-transcriptional role in sympathetic axons that complements its role stimulating regeneration associated genes.
Materials and methods
Materials
Matrigel™ was purchased from BD Biosciences (San Jose, CA). Fetal bovine serum was purchased from ATCC (Manassas, VA). Goat serum was from Jackson Immunoresearch laboratories (West Grove, PA) CNTF was from PreproTech (Rocky Hills, NJ). Nerve growth factor (NGF) was purchased from Austral Bioloicals (San Ramon, CA). Dispase was purchased from Boehringer Mannheim (Indianapolis, IN). Collagenase type II was purchased from Worthington Biochemicals (Freehold, NJ). Nitrocellulose membranes were from Schleicher & Schuell (Dassel, Germany). Protease inhibitor cocktail tablets were from Roche Applied Science (Indianapolis, IN). Bovine Serum Albumin-Fraction V (BSA) was from Thermo Fischer Scientific (Waltham, MA). UO126, PMSF (phenylmethylsulfonyl fluoride), Phosphatase Inhibitor cocktails (#2 & #3), and poly-L-lysine were from Sigma-Aldrich (St. Louis, MO). Maxi Prep Kits were from Quigen (Valencia, CA). SuperSignal Dura and bicinchoninic acid (BCA) protein assay kit Cat# 23227 were from Thermo Scientific (Rockford, IL). STAT3i V/Stattic (phospho-STAT3 inhibitor) was from Calbiochem (Darmstadt, Germany). Galiellalactone (STAT3 DNA binding inhibitor) was from Santa Cruz Biotechnology (Santa Cruz, CA). 4x sample buffer and 20x reducing agent were from Bio-Rad (Hercules, CA). Poly-D-Lysine/Laminin Coated Glass Coverslips (Round 12 mm No. 1 German Glass) and mouse type IV collagen were from BD Biosciences (San Jose, CA).
Antibodies
βIII-Tubulin TUJ1 mouse monoclonal antibody was from Covance (Princeton, NJ). Total STAT3 (#9132), phospho-STAT3 (Ser727) (#9134), phospho-STAT3 (Tyr705) (#9131), Total ERK1/2 (#9102), phospho-ERK1/2 (#9101), STAT3 (79D7) Rabbit mAb (sepharose bead conjugate) #4368, and Rabbit (DA1E) mAb isotype control (sepharose bead conjugate) #3423 were all from Cell Signaling Technology (Danvers, MA). Rabbit anti-Tyrosine Hydroxylase (TH) was from Millipore (Bedford, MA). Species-specific secondary antibodies conjugated to horseradish peroxidase were from Pierce (Rockford, IL). Alexa Fluor® 488-conjugated goat anti-rabbit and Alexa Fluor® 568-conjugated goat anti-mouse secondary antibodies were from Jackson Immunoresearch Laboratories (West Grove, PA).
Animals
Pregnant adult Sprague-Dawley rats were obtained from Charles River Laboratories (Wilmington, MA). Wild type C57BL/6J mice were obtained from Jackson Laboratories West (Sacramento, CA). STAT3lox/DBH-Cre mice (referred to as STAT3 KO mice) and gp130lox/DBH-Cre mice (referred to as gp130 KO mice) (Stanke et al, 2006) were acquired from Hermann Rohrer (Max-Planck Institute for Brain Research, Frankfurt am Main). All mice were kept on a 12h:12h light-dark cycle with ad libitum access to food and water. Age and gender-matched male and female mice 12–18 weeks old were used for surgeries, while ganglia from neonatal mice were used for explants and dissociated cultures. All procedures were approved by the OHSU Institutional Animal Care and Use Committee and comply with the Guide for the Care and Use of Laboratory Animals published by the National Academies Press (8th edition).
Cell culture
Sympathetic neurons were dissociated from superior cervical ganglia (SCG) of newborn rat pups or mouse pups as described (Pellegrino et al, 2011). Neurons were cultured in C2 media (DMEM/F12 1:1, BSA 0.5 mg/ml, L-glutamate 1.4 mM, selenium 30 nM, transferrin 10 μg/ml, insulin 10μg/ml) supplemented with 100U/ml penicillin G, 100 μg streptomycin sulfate, and 10 or 50 ng/ml NGF). Cultures were maintained at 37° C in 5% CO2 in C2 media supplemented with10 ng/ml NGF (transfection experiments) or 50 ng/ml NGF (immunocytochemistry). Half the media was changed every other day as needed. The duration and doses are noted in figure legends. PC12 cells were a gift from Philip J.S. Stork (Vollum Institute, OHSU, Portland, OR). PC12 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 100U/ml penicillin G, and 100 μg/ml streptomycin sulfate at 37° in 5% CO2. Cells were starved in 1% FBS for 3 hrs prior to all stimulations.
Plasmids and transfection
STAT3 constructs [Stat3-WT (pcDNA3), Stat3-WT Flag (pRc/CMV), Stat3-S727A (pRc/CMV), Stat3-Y705F-Flag (pRc/CMV)] were from Addgene (Cambridge, MA; deposited by Jim Darnell, The Rockefeller University). The pcDNA 3.1 expression vector and Emerald GFP (pcDNA6.2-EmGFP) were from Invitrogen-Life technologies (Grand Island, NY). Dissociated SCG neurons from neonatal STAT3 KO mice were co-transfected with the STAT3 constructs and GFP using the Neon™ Transfection System from Invitrogen-Life technologies (Grand Island, NY). Neurons were electroporated per manufacturer’s instructions: pulse voltage (1500 V), pulse width (20 ms), and pulse number (1). Equal numbers of neurons were transfected with equal amounts of DNA in each experiment. In all transfection experiments, neurons were cultured in C2 media supplemented with NGF (10ng/ml) and CNTF (100ng/ml). Axons of GFP-expressing neurons were measured at 40 hrs post-transfection using NIS-Elements AR 3.0.
Explant axon outgrowth analysis
SCG explants from P1 rat or mouse pups were explanted into Matrigel™ using C2 media supplemented with NGF (2ng/ml). Axon outgrowth was visualized 18 hr after plating (time 0). Images were captured at time 0 and then 6 hrs later and axon length was measured using NIS-Elements AR 3.0. Multiple sites of growth (3–6) were measured for each ganglion and averaged. A growth rate (μm/hr) was calculated for the 6 h window from a minimum of 3 explants per condition in each experiment, and experiments were repeated at least three times with similar results. In explants experiments, STAT3 phosphorylation was stimulated on serine by exogenous NGF and on tyrosine by endogenous LIF from ganglionic non-neuronal cells (Sun et al, 1994).
Immunocytochemistry
Neonatal sympathetic neurons were plated onto 12mm Poly-D-Lysine/Laminin coated glass coverslips (BD Biosciences), and at 4 DIV neurons were treated with NGF or CNTF as described in figure legends. Neurons were fixed in 4% paraformaldehyde-4% sucrose (10min), permeabilized with 0.3% Triton X-100 in phosphate buffered saline, pH 7.5 (PBST) for 5 min, and blocked with 10% BSA (30min). Cells were washed in PBST 3×10 minutes. Neurons were labeled with primary antibodies diluted in PBST/3% BSA (Total STAT3, 1:50), (Phospho-Serine 727 STAT3, 1:50) and (βIII-Tubulin, 1:500). Neurons were washed 3×10 minutes with PBST and stained with secondary antibodies diluted 1:500 in PBST/3%BSA. Hoechst 34580 Stain (0.5 μg/ml) was added to the last wash (3×10 minutes) to visualize nuclei. Secondary antibodies alone were performed to control for non-specific staining and signal was negligible. Imaging was performed on a LSM710 laser scanning confocal microscope from Carl Zeiss MicroImaging (Thornwood, NY) and digital images were processed using ZEN, LSM Image, ImageJ and/or Photoshop.
Myocardial Infarction by ischemia-reperfusion
Ischemia-reperfusion was carried out as previously described (Alston et al, 2011; Parrish et al, 2009). All surgical procedures were performed under aseptic conditions using isoflurane anesthesia. The left anterior descending coronary artery (LAD) was ligated for 30 minutes before the coronary ligature was released. Reperfusion was confirmed by visible epicardial hyperemia. Buprenorphine (0.1mg/kg) was administered as needed to ensure the animals were comfortable following surgery. Sham animals underwent the same procedure except for ligation of the LAD. Animals were sacrificed 24 hrs or 3 days after surgery for immunohistochemistry.
Immunohistochemistry and innervation density
Hearts were fixed for 1h in 4% paraformaldehyde, rinsed in PBS, cryoprotected in 30% sucrose and 10 μm transverse sections thaw-mounted onto charged slides. Sections were stained for tyrosine hydroxylase exactly as described in (Lorentz et al, 2010). Innervation density was determined by threshold discrimination using ImageJ as previously described (Lorentz et al, 2010). Three sections at least 150 μm apart were analyzed from each heart and averaged together. Each section was analyzed by two independent observers, and the data shown are the average of both determinations. Fiber density was quantified in 4–6 hearts from each experimental group, focusing on the region approximately 500–900 μm from the edge of the infarct. Fiber density from sham animals were quantified in the corresponding region of the left ventricle.
Western Blotting
Cells or tissue were placed on ice and lysed in RIPA buffer (1% Triton-X 100, 1%sodium deoxycholate, 0.2% sodium docecyl sulfate, 125 mM NaCl, 50 mM Tris pH 8.0, 10% glycerol, 1 mM NaF, 1 mM PMSF, 1x phosphatase inhibitor cocktail #2 & #3, 1x complete protease inhibitor cocktail EDTA-free). Samples were size fractionated on 4%-12% Bis-Tris acrylamide denaturing gels and transferred to nitrocellulose membranes for blotting. Membranes were blocked in 3% nonfat dry milk in TBST (100 mM NaCl, 10 mM Tris, and 0.1% Tween 20, pH 7.4) for 30 minutes and incubated at 4°C overnight in antibodies against STAT3, STAT3 (pS727), STAT3 (pY705), ERK1/2, and Phospho-ERK1/2 diluted at 1:1000 in TBST with 3% BSA. After washing in TBST 3 times for 5 minutes, blots were incubated for 1 hr at room temperature with the appropriate secondary antibody diluted 1:5000 in TBST with 3% nonfat dry milk. Blots were washed as before and immunoreactive bands were detected via chemiluminescence. Bands were captured by a −40°C CCD camera and total band densities quantified using Labworks 4.0 Software.
Real-time PCR
Real-time PCR was performed as previously described (Pellegrino et al, 2011). For in vitro experiments, RNA was isolated from neonatal cultured rat neurons using Cells to cDNA (Ambion) and reverse transcribed. For MI experiments, stellate ganglia were harvested 3 days after sham or ischemia-reperfusion surgery and stored immediately in RNAlater. RNA was isolated from individual stellate ganglia using the Ambion RNAqueous micro kit, and 200 ng of total RNA was reverse transcribed and diluted for use. All real-time PCR reactions were performed using the 7500 Real-Time PCR System from ABI/Life Technologies. Pre-validated TaqMan® primers to STAT3, stathmin (STMN1), SCG10 (STMN2), SCLIP (STMN3), RB3 (STMN4), and GAPDH were used with TaqMan® Universal PCR Master mix from ABI/Life Tech. For the PCR amplification, 4 μl of RT reactions (representing 5 ng of RNA template) were used in a total volume of 20 μl, and each sample was assayed in duplicate. Standard curves were generated from each primer set from known amounts of sympathetic neuron RNA. Values for STAT3, Stathmin, SCG10/STMN2, SCLIP/STMN3, RB3/STMN4 were normalized to GAPDH from the same sample.
Immunoprecipitation
Dissociated sympathetic neurons were cultured for 3 days prior to treatment. Neurons were treated with vehicle (DMSO) or Stattic (20μm) for 10 minutes prior to stimulation with NGF (100 ng/ml) and CNTF (150 ng/ml) for 15 minutes to induce phosphorylation of both Y705 and S727 on STAT3. Cells were washed with cold PBS and lysed in RIPA buffer. Lysates were rocked at 4°C for 15 minutes and protein concentrations were determined using a BCA protein assay. Equal amounts of protein and 10μl of STAT3 antibody-sepharose bead conjugate or isotype control antibody-sepharose bead conjugate in equal volumes of RIPA buffer were immunoprecipitated overnight at 4°C on a Nutator. The following morning, immunoprecipitations (IPs) were spun down at 4°C, 13,000g for 2 minutes and washed with RIPA buffer. Washes were repeated 4 times and IPs resuspended in sample buffer (+reducing agent). Samples were immediately subjected to Western blotting or frozen at −20°C.
Statistics
Student’s t-test was used for a single comparison between two groups. One-way ANOVA with a Newman-Keuls post-test was used to compare multiple groups. Two-way ANOVA with a Bonferroni post-test was used to compare across genotypes and surgical groups. Statistics were carried out using Prism 5.0 (Graphpad Software).
Results
STAT3 enhances sympathetic axon regeneration
STAT3 is involved in axon regeneration in several types of neurons (Miao et al, 2006; Qiu et al, 2005; Smith et al, 2009), and we asked if STAT3 was required for sympathetic axon regeneration. To address this question, we cultured SCG explants from wild-type mice and mice whose sympathetic neurons lack STAT3 (Figure 1A–B). We compared growth rates from STAT3 KO and wild-type ganglia and discovered that axon growth was impaired in neurons lacking STAT3 (Figure 1C).
Figure 1.
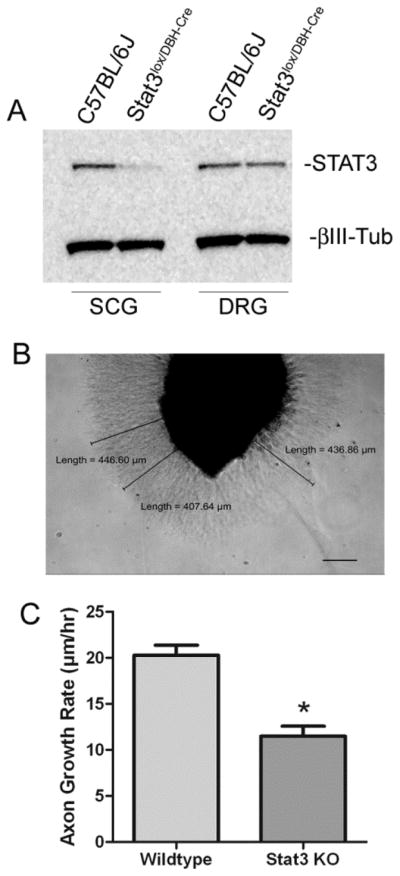
Sympathetic neurons that lack STAT3 have impaired axon growth. (A) SCG and DRG from adult mice were harvested and equal amounts of protein lysate were analyzed for western blotting. A representative blot for STAT3 and βIII-tubulin (βIII-Tub) is shown. STAT3 is absent from the SCG but not DRG of STAT3lox/DBH-Cre mice. The minor residual STAT3 in SCG is probably due to ganglionic non-neuronal cells. (B) A WT SCG explant is pictured 18 hrs after plating, with representative axon length measurements and scale bar (200μm). (C) STAT3 KO explants have reduced axon growth compared to C57BL/J6 explants (wildtype). Data are mean ± SEM from a single experiment (wildtype N=6, STAT3 KO N=5, *p<0.05), and are representative of 3 independent experiments.
NGF stimulates serine phosphorylation of STAT3 in sympathetic axons via ERK1/2 pathway
Ng and colleagues made the surprising discovery that NGF stimulates serine phosphorylation of STAT3 in PC12 cells, and that serine phosphorylation of STAT3 was required for neurite outgrowth (Ng et al, 2006b). We were interested in whether NGF also stimulates serine phosphorylation of STAT3 in sympathetic neurons, and we tested that using PC12 cells (Figure 2A) as a positive control. Acute NGF stimulation of dissociated sympathetic neurons increased phosphorylation of STAT3 on S727 but not Y705 (Figure 2B). Control levels of serine phosphorylated STAT3 were somewhat variable between individual experiments since neurons were cultured in low levels of NGF. However, acute NGF stimulation reproducibly increased serine phosphorylation of STAT3 in all experiments (quantification in Supplementary Figure 1). This contrasts with the classic tyrosine phosphorylation that is stimulated by treating sympathetic neurons with a cytokine like CNTF (Figure 2C). Thus, in sympathetic neurons, NGF stimulates STAT3 phosphorylation on serine (S727) but not tyrosine, while CNTF stimulates phosphorylation on tyrosine (Y705) but not serine.
Figure 2.

NGF stimulates S727 phosphorylation of STAT3 via ERK1/2. (A) Western blots of PC12 cells stimulated with NGF (100ng/ml) for the indicated time points. NGF stimulated S727 phosphorylation of STAT3 (pS727), but not Y705 phosphorylation (pY705). Total STAT3 protein levels were unchanged (STAT3). NGF stimulated phosphorylation of ERK1/2 (pERK1/2) at all time points, but total levels of ERK1/2 were unchanged (ERK1/2). (B) A representative western blot of sympathetic neurons stimulated with NGF (125ng/ml) for the indicated time points ± vehicle (DMSO) or U0126. NGF stimulated phosphorylation of S727 (pS727) but not Y705. Pretreatment with UO126 blocked NGF phosphorylation of STAT3 on S727 and phosphorylation of ERK1/2 (pERK1/2). Total STAT3 and ERK1/2 were unchanged. (C) Sympathetic neurons were stimulated with CNTF (150ng/ml) for the indicated time points and blotted for phosho-STAT3. CNTF stimulated phosphorylation of pY705 (pY705) but not S727. (D) Sympathetic explants were treated with vehicle (DMSO), Bis-II (100nM) to inhibit PKC, or UO126 (40μM) to inhibit ERK1/2 activation, and axon growth rate quantified after 18 hours in culture. Data are mean ±SEM, N=3-6 ganglia per group, ***p<0.001 Vehicle vs UO126. Each experiment was repeated at least 3 times.
NGF-induced serine phosphorylation of STAT3 requires ERK1/2 activation in PC12 cells (Ng et al, 2006b), and we asked if ERK1/2 was required for NGF stimulation of S727 phosphorylation in sympathetic neurons. We pretreated cells with vehicle or the MEK1/2 inhibitor UO126, to inhibit activation of ERK1/2. Blocking ERK1/2 activation abolished NGF-stimulated phosphorylation of S727 in sympathetic neurons (Figure 2B), and blunted axon outgrowth in sympathetic ganglion explants (Figure 2D). In contrast, inhibiting NGF-stimulated protein kinase C signaling had no effect on axon extension. These data suggest that NGF stimulates phosphorylation of STAT3 on S727 via an ERK1/2-dependent pathway, and that serine-phosphorylated STAT3 plays a role in sympathetic outgrowth.
STAT3-dependent regeneration has a non-transcriptional component
STAT3 is proposed to promote axon outgrowth primarily through its actions as a transcription factor (Bareyre et al, 2011; Ben-Yaakov et al, 2012; Ng et al, 2006b). To test the role of STAT3 stimulated gene transcription in sympathetic regeneration, we treated cells for 6 hours with Galiellalactone (Gal-Lac) to block STAT3 DNA binding (Weidler et al, 2000), and measured axon growth. Explants contain endogenous LIF, which is produced by glial cells upon removal of the ganglion (Sun et al, 1994), and NGF was added to the explants, thus stimulating phosphorylation of STAT3 on both tyrosine and serine residues. Gal-Lac treated explants had significantly reduced growth compared to vehicle treated explants (Figure 4A), indicating the loss of STAT3-dependent gene transcription reduced axon outgrowth. The inhibitory effect of Gal-Lac on DNA binding was confirmed by quantifying mRNA for the STAT3-dependent gene SOCS3 (suppressor of cytokine signaling 3) (Supplementary Figure 2A). However, inhibiting phosphorylation of both S727 and Y705 on STAT3 (Fig. 3A) with the compound Stattic (Schust et al, 2006) resulted in a complete block in axon outgrowth (Figure 3B). Both inhibitors (Gal-Lac and Stattic) were used at lower concentrations than published previously in hippocampal neurons (Nicolas et al, 2012). Stattic, like Gal-Lac, blocked transcription of SOCS3 mRNA (Supplementary Figure 2B), but inhibiting STAT3 phosphorylation had a much bigger effect on axon outgrowth than preventing DNA binding. These data suggest that phosphorylated STAT3 has both transcriptional (Vehicle vs Gal-Lac) and non-transcriptional (Gal-Lac vs Stattic) effects that stimulate sympathetic axon regeneration.
Figure 4.

S727-phosphorylated STAT3 is present throughout the cell body and in processes (A) Sympathetic neurons maintained in 10ng/ml NGF were treated for 5 days with CNTF (150ng/ml). Total STAT3 was still abundant in axons and growth cones after long-term CNTF treatment (LT-CNTF). (B) Sympathetic neurons were treated with NGF (125ng/ml,) or CNTF (150ng/ml) for 15 minutes (CON, NGF, CNTF) and stained for total STAT3 (green) and βIII-Tubulin (red). Nuclei were labeled with Hoechst 34580 (blue in merged). CNTF stimulated translocation of STAT3 from the cell body to the nucleus, but NGF did not. (C, D) Sympathetic neurons were treated for 15 min with 125 ng/ml NGF and stained for S727-phosphorylated STAT3 (green), and βIII-Tubulin (red). Nuclei were labeled with Hoechst 34580 (blue in merged). Serine phosphorylated STAT3 was present throughout the cell body, nucleus (C), and in processes including growth cones (D). All scale bars =5μm.
Figure 3.

STAT3 phosphorylation and DNA binding are required for sympathetic axon outgrowth. (A) Sympathetic explants were treated with vehicle (DMSO), Gal-Lac (20μm), or Stattic (20μm) prior to axon outgrowth analysis. Three experiments (each assayed in triplicate) were averaged and are shown as percent % control. Data are expressed as mean ± SEM, **p<0.01. (B) Dissociated sympathetic neurons were pretreated with Stattic at the indicated concentrations. A representative western blot shows NGF (5 minutes, 125ng/ml) stimulated pS727 (top panels) and CNTF (5 minutes, 150ng/ml) stimulated pY705 (bottom panels). At all Stattic concentrations tested, phosphorylation of both S727 and Y705 on STAT3 was blocked. Total levels of STAT3 were unchanged.
Localization of serine phosphorylated STAT3
Numerous studies have identified tyrosine phosphorylated STAT3 in axons, both in vitro and in vivo, with a focus on retrograde signaling by STAT3 as a transcription factor (Lee et al., 2004; Bareyre et al, 2011; Selvaraj et al, 2012; O’Brien & Nathanson, 2007; Ben-Yaakov et al, 2012). In sympathetic neurons, gp130 cytokines can stimulate retrograde transport of Y705-phosphorylated STAT3 from distal axons to cell bodies, in addition to stimulating nuclear translocation of STAT3 within the cell body (O’Brien & Nathanson, 2007). Given the retrograde trafficking of STAT3 in sympathetic neurons, we asked if prolonged exposure to CNTF over several days led to redistribution of STAT3 from growth cones to the nucleus, and found that STAT3 remained abundant throughout neuronal processes (Figure 4A).
Less is known about the localization of serine phosphorylated STAT3, but Ng and colleagues detected S727-phosphorylated STAT3 in neurites and growth cones of PC12 cells (Ng et al, 2006b), while another study localized S727-STAT3 to mitochondria in PC12 cells (Zhou & Too, 2011). Using confocal microscopy, we examined the distribution of STAT3 and S727-phosphorylated STAT3 in sympathetic neurons after acute treatment with NGF or CNTF. As expected, STAT3 in the cell body translocated to the nucleus after CNTF treatment (Figure 4B). In contrast, NGF treatment stimulated minimal change in nuclear STAT3 compared to untreated cells (Figure 4B). We also examined the distribution of S727-phosphorylated STAT3 in neurons after treatment with NGF, and found phospho-S727 STAT3 in the nucleus, but also significant phospho-S727 staining in processes and growth cones (Figure 4C, D).
Serine and Tyrosine STAT3 phosphorylation are both required for STAT3-dependent axon outgrowth
Since our explant experiments suggested that inhibiting STAT3 phosphorylation on both tyrosine and serine blocked axon outgrowth, we wanted to determine the role of individual phosphorylation events in axon elongation. In order to test residues S727 and Y705 individually in axon outgrowth, we used sympathetic neurons lacking STAT3. We co-transfected dissociated STAT3 KO neurons with GFP and either vector alone or a STAT3 expression construct (STAT3-WT, STAT3-Y705F, or STAT3-S727A) (Figure 5A). Western blots confirmed similar expression of the different transfected STAT3 constructs (Supplementary Figure 3). We cultured neurons in the presence of NGF and CNTF to stimulate serine and tyrosine phosphorylation, respectively, and measured the longest process of GFP-expressing neurons. Wild-type STAT3 (STAT3-WT) increased process length significantly, but mutants of STAT3 deficient for either tyrosine (STAT3-Y705F) or serine (STAT3-S727A) phosphorylation did not increase process length compared to vector transfected neurons (Figure 5B–C). The small number of STAT3 KO cells available per experiment allowed for only 3 conditions in each transfection experiment. To confirm that STAT-WT was stimulating a similar amount of growth across all of our experiments, we normalized growth as percent of control, with empty vector set as 100%. We observed a consistent increase in axon length in STAT3-WT versus vector transfected neurons when all experiments were averaged together (Figure 5D). Likewise, transfection of either S727A or Y705F-STAT3 alone consistently had no effect on process outgrowth compared to vector transfected neurons (Figure 5D).
Figure 5.

Maximal sympathetic axon outgrowth requires serine (727) and tyrosine (705) phosphorylation of STAT3. (A) Images of sympathetic neurons from STAT3 KO mice that have been co-transfected with GFP and either pcDNA3.1 (Vec) or wildtype STAT3 (WT). Scale bar=20μm. Cells were cultured in NGF (10ng/ml) and CNTF (100ng/ml) for all transfection experiments (B) Sympathetic neurons were co-transfected with GFP and either wildtype STAT3 (WT), or STAT3 with Y705 mutated to phenylalanine (Y705F). Axon lengths of GFP expressing neurons were measured 40 hours post-transfection. Data are mean ± SEM from a single experiment N=25–35 cells/group, ***p<0.001, Vector vs WT. Similar results were obtained in 4 independent experiments. (C) Neurons were co-transfected with GFP and wildtype STAT3 (WT), or STAT3 with S727 mutated to alanine (S727A). Data are mean ± SEM from a single experiment, N=20–22 cells/group, ***p<0.001, Vector vs WT. Similar results were obtained in 4 independent experiments. (D) Data from all transfection experiments were normalized to the vector alone control and graphed together. Axon length in STAT3-WT transfected neurons was consistent across all experiments. Similarly, axon length in STAT3 mutant transfected neurons was consistently similar to vector control. Data are mean ± SEM. N=6 experiments (3 for serine mutant and 3 for tyrosine mutant). ***p<0.001, Vector vs STAT3-WT (tyr), **p<0.01, Vector vs STAT3-WT (ser). NS= no significant difference.
GP130 signaling is required for sympathetic axon regeneration in vivo
Our transfection data confirmed that NGF-stimulated serine phosphorylation of STAT3 was important for axon outgrowth in sympathetic neurons, just as it is important in PC12 cells and cortical neurons (Ng et al, 2006b). Our data also revealed, however, that phosphorylation of tyrosine 705 was required for maximum sympathetic process outgrowth in vitro. To determine if cytokine-induced phosphorylation of STAT3 was required for sympathetic axon regeneration in vivo, we used mice whose sympathetic neurons lack gp130 (gp130 KO). The gp130 cytokine receptor is used by all LIF- related cytokines (Levy & Darnell, 2002; Taga, 1996), and we previously found that sympathetic neurons lacking gp130 have no detectable tyrosine phosphorylation of STAT3 after nerve injury in vivo (Habecker et al, 2009), or after cytokine treatment in vitro (Shi et al., 2012). We first quantified axon outgrowth in SCG explants from wild-type mice and mice whose sympathetic neurons lacked either gp130 or STAT3. We found that axon growth was impaired similarly in neurons lacking gp130 and STAT3 (Figure 6A). STAT3 expression can be regulated by gp130 cytokines (Ichiba et al, 1998), raising the possibility that the increased axon regeneration in wild-type hearts was due to higher levels of STAT3, rather than altered phosphorylation. We examined STAT3 levels in adult ganglia from unoperated gp130 KO mice and found they were similar to STAT3 levels in wild-type ganglia (Figure 6B).
Figure 6.

Decreased nerve regeneration in sympathetic neurons lacking gp130 KO. A) Neuronal gp130 KO (gp130 KO) and neuronal STAT3 KO (STAT3 KO) explants have reduced axon growth compared to C57BL/J6 (WT) ganglia. Data are mean ± SEM from a single experiment (n=3–6, *p<0.05), and are representative of 3 independent experiments. B) A representative western blot of adult SCG from C57BL/J6 (WT) and gp130lox/DBH-Cre (gp130 KO) mice showing total STAT3 (top panel), TH (middle panel), and GAPDH (bottom panel) protein. C,D) Sympathetic innervation identified by TH staining in WT (C) and neuronal gp130 KO (D) left ventricle 3 days after ischemia-reperfusion. Pictures are from the peri-infarct region approximately 500–900 μm from the infarct. E) Quantification of sympathetic innervation density in sham and peri-infarct left ventricle 24 hours and 3 days after ischemia-reperfusion. Data are mean±SEM, n=4–6 animals per group, *p<0.05 WT vs. KO. (F) Stat3 mRNA was quantified by real-time PCR in stellate ganglia and normalized to GAPDH mRNA in the same sample. Stat3 mRNA was not altered by the absence of gp130 (gp130 KO), or by myocardial infarction (3d MI). Data are mean ± SEM, n=4 mice/group.
To examine axon regeneration in vivo, we carried out cardiac ischemia-reperfusion surgery in WT and gp130 KO mice, and quantified sympathetic fiber density in the peri-infarct heart 24 hrs and 3 days after surgery. At 3 days post-MI we detected a significant deficit of sympathetic nerve sprouting in the peri-infarct region of gp130 KO hearts (6D, E) compared to WT hearts (Figure 6C, E). Furthermore, stat3 mRNA expression in the stellate ganglia, which contain the majority of cardiac-projecting sympathetic neurons, was identical in both genotypes 3 days after MI (Figure 6F). These data suggest the lack of Y705-phosphorylated STAT3 in the neuronal gp130 KO animals plays a role in the reduced nerve sprouting observed after myocardial infarction.
Separate pools of STAT3 mediate sympathetic axon outgrowth
Our data suggest transcriptional and non-transcriptional effects of STAT3 on sympathetic axon regeneration. Therefore, we postulated that there may be two distinct pools of STAT3 required for regeneration - a tyrosine phosphorylated pool and a serine phosphorylated pool - and that a single STAT3 protein need not be phosphorylated on both sites. To test this, we co-transfected sympathetic neurons with different amounts of WT-STAT3, or with both of the phosphorylation-deficient mutant STAT3 constructs (S727A and Y705F). Although individual phospho-mutants were unable to enhance growth compared to vector alone (Figure 5), the expression of both mutants simultaneously was able to rescue axon growth in STAT3 KO neurons to the level seen in STAT3-WT transfected neurons (Figure 7). These data suggest that both phosphorylation events are required for maximal STAT3-dependent axon regeneration, but the same STAT3 protein does not need to be phosphorylated on both sites.
Figure 7.
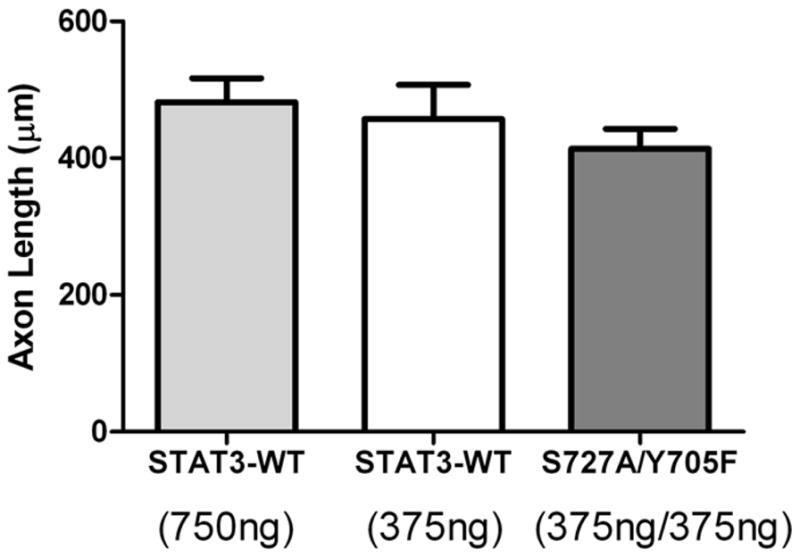
Rescue of axon regeneration by co-transfection of both STAT3 mutants. STAT3 KO neurons were transfected with differing amounts of wildtype STAT3 (STAT3-WT) or a combination of STAT3-S727A and STAT3-Y705F. All groups were co-transfected with GFP and had equal total amounts of DNA transfected by adding vector (pcDNA3.1). Neurons transfected with the two mutants together generated axons the same length as neurons transfected with STAT3-WT. Data are mean ± SEM, N=14–30 cells/group. Identical results were obtained in 3 separate experiments.
STAT3 interacts with microtubules in sympathetic neurons
Recently, STAT3 has been shown to interact with elements of the cytoskeleton in cancer cells to promote migration and metastasis (Walker et al, 2010). Similar interactions have been observed in pmn (progression motor neuronopathy) mutant motoneurons, where tyrosine phosphorylated STAT3 interacts with stathmin to stabilize microtubule dynamics and prevent axon degeneration in axons with microtubule instability (Selvaraj et al, 2012). We hypothesized that STAT3 might also interact with βIII tubulin or alter stathmin-related proteins in sympathetic neurons that have normal microtubule stability, in order to promote axon elongation rather than prevent axon degeneration. To test if STAT3 interacted with the neuronal cytoskeleton, we carried out immunoprecipitation experiments. Dissociated sympathetic neurons were treated with vehicle or Stattic prior to treatment with both NGF and CNTF. Neurons were harvested, STAT3 was immunoprecipitated, and samples were blotted for βIII-tubulin which is a component of the axon cytoskeleton. Immunoprecipitation of STAT3 pulled down βIII-tubulin in vehicle and stattic-treated cells, with more βIII-tubulin pulled down from stattic treated cells which have unphosphorylated STAT3 (Figure 8A). This suggests that STAT3 interacts with βIII-tubulin in sympathetic neurons, and that this interaction is modified by STAT3 phosphorylation. This interaction with the cytoskeleton may underlie the non-transcriptional role of STAT3 in sympathetic axon regeneration.
Figure 8.

STAT3 interacts with the neuronal cytoskeleton. (A) Dissociated neurons were pretreated with DMSO vehicle (Veh) or 20μm Stattic and then stimulated with NGF (100ng/ml) and CNTF (150ng/ml). STAT3 was immunoprecipitated with STAT3 Ab sepharose bead conjugate or isotype control sepharose bead conjugate IgG control (IgG). 10% input of lysate is shown. βIII-Tubulin (Tub) and STAT3 were identified by western blot. Tubulin was precipitated together with STAT3. (left panel) Optical Density (O.D.) quantification of immunoprecipitated βIII-Tubulin averaged from 3 independent experiments. Data are mean ±SEM, *p<0.05.
Given the recent observation that STAT3 modulates microtubule dynamics in dystrophic motor neurons (Selvaraj et al, 2012) and metastatic cancer (Walker et al, 2010) through interactions with stathmin, we examined expression of stathmin and related genes in sympathetic neurons. We detected mRNA encoding all four stathmin-related genes (stathmin, SCG10/stathmin 2, SCLIP/stathmin3, and RB3/stathmin4) in cultured sympathetic neurons using real-time PCR. SCLIP and RB3 mRNA were more abundant than stathmin or SCG10 mRNA (Figure 8B). Therefore, we quantified SCLIP and RB3 mRNA in the stellate ganglia 3 days after MI, when WT neurons exhibit sprouting in the peri-infarct myocardium and gp130 KO neurons do not. No differences were observed between the genotypes in sham operated animals, but SCLIP and RB3 mRNA were both significantly lower 3 days after MI in neurons lacking gp130 compared to WT neurons (Figure 8C-D). This suggests that MI-induced cytokines acting via gp130 regulate expression of at least two stathmin-related genes in cardiac sympathetic neurons, and raises the possibility that stathmin family members are involved in sympathetic axon regeneration in vivo.
Discussion
gp130 signaling is required for NGF-dependent nerve sprouting after myocardial infarction
Sympathetic nerve regeneration has been characterized in many peripheral tissues, and two general categories of regeneration have been identified: NGF-independent, as seen in the regrowth of transected nerves back to skin (Gloster & Diamond, 1995), and NGF-dependent, as seen in the collateral sprouting of sympathetic axons within injured skin (Gloster & Diamond, 1992; Gloster & Diamond, 1995) or damaged myocardium (Gardner & Habecker, 2013; Hasan et al, 2006). Neurotrophins and inflammatory cytokines are often present together following injury, and we were interested in potential interactions between these two sets of factors with regard to sympathetic axon regeneration. Previous studies in sympathetic neurons suggested that NGF antagonized the effect of gp130 cytokines on expression of regeneration associated genes (Corness et al, 1998; Ng et al, 2003; Rajan et al, 1998; Shadiack et al, 2001; Shadiack et al, 1998). We investigated neurotrophin-cytokine interactions in vivo by quantifying NGF-dependent nerve sprouting in sympathetic neurons lacking gp130, and found that gp130 activation was required for sympathetic sprouting in the heart after myocardial infarction. This was consistent with the lack of a conditioning lesion effect in sympathetic neurons lacking gp130 (Hyatt Sachs et al, 2010), but inconsistent with the observation that adult SCG explants from gp130 KO mice exhibited a similar, albeit limited, degree of growth compared to wildtype ganglia (Hyatt Sachs et al, 2010). A critical difference between the studies was the presence or absence of NGF. We investigated an injury characterized by inflammation combined with high NGF, while the axotomy/conditioning lesion study examined an injury characterized by inflammation combined with the loss of NGF (Hyatt Sachs et al, 2007). Thus, the first unexpected and novel finding of our study is that cytokine activation of gp130 is required for NGF-stimulated nerve sprouting.
The role of STAT3 in sympathetic axon regeneration
Neurotrophins and cytokines stimulate phosphorylation of STAT3 on two different sites in sympathetic neurons: S727 and Y705, respectively. STAT3 is implicated in axon growth in other types of neurons (Bareyre et al, 2011; Miao et al, 2006; Ng et al, 2006b; Shin et al, 2012; Smith et al, 2009; Sun et al, 2011), and the availability of mice whose sympathetic neurons lacked STAT3 gave us a unique opportunity to investigate the role of these phosphorylation sites in sympathetic axon outgrowth. Our initial studies in wildtype neurons revealed that inhibiting phosphorylation of both Y705 and S727 completely blocked axon outgrowth. In contrast, preventing STAT3 DNA binding with Gal-Lac or blocking S727 phosphorylation with U0126 only inhibited axon outgrowth by 50% during the same 6 hour time window. This suggested that STAT3 had two distinct roles – one as a transcription factor and a second that did not involve DNA binding. Previous studies showed that cytokines stimulate the retrograde transport and DNA binding of phospho-Y705 STAT3 in sympathetic neurons (O’Brien & Nathanson, 2007), while our immunohistochemistry revealed S727-phosphorylated STAT3 was abundant throughout processes and growth cones. We were surprised to find that mutating either S727 or Y705 was sufficient to disrupt STAT3-dependent process outgrowth in neurons grown in media containing neurotrophins and cytokines. Adding the two mutants into the same cell fully recapitulated the axon outgrowth seen with wildtype STAT3. This suggests that there are two distinct pools of STAT3 in sympathetic neurons exposed to both neurotrophins and cytokines – phospho-S727 and phospho-Y705 – and while both phosphorylation events are required for axon regeneration, an individual STAT3 protein need not be phosphorylated on both sites. Tyrosine phosphorylation is especially critical for gene transcription, but S727 phosphorylation can also impact gene expression (Ng et al., 2006b). Likewise, serine-phosphorylated STAT3 is abundant in NGF-stimulated axons, but tyrosine-phosphorylated STAT3 is also in axons and plays a role in microtubule stability under some conditions (Selvaraj et al, 2012). Thus, the two functional pools of STAT3 may not be physically segregated to distinct sub-domains of the neuron.
The role of STAT3 in nerve regeneration has also been examined in the context of sensory nerves, where it is critical for the initiation of regeneration 2-4 days after injury (Bareyre et al, 2011). However, STAT3-independent growth occurs during development (Zhou & Snider, 2006), and 7–8 days after a peripheral lesion (Bareyre et al, 2011) suggesting that within the same type of neuron different mechanisms can control growth over time. We quantified sympathetic fiber density in the heart 24 hours and 3 days after MI, when we predicted nerve sprouting would be STAT3-dependent. The lack of sympathetic regeneration seen in gp130 KO neurons 3 days after cardiac injury is consistent with the lack of sensory regeneration in STAT3 KO neurons 2–4 days after injury (Bareyre et al, 2011). It will require further investigation to determine if STAT3-independent growth occurs in cardiac sympathetic neurons at later time points. The presence of STAT3-dependent and STAT3-independent growth pathways within the same cell may explain why sympathetic neurons lacking STAT3 exhibited some axon outgrowth in vitro, although they exhibited significantly greater process outgrowth after addition of wildtype STAT3.
Non-transcriptional mechanisms of STAT3-dependent axon outgrowth
Although it is not clear how STAT3 acts in sympathetic axons to enhance elongation, several recent studies suggest potential mechanisms. First, experiments in NGF-treated PC12 cells found that S727-phosphorylated STAT3 is associated with mitochondria, where it alters production of reactive oxygen species (ROS) and impacts neurite outgrowth (Zhou & Too, 2011). Second, STAT3 plays a role in the metastasis of some cancers by antagonizing the depolymerization activity of stathmin (Ng et al, 2006a). Stathmin and related proteins like SCLIP sequester free tubulin and increase microtubule catastrophe (Curmi et al, 1999), so that STAT3 binding to stathmin-related proteins promotes microtubule formation (Verma et al, 2009). Furthermore, Y705-phosphorylated STAT3 interacts with tubulin and stathmin in dystrophic embryonic motor neurons that contain unstable microtubules, stabilizing the microtubules and preventing axon degeneration (Selvaraj et al, 2012). The role for cytokine-phosphorylated STAT3 in preventing axon degeneration was particularly interesting, since tubulin and stathmin-related proteins are also involved in axon elongation. Cytokine activation of STAT3 had no effect on axon length in control motor neurons (Selvaraj et al, 2012), raising the possibility that STAT3 only plays an important role in neurodegeneration and not axon regeneration. However, our data suggest that in sympathetic neurons, phosphorylation of STAT by both neurotrophins and cytokines has a significant impact on axon outgrowth. Our data show an interaction between STAT3 and βIII-tubulin in sympathetic neurons under normal (not dystrophic) conditions, and reveal that gp130 cytokines regulate expression of the stathmin-related genes SCLIP and RB3 in the sympathetic neurons projecting to the heart after myocardial infarction. Decreased expression of these genes in gp130 KO neurons, which exhibited impaired axon sprouting after MI, is consistent with work showing these genes are most highly expressed during axon elongation (Curmi et al, 1999; Gavet et al, 1998; Ozon et al, 1999). The exact nature of interactions between STAT3, mitochondria, microtubules, and stathmin family members is yet to be determined, but our data support a role for STAT3 in axons where it may regulate microtubule dynamics and/or production of ROS by mitochondria.
Concluding remarks
In summary, we investigated the role of STAT3 in sympathetic axon regeneration, and found that STAT3 phosphorylation by neurotrophin and inflammatory cytokines is required for maximal axon outgrowth. We show that cytokine signaling is necessary for the NGF-induced sympathetic nerve sprouting in the heart after MI, and provide evidence that STAT3 plays a non-transcriptional role in axons that complements its role stimulating regeneration associated genes.
Supplementary Material
Acknowledgments
This work was supported by R01 HL068231 & HL093056 (BAH) and NINDS P30 Center Grant (Jungers Center-OHSU). The authors thank Diana Parrish, Eric Alston, and Dr. Xiao Shi for technical assistance, and thank Dr. Richard Zigmond and Jon Niemi for comments on the manuscript. After this work was completed, we were informed by Addgene (Cambridge, MA) that STAT3-WT (pcDNA3), a construct we purchased from them, had a mutation in the open reading frame resulting in an amino acid change (E16K). We compared results to another plasmid (STAT3-WT Flag pRc/CMV) and saw no difference.
Abbreviations
- STAT3
signal transducer and activator of transcription 3
- LIF
leukemia inhibitory factor
- CNTF
ciliary neurotrophic factor
- DBH
dopamine-β-hydroxylase
- MI
myocardial infarction
- NGF
nerve growth factor; brain-derived neurotrophic factor
- TrkA
tropomyosin-related receptor kinase A
- gp130
glycoprotein 130
Footnotes
Conflict of Interest
The authors declare they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe T, Morgan DA, Gutterman DD. Protective role of nerve growth factor against postischemic dysfunction of sympathetic coronary innervation. Circulation. 1997;95:213–220. doi: 10.1161/01.cir.95.1.213. [DOI] [PubMed] [Google Scholar]
- Adler R. Ciliary neurotrophic factor as an injury factor. Curr Opin Neurobiol. 1993;3:785–789. doi: 10.1016/0959-4388(93)90154-q. [DOI] [PubMed] [Google Scholar]
- Alston EN, Parrish DC, Hasan W, Tharp K, Pahlmeyer L, Habecker BA. Cardiac ischemia-reperfusion regulates sympathetic neuropeptide expression through gp130-dependent and independent mechanisms. Neuropeptides. 2011;45:33–42. doi: 10.1016/j.npep.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama T, Takimoto Y, Pennica D, Inoue R, Shinoda E, Hattori R, Yui Y, Sasayama S. Augmented expression of cardiotrophin-1 and its receptor component, gp130, in both left and right ventricles after myocardial infarction in the rat. J Mol Cell Cardiol. 2000;32:1821–1830. doi: 10.1006/jmcc.2000.1218. [DOI] [PubMed] [Google Scholar]
- Barber MJ, Mueller TM, Henry DP, Felten SY, Zipes DP. Transmural myocardial infarction in the dog produces sympathectomy in noninfarcted myocardium. Circulation. 1983;67:787–796. doi: 10.1161/01.cir.67.4.787. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Garzorz N, Lang C, Misgeld T, Buning H, Kerschensteiner M. In vivo imaging reveals a phase-specific role of STAT3 during central and peripheral nervous system axon regeneration. Proc Natl Acad Sci U S A. 2011;108:6282–6287. doi: 10.1073/pnas.1015239108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Yaakov K, Dagan SY, Segal-Ruder Y, Shalem O, Vuppalanchi D, Willis DE, Yudin D, Rishal I, Rother F, Bader M, Blesch A, Pilpel Y, Twiss JL, Fainzilber M. Axonal transcription factors signal retrogradely in lesioned peripheral nerve. EMBO J. 2012;31:1350–1363. doi: 10.1038/emboj.2011.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferty WB, Gardiner NJ, Gavazzi I, Powell J, McMahon SB, Heath JK, Munson J, Cohen J, Thompson SW. Leukemia inhibitory factor determines the growth status of injured adult sensory neurons. J Neurosci. 2001;21:7161–7170. doi: 10.1523/JNEUROSCI.21-18-07161.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corness J, Stevens B, Fields RD, Hokfelt T. NGF and LIF both regulate galanin gene expression in primary DRG cultures. Neuroreport. 1998;9:1533–1536. doi: 10.1097/00001756-199805110-00053. [DOI] [PubMed] [Google Scholar]
- Curmi PA, Gavet O, Charbaut E, Ozon S, Lachkar-Colmerauer S, Manceau V, Siavoshian S, Maucuer A, Sobel A. Stathmin and its phosphoprotein family: general properties, biochemical and functional interaction with tubulin. Cell Struct Funct. 1999;24:345–357. doi: 10.1247/csf.24.345. [DOI] [PubMed] [Google Scholar]
- Ekstrom PA, Kerekes N, Hokfelt T. Leukemia inhibitory factor null mice: unhampered in vitro outgrowth of sensory axons but reduced stimulatory potential by nerve segments. Neurosci Lett. 2000;281:107–110. doi: 10.1016/s0304-3940(00)00816-8. [DOI] [PubMed] [Google Scholar]
- Fischer P, Hilfiker-Kleiner D. Survival pathways in hypertrophy and heart failure: the gp130-STAT axis. Basic Res Cardiol. 2007;102:393–411. doi: 10.1007/s00395-007-0674-z. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- Gardner RT, Habecker BA. Infarct-Derived Chondroitin Sulfate Proteoglycans Prevent Sympathetic Reinnervation after Cardiac Ischemia-Reperfusion Injury. J Neurosci. 2013 doi: 10.1523/JNEUROSCI.5866-12.2013. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavet O, Ozon S, Manceau V, Lawler S, Curmi P, Sobel A. The stathmin phosphoprotein family: intracellular localization and effects on the microtubule network. J Cell Sci. 1998;111 (Pt 22):3333–3346. doi: 10.1242/jcs.111.22.3333. [DOI] [PubMed] [Google Scholar]
- Ghilardi JR, Freeman KT, Jimenez-Andrade JM, Coughlin KA, Kaczmarska MJ, Castaneda-Corral G, Bloom AP, Kuskowski MA, Mantyh PW. Neuroplasticity of sensory and sympathetic nerve fibers in a mouse model of a painful arthritic joint. Arthritis Rheum. 2012;64:2223–2232. doi: 10.1002/art.34385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glebova NO, Ginty DD. Growth and survival signals controlling sympathetic nervous system development. Annu Rev Neurosci. 2005;28:191–222. doi: 10.1146/annurev.neuro.28.061604.135659. [DOI] [PubMed] [Google Scholar]
- Gloster A, Diamond J. Sympathetic nerves in adult rats regenerate normally and restore pilomotor function during an anti-NGF treatment that prevents their collateral sprouting. J Comp Neurol. 1992;326:363–374. doi: 10.1002/cne.903260305. [DOI] [PubMed] [Google Scholar]
- Gloster A, Diamond J. NGF-dependent and NGF-independent recovery of sympathetic function after chemical sympathectomy with 6-hydroxydopamine. J Comp Neurol. 1995;359:586–594. doi: 10.1002/cne.903590406. [DOI] [PubMed] [Google Scholar]
- Gwechenberger M, Mendoza LH, Youker KA, Frangogiannis NG, Smith CW, Michael LH, Entman ML. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546–551. doi: 10.1161/01.cir.99.4.546. [DOI] [PubMed] [Google Scholar]
- Habecker BA, Sachs HH, Rohrer H, Zigmond RE. The dependence on gp130 cytokines of axotomy induced neuropeptide expression in adult sympathetic neurons. Dev Neurobiol. 2009;69:392–400. doi: 10.1002/dneu.20706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan W, Jama A, Donohue T, Wernli G, Onyszchuk G, Al-Hafez B, Bilgen M, Smith PG. Sympathetic hyperinnervation and inflammatory cell NGF synthesis following myocardial infarction in rats. Brain Res. 2006;1124:142–154. doi: 10.1016/j.brainres.2006.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker-Kleiner D, Shukla P, Klein G, Schaefer A, Stapel B, Hoch M, Muller W, Scherr M, Theilmeier G, Ernst M, Hilfiker A, Drexler H. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation. 2010;122:145–155. doi: 10.1161/CIRCULATIONAHA.109.933127. [DOI] [PubMed] [Google Scholar]
- Hiltunen JO, Laurikainen A, Vakeva A, Meri S, Saarma M. Nerve growth factor and brain-derived neurotrophic factor mRNAs are regulated in distinct cell populations of rat heart after ischaemia and reperfusion. J Pathol. 2001;194:247–253. doi: 10.1002/path.878. [DOI] [PubMed] [Google Scholar]
- Homs J, Ariza L, Pages G, Udina E, Navarro X, Chillon M, Bosch A. Schwann cell targeting via intrasciatic injection of AAV8 as gene therapy strategy for peripheral nerve regeneration. Gene Ther. 2011;18:622–630. doi: 10.1038/gt.2011.7. [DOI] [PubMed] [Google Scholar]
- Hyatt Sachs H, Rohrer H, Zigmond RE. The conditioning lesion effect on sympathetic neurite outgrowth is dependent on gp130 cytokines. Exp Neurol. 2010;223:516–522. doi: 10.1016/j.expneurol.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt Sachs H, Schreiber RC, Shoemaker SE, Sabe A, Reed E, Zigmond RE. Activating transcription factor 3 induction in sympathetic neurons after axotomy: response to decreased neurotrophin availability. Neuroscience. 2007;150:887–897. doi: 10.1016/j.neuroscience.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Ichiba M, Nakajima K, Yamanaka Y, Kiuchi N, Hirano T. Autoregulation of the Stat3 gene through cooperation with a cAMP-responsive element-binding protein. The Journal of biological chemistry. 1998;273:6132–6138. doi: 10.1074/jbc.273.11.6132. [DOI] [PubMed] [Google Scholar]
- Inoue H, Zipes DP. Time course of denervation of efferent sympathetic and vagal nerves after occlusion of the coronary artery in the canine heart. Circ Res. 1988;62:1111–1120. doi: 10.1161/01.res.62.6.1111. [DOI] [PubMed] [Google Scholar]
- Ip NY, Nye SH, Boulton TG, Davis S, Taga T, Li Y, Birren SJ, Yasukawa K, Kishimoto T, Anderson DJ, et al. CNTF and LIF act on neuronal cells via shared signaling pathways that involve the IL-6 signal transducing receptor component gp130. Cell. 1992;69:1121–1132. doi: 10.1016/0092-8674(92)90634-o. [DOI] [PubMed] [Google Scholar]
- Lee N, Neitzel KL, Devlin BK, MacLennan AJ. STAT3 phosphorylation in injured axons before sensory and motor neuron nuclei: potential role for STAT3 as a retrograde signaling transcription factor. J Comp Neurol. 2004;474:535–545. doi: 10.1002/cne.20140. [DOI] [PubMed] [Google Scholar]
- Leibinger M, Muller A, Andreadaki A, Hauk TG, Kirsch M, Fischer D. Neuroprotective and axon growth-promoting effects following inflammatory stimulation on mature retinal ganglion cells in mice depend on ciliary neurotrophic factor and leukemia inhibitory factor. J Neurosci. 2009;29:14334–14341. doi: 10.1523/JNEUROSCI.2770-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Liu RY, Snider WD. Different signaling pathways mediate regenerative versus developmental sensory axon growth. J Neurosci. 2001;21:RC164. doi: 10.1523/JNEUROSCI.21-17-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorentz CU, Alston EN, Belcik T, Lindner JR, Giraud GD, Habecker BA. Heterogeneous ventricular sympathetic innervation, altered beta-adrenergic receptor expression, and rhythm instability in mice lacking the p75 neurotrophin receptor. American journal of physiology Heart and circulatory physiology. 2010;298:H1652–1660. doi: 10.1152/ajpheart.01128.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuarrie IG, Grafstein B. Axon outgrowth enhanced by a previous nerve injury. Arch Neurol. 1973;29:53–55. doi: 10.1001/archneur.1973.00490250071008. [DOI] [PubMed] [Google Scholar]
- Meloni M, Caporali A, Graiani G, Lagrasta C, Katare R, Van Linthout S, Spillmann F, Campesi I, Madeddu P, Quaini F, Emanueli C. Nerve growth factor promotes cardiac repair following myocardial infarction. Circ Res. 2010;106:1275–1284. doi: 10.1161/CIRCRESAHA.109.210088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao T, Wu D, Zhang Y, Bo X, Subang MC, Wang P, Richardson PM. Suppressor of cytokine signaling-3 suppresses the ability of activated signal transducer and activator of transcription-3 to stimulate neurite growth in rat primary sensory neurons. J Neurosci. 2006;26:9512–9519. doi: 10.1523/JNEUROSCI.2160-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro X, Kennedy WR. The effect of a conditioning lesion on sudomotor axon regeneration. Brain Res. 1990;509:232–236. doi: 10.1016/0006-8993(90)90547-o. [DOI] [PubMed] [Google Scholar]
- Ng DC, Lin BH, Lim CP, Huang G, Zhang T, Poli V, Cao X. Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J Cell Biol. 2006a;172:245–257. doi: 10.1083/jcb.200503021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng YP, Cheung ZH, Ip NY. STAT3 as a downstream mediator of Trk signaling and functions. The Journal of biological chemistry. 2006b;281:15636–15644. doi: 10.1074/jbc.M601863200. [DOI] [PubMed] [Google Scholar]
- Ng YP, He W, Ip NY. Leukemia inhibitory factor receptor signaling negatively modulates nerve growth factor-induced neurite outgrowth in PC12 cells and sympathetic neurons. The Journal of biological chemistry. 2003;278:38731–38739. doi: 10.1074/jbc.M304623200. [DOI] [PubMed] [Google Scholar]
- Nicolas CS, Peineau S, Amici M, Csaba Z, Fafouri A, Javalet C, Collett VJ, Hildebrandt L, Seaton G, Choi SL, Sim SE, Bradley C, Lee K, Zhuo M, Kaang BK, Gressens P, Dournaud P, Fitzjohn SM, Bortolotto ZA, Cho K, Collingridge GL. The Jak/STAT pathway is involved in synaptic plasticity. Neuron. 2012;73:374–390. doi: 10.1016/j.neuron.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JJ, Nathanson NM. Retrograde activation of STAT3 by leukemia inhibitory factor in sympathetic neurons. J Neurochem. 2007;103:288–302. doi: 10.1111/j.1471-4159.2007.04736.x. [DOI] [PubMed] [Google Scholar]
- Ozon S, El Mestikawy S, Sobel A. Differential, regional, and cellular expression of the stathmin family transcripts in the adult rat brain. J Neurosci Res. 1999;56:553–564. doi: 10.1002/(SICI)1097-4547(19990601)56:5<553::AID-JNR11>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Parrish DC, Alston EN, Rohrer H, Hermes SM, Aicher SA, Nkadi P, Woodward WR, Stubbusch J, Gardner RT, Habecker BA. Absence of gp130 in dopamine beta-hydroxylase-expressing neurons leads to autonomic imbalance and increased reperfusion arrhythmias. American journal of physiology Heart and circulatory physiology. 2009;297:H960–967. doi: 10.1152/ajpheart.00409.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MJ, Parrish DC, Zigmond RE, Habecker BA. Cytokines inhibit norepinephrine transporter expression by decreasing Hand2. Mol Cell Neurosci. 2011;46:671–680. doi: 10.1016/j.mcn.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Cafferty WB, McMahon SB, Thompson SW. Conditioning injury-induced spinal axon regeneration requires signal transducer and activator of transcription 3 activation. J Neurosci. 2005;25:1645–1653. doi: 10.1523/JNEUROSCI.3269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan P, Gearan T, Fink JS. Leukemia inhibitory factor and NGF regulate signal transducers and activators of transcription activation in sympathetic ganglia: convergence of cytokine- and neurotrophin-signaling pathways. Brain Res. 1998;802:198–204. doi: 10.1016/s0006-8993(98)00611-8. [DOI] [PubMed] [Google Scholar]
- Rao MS, Sun Y, Escary JL, Perreau J, Tresser S, Patterson PH, Zigmond RE, Brulet P, Landis SC. Leukemia inhibitory factor mediates an injury response but not a target-directed developmental transmitter switch in sympathetic neurons. Neuron. 1993;11:1175–1185. doi: 10.1016/0896-6273(93)90229-k. [DOI] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB American Heart Association Statistics C, Stroke Statistics S. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruit KG, Osborne PA, Schmidt RE, Johnson EM, Jr, Snider WD. Nerve growth factor regulates sympathetic ganglion cell morphology and survival in the adult mouse. J Neurosci. 1990;10:2412–2419. doi: 10.1523/JNEUROSCI.10-07-02412.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Selvaraj BT, Frank N, Bender FL, Asan E, Sendtner M. Local axonal function of STAT3 rescues axon degeneration in the pmn model of motoneuron disease. J Cell Biol. 2012;199:437–451. doi: 10.1083/jcb.201203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadiack AM, Sun Y, Zigmond RE. Nerve growth factor antiserum induces axotomy-like changes in neuropeptide expression in intact sympathetic and sensory neurons. J Neurosci. 2001;21:363–371. doi: 10.1523/JNEUROSCI.21-02-00363.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadiack AM, Vaccariello SA, Sun Y, Zigmond RE. Nerve growth factor inhibits sympathetic neurons’ response to an injury cytokine. Proc Natl Acad Sci U S A. 1998;95:7727–7730. doi: 10.1073/pnas.95.13.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Habecker BA. gp130 cytokines stimulate proteasomal degradation of tyrosine hydroxylase via extracellular signal regulated kinases 1 and 2. J Neurochem. 2012;120:239–247. doi: 10.1111/j.1471-4159.2011.07539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Cho Y, Beirowski B, Milbrandt J, Cavalli V, DiAntonio A. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron. 2012;74:1015–1022. doi: 10.1016/j.neuron.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker SE, Sachs HH, Vaccariello SA, Zigmond RE. A conditioning lesion enhances sympathetic neurite outgrowth. Exp Neurol. 2005;194:432–443. doi: 10.1016/j.expneurol.2005.02.023. [DOI] [PubMed] [Google Scholar]
- Smith DS, Skene JH. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J Neurosci. 1997;17:646–658. doi: 10.1523/JNEUROSCI.17-02-00646.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PD, Sun F, Park KK, Cai B, Wang C, Kuwako K, Martinez-Carrasco I, Connolly L, He Z. SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron. 2009;64:617–623. doi: 10.1016/j.neuron.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci. 2001;24:1217–1281. doi: 10.1146/annurev.neuro.24.1.1217. [DOI] [PubMed] [Google Scholar]
- Stanke M, Duong CV, Pape M, Geissen M, Burbach G, Deller T, Gascan H, Otto C, Parlato R, Schutz G, Rohrer H. Target-dependent specification of the neurotransmitter phenotype: cholinergic differentiation of sympathetic neurons is mediated in vivo by gp 130 signaling. Development. 2006;133:141–150. doi: 10.1242/dev.02189. [DOI] [PubMed] [Google Scholar]
- Sun F, Park KK, Belin S, Wang D, Lu T, Chen G, Zhang K, Yeung C, Feng G, Yankner BA, He Z. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature. 2011;480:372–375. doi: 10.1038/nature10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Rao MS, Zigmond RE, Landis SC. Regulation of vasoactive intestinal peptide expression in sympathetic neurons in culture and after axotomy: the role of cholinergic differentiation factor/leukemia inhibitory factor. J Neurobiol. 1994;25:415–430. doi: 10.1002/neu.480250407. [DOI] [PubMed] [Google Scholar]
- Taga T. Gp130, a shared signal transducing receptor component for hematopoietic and neuropoietic cytokines. J Neurochem. 1996;67:1–10. doi: 10.1046/j.1471-4159.1996.67010001.x. [DOI] [PubMed] [Google Scholar]
- Taga T, Kishimoto T. Gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol. 1997;15:797–819. doi: 10.1146/annurev.immunol.15.1.797. [DOI] [PubMed] [Google Scholar]
- Vaseghi M, Lux RL, Mahajan A, Shivkumar K. Sympathetic stimulation increases dispersion of repolarization in humans with myocardial infarction. American journal of physiology Heart and circulatory physiology. 2012;302:H1838–1846. doi: 10.1152/ajpheart.01106.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma NK, Dourlat J, Davies AM, Long A, Liu WQ, Garbay C, Kelleher D, Volkov Y. STAT3-stathmin interactions control microtubule dynamics in migrating T-cells. The Journal of biological chemistry. 2009;284:12349–12362. doi: 10.1074/jbc.M807761200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SR, Chaudhury M, Nelson EA, Frank DA. Microtubule-targeted chemotherapeutic agents inhibit signal transducer and activator of transcription 3 (STAT3) signaling. Mol Pharmacol. 2010;78:903–908. doi: 10.1124/mol.110.066316. [DOI] [PubMed] [Google Scholar]
- Weidler M, Rether J, Anke T, Erkel G. Inhibition of interleukin-6 signaling by galiellalactone. FEBS Lett. 2000;484:1–6. doi: 10.1016/s0014-5793(00)02115-3. [DOI] [PubMed] [Google Scholar]
- Wernli G, Hasan W, Bhattacherjee A, van Rooijen N, Smith PG. Macrophage depletion suppresses sympathetic hyperinnervation following myocardial infarction. Basic Res Cardiol. 2009;104:681–693. doi: 10.1007/s00395-009-0033-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FQ, Snider WD. Intracellular control of developmental and regenerative axon growth. Philos Trans R Soc Lond B Biol Sci. 2006;361:1575–1592. doi: 10.1098/rstb.2006.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Too HP. Mitochondrial localized STAT3 is involved in NGF induced neurite outgrowth. PLoS One. 2011;6:e21680. doi: 10.1371/journal.pone.0021680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Chen LS, Miyauchi Y, Miyauchi M, Kar S, Kangavari S, Fishbein MC, Sharifi B, Chen PS. Mechanisms of cardiac nerve sprouting after myocardial infarction in dogs. Circ Res. 2004;95:76–83. doi: 10.1161/01.RES.0000133678.22968.e3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.