

Abstract
Microglia are ramified cells that serve as central nervous system (CNS) guardians, capable of proliferation, migration, and generation of inflammatory cytokines. In non-pathological states, these cells exhibit ramified morphology with processes intermingling with neurons and astrocytes. Under pathological conditions, they acquire a rounded amoeboid morphology and proliferative and migratory capabilities. Such morphological changes require cytoskeleton rearrangements. The molecular control points for polymerization states of microtubules and actin are still under investigation. Caveolins (Cav), membrane/lipid raft proteins, are expressed in inflammatory cells, yet the role of Caveolin isoforms in microglia physiology is debatable. We propose caveolins provide a necessary control point in the regulation of cytoskeletal dynamics, and thus investigated a role for caveolins in microglia biology. We detected mRNA and protein for both Cav-1 and Cav-3. Cav-1 protein was significantly less and localized to plasmalemma (PM) and cytoplasmic vesicles (CV) in the microglial inactive state, while the active (amoeboid-shaped) microglia exhibited increased Cav-1 expression. In contrast, Cav-3 was highly expressed in the inactive state and localized with cellular processes and perinuclear regions and was detected in active amoeboid microglia. Pharmacological manipulation of the cytoskeleton in the active or non-active state altered caveolin expression. Additionally, increased Cav-1 expression also increased mitochondrial respiration, suggesting possible regulatory roles in cell metabolism necessary to facilitate the morphological changes. The present findings strongly suggest that regulation of microglial morphology and activity are in part due to caveolin isoforms, providing promising novel therapeutic targets in CNS injury or disease.
Keywords: caveolin, cytoskeleton, mitochondria, microglial activation, neuroinflammation
INTRODUCTION
Microglia, resident central nervous system (CNS) macrophages, play an important role in the response to injury, infection and aging. Once considered to be “resting” or inactive cells, their ability to migrate and sense markers of injury and stress in the extracellular environment has led to a new understanding and appreciation of glia in response to CNS injury (Nimmerjahn et al., 2005). The central manifestation of the microglial response is to alter their morphology and migration to the site of injury, proliferate, and secrete cytotoxic or trophic molecules (i.e., the classical M1 macrophage type response). Chronically “activated” microglia result in neuroinflammation, typically exhibited in disease states such as Alzheimer’s disease (AD), Parkinson’s disease (PD) or multiple sclerosis (MS) (Glass et al., 2010). Though the M1 type activation of microglia has been extensively studied (Monif et al., 2010), reversal back to an inactive state has only recently been investigated (Larson et al., 2010; Lee et al., 2008; Watson et al., 2010)
Caveolae, a subset of membrane/lipid rafts (MLR), are 50–70 nm plasmalemmal invaginations, highly enriched in sphingolipids and cholesterol, and the cholesterol binding protein caveolin; the latter of which serves to scaffold and dock many signaling proteins (Head and Insel, 2007; Patel et al., 2008). Three isoforms of caveolin (Cav-1, Cav-2 and Cav-3) are expressed in a variety of cells, with Cav-1 and Cav-2 predominantly expressed as heterodimers in vascular tissue, adipocytes and fibroblasts, while Cav-3 is highly expressed as a homodimer in muscle tissues (Chidlow and Sessa, 2010).
In regards to the CNS, all three isoforms have been described (Shin et al., 2005; Silva et al., 2007). Neurons, which are devoid of morphological caveolae, do express all three isoforms (Stern and Mermelstein, 2010). Astrocytes, neuronal supporting glia, show morphological caveolae and express all three isoforms (Ikezu et al., 1998). Peripheral macrophages have been found to express Cav-1 and Cav-2 (Kiss et al., 2002), yet microglial expression of caveolin has not been thoroughly investigated. Although Marella and colleagues (Marella et al., 2002) detected Cav-1 in a human microglia cell line, other reports have shown no Cav-1 expression in BV2 cells, a well-documented mouse microglia cell line (Park et al., 2009). Thus, the functional role of caveolin in microglia is still unknown.
Caveolins serve structural and metabolic roles in cells (Fridolfsson et al., 2012); two processes that may be key to microglial activation and inactivation. Polymerization and depolymerization of actin and tubulin are key regulators of cell morphology, organelle and vesicular transport, and cell migration. Cav-1 localization is dynamically regulated by both actin and tubulin in CHO cells (Mundy et al., 2002). An ultrastructural localization study of Cav-3 in splenic endothelial cells demonstrated an association with stress fibers determined to be mainly composed of actin (Uehara and Miyoshi, 2002). Regulation of the cell cytoskeleton directly by caveolins has been studied in other cell types for both Cav-1 (Shajahan et al., 2007) and Cav-3 (Head et al., 2006). With increasing awareness of the role of chronic inflammation after CNS injury or in neurodegenerative states (Cunningham, 2013; Nolan et al., 2013; Shultz et al., 2013), we investigated potential functional roles for Cav-1 and Cav-3 in regulation of microglial cell structure and metabolism. In this report, we define the expression of Cav-1 and Cav-3 in BV2 cells and in primary mouse derived microglia (PMG). Caveolins, which provide scaffolds for receptor signaling cascades as well as cytoskeletal tethering points at the PM, may serve an essential role for regulating early changes, both morphological and biochemical, in transitioning cell states between activation and inactivation of microglia. We have further defined potential key roles for caveolin in the regulation of mitochondrial function and cytoskeletal polymerization. Therefore, caveolin isoforms may represent potential regulators of microglial function and may also serve as a target for genetic or pharmacological manipulation in a variety of disease settings.
MATERIALS and METHODS
Reagents, antibodies, vectors and primers
Cholera toxin (CTx)-488 and 594; phalloidin-488 and 594; GSA (Griffonia simplificia)-488, DAPI and all secondary fluorescent antibodies were purchased from Invitrogen (Carlsbad, CA). Polyclonal antibodies used were, Cav-1 (D46G3, Cell Signaling), Cav-1 (C4490, Sigma), β-tubulin (sc-9104, Santa Cruz Biotechnology), flotillin-1 (ab50671, Abcam), flotillin-2 (#3436S, Cell Signaling) and goat pAb actin 1–19 (sc-1616, Santa Cruz), Iba1 (#019–19741, Wako Pure Chemicals Ltd). Monoclonal antibodies included Cav-3 (sc29944; Santa Cruz Biotechnology), Cav-2 (#79382, Abcam), IL-1β (#9722, Abcam) and HSP90 (rat mAb) (SPA-835, Stressgen). The MTT cell proliferation assay (#10009365) was from Cayman Chemical Co., Ann Arbor, MI. Products from Qiagen-SABiosciences, Valencia, CA were RNeasy kits, mouse specific caveolin PCR primers (Quantitech #QT0024825), RT2 First Strand cDNA kit (#330401) and SYBRGreen® Mastermix (#330500). DMEM (#11965) and Opti-MEM (#31985) medias were from Gibco-Invitrogen, Carlsbad, CA. Specialized serum free media, (ADCF-MAb #SH30340.01) was purchased from HyClone-Thermo Scientific, Rockford, IL. Primary cells were isolated using a Papain dissociation kit (#3150) from Worthington, Lakewood, NJ. Cells were transfected with mouse specific siRNA pools of 2–5 target specific 19–25 nt pools (Cav-1 sc-29942 and Cav-3 sc-29944; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) with Lipofectamine 2000® (#11668–019; Invitrogen, Carlsbad, CA). Reagents for cytoskeletal experiments were purchased from Calbiochem, La Jolla, CA (jasplakinolide #420127, nocodazole #487928, cytochalasin D #250255, paclitaxel #580555) and Cayman Chemical Co., Ann Arbor, MI (latrunculin B #10010631). Cell fractionation experiments were performed with Qproteome Cell Compartment Kit (cat. # 37502) from Qiagen-SA Biosciences, Valencia, CA. Lipopolysaccharide (LPS) (cat. # L2654) was from Sigma, St. Louis, MO. Electron microscopy transition fluid, HPMA (2- hydroxypropyl methacrylate #16900) was purchased from EMS, Hatfield, MA, and embedding kit was LX112 (#21210; Ladd, Willston, VT). Specific antisense morpholino contructs for mouse Cav-1 and Cav-3 were designed and manufactured, along with control morpholinos and Endo-porter® by Gene Tools, LLC (Philomath, OR). Microtubule stability was assessed with BK038 kit from Cytoskeleton, Inc (Denver, CO).
Cell Culture
BV2 cells were a generous gift from Dr. Christopher Glass. Cells were continually grown and passaged in Opti-Mem 1% FBS to maintain a moderate rate of growth and minimal activation. 1–2 days before experimentation, cells were split into appropriate culture dishes, allowed to adhere and then washed with PBS followed by a media change. Treatments, for all experiments, were a minimum of 24–48 hours after media changes. Primary cultures were obtained from post-natal day 3 mouse pups. Mixed glia were separated from neurons according to manufacturer’s instructions and grown to confluence in T-75 flasks. Overlaying microglia were shaken for 2–4 hours at 200 rpm to separate from astrocytes and plated on glass coverslips or 12 well dishes (Giulian and Baker, 1986).
Immunofluorescence (IF) Microscopy
BV2 cells and PMG were seeded on uncoated glass coverslips, allowed to grow in appropriate media for two days then fixed with 2% paraformaldehyde in 0.1M PO4 buffer. After washing in PBS, cells were blocked with 10% goat serum, 0.1% Triton X-100 and 0.7% glycine for 15 minutes. Coverslips were either labeled for 2–3 hours overnight at 4°C in primary antibodies followed by 2 hours of incubation in secondary antibodies with DAPI, PHL or GSA. They were washed in PBS, mounted with an anti-fade solution and imaged on an Axiophot scope (Zeiss). When appropriate, matched exposures were obtained. All other images were exposure and saturation optimized. For quantitation of morphology or mitotic structures, random 10x –25x fields were taken (minimum =5).
MTT Cell Proliferation Assay
BV2 cells were seeded and diluted serially in 96 well plates, with an initial seeding density of 10,000 cells/well. After 4 days of growth in appropriate media, 10μl of MTT reagent was added according to manufacturer’s directions. After 4 hours at 37°C, media was carefully removed and the MTT crystals dissolved. The plates were read at 570nm on a Tecan Infinite M200 plate reader.
Electron Microscopy
After 2 days of growth in 60mm dishes, cells were washed and fixed with 4% paraformaldehyde, 1% gluteraldehyde, 5% sucrose in 0.1M cacodylate buffer pH 7.2. After 1% OsO4 in 0.1M cacodylate buffer for 1 hour, followed by 1 hour in uranyl acetate (UA) in 10% ethanol, cells were dehydrated through a standard ethanol series, infiltrated, using HPMA as a transitional media and finally embedded as monolayers with LX112. Pieces were mounted on blocks, thin sectioned to 70nm, placed on Cu grids, contrasted with Pb and UA and examined on a JEOL CX100 scope.
Mitochondrial function assays
Oxygen Consumption Rate (OCR) and Extra-Cellular Acidification Rate (ECAR) were measured in cell culture using an XF24 Extracellular Flux Analyzer (Seahorse Bioscience). BV2 cells were suspended in appropriate growth medium and normalized to 2.0 x 105 cells/ml. 100μl aliquots of cell suspension were transferred to an uncoated 24- well XF plate, whereupon the plate was incubated at 37°C and 5% CO2 for 1–2 hours to allow cell adherence. After incubation, 400μl of additional growth medium was added to each well for overnight incubation. “Flux Pak” cartridge (containing O2 and H+ sensitive fluorophores) was hydrated in XF Calibrant solution overnight at 37°C and ambient atmosphere. The following day, cells were gently washed and then placed into 675μl of XF Assay medium (bicarbonate-free DMEM; supplemented with 25mM glucose, 2mM pyruvate), followed by 1 hour incubation of the plate in an ambient atmosphere, 37°C incubator to allow CO2 de-gassing. Following the Seahorse MitoStress Test® metabolic profiling protocol, mitochondrial complex inhibitor solutions were prepared and loaded into the hydrated Flux Pak as such: Port A: 75μl oligomycin A ([stock]=10μM, 1% DMSO), Port B: 83.3μl FCCP ([stock]=20μM, 1% DMSO), and Port C: 92.5μl rotenone - antimycin A cocktail (each [stock]=10μM, 1% EtOH). Loaded Flux Pak was calibrated for 20 minutes prior to adding de-gassed cell plate and conducting MitoStress Test protocol (baseline respiration t=~0–30min, Port A inject t=~30min, Port B inject t=60~min, Port C inject t=~90min).
RNA Isolation and RT-PCR
Total cellular RNA was isolated according to manufacturer’s directions by RNeasy columns (Qiagen). Concentration and purity was assessed with Implen® Pearl nanospectrophotometer. For semiquantitative RT-PCR, equal amounts of total RNA were reverse transcribed and equal volumes of cDNA used for PCR. Products were analyzed on 1% TAE agarose gels. RNA to be used for quantitative real time PCR was further characterized by electropherogram spectroscopy to determine 18 and 28S ribosomal bands. A ratio of 28S/18S below 2.0 was discarded. RNA used had ratios ranging from 2.5–3.0. Real time PCR was analyzed by ΔΔCT methods using GAPDH to normalize Cav-1 and actin to normalize Cav-3 mRNA. Products were generated and quantitated using SYBR-green® and Bio-Rad real time PCR thermocycler (CFX96 C1000).
Cell Lysates and Western Blotting
Cells were lysed in buffer containing 50mM Tris-HCl, 150mM NaCl, 5mM EDTA, 0.2% Triton X-100 and 0.2% deoxycolate pH 7.4 (DXY buffer), for Cav-1 or a bicarbonate high pH buffer (150mM Na2CO3, 1mM EDTA pH 13), to disrupt cellular cytoskeleton and release any associated Cav-3, sonicated and normalized by Bradford protein assay. 2–5μg of protein were electrophoresed on 10% SDS-PAGE gels and transferred to PDVF membranes. Membranes were incubated overnight at 4°C in blocking solution and later imaged on a UVP imaging system. Protein loading was assessed with GAPDH.
Sucrose Density Fractions
Cells were lysed using bicarbonate lysis buffer with protease and phosphatase inhibitors. Cell lysate was sonicated in 1.5ml microfuge tubes 3 cycles for 15 seconds on ice. Sucrose was dissolved in MBS buffer to prepare 80%, 35% and 5% solutions (28). Sucrose gradients were made by adding 1ml of 80%, followed by 1ml of sonicated sample with brief vortexing, followed by 6ml of 35% sucrose, followed by 4ml of 5% sucrose. Gradients were spun in an ultracentrifuge using a SW-41 rotor at 39000 rpm at 4°C for 3 hours. Fractions (1ml) were collected from the top of each tube starting at 4ml to 12ml. Fractions were further analyzed using WB.
Cell fractionation assay
For Qproteome compartmentation assays, cells were pelleted and extracted according to manufacturer directions. Defined fractions were collected and electrophoresed for WB analysis.
Cav-1 and Cav-3 Gene Silencing and Overexpression
BV2 cells were plated in 24 well plates and grown in appropriate media, without antibiotics, to 70–80% confluence. siRNA’s were prepared and complexed to Lipofectamine 2000®, according to manufacturer protocol. siRNA was dripped into wells avoiding swirling and incubated with the cells for 48 hours. Cells were lysed in DXY buffer and analyzed by WB. PCMG were seeded in 24 well plates and allowed to grow for 48–72 hours and washed to remove any floating debris generated by mechanical shaking. Cells were treated according to Gene Tool® instructions. Antisense morpholinos were heated to 65°C for 10 minutes, added to wells at 5μM, followed by Endo-porter®, swirled gently and returned to incubator for 48 hours. For Adv-mediated overexpression, viral constructs for Adv-Cav-1, Adv-GFP and Adv-Cav-3 were directly added to cells in D10% at 70–80% confluence at 106 PFU/μl and allowed to express for 24 hours.
RESULTS
In vitro model of microglial activation and inactivation
Microglia activation has been defined by proliferative potential, morphologic criteria, metabolic states, differential transcriptional regulation, and inflammatory markers (van Rossum and Hanisch, 2004). Cells were grown in either serum-free (HyC, homoestasis) or high serum conditions (D10%, constitutively active) prior to analysis of cellular morphology. GSA (Griffonia simplicifolia) lectin (red) and phalloidin (PHL-green) immunoflourescence (IF) labeling confirmed both the proliferation and morphological changes associated with culturing in the serum-free condition (HyC) or in 10% FBS (D10%) (Fig. 1A). HyC cells are highly processed and less numerous than D10% cells indicative of microglial inactivation and activation respectively. To quantify the morphological difference, cell fields (10 per group), were counted for processed cells from the GSA-PHL coverslips. HyC cells averaged 65–70% processed, while less than 10% of D10% cells exhibited extended processes (Fig. 1B). MTT assays assessed the proliferation of cells grown with high serum (D10%) or serum-free (HyC) media (Fig. 1C). Statistically significant growth differences were seen between HyC and D10% cells, with HyC grown cells numbering 40% less than D10% (**p=0.007). Using cholera toxin (Ctx-green) as a marker for membrane/lipid rafts (MLR), BV2 cells were assessed for presence of MLR by IF microscopy (Fig. 1D). Cells grown in either media display Ctx labeled MLR. The cytoplasmic protein and microglial marker Iba1 (red) was used as a control for PM localization (Fig. 1E). By switching BV2 cells from an activation state/growth promoting media (D10%) to an inactivated and proliferation suppressing media (HyC), a model system has been established to study the transition from an inactive-homeostatic state to a constitutively active and highly proliferative cell phenotype.
Figure 1. BV2 cells proliferation and morphology are altered with or without FBS.

A. Phalloidin (PHL-green) and GSA-lectin (red) labeling of cells grown in Hyclone; ADCF-mAb (HyC-serum free) media and cells grown in 10% FBS (D10%). B. Quantification of the IF shows 85% of HyC cells are processed while 90% of D10% cells are round. C. MTT proliferation assay shows significantly increased rate of proliferation of D10% cells compared to HyC 4 days after seeding. Assay was performed a minimum of 3 times. D–E. HyC or D10% cells labeled with cholera toxin (Ctx-green) to stain PM lipid rafts. HyC cells are elongated and processed (left panels) while D10% cells are rounded (i, ii). Ctx specificity for PM was tested with the cytoplasmic microglia marker, Iba1 (E). Magnification bars = 10μM. Statistical analysis was performed with student’s paired t-test. F. TEM analysis of BV2 cells. Small A (a) and C are D10% grown cells. B (b) and D are HyC grown cells. A. Numerous mitochondria (M) and a dense network of rough endoplasmic reticulum (RER) is evident, typical of highly proliferative cells. Inset shows a representative caveolae at the PM from these cells. B. HyC grown cells have minimal mitochondria, and reduced RER. Large cytoplasmic vacuoles (V), some with vesicles inside or entering (arrows) are present indicating these cells are still able to internalize and process extracellular proteins. Inset shows typical HyC caveolae. (Arrow) marks a potential vesicle fusion with a cytoplasmic vacuole. C and D show representative vesicle uptake in either culture condition. Both clathrin coated vesicles (CCV) and morphological caveolae are seen in both images (arrowheads = CCV; arrows = caveolae). One degradative multi-vesicular body (MVB) is seen in D. * magnification bar = 200nm ** magnification bar = 50nm. G. O2 consumption rate (OCAR) is enhanced in BV2 cells grown with D10% media suggesting higher mitochondrial respiratory capacity than HyC cells. Glycolysis, measured by extracellular acidification rate (ECAR), is also higher at baseline in D10% cells compared to HyC.
Activation/inactivation of microglia results in distinct ultrastructural and metabolic profiles
Further morphological analysis was performed using routine electron microscopy to determine presence of morphological caveolae and to assess any ultrastructural difference between the two conditions (Fig. 1F: small A–D). Caveolae and clathrin coated pits (CCP) were localized to the PM in both conditions with similar distribution and localization. D10% grown cells had very dense endoplasmic reticulum and more abundant mitochondria (M) (Fig. 1F, A), suggestive of highly metabolic cells. HyC grown cells (Fig. 1F, B) contained a greater percentage of empty vacuoles and vacuoles with fusing vesicles and less numerous, smaller mitochondria, indicating a less active metabolic state. C and D are higher resolution images of PM areas focused on both CCP and caveolae in either condition. Metabolic activity was assessed with the Seahorse Bioscience metabolic flux analyzer. Both oxygen consumption rate (OCR, Fig. 1G) and extracellular acidification rate (ECAR, Fig. 1G), measures of mitochondrial respiration and glycolysis, respectively, were enhanced in D10% verses HyC grown cells. Such data suggest that the morphologic features observed in cells under various growth conditions impact metabolic function.
Cav-1 and -3 are differentially expressed with culture conditions and localize to discrete intracellular domains
IF microscopy of the BV2 cells grown in the different media exhibit different morphology, and exhibit different expression levels and localization of Cav-1 and Cav-3 (Fig. 2A). HyC grown BV2 have low levels of Cav-1 (top left), while serum activated cells (D10%) have higher levels and show distinct punctate staining along the PM (left panels Fig. 2A). The middle columns of Fig. 2A provide the first evidence for the expression of Cav-3, normally associated with skeletal and cardiac muscle, in myeloid derived macrophage-like cells. In the highly processed cells (HyC), Cav-3 staining was associated with the cytosol, in perinuclear regions, as well as along cellular processes, the latter suggestive of a cytoskeletal distribution. Furthermore, both the perinuclear and total expression of Cav-3 was decreased as the cells transitioned from inactive to an active state. We further assessed Cav isoforms in primary microglia (PMG) from neonatal mice (Fig. 2B). IF analysis of isolated PMG grown with either HyC or D10% and stained for Cav-1 and Cav-3 revealed similar findings seen in BV2 cells: in HyC media, PMG display extended processes with enhanced Cav-3 expression (red); D10% cells were more rounded or flattened with Cav-1 expression predominantly (green) at the PM and in the cytosol. However, in contrast to the highly compartmented expression in BV2 cells, some co-localization was detected in perinuclear regions in both media. To confirm these results, we used PCR in conjunction with Western blot (WB) assays to measure changes in caveolin isoform expression. HyC cells have 1.5 fold less Cav-1 mRNA (p<0.05, n=3 experiments) than D10% cells, but a 1.1 fold increase in Cav-3 mRNA (p<0.02, n=3 experiments) (Fig. 2C) in BV2 cells. Fig. 2C (right) shows qPCR products from wild type (WT) PMG grown in D10% for Cav-1 and Cav-3. The efficiency of Cav-1 and Cav-3 primers used for BV2 qPCR was not sufficient when used for PMG to acquire statistically significant quantitations but a reduction in Cav-1 mRNA was found for HyC to D10% (−1 ± 0.16 S.E.M ΔΔCT n=4) and an increase in mRNA for Cav-3 was found for HyC to D10% (+1.74 ± 0.31 S.E.M ΔΔCT n=4). Protein expression and antibody specificity was determined by WB for WT, Cav-1 and Cav-3 KO PMG lysates, all grown in D10% to demonstrate antibody specificity (Fig. 2D). In BV2 cell lysates, Cav-1 showed the lowest expression in the inactive state (i.e., HyC); in contrast Cav-3 expression remained elevated (Fig. 2E). When PMG are cultured similarly to BV2 cells, HyC conditions also resulted in decreased Cav-1 and increased Cav-3 expression (Fig. 2F).
Figure 2. Caveolin isoform expression is cell state specific in both BV2 microglia and neonatal mouse primary microglia cultures (PMG).

A. IF analysis of BV2 cells expression of Cav-1 (left panels) and Cav-3 (middle panels). Cav-1 is minimally expressed as small fine cytoplasmic and PM vesicles in HyC cells and in dense PM patches in D10%. Cav-3 is associated with long processes seen in HyC (top) and also localized in a dense perinuclear region. Lesser cytoplasmic expression and less frequent perinuclear localization are seen in D10% cells (bottom). B. IF analysis of PMG showing highly processed, elongated cells in HyC with increased expression of Cav-3 (red – top panels). D10% cells are rounded with significant Cav-1 label (green) at the PM and in the cytosol. Higher magnification more clearly demonstrates intracellular localization of Cav-3 compared to Cav-1. Magnification = 10μm. C. qPCR analysis of HyC or D10% BV2 cells shows the same pattern for RNA transcripts as protein expression between conditions. Cav-1 mRNA is increased in D10% cells and Cav-3 mRNA is increased in HyC. PCR was normalized with 28S ribosomal RNA and statistically significant ΔΔCT are graphed below (n=3 separate experiments). qPCR (right) products (n=2 experiments) from D10% grown PMG show distinct bands for Cav-1 and Cav-3 with normalizing actin and GAPDH. D. WB analysis of WT, Cav-1 and Cav-3 KO PMG demonstrate specificity of antibodies used and expression of Cav-1 and Cav-3 protein. E. WB analysis, of caveolin isoforms in model conditions, in BV2 cells (left) and PMG (F) demonstrate Cav-1 increases with D10% culture conditions. Cav-3 expression is highest in quiescent HyC cells. Lanes represent cells grown and analyzed separately and blots were normalized with GAPDH (n=3). Statistically significant differences are graphed (right). WT PMG grown in HyC or D10% show similar expression of caveolin as BV2 cells. G. Cav-3 and Cav-1 expression is modulated by phenotype state and can be switched when the conditions change. Cav-3 expression decreases within 4 hours of FBS treatment in HyC cells. Cav-1 expression decreases more slowly when FBS is removed and cells revert to a more resting phenotype (D10% to HyC switch).
Microglia in vivo must be able to both activate and inactivate in response to a changing environment. To determine if the results of cells grown in either condition were reversible, cells grown 2 days in each media were washed and replaced with the opposite condition (Fig. 2G). Cells were lysed and subjected to WB analysis for Cav-3 down-regulation and Cav-1 up-regulation in the switch from HyC to D10%, or for Cav-1 down-regulation and Cav-3 up-regulation for a D10% to HyC switch. Cav-3 expression decreased within 4 hours in HyC to D10% cells but the decrease in overall Cav-1 expression in D10% to HyC cells was more gradual, indicating difficulty of reversing activated cells to an inactive phenotype. Such data also suggest that Cav-1 at the PM is not recycled as rapidly under steady state conditions. Cav-1 can be held stationary within the MLR domain or dynamically exchanged from a multi-vesicular pool of stored Cav-1 by a described “kiss and run” mechanism to maintain a steady pool of available protein (Pelkmans and Zerial, 2005) at the PM. This pool would need to be depleted by increased signaling or increased degradation, which is time-dependent.
Subcellular distribution analysis suggest preferential localization of Cav-1 to MLR whereas Cav-3 associates with the cytoskeleton
To confirm subcellular localization by biochemical analysis, we performed sucrose density fractionations to isolate MLR. Localization of caveolin in Cav-enriched membrane fractions (i.e., MLR) was confirmed by Cav-1 localization to the buoyant fraction 5 (F5) in both HyC and D10% cells (Fig. 3A), indicating association with MLR. Considerably less Cav-3 was found in MLR, with a shift to non-caveolar membrane fractions (F11–12) especially in the inactive state (HyC). To test whether this switch in Cav isoforms was specific to Cav, a second MLR associated protein, flotillin, was examined, by WB. Flotillin has similar scaffolding properties and is inserted into the PM by cysteine palmitoylation, similar to Cav. Figure 3B shows an interesting switch for the two isoforms. Flotillin-1 was increased in D10% and flotillin-2 increased in HyC cells, indicating changing compositions of MLR within these two conditions. WB analysis was performed to determine expression differences of the two major cytoskeletal proteins with differing conditions in BV2 cells (Fig. 3C). D10%, which has higher Cav-1 expression, exhibited higher tubulin expression. Conversely, HyC cells, which have higher Cav-3 expression, show increased actin expression. To further define caveolin localization, BV2 and PMG cells grown in the two media conditions were fractionated to determine the subcellular distributions (Fig. 3D–E) of caveolins. Cav-3 was highly abundant in the cytoskeletal fraction (lane 4 in all blots) in both HyC and D10% media, in accordance with the observed non-membrane localization. Cav-1, however, was localized to the defined membrane fraction (lane 2 in all blots), although in PMG cells a significant pool of Cav-1 was also found in the cytoskeletal fractions, along with a shift of tubulin from membrane fractions in BV2 cells to cytoskeletal fractions in the PMG. Actin was found in all fractions in both BV2 and PMG. These data suggest a connection between caveolins and key cytoskeletal proteins.
Figure 3. Cav-1 and Cav-3 are associated with cytoskeletal fractions in BV2 and PMG.

A. Detergent resistant fractions (DRM) from HyC and D10% cells – top panel shows Cav-1 is found in DRM f4–6. Bottom panel for Cav-3 shows a minimal amount of Cav-3 protein in f5 with the majority of detectable Cav-3 in the heaviest non-lipid raft fractions (f11–12). B. HyC cells have less flotillin-1 and increased flotillin-2 compared to D10% cells. C. In a semi-quantitive assay based on total protein, HyC cells show decreased tubulin and slightly increased actin whereas D10% cells show the converse pattern. Blots are normalized to HSP90, since GAPDH was too abundant for accurate normalization on the stripped blots. D. Qproteome cell compartmentation assay was used to determine Cav-1 and Cav-3 cellular distribution between cytosol, membrane, nuclear and cytoskeletal compartments in BV2 cells – HyC (right) D10% (left). In either condition, Cav-1 distributes to the membrane component (lane 2). Cav-3 is predominantly distributed to the cytoskeleton component (lane 4). Tubulin is confined to cytosolic and membrane fractions, while actin is distributed across the fractions. E. Qproteome cell compartmentation assay was used to determine Cav-1 and Cav-3 cellular distribution between cytosol, membrane, nuclear and cytoskeletal compartments in PMG – HyC (right) D10% (left). Cav-1 is found in membrane fractions, cytosolic fractions and also distributes to the heavy cytoskeletal fractions, along with tubulin, unlike the exclusively membrane localized Cav-1 in BV2. Cav-3 is found exclusively in the cytosol and cytoskeletal fractions. As with BV2 cells, actin is found in all fractions, but a shift to cytosolic fractions is seen in the D10% cells.
Cytoskeletal disruption and stabilization differentially regulated caveolin isoform expression
Tubulin and Cav-1 were detected in membrane fractions in BV2 cells (Fig. 3B); therefore, we used microtubule disrupting (nocodazole, Nocz) or stabilizing agents (paclitaxel, Taxol) to determine the impact of microtubule dynamics on Cav-1 expression in BV2 cells (Fig. 4A). When HyC cells were treated with Nocz (10 μM), the cells rounded, lost processes and Cav-1 (green) expression increased over 4 hours at or near the PM. Tubulin also became more intense proximal to the PM; these findings were confirmed by WB (Fig. 4C - top panels). D10% cells had a reduction of Cav-1 (green) as detected by both IF and WB after 4 hours of Taxol (0.5 μM)(Fig. 4A, C). With an apparent cytoskeletal connection by both compartmentation and IF studies for Cav-3 and no expression change seen with microtubule disrupting agents, we depolymerized or stabilized F-actin with cytochalasin D (CytoD) or jasplakinolide (Jasp) respectively and assessed Cav-3 expression/localization (Fig. 4B). When HyC cells were treated with CytoD (1μg/ml) for 4 hours and labeled with Cav-3 (red) and PHL (green), the actin cytoskeleton and Cav-3 did not colocalize. However, as the cells progressively collapsed inward, the expression of Cav-3 decreased while actin remained at the vestiges of the former processes. Fig. 4D (left) confirmed the decreased Cav-3 expression by WB (left). The converse is seen when D10% cells are treated with Jasp (0.5 μM) to stabilize F-actin. At time 0, actin is localized both intracellularly and cortically, but redistributed to discrete PM areas at 4 hours, which are distinct from an increasingly strong intracellular Cav-3 expression. Jasp treatment increased Cav-3 expression in D10% cells (Fig. 4D right).
Figure 4. Depolymerizing or stabilizing cytoskeletal proteins alter expression and localization of Cav-1 and Cav-3 in BV2 cells and PMG.

A. Microtubules were either depolymerized with 10μM nocodazole (Nocz) to disrupt processes in HyC cells or stabilized with 0.5μM pacliltaxel (Taxol) in D10% cells. Tubulin (red) appears mainly associated with PM domains, with limited staining seen through the processes, consistent with the compartmentation experiments of Fig. 4A. Cav-1 (green) expression shows a reduction over time at 4 hours in D10% cells while HyC cells increase the expression of Cav-1 and at 4 hours resemble the initial time 0 of D10% cells. B. Actin was either depolymerized with 1μg/ml cytochalasin D (CytoD) to disrupt processes in HyC cells or stabilized with 0.5mM jasplakinoide (Jasp) in D10% cells. Actin, stained with PHL (green), localizes to processes and in globular bundles in HyC cells and mainly as cortical or perinuclear actin in D10% cells. Cav-3 (red) does not colocalize with actin in either condition, indicating discrete domain separation. HyC cells show a loss of Cav-3 as actin becomes increasingly concentrated to shrinking processes. In D10% cells, expression of Cav-3 increases as actin becomes more concentrated intracellularly. C–D. WB analysis of all treatments confirms the expression patterns seen by IF. Cav-1 expression increases within 2 hours in the HyC cells treated with Nocz and decreases by 2 hours in D10% cells treated with Taxol (C). Minimal changes were seen for Cav-3 following microtubule toxin treatment. Cav-3 expression decreases in HyC treated with CytoD within 4 hours and increases in D10% treated with Jasp at 4 hours (D), with minimal changes in Cav-1 seen following CytoD treatment. Magnification bar = 10μm. E–H. Results of IF and WB analysis, of PMG cells, treated with cytoskeletal toxins. E. HyC grown cells treated with 10μM Nocz do not lose their processes but show an increase in Cav-1 expression both at the PM (arrowheads) and in intracellular domains. Cav-3 (red) remains in processes. WB analysis mirrors the IF with Cav-1 (top) increasing by 4 hours and Cav-3 unchanged. F. D10% cells treated with 0.5μM Jasp result in the greatest morphological changes. Nuclei become eccentric (arrows), Cav-3 expression increases and condenses in intracellular domains. Cav-1 remains at the PM (arrowhead) and expression is largely unchanged. WB analysis shows a similar expression pattern as the IF, with Cav-3 (top) increasing by 2 hours. Insets are untreated (UT) 0 minute cells showing typical Cav-1 distribution in HyC cells and typical Cav-3 distribution in D10% cells. G. When HyC cells are treated with CytoD, unlike BV2 cells, neither isoform expression is altered, but localization of Cav-1 shifts from PM and intracellular domains to a perinuclear ring (arrows). H. Stabilizing microtubules in D10% cells increases Cav-1 expression and increases PM localized Cav-1 positive vesicles (arrows) in denser patched areas. Nuclei remain centric (N) and Cav-3 expression is unchanged. Magnification bar = 10μm. I. When G-monomers are sequestered with 0.5μM LatB, in PMG, Cav-3 decreases by 24 hours in HyC cells and increases within 4 hours in D10% cells.
With a slightly different cellular distribution of tubulin seen in BV2 cells, PMG cells responded differently to the same reagents (Fig. 4E–H). HyC PMG treated with Nocz and D10% PMG treated with Jasp had the same increases in caveolin isoforms as BV2 cells. Nocz increased Cav-1 expression and Jasp increased Cav-3 (Fig. 4E, F). However, CytoD treatment resulted in Cav-1 collapsing to a perinuclear ring, with no vestiges of PM labeling remaining (Fig. 4G) while there was no expression change of either isoform by WB. Taxol increased expression of Cav-1 and also increased the PM localization into dense patches in D10% PMG (Fig. 4H). Treatment of BV2 cells with latrunculin B (LatB, 0.5μM), which sequesters G-actin monomers, resulted in no caveolin expression change, but a longer LatB treatment (24 hour) did differentially change Cav-3 in PMG (Fig. 4I). PMG, unlike BV2 cells, are not highly proliferative in culture, unless grown with astrocytes or in astrocyte conditioned media, so LatB did not significantly increase cell numbers as in BV2 cells (Fig. 5B). Sequestration of G-actin in CHO cells causes an immediate increase in Cav-1 positive vesicles trafficking to perinuclear domains (Mundy et al., 2002). Therefore, we postulate that in PMG this potential loss of PM Cav-1 shifts tubulin and actin ratios. Whereas in BV2 cells, this ratio change increases cell proliferation (D10%). In PMG, Cav-3 is potentially degraded (HyC) or expression upregulated (D10%) to return ratios to previous homeostatic conditions. Therefore, in both cell systems, there appears to be tubulin/Cav-1 or actin/Cav-3 regulation.
Figure 5. Cav-3 localizes to mitotic structures in cells, either induced to proliferate with LatB or in proliferative cultures.

A. Following 8 hours of 0.5μM LatB treatment, HyC cells round, lose filamentous actin structure, and both caveolin isoforms concentrate in opposite polar ends to shrinking nuclei. Cav-3 concentrates in intracellular regions. D10% cells show cellular enlargement and increased numbers of mitotic cells. The mitotic figures seen in D10% cell have specific Cav-3 labeling on the mitotic spindle (*) and perinuclear areas (arrows) colocalize with actin. Insets are the corresponding Cav-1 labeled cells with a different distribution from Cav-3. B. Sequestering G-actin with 0.5μM LatB in either condition effects the survival or proliferation of the cells. An MTT assay quantified the increase in proliferation of LatB treated D10% cells and the probable cell death or retarded growth seen in HyC cells after 3 days of growth. Assay was repeated 3 times. * p<0.05 **p<0.02 by student’s paired t-test. C. The number of mitotic figures was quantified by IF. Total number of cells and total number of mitotic figures per 25x field was determined. In the UT samples, an average of less than 6 were seen, while an average of 11 were counted after 8 hrs of LatB treatment. D. In mixed glia cultures, both astrocytes and microglia are evident. Cav-2 labeling (red) is abundant in morphologically identifiable astrocytes (large nuclei-DAPI) and localized to mainly PM in morphologically identifiable microglia (*)(left top panel). Cav-3 (green) is highly expressed in these cells, with cytoplasmic and perinuclear distribution. Cav-3 shows novel distribution to MTOC in actively dividing putative PMG cells in mixed cultures, where such cells are more abundant. The last panel is a PMG, in a pure culture from D10% grown cells, with Cav-3 (red) clearly evident during mitotic events, representing a novel localization for a normally PM associated protein, while Cav-1 (green) remains intracellular. Magnification bars = 10μm.
Cav-3 associates with nuclear cytoskeleton to regulate microglial proliferation
BV2 cells were treated with 0.5 μM LatB, for 1 and 4 hours, to sequester globular G-actin and prevent filamentous F-actin formation. BV2, in either media, display a rounded or condensed morphology with a reduction in PHL fluorescence (Fig. 5A). However, Cav-3 (top panels) showed clear differences in subcellular distribution after 4 hours of treatment. In HyC cells, nuclei became highly polarized, similar to Jasp treated D10% PMG, and Cav-3 was distributed to opposite polar domains; Cav-1 appeared similar to Cav-3 distribution (inset). Actin was only detected on PM patches with an obvious lack of association with processes, whereas, nuclei remained centric in D10% conditions (Fig. 5A-right 2 panels) and Cav-3 began to associate with perinuclear mitotic organizing centers (MTOC; small arrows) and mitotic structures (*). Cav-1 did not localize to spindles (inset) and maintained a similar distribution to that exhibited in untreated cells. To determine if the loss of G-actin also directly increases BV2 proliferation, MTT assays were performed as previously described (Fig. 5B) with 3 days of LatB treatment (0.5 μM). LatB significantly (**p=0.005) increased D10% grown cell proliferation over untreated cells and either slowed HyC growth or induced cell death. Equal numbers of seeded D10% cells were treated for 8 hours with LatB or vehicle followed by assessment of percentage of mitotic figures per field (Fig. 5C). LatB averaged 11–12 clearly dividing cells, while untreated (UT) averaged 5. Fig. 5D provides evidence in proliferating mixed glia cultures and PMG of the association of Cav-3 with both MTOC and mitotic structures, suggesting the possibility that Cav-3 may be an attractive target to attenuate inflammation by preventing the proliferation of activated microglia.
Caveolin knockdown attenuates the inflammatory response induced by LPS in microglia, potentially by mediating intracellular trafficking
Microglia IL-1β response to LPS is well documented, and can be mediated by Cav-1 in macrophages (Tsai et al., 2011). After 10 ng/ml LPS stimulation, both HyC and D10% BV2 cells have increased Cav-1 expression (Fig. 6A left). Although pro-IL-1β (IL-1β) response was robust in either treatment, HyC cells have slightly attenuated expression most likely due to the initial lower level of PM localized Cav-1 and a lack of LPS-binding protein (LBP) in the media. For comparison, similar experiments were performed with PMG (Fig. 6A right). Following 10 ng/ml LPS stimulation, Cav-1 expression increased and a similar attenuation of IL-1β in WT PMG HyC cells was seen. The upregulation of Cav-1, following LPS stimulation, has lead to the suggestion of the association of LPS signaling/trafficking with PM localized Cav-1 (Lei and Morrison, 2000). Experiments to understand potential signaling defects downstream from LPS-TLR4 interactions resulting from Cav isoform loss were performed. Initial dose response experiments using siRNA to decrease either Cav-1 or Cav-3 were performed to achieve at least a 50% reduction in expression (Fig. 6B left). BV2 cells increased expression of IL-1β at 4–8 hours of stimulation. siRNA for Cav-1 resulted in an attenuation of IL-1β expression, with maximal expression seen at 8 hours (Fig. 6B -right). While siRNA for Cav-3 treated cells reached maximal but attenuated IL-1β expression within 4 hours, similar to untreated (UT) and SCR treated cells.
Figure 6. Altered signaling, cell migration after injury and mitochondrial functional are dependent on specific caveolin isoform expression following LPS stress.

A. WB baseline data of BV2 and PMG treated with 10ng/ml LPS for 4 and 8 hours. Cav-1 expression increases while Cav-3 does not. IL-1β (33kDa: IL-1β) response to LPS is attenuated when cells are grown without serum at 8 hours. Blots are normalized to GAPDH. B. Left panels are knockdown control WB for corresponding right panels of knockdown functional consequences following LPS treatment with at least a 50% reduction in expression of caveolin. siRNA treated cells were further analyzed by 10ng/ml LPS treatment and WB for IL-1β response. Both siCav-1 and siCav-3, in either BV2 or PMG, show attenuation of maximal IL-1β expression evident as early as 4 hours. For WT PMG cells, antisense morpholinos (mrpl), specific stable oligos, that bind to complementary sequences, for either mRNA Cav-1 or Cav-3 were designed. VC = DMSO vehicle; SCR = mrpl control; mCav-1 5uM and mCav-3 5uM. Each experiment was repeated n=3 and blots are representative of results. C. A wound healing/migration assay was used to assess BV2 migration following siRNA at 0 min, 8 hours and 24 hours. Plates were fixed, stained with methylene blue and imaged. D. Comparison of oxygen consumption rate (OCR; left panels) and extracellular cellular acidification rate (ECAR; right panels) measurements for BV2 cells treated with siRNAfor gene knockdown of Cav-1 and Cav-3. Scrambled siRNA (SCR)) was used as a control. Comparisons of oxygen consumption rate (OCR; left panel bottom) and extracellular cellular acidification rate (ECAR; right panel bottom) measurements for BV2 cells treated with adenovirus for gene overexpression of Cav-1 and Cav-3. GFP adenoviral vectors were used for controls. (*; p≤ 0.05)
For comparison, similar silencing experiments were performed with PMG. The siRNA used for BV2 silencing proved cytotoxic and activating for PMG, so a less immunogenic anti-sense morpholino approach was utilized (Fig. 6B left). A similar attenuation of IL-1β response to LPS with either anti-sense morpholino Cav-1 (mCav-1) or anti-sense morpholino Cav-3 (mCav-3) was observed (Fig. 6B right). MLR have been shown to be critical in TLR4 signaling via the TLR4-MyD88 complex implicating an interaction with Cav-1 in downstream signaling events critical to cell inflammatory responses (Mirza et al., 2010; Triantafilou et al., 2002; Wang et al., 2009). Therefore, a reduction in Cav-1 should result in delayed or truncated intracellular signaling, supporting previous findings (Lei and Morrison, 2000). Silencing Cav-3, however, also results in attenuation of the enhanced IL-1β expression, indicating downstream messengers may have been affected, possibly by disruption of critical cytoskeletal interactions during initial signaling events.
Caveolin regulates migration and metabolism in microglia
Cell migration differences were studied by a wound-healing assay in caveolin knockdown BV2 cells. PMG cells show limited migratory abilities by time-lapse microscopy, at rest (Movie S1 in supplementary material), or in wound healing assays (Movie S3) but BV2 cells have robust migratory responses without wounding (Movie S2) or post wounding (Figure S1 and Movie S4), so were chosen for this assay. Fig. 6C shows BV2 cells 8 hours post wounding, when approximately 50% of the wounded area is filled in by the three treatment groups: siSCR, siCav-1 and siCav-3. However, at 24 hours, siCav-1 cells have about the same cell number, indicating reduced overall migration. siCav-3 cells have an overabundance of migrated cells, suggesting an overall increase in actin polymerization, a requirement for cell migration. Polymerized F-actin forms the typical fan-shaped expanding lamellae seen during the migratory phase (Movie S4) and has been characterized as a superstructure composed of “podosomes” rich in F-actin, phosphotyrosine residues, tyrosine kinases and phosphorylated Cav-1 in migrating microglia (Vincent, et al., 2012).
In order to assess the effect of altered expression levels of Cav-1 and Cav-3 isoforms upon the metabolic state of microglia, BV2 cells cultured in D10% media were challenged with the Mitochondrial Stress Test® assay using the XF24 Extracellular Flux Analyzer (Seahorse Biosciences). As depicted in Fig. 6D, both oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured simultaneously in BV2 cells subjected to siRNA gene knockdown (left top and bottom) or adenovirus-mediated gene overexpression (right top and bottom) of Cav-1 and Cav-3. Comparative analysis of OCR readings between knockdown and overexpressing cells showed that knockdown cells of both Cav-1 and Cav-3 had significantly lower mitochondrial respiration profiles as compared to scrambled siRNA controls (p≤ 0.05), and inversely that overexpressing cells had significantly higher respiration patterns as compared to GFP controls (p≤ 0.05). This data suggests that the degree of caveolin protein expression in BV2 cells is directly correlated with overall metabolic functional capacity in glial mitochondria. Interestingly, when comparing ECAR measurements in the siRNA treated cells (left bottom), we observed that only Cav-1 knockdown cells had a significantly lowered ECAR profile in both baseline conditions and in response to mitochondrial stress, implicating that Cav-1 is a more critical regulator of background cellular signaling and metabolic activity in response to metabolic stress. Conversely, for adenoviral overexpression, ECAR levels in Cav-3 BV2 cells were observed to be significantly higher at both stressed and unstressed conditions, signifying an increase in global metabolic flux for Cav-3 expressing cells.
Altered expression of Cav changes tubulin and actin expression and further results in distinct morphological changes
Expression of β tubulin and actin in BV2 cells, following either knockdown or Adv-mediated overexpression of Cav, was assessed by semi-quantitative WB analysis (Fig 7A–C). β-tubulin (hereafter termed tubulin) has 2 distinct molecular weights in our cells, when lysed with a proprietary microtubule stabilizing buffer: the described 55kD and a slightly higher weight band of approximately 75kD, which we hypothesize is a post-translationally modified (PTM) form with either polyglycylation or polyglutamination side chains, both common modifications indicating increased stability (reviewed by Janke and Bulinski, 2011). When Cav-1 is knocked down in D10% cells, tubulin appears to have increased stability, substantiating the decrease in Cav-1 seen following microtubule stabilization with Taxol in BV2 cells (Fig. 4A,C). Overexpression of Cav-1 by Adv infection (Fig. 7B) results in a loss of the 75kD band and a reduction in tubulin expression overall, leading us to conclude that Cav-1 may sequester free tubulin and prevent polymerization. Actin expression is slightly increased with siCav-3 treatment (Fig. 7C) but assessment of F/G ratios would provide a more precise measure of the exact changes. Adv-mediated overexpression of Cav-3 showed only very slight actin expression decreases with less cytoskeletal associated Cav-3 and more cytosolic pools, indicating possible mis-localizations (data not shown). WB analysis was performed using WT and KO PMG to assess cytoskeletal proteins (Fig 7D, E). In Cav-1KO microglia, both tubulin and actin are increased over WT, with greatly increased 75kD bands. Cav-3KO cells had higher levels of actin expression over WT and on the tubulin WB, no 75kD was detected and a prominent lower band seen, indicating the lack of PTM or less stabilized microtubules overall. When these cells were stained for actin (PHL-green) and tubulin (red), more highly polymerized forms of both actin and tubulin are evident in Cav-1KO. Cav-3KO cells show non-uniform sizes with large pools of G-actin and punctated tubulin, reminiscent of less stabilized form for both proteins.
Figure 7. Cav-1 and Cav-3 negatively regulate tubulin and actin expression.

A. WB of BV2 tubulin following siCav-1 treatment and lysis in a microtubule-stabilizing buffer. A distinct upper band at approximately 75kD is increased with loss of Cav-1. B. WB of BV2 tubulin after Adv-Cav-1 overexpression. The 75kD band is not evident and overall expression decreases compared to Adv-GFP controls. C. Actin levels are slightly elevated in BV2 following siCav-3 treatment, similar to (E) below. D. In Cav-1KO PMG, the 75kD band is highly expressed along with increased 55kD (2.5 fold increase). Actin is also increased over WT controls. E. As in (C), actin levels are increased in Cav-3KO (2 fold increase). A lower molecular weight band of tubulin is detected, which is only slightly resolved in WT, indicating possible changes in post-translational modifications or less polymerization. F. WT microglia (small arrows) display strongly polymerized MT (arrowhead) and regions of punctated tubulin associated with processes, along with both globular (G-actin) and filamentous (F-actin) structures, representing actively remodeling cells. Cav-1KO and Cav-3KO PMG have altered cellular structures. Both subcortical MT and proximal F-actin, are prominent features of highly stellate Cav-1KO PMG (arrowheads). Both overall size and nuclear morphology are affected in Cav-3KO PMG. Cav-3KO PMG (small arrows) demonstrate the size heterogeneity of the culture and the increased punctated tubulin and actin (*). Mag bar = 10μM.
DISCUSSION and CONCLUSIONS
Microglial activation is a tightly regulated and self-limiting process in vivo, with external signals coordinating the following steps: process retraction, increased transcriptional synthesis, migration, phagocytosis, and cell fate (i.e., apoptosis). Our initial goal for these experiments was to design a cell culture model system of microglial activation/inactivation states. In the process of defining such a model we have uncovered a unique Cav isoform switch. In our defined experimental conditions in BV2 cells, we have found that putative inactive (homeostatic) cells (HyC) express high levels of Cav-3 in intracellular domains and reduced amounts of Cav-1 while constitutively active cells (D10%) express much lower levels of Cav-3 but increased Cav-1, especially at the PM. These observations were also confirmed in PMG. This differential expression was mirrored, by a second MLR associated protein, flotillin. The expression of the two isoforms of flotillin, flotillin-1 and flotillin-2, is independently regulated and not thought to follow Cav expression (Volonte et al., 1999). But in our model system, it appears that flotillin expression is dependent upon Cav isoforms expression. Flotillin-1, increased in D10%, is known to regulate cell signaling (insulin receptor) (Baumann et al., 2000) and cell proliferation (Santamaria et al., 2005). In live cell imaging experiments with flotillin-2-GFP, hematopoietic cells keep their flotillin localization to tips of the leading edge during migration, but this polarity is lost after actin destabilization (Rajendran et al., 2009). This suggests that microglia may use MLR as a common mechanism to segregate signaling and cytoskeletal regulatory proteins during the transition between homeostatic or active states.
A previously published paper (Park et al., 2009) reported that BV2 cells did not express Cav-1, yet we detected both mRNA and protein for Cav-1 and Cav-3. We have used a different Cav-1 antibody and optimized lysis buffers for our experiments, although BV2 clone differences remains a plausible explanation for our conflicting results. Determining the localization of Cav-1 to PM domains is not surprising due to previously published studies of Cav-1-mediated signaling for many receptors (Panneerselvam et al., 2012; Williams and Lisanti, 2004). And the finding that Cav-1 is not rapidly degraded or removed from the PM during the D10%-HyC switch demonstrates the importance of this Cav in inflammatory phenotypes and also points towards the difficulty in achieving resolution of existing inflammation during injury or disease by a strict knockdown approach. As the two Cav isoforms appear to function cooperatively, a two-step approach, by reducing Cav-1 and increasing Cav-3 expression might accelerate the repression of the inflammatory phenotype. Localization of Cav-3, traditionally thought to be specific to cardiac and skeletal muscle, to a non-canonical intracellular domain is a novel finding. This observation was confirmed by cell compartment fractionation assays, which show Cav-1 distribution to membrane and cytoskeletal fractions, while Cav-3 was confined to the cytoskeleton and cytosolic compartments. By treating our differentially cultured cells with cytoskeletal disrupting agents, we establish putative connections between tubulin stability with Cav-1 expression and actin polymerization with Cav-3 expression and localization. Furthermore, by sequestering G-actin monomers with LatB in BV2 cells, we demonstrate Cav-3 specific localization to the mitotic spindle of dividing cells, indicating a possible mechanistic role in mitosis and cytokinesis of microglia cells.
Regulation of the cell cytoskeleton by caveolins has been studied in other cell types for both Cav-1 (Shajahan et al., 2007) and Cav-3 (Head et al., 2006). The actin cytoskeleton has been shown to post-translationally modify iNOS and therefore regulate nitric oxide production following LPS challenge in macrophages (Eswarappa et al., 2008). Recently, a link between ezrin-radixin-moesin (ERM proteins) cytoskeletal linkers and microglia activation following a traumatic brain injury has been described (Moon et al., 2011). In that study, limited expression of ERM proteins was found in normal tissue and high expression was detected in Iba1+ cells following injury, implicating a critical role for the cytoskeleton in the injury response. Studies of ameboid microglia treated with the GluR1 agonist kainite (KA), a receptor regulated by Cav-1, results in rapid actin changes, associated with increased phagocytosis (Christensen et al., 2006). However, this study was hampered by the lack of morphologically quiescent microglia with high levels of Cav-3 expression, which we have shown to interact with the actin cytoskeleton. 2DE map analysis, using BV2 cells treated with Aβ25–35, localized an acidic cytosolic actin species upregulated over nontreated cells, which has been postulated to be associated with the initial cytoskeletal dynamic changes needed for conversion to a phagocytic phenotype (Di Francesco et al., 2012). A newly emerging importance for defective actin function in microglia has been described for Huntington disease (HD). Microglia from early, non-symptomatic, HD model mice, are compromised in their migrational abilities, as mutant Huntington protein (Htt) interferes with cofilin-actin interactions (Kwan et al., 2012).
An important observation, from this study, is that constitutively active microglia (D10%) have increased expression of Cav-1, with enhanced mitochondrial respiration and glycolysis, indicative of a higher metabolic states. This functional change may be facilitated by structural changes as well, as active cells appeared to be more densely packed and had enhanced number of mitochondria. We have recently shown in cancer cells that upregulation of Cav-1 leads to enhanced mitochondrial function, biogenesis, and stress adaptation (Fridolfsson et al., 2012). It appears the relationship between caveolin and mitochondria is highly conserved and may be a common means of stress adaptation in numerous cell types.
Normal aging results in a reduction in Cav-1 expression in the CNS (Head et al., 2010). Cav-1KO mice have increased cerebral injury following ischemia (Jasmin et al., 2007), and work from our laboratory (Head et al., 2010) details an Alzheimer’s Disease-like pathology associated with young adult Cav-1KO mice (3 months of age). Furthermore, these mice exhibit increased astrogliosis and increased numbers of Iba1+ microglia in the hippocampus. To date however, there are no studies on the pathogenic consequences of Cav-3 loss in the brain, probably because Cav-3 role within the CNS is still undefined. Although both neurons (Luoma et al., 2008) and astrocytes (Silva et al., 2007) express Cav-1 and Cav-3 (Stern and Mermelstein, 2010), expression of these isoforms in a microglia is a novel concept. In macrophages, either Cav-1 deletion or disruption of caveolae by the cholesterol chelator methyl-β-cyclodextrin (MβCD), leads to defective phenotypes (Cuschieri, 2004; Tsai et al., 2011). Aged microglia display dystrophic, severely fragmented, processes in vivo (Streit et al., 2009) and this dystrophia has been proposed as an alternative hypothesis for the pathogenesis of many neurodegenerative diseases. Initial intracellular signaling cascades mediated by caveolae and Cav-1 need an intact and functional cytoskeletal arrangement. Following ligand binding to receptor, caveolae pinch off the PM, and are transported via microtubules (Mundy et al., 2002). Our data suggests that besides a direct receptor-mediated signaling role, Cav-1 also acts as a microtubule-associated protein (MAP) to negatively regulate localized MT polymerization within microglia cells, a previously un-described and novel function. In coordination with Cav-3 maintaining a useable pool of localized G-actin monomers, microglia can maintain functionally quiet states, with long processes moving and sampling the environment, but can rapidly change to an active functional state once local external signals are detected (summarized in Fig. 8 – schematic diagram). From our data, we propose a model of cytoskeletal regulation at the localized PM, which ultilizes the scaffolding capabilities of caveolins to regulate polymerization states of microtubules and actin. Although the exact molecular binding sites or other potential binding partners for either the proposed Cav-1: “free/non-polymerized” tubulin and Cav-3: G-actin interactions, needs to be further studied in depth; we believe this model is a starting point to design future experiments.
Figure 8. Schematic representation of caveolin-mediated cytoskeletal regulation in microglia cells.
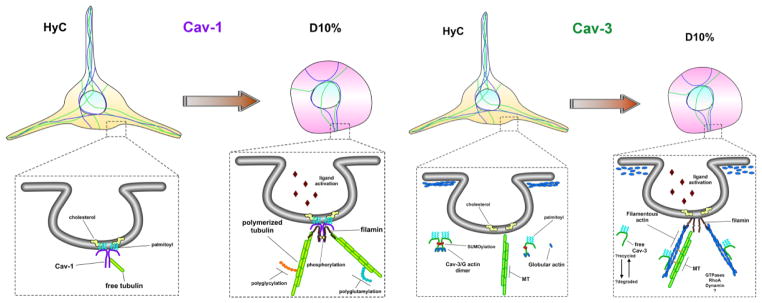
In quiescent microglia, represented by our HyC condition, Cav-1 resides within the inner leaflet of the PM, anchored to cholesterol via palmitoylated cysteine residues and interacts with free-non-polymerized tubulin to maintain a readily available pool, while the majority of cellular tubulin is polymerized to maintain the long and active sentinel processes. Once caveolae-mediated receptors bind ligand, represented by our D10% condition, free tubulin is displaced, by another microtubule binding partner, possibly filamin, which tethers the now polymerizing microtubules to the internalizing caveolae, facilitating downstream signaling cascades. Cav-3, on the other hand, is never localized to the PM and remains within the cytosol, as monomers or dimers, or bound to actin cytoskeletal compartments. Unbound free cytosolic Cav-3 interacts with, and maintains a pool of G-actin monomers, near the inner leaflet of the PM, which is structurally maintained by a dense cortical actin network, represented by HyC cells. Once caveolae-mediated receptors bind ligand, the G-actin is released by the cytosolic Cav-3 or directly incorporated, along with Cav-3, into the rapidly nucleating actin needed for internal transport. The released cytosolic Cav-3 is either degraded or is available to interact with the rapidly denucleating cortical actin network G-actin monomers, to resupply the localized environment for further signaling.
In conclusion, we describe novel and emerging roles for both Cav-1 and Cav-3 in microglial cell physiology. Cav-1, besides mediating signaling events at the PM, acts as a localized negative regulator for microtubule stability and enhances mitochondrial function. While Cav-3 acts as a localized negative regulator of actin polymerization by potentially sequestering G-actin monomers and has no direct role in mediating cell signaling from caveolae: a previously defined functional role in striated muscle. Future experiments, designed to explore the relationships with other cytoskeletal associated proteins, such as filamin, or cytoskeletal regulators, such as RhoA or Rac1, and Cav isoforms are currently planned. Observations from this work also suggest novel therapeutic targets, using glial specific morpholinos or adeno-associated viruses (AAV) to modulate expression levels of either Cav-1, by suppressing expression to decrease inflammation or augmenting Cav-3 to retain the surveillance functions in microglia, for a variety of neurodegenerative pathologies.
Supplementary Material
Acknowledgments
This work was supported by grants from AP Giannini Foundation (HNF); NIH HL091071 (HHP), HL107200 (HHP and DMR), NS073653 (BPH), and HL081400 (DMR); VA Merit BX001225 (BPH) and BX000783 (DMR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baumann CA, Ribon V, Kanzaki M, Thurmond DC, Mora S, Shigematsu S, Bickel PE, Pessin JE, Saltiel AR. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature. 2000;407:202–207. doi: 10.1038/35025089. [DOI] [PubMed] [Google Scholar]
- Chidlow JH, Jr, Sessa WC. Caveolae, caveolins, and cavins: complex control of cellular signalling and inflammation. Cardiovascular research. 2010;86:219–225. doi: 10.1093/cvr/cvq075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen RN, Ha BK, Sun F, Bresnahan JC, Beattie MS. Kainate induces rapid redistribution of the actin cytoskeleton in ameboid microglia. Journal of neuroscience research. 2006;84:170–181. doi: 10.1002/jnr.20865. [DOI] [PubMed] [Google Scholar]
- Cunningham C. Microglia and neurodegeneration: The role of systemic inflammation. Glia. 2013;61:71–90. doi: 10.1002/glia.22350. [DOI] [PubMed] [Google Scholar]
- Cuschieri J. Implications of lipid raft disintegration: enhanced anti-inflammatory macrophage phenotype. Surgery. 2004;136:169–175. doi: 10.1016/j.surg.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Eswarappa SM, Pareek V, Chakravortty D. Role of actin cytoskeleton in LPS-induced NF-kappaB activation and nitric oxide production in murine macrophages. Innate immunity. 2008;14:309–318. doi: 10.1177/1753425908096856. [DOI] [PubMed] [Google Scholar]
- Fridolfsson HN, Kawaraguchi Y, Ali SS, Panneerselvam M, Niesman IR, Finley JC, Kellerhals SE, Migita MY, Okada H, Moreno AL, Jennings M, Kidd MW, Bonds JA, Balijepalli RC, Ross RS, Patel PM, Miyanohara A, Chen Q, Lesnefsky EJ, Head BP, Roth DM, Insel PA, Patel HH. Mitochondria-localized caveolin in adaptation to cellular stress and injury. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2012;26:4637–4649. doi: 10.1096/fj.12-215798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head BP, Insel PA. Do caveolins regulate cells by actions outside of caveolae? Trends in cell biology. 2007;17:51–57. doi: 10.1016/j.tcb.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Head BP, Patel HH, Roth DM, Murray F, Swaney JS, Niesman IR, Farquhar MG, Insel PA. Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. The Journal of biological chemistry. 2006;281:26391–26399. doi: 10.1074/jbc.M602577200. [DOI] [PubMed] [Google Scholar]
- Head BP, Peart JN, Panneerselvam M, Yokoyama T, Pearn ML, Niesman IR, Bonds JA, Schilling JM, Miyanohara A, Headrick J, Ali SS, Roth DM, Patel PM, Patel HH. Loss of caveolin-1 accelerates neurodegeneration and aging. PloS one. 2010;5:e15697. doi: 10.1371/journal.pone.0015697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikezu T, Ueda H, Trapp BD, Nishiyama K, Sha JF, Volonte D, Galbiati F, Byrd AL, Bassell G, Serizawa H, Lane WS, Lisanti MP, Okamoto T. Affinity-purification and characterization of caveolins from the brain: differential expression of caveolin-1, -2, and -3 in brain endothelial and astroglial cell types. Brain research. 1998;804:177–192. doi: 10.1016/s0006-8993(98)00498-3. [DOI] [PubMed] [Google Scholar]
- Jasmin JF, Malhotra S, Singh Dhallu M, Mercier I, Rosenbaum DM, Lisanti MP. Caveolin-1 deficiency increases cerebral ischemic injury. Circulation research. 2007;100:721–729. doi: 10.1161/01.RES.0000260180.42709.29. [DOI] [PubMed] [Google Scholar]
- Kiss AL, Turi A, Muller N, Kantor O, Botos E. Caveolae and caveolin isoforms in rat peritoneal macrophages. Micron. 2002;33:75–93. doi: 10.1016/s0968-4328(00)00100-1. [DOI] [PubMed] [Google Scholar]
- Kwan W, Trager U, Davalos D, Chou A, Bouchard J, Andre R, Miller A, Weiss A, Giorgini F, Cheah C, Moller T, Stella N, Akassoglou K, Tabrizi SJ, Muchowski PJ. Mutant huntingtin impairs immune cell migration in Huntington disease. The Journal of clinical investigation. 2012;122:4737–4747. doi: 10.1172/JCI64484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson KC, Lipko M, Dabrowski M, Draper MP. Gng12 is a novel negative regulator of LPS-induced inflammation in the microglial cell line BV-2. Inflammation research : official journal of the European Histamine Research Society … [et al.] 2010;59:15–22. doi: 10.1007/s00011-009-0062-2. [DOI] [PubMed] [Google Scholar]
- Lee JK, McCoy MK, Harms AS, Ruhn KA, Gold SJ, Tansey MG. Regulator of G-protein signaling 10 promotes dopaminergic neuron survival via regulation of the microglial inflammatory response. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:8517–8528. doi: 10.1523/JNEUROSCI.1806-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei MG, Morrison DC. Differential expression of caveolin-1 in lipopolysaccharide-activated murine macrophages. Infection and immunity. 2000;68:5084–5089. doi: 10.1128/iai.68.9.5084-5089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luoma JI, Boulware MI, Mermelstein PG. Caveolin proteins and estrogen signaling in the brain. Molecular and cellular endocrinology. 2008;290:8–13. doi: 10.1016/j.mce.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marella M, Lehmann S, Grassi J, Chabry J. Filipin prevents pathological prion protein accumulation by reducing endocytosis and inducing cellular PrP release. The Journal of biological chemistry. 2002;277:25457–25464. doi: 10.1074/jbc.M203248200. [DOI] [PubMed] [Google Scholar]
- Mirza MK, Yuan J, Gao XP, Garrean S, Brovkovych V, Malik AB, Tiruppathi C, Zhao YY. Caveolin-1 deficiency dampens Toll-like receptor 4 signaling through eNOS activation. The American journal of pathology. 2010;176:2344–2351. doi: 10.2353/ajpath.2010.091088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monif M, Burnstock G, Williams DA. Microglia: proliferation and activation driven by the P2X7 receptor. The international journal of biochemistry & cell biology. 2010;42:1753–1756. doi: 10.1016/j.biocel.2010.06.021. [DOI] [PubMed] [Google Scholar]
- Moon Y, Kim JY, Choi SY, Kim K, Kim H, Sun W. Induction of ezrin-radixin-moesin molecules after cryogenic traumatic brain injury of the mouse cortex. Neuroreport. 2011;22:304–308. doi: 10.1097/WNR.0b013e3283460265. [DOI] [PubMed] [Google Scholar]
- Mundy DI, Machleidt T, Ying YS, Anderson RG, Bloom GS. Dual control of caveolar membrane traffic by microtubules and the actin cytoskeleton. Journal of cell science. 2002;115:4327–4339. doi: 10.1242/jcs.00117. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Nolan YM, Sullivan AM, Toulouse A. Parkinson’s disease in the nuclear age of neuroinflammation. Trends in molecular medicine. 2013;19:187–196. doi: 10.1016/j.molmed.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Panneerselvam M, Patel HH, Roth DM. Caveolins and heart diseases. Advances in experimental medicine and biology. 2012;729:145–156. doi: 10.1007/978-1-4614-1222-9_10. [DOI] [PubMed] [Google Scholar]
- Park JY, Kim KS, Lee SB, Ryu JS, Chung KC, Choo YK, Jou I, Kim J, Park SM. On the mechanism of internalization of alpha-synuclein into microglia: roles of ganglioside GM1 and lipid raft. Journal of neurochemistry. 2009;110:400–411. doi: 10.1111/j.1471-4159.2009.06150.x. [DOI] [PubMed] [Google Scholar]
- Patel HH, Murray F, Insel PA. G-protein-coupled receptor-signaling components in membrane raft and caveolae microdomains. Handbook of experimental pharmacology. 2008:167–184. doi: 10.1007/978-3-540-72843-6_7. [DOI] [PubMed] [Google Scholar]
- Pelkmans L, Zerial M. Kinase-regulated quantal assemblies and kiss-and-run recycling of caveolae. Nature. 2005;436:128–133. doi: 10.1038/nature03866. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Beckmann J, Magenau A, Boneberg EM, Gaus K, Viola A, Giebel B, Illges H. Flotillins are involved in the polarization of primitive and mature hematopoietic cells. PloS one. 2009;4:e8290. doi: 10.1371/journal.pone.0008290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamaria A, Castellanos E, Gomez V, Benedit P, Renau-Piqueras J, Morote J, Reventos J, Thomson TM, Paciucci R. PTOV1 enables the nuclear translocation and mitogenic activity of flotillin-1, a major protein of lipid rafts. Molecular and cellular biology. 2005;25:1900–1911. doi: 10.1128/MCB.25.5.1900-1911.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shajahan AN, Wang A, Decker M, Minshall RD, Liu MC, Clarke R. Caveolin-1 tyrosine phosphorylation enhances paclitaxel-mediated cytotoxicity. The Journal of biological chemistry. 2007;282:5934–5943. doi: 10.1074/jbc.M608857200. [DOI] [PubMed] [Google Scholar]
- Shin T, Kim H, Jin JK, Moon C, Ahn M, Tanuma N, Matsumoto Y. Expression of caveolin-1, -2, and -3 in the spinal cords of Lewis rats with experimental autoimmune encephalomyelitis. Journal of neuroimmunology. 2005;165:11–20. doi: 10.1016/j.jneuroim.2005.03.019. [DOI] [PubMed] [Google Scholar]
- Shultz SR, Bao F, Weaver LC, Cain DP, Brown A. Treatment with an anti-CD11d integrin antibody reduces neuroinflammation and improves outcome in a rat model of repeated concussion. Journal of neuroinflammation. 2013;10:26. doi: 10.1186/1742-2094-10-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva WI, Maldonado HM, Velazquez G, Garcia JO, Gonzalez FA. Caveolins in glial cell model systems: from detection to significance. Journal of neurochemistry. 2007;103(Suppl 1):101–112. doi: 10.1111/j.1471-4159.2007.04712.x. [DOI] [PubMed] [Google Scholar]
- Stern CM, Mermelstein PG. Caveolin regulation of neuronal intracellular signaling. Cellular and molecular life sciences : CMLS. 2010;67:3785–3795. doi: 10.1007/s00018-010-0447-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta neuropathologica. 2009;118:475–485. doi: 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou M, Miyake K, Golenbock DT, Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. Journal of cell science. 2002;115:2603–2611. doi: 10.1242/jcs.115.12.2603. [DOI] [PubMed] [Google Scholar]
- Tsai TH, Chen SF, Huang TY, Tzeng CF, Chiang AS, Kou YR, Lee TS, Shyue SK. Impaired Cd14 and Cd36 expression, bacterial clearance, and Toll-like receptor 4-Myd88 signaling in caveolin-1-deleted macrophages and mice. Shock. 2011;35:92–99. doi: 10.1097/SHK.0b013e3181ea45ca. [DOI] [PubMed] [Google Scholar]
- Uehara K, Miyoshi M. Localization of caveolin-3 in the sinus endothelial cells of the rat spleen. Cell and tissue research. 2002;307:329–336. doi: 10.1007/s00441-002-0516-0. [DOI] [PubMed] [Google Scholar]
- van Rossum D, Hanisch UK. Microglia. Metabolic brain disease. 2004;19:393–411. doi: 10.1023/b:mebr.0000043984.73063.d8. [DOI] [PubMed] [Google Scholar]
- Volonte D, Galbiati F, Li S, Nishiyama K, Okamoto T, Lisanti MP. Flotillins/cavatellins are differentially expressed in cells and tissues and form a hetero-oligomeric complex with caveolins in vivo. Characterization and epitope-mapping of a novel flotillin-1 monoclonal antibody probe. The Journal of biological chemistry. 1999;274:12702–12709. doi: 10.1074/jbc.274.18.12702. [DOI] [PubMed] [Google Scholar]
- Wang XM, Kim HP, Nakahira K, Ryter SW, Choi AM. The heme oxygenase-1/carbon monoxide pathway suppresses TLR4 signaling by regulating the interaction of TLR4 with caveolin-1. J Immunol. 2009;182:3809–3818. doi: 10.4049/jimmunol.0712437. [DOI] [PubMed] [Google Scholar]
- Watson MB, Costello DA, Carney DG, McQuillan K, Lynch MA. SIGIRR modulates the inflammatory response in the brain. Brain, behavior, and immunity. 2010;24:985–995. doi: 10.1016/j.bbi.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Williams TM, Lisanti MP. The caveolin proteins. Genome biology. 2004;5:214. doi: 10.1186/gb-2004-5-3-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.