

Abstract
Hereditary gingival fibromatosis (HGF) is a rare, autosomal dominant form of gingival overgrowth. Affected individuals have a benign, slowly progressive, nonhemorrhagic, fibrous enlargement of the oral masticatory mucosa. Genetic loci for autosomal dominant forms of HGF have been localized to chromosome 2p21-p22 (HGF1) and chromosome 5q13-q22 (HGF2). To identify the gene responsible for HGF1, we extended genetic linkage studies to refine the chromosome 2p21-p22 candidate interval to ∼2.3 Mb. Development of an integrated physical and genetic map of the interval identified 16 genes. Sequencing of these genes, in affected and unaffected HGF1 family members, identified a mutation in the Son of sevenless–1 (SOS1) gene in affected individuals. In this report, we describe the genomic structure of the SOS1 gene and present evidence that insertion of a cytosine between nucleotides 126,142 and 126,143 in codon 1083 of the SOS1 gene is responsible for HGF1. This insertion mutation, which segregates in a dominant manner over four generations, introduces a frameshift and creates a premature stop codon, abolishing four functionally important proline-rich SH3 binding domains normally present in the carboxyl-terminal region of the SOS1 protein. The resultant protein chimera contains the wild-type SOS1 protein for the N-terminal amino acids 1–1083 fused to a novel 22–amino acid carboxyl terminus. Similar SOS1 deletion constructs are functional in animal models, and a transgenic mouse construct with a comparable SOS1 chimera produces a phenotype with skin hypertrophy. Clarification of the functional role of this SOS1 mutant has implications for understanding other forms of gingival fibromatosis and corrective gingival-tissue management.
Introduction
Hereditary gingival fibromatosis (HGF) is a genetically heterogeneous overgrowth condition characterized by a benign, slowly progressive, nonhemorrhagic, fibrous enlargement of maxillary and mandibular keratinized gingiva (MIM 135300). Keratinized oral gingiva constitutes a developmentally unique and functionally important component of the masticatory mucosa surrounding the teeth. Insufficient gingiva can predispose to chronic infection, pathologic recession, and oral morbidity (Smith 1997). Chronic infection of the gingival-tooth interface can result in periodontitis, an important oral pathology having potential systemic consequences (Scannapieco 1998). Too much oral gingiva is associated with improper tooth eruption resulting in malocclusion. The severity of gingival overgrowth in HGF varies among affected individuals and, in severe cases, can prevent tooth eruption, resulting in functional and aesthetic problems (Raeste et al. 1978). Treatment by surgical resection is, in some cases, followed by a regrowth of the gingival tissues. HGF may be transmitted as a Mendelian trait, and both autosomal dominant and autosomal recessive transmission have been reported (Jorgenson and Cocker 1974). Linkage studies have localized loci for isolated, nonsyndromic autosomal dominant forms of gingival fibromatosis to chromosome 2p21-p22 (Hart et al. 1998; Xiao et al. 2000) and to chromosome 5q13-q22 (Xiao et al. 2001). In addition to nonsyndromic forms, gingival fibromatosis has been associated with a variety of syndromic manifestations. Chromosomal anomalies reported for syndromic forms of gingival fibromatosis include duplications, deletions, and/or other anomalies of chromosomes 2p13-16 (Fryns 1996; Shashi et al. 1999), 4q (MIM 252500), 8 (MIM 266270), 14q (Rivera et al. 1992), 19p (MIM 246200), 19q (MIM 248500), and Xq (Macias-Flores et al. 1984). Gingival fibromatosis is also an unwanted side effect of several pharmacological agents, including dilantin, cyclosporin, and several calcium-channel blockers (reviewed in Hassell and Hefti 1991). This drug-induced overgrowth response varies between individuals and may reflect an underlying, genetically determined susceptibility (Seymour et al. 1996).
Two groups have previously linked autosomal dominant HGF to an overlapping interval of chromosome 2p (Hart et al. 1998; Shashi et al. 1999; Xiao et al. 2000). As part of our ongoing efforts to identify the genetic basis for gingival fibromatosis, we developed a physical map spanning the HGF1 candidate interval, permitting us to integrate and localize 32 genetic markers and 33 genes to the interval (M.C.G. and T.C.H., unpublished data). In the present study, we describe the refinement of the HGF1 locus to a 2.3-Mb interval by extension of the pedigree and genotyping additional markers in the region. Sequence analysis of 6 known candidate genes, as well as 10 additional, previously uncharacterized genes within the refined interval, permitted us to identify a mutation, in the Son of sevenless–1 (SOS1) gene, that segregated with the HGF phenotype. In this report, we describe the genomic organization of the SOS1 gene and present evidence that a single-nucleotide–insertion mutation in codon 1083 of the SOS1 gene is the cause of HGF1 in humans.
Subjects and Methods
Pedigree and Diagnosis
A large, multigenerational Brazilian family segregating HGF as a highly penetrant autosomal dominant trait was identified by proband ascertainment. Linkage studies of part of this family led to the original linkage of HGF1 to chromosome 2p21 (Hart et al. 1998). Individuals examined for the current study are shown in figure 1figure 1. All participating family members provided informed consent to protocols approved by institutional review boards at the University of Taubate and the University of Pittsburgh. All individuals received oral and dental exams and, on the basis of the presence or absence of gingival enlargement, were classified as affected or unaffected, as described elsewhere (Hart et al. 1998). All individuals were also asked about exposure to prescription and nonprescription medications and, specifically, about exposure to medications associated with gingival overgrowth, including phenytoin, cyclosporin, and calcium-channel blockers.
Figure 1.

Pedigree of Brazilian family with hereditary gingival fibromatosis. Affected individuals are indicated by blackened symbols. Circles denote females, and squares denote males; a slash through a symbol denotes a deceased individual. Clinically unaffected individuals are indicated by un unblackened symbol. A clinical diagnosis of unknown or uncertain diagnosis is denoted by a question mark (?) within the symbol. The proband is indicated by the arrow (↗). The flattened oval symbol indicates individuals who participated in this study.
Figure 1.

Pedigree of Brazilian family with hereditary gingival fibromatosis. Affected individuals are indicated by blackened symbols. Circles denote females, and squares denote males; a slash through a symbol denotes a deceased individual. Clinically unaffected individuals are indicated by an unblackened symbol. A clinical diagnosis of unknown (or an uncertain diagnosis) is denoted by a question mark (?) within the symbol. The proband is indicated by the arrow (↗). The flattened oval symbol indicates individuals who participated in this study.

DNA-Marker Analysis
Genomic DNA was extracted from peripheral blood by standard methods using the QIAamp blood kit (Qiagen). Available family members were genotyped for STRP-type genetic markers spanning the candidate interval. In addition to 11 previously reported STRP loci, 21 novel STRP loci were developed from a 6-Mb physical map of the interval (M.C.G. and T.C.H., unpublished data). These marker loci were PCR amplified by use of fluorescence-labeled primers, permitting genotyping by conventional methods (Zhang et al. 2001). PCR products were detected by an ABI377 fluorescent sequencer and were analyzed by GENESCAN 2.1 (Applied Biosystems). Alleles were determined, and genotype data were entered into the pedigree file of the LINKAGE software package (Lathrop and Lalouel 1984).
Parametric Linkage Calculations: LOD Scores
Sublocalization of the candidate interval for HGF1 on chromosome 2p21-p22 was achieved by means of genetic linkage studies. We calculated two-point LOD scores in the extended kindred, using the Elston-Stewart algorithm (Elston and Stewart 1971), employing the LINKAGE program (Lathrop and Lalouel 1984) with updates to speed calculations (VITESSE and FASTLINK; see Cottingham et al. 1993; Terwilliger and Ott 1994; O'Connell and Weeks 1995). The LOD-score analyses assumed a genetic model for HGF, with autosomal dominant transmission, disease-allele frequency of 0.0001, 0%–1% phenocopy rates, and 95% penetrance.
Multipoint LOD-Score Calculations
The Markov chain–Monte Carlo (MCMC) algorithm, as implemented in the computer program SimWalk2 (Sobel and Lange 1996), was used for the multipoint LOD-score calculations.
Analysis of Genes within the Candidate Interval, Identification of the SOS1 Genomic Region, and SOS1 Exon Sequencing
Using bioinformatic approaches, we identified, assembled, and aligned 64 BACs, to develop a 6-Mb integrated physical and genetic map spanning the HGF1 candidate-gene region on chromosome 2p21-p22 (Zhang et al., in press; M.C.G. and T.C.H., unpublished data). By means of linkage analyses, we refined the HGF1 candidate interval to a 2.3-Mb interval. We localized six known genes to this region. Additionally, using an integrated bioinformatic and bench lab approach, we identified 10 previously uncharacterized genetic loci within the interval. All exons and intron-exon boundaries of these 16 genes were analyzed by sequence analysis. For these analyses, genomic DNA from three HGF1 affected individuals and from two unaffected (control) individuals were sequenced. Details for sequencing of all genes, including their GenBank accession numbers, will be reported separately (Zhang et al., in press; M.C.G. and T.C.H., unpublished data). The genomic structure of the SOS1 gene was determined bioinformatically and was confirmed by sequence analysis. To determine the genomic organization for the entire SOS1 gene, all available SOS1 mRNA and EST data were aligned to identify any possible splice variants. Exon 1 was identified from the dbEST data (NCBI dBEST Web site), since all available mRNA information began at the ATG start site in exon 2. Primers were designed with Oligo 4.02 (National Biosciences) to amplify each exon, including exon-intron boundaries for sequence analysis (table 1).
Table 1.
Primer Sets for Exonic Amplification of Human SOS1 Gene
|
Primer (5′→3′) |
|||||
| Exona | Forward | Reverse | Size(bp) | PCRConditionb | GenBankAccessionNumber |
| 01–02 | CTTGCGTTTCGGAGTCCCAACTAC | AAGGGCAAAGCATTCACCTCTCAG | 806 | C | AF441466 |
| 03 | GGGTTGAGAACTCCTGACTTCTAG | TTTCCCTGTTCACTGACATTACAA | 659 | B | AF441467 |
| 04 | TGTGATATTCCCCCTAGATAATAG | CCCTTCTCACCACATAAATCTCTG | 515 | B | AF441468 |
| 05 | CATTTGCTCCCTTCCCCAAACTTG | GATCTTCCCCAACACATAATACAA | 464 | A | AF441469 |
| 06 | CCAAGTCAGGGAAATTAAAGACAG | TGGAGTACATGGAGAATTCTGTGA | 761 | A | AF441470 |
| 07 | AATACAGCCTCACTGAATTAATGT | CCTTGGCGGTATCTGTCTTTCTAT | 409 | A | AF441471 |
| 08-09 | TAGTCGTGCCCCATAATTAAATCT | TGTGCAGGGTACTCACACAATAAT | 462 | A | AF441472 |
| 10 | ATTACTGCCTGTCACAAGATATAA | TCTAAAAGACCAGGCTTGTCACTA | 501 | A | AF441473 |
| 11 | AGTAGCCAAGATAGAAATAATCAT | TCAAGCATCCTTTCCAGTGTACTC | 1,230 | A | AF441474 |
| 11S | AGGAGCCAAACATGAGAGACACAT | AGATCCCAATAATGATGATTTACT | |||
| 12 | GTGTGCAGAGGGCAAGAAATAAGT | AGGGTAATTCCAGAAAAACTGACT | 731 | A | AF441475 |
| 13-14 | AGTCCCGTAACTTTATAGTCACAT | TTACTGAGCCCCAATGACATCAAT | 911 | A | AF441476 |
| 13-14S | GCGGTAAGCATTAAATAAATGAAG | ACCGCAGTTGCACAGGCTGTATAT | |||
| 15 | GAACAATGGGATGGACAGTCTCTA | TGCCTGGCCTTATTACTAGACACT | 656 | A | AF441477 |
| 16 | TAAGGAAATATGCATAATTACACT | CTGCACTCCAGCCTATGTGACAGA | 418 | A | AF441478 |
| 17 | CTATCAGTCACCCTGAATGTGTCT | GCTTAGGCTGGGACCTGTGAATAC | 524 | A | AF441479 |
| 18 | GTTGTATTTGGGCGTTTCTGTTAG | AAGGCACAGCAGGCACACTATGAT | 586 | A | AF441480 |
| 19–20 | GTGACGAAGTTATTCTTAAAGTTC | TTTGGCTGGGCACTGACAAGTAAC | 1,400 | A | AF441481 |
| 19–20S | GCAACTGAGATGGTACAGTGTAAT | CATCTTTGATATCAGGATTTACTC | |||
| 19–20S | ATCAGCCTTACTGTTTACGAGTAG | ||||
| 21 | AGGGCTTTAGCAAAATAGAATGTT | ACTTGCAGATTTTAAGACTGATCT | 776 | A | AF441482 |
| 22 | TGCTTGAATTGGGTTTCTACAT | AATGCTGCCAGACCCAAGAAGAGT | 445 | A | AF441483 |
| 23 | CACTGTCTCCCAGACATCTGAGAA | TTAGGTTTACAAGGGTCTTGAATG | 654 | A | AF441484 |
| 24 | CCAGGAAGTTGAGGCTACAGTGAG | TCGGTCTTTCCATATTCTAAACTG | 944 | D | AF441485 |
| 24S | CGGCCCAGCAATGGAATGAAGGTC | ||||
S denotes primers used in sequencing reactions.
The standard PCR amplification for each exon contains: Taq (0.025 U/μl), 1× PCR buffer, 25 nM each dNTP, and Mg as follows: A = 4.0 mM Mg, 95-2′ + 94-30″/55-30″/72-2′ 35X + 72C-10′, no enhancer. B = 3.0 mM Mg, 95-2′ + 94-30″/55-30″/72-2′ 35X + 72C-10′, no enhancer. C = 1.5 mM Mg, 95-2′ + 94-30″/55-30″/72-2′ 35X + 72C-10′, 20% enhancer. D = 1.5 mM Mg, 95-2′ + 94-30″/55-30″/72-2′ 35X + 72C-10′, 5% enhancer.
PCR amplification of the SOS1 gene was performed as indicated in table 1, with or without Enhancer (Life Technology). Amplified DNA was purified with the QIAquick PCR Purification Kit (Qiagen) and was sequenced using the BigDyeTerminator Cycle Sequencing Kit and ABI 3700 or 310 DNA analyzer (Applied Biosystems). Sequencing was performed by use of the amplification primers and appropriate sequencing primers, as indicated in table 1. Sequence analysis was performed with Sequencher 4.1 software (GeneCodes). Genomic DNA from three affected and two unaffected family members was evaluated for the initial sequencing. Any polymorphism detected was evaluated for possible linkage to HGF1 in a larger segment of the family.
RNA Verification of Wild-Type and Mutant SOS1
Peripheral venous blood was obtained by standard venipuncture from affected individuals, and white blood cells were isolated by Ficoll gradient separation, according to the manufacturer’s instructions (Pharmacia). Gingival tissues were collected from two affected individuals during routine surgery for removal of the overgrown gingival tissue. Total RNA was purified from both white blood cells and homogenized gingival tissues by use of Trizol solution (Life Technology). Approximately 1 μg of total RNA from each tissue was reverse transcribed using the Reverse Transcription System (Promega), with both oligo dT and random primers. The cDNA products were used as templates for a partial amplification of SOS1 mRNA exon 21–24 (808 bp) and exon 19-22 (501 bp) with primer sets U3174/L3958 (5′-GGAGCCAAGGAAAATTAGTTATAG-3′/5′-CCAACAGTGGTGGTCCATCTCTGT-3′) and U2831/L3310 (5′-ACCCTGAGGTCCTAAAAAGACATG-3′/5′-GCTCGAATGATCGGAATCAAATAC-3′), respectively. The PCR amplification was performed with Taq DNA polymerase, at an annealing temperature of 60°C. The amplified products were purified and sequenced as described above. The DNA sequences were aligned with the human SOS1 cDNA in GenBank (accession number 306777). To further confirm the mutant allele of the SOS1 cDNA, the amplified PCR products were subsequently cloned directly into the pGEM-Teasy vector (Promega), and their sequences were confirmed by automated DNA sequencing.
Screening for the SOS1 Exon 21 Insertion Mutation
Exon 21 of the SOS1 gene was sequenced in 78 available family members. To determine if the C insertion in codon 1083 represented a SOS1 polymorphism present in the general population, 208 chromosomes from clinically unaffected white and Chinese controls (104 chromosomes from each population) were also analyzed.
Western Blot Analysis
Total protein was extracted from gingival tissue by homogenization in lysis buffer (1% SDS, 1.0 mM sodium ortho-vanadate, 10 mM Tris, pH 7.4). Cellular lysates were electrophoresed on 6% SDS-polyacrylamide gels. Following electrophoresis, separated proteins were electroblotted onto polyvinylidine difluoride membrane. The membrane was blocked in blocking buffer (5% dry milk in phosphate-buffered saline) for 1 h at room temperature and then was incubated with 1 μg/ml primary antibody in blocking buffer for 3 h. After rinsing, the membrane was incubated with peroxidase-conjugated secondary antibody for 1 h followed by chemiluminescence detection (NEN). Primary antibodies were rabbit anti-mouse Sos1, a polyclonal generated by immunization with a synthetic peptide corresponding to amino acids 1241–1260 in the C-terminus of the mouse Sos1 and the monoclonal anti-mouse Sos1, directed against the N-terminus of mouse Sos1 (BD Biosciences). The primary antibodies are known to cross-react with human SOS1 (170 kDa). The N-terminal antibody also cross-reacts with human SOS2 (150 kDa). The secondary antibodies were goat anti-rabbit and goat anti-mouse for the polyclonal and monoclonal primaries, respectively.
Results
Clinical Findings
An extended, multigenerational kindred segregating HGF was studied. Ninety-two family members were clinically examined, and blood was available from 82 family members for DNA analysis. Photographs, medical records, and family histories permitted a diagnosis in 14 deceased individuals. Thirty-eight individuals were diagnosed as affected, and 40 family members were diagnosed as unaffected (fig. 1fig. 1). A diagnosis could not be determined in five individuals. Two individuals, V-2 and V-34, reported a history of taking dilantin. Because of the association with gingival overgrowth in 15%–30% of individuals taking dilantin, both individuals were given a diagnosis of unknown. A diagnosis could not be determined in three additional individuals: two were children under the age of 6 years (VI-2 and VI-4), and one was an edentulous adult (III-23). No family members reported a history of hearing loss or epilepsy, and hypertrichosis was not observed in any individuals. All family members were otherwise healthy and of normal intelligence. HGF was present in all generations, male-to-male transmission was observed, and there were approximately equal proportions of affected males and females, findings consistent with autosomal dominant transmission of the HGF phenotype. Clinical findings are consistent with a diagnosis of isolated, nonsyndromic hereditary gingival fibromatosis.
Development of a Physical Map of the HGF1 Candidate Interval
A 6-Mb integrated physical and genetic map spanning the HGF1 candidate interval was developed (M.C.G. and T.C.H., unpublished data). This map permitted identification and localization of 32 STRP-type genetic markers and 33 genes to the interval. The integration of polymorphic genetic markers and genes in the interval permitted us to systematically refine the region segregating with the HGF1 phenotype. A schematic of the region containing the smallest genetic interval segregating with the HGF1 phenotype in the Brazilian kindred studied is shown in figure 2. The refined interval is 2.3 Mb, flanked by the STRP markers 92F3-01 and D2S2238. Also indicated in figure 2 is the centromeric portion of the HGF candidate interval reported by Xiao et al. (2000), which extends in a telomeric direction from D2S2163. This refined candidate interval contained 6 known genes and 10 novel genes (table 2).
Figure 2.
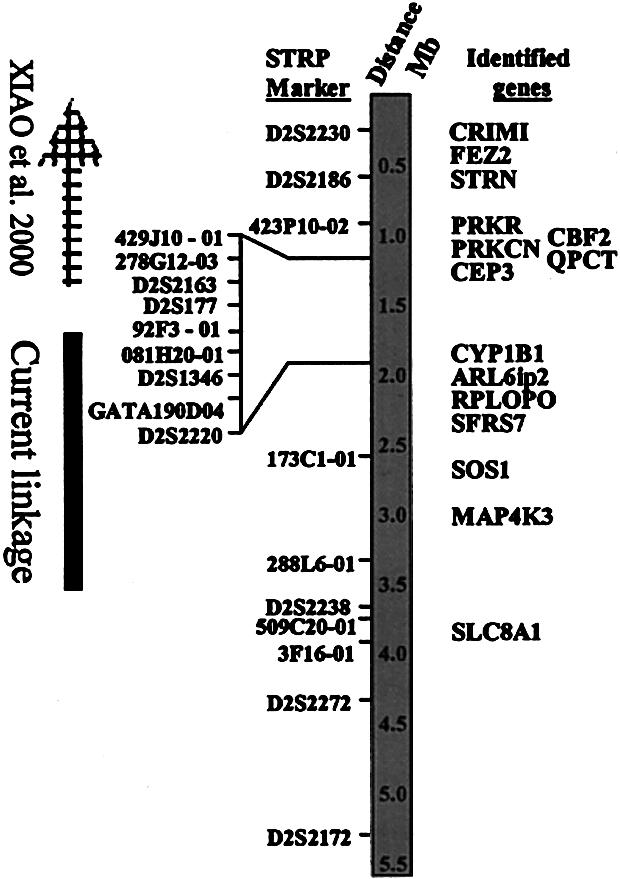
An indication of the relative positioning of STRP markers used in our analysis and genes with known functions identified in the region. All genes from CEP3 to SLC8A1 were sequenced in our search. CRIM1 = cysteine-rich motor neuron 1 (locus 51232); FEZ2 = fasciculation and elongation protein zeta 2 (locus 9637); STRN = striatin (locus 6801); PRKR = protein kinase, interferon-inducible double-stranded RNA dependent (locus 5610); CBF2-CCAAT = box binding transcription factor (locus 10153); PRKCN = protein kinase C, nu (locus 23683); QPCT = glutaminyl-peptide cyclotransferase (locus 25797); CEP3 = cdc42 effector protein 3 (locus 10602); CYP1B1 = cytochrome P450, subfamily 1, polypeptide 1 (locus 1545); ARL6ip2 = ADP-ribosylation-like factor 6 interacting protein 2 (locus 64225); RPLPO = ribosomal protein, acidic, P0 (locus 6175); SFRS7 = splicing factor, arginine/serine-rich 7 (locus 6432); SOS1 = son of sevenless (D. melanogaster) homolog 1(locus 6654); MAP4K3 = mitogen-activated protein kinase kinase kinase kinase 3 (locus 8491); and SLC8A1 = solute carrier family 8 (sodium/calcium exchanger), member 1 (locus 6546). All loci can be accessed through LocusLink. The total region covered corresponds to 5,500,000 bases.
Table 2.
Genes Identified within the Refined 2.3-Mb HGF1 Candidate Interval on 2p21-22[Note]
| ID | Name | Locus ID | mRNAAccessionNumber | GenBankAccessionNumber |
| CEP3 | Cdc42 effector protein 3 | 10602 | NM_006449 | |
| BLOCK 18 | Unknown | AK057516 | AF450120-AF450129 | |
| CYP1B1 | Cytochrome P450, subfamily I (dioxin-inducible), polypeptide 1 | 1545 | NM_000104 | AF450130-AF450132 |
| ARL6ip2 | ADP-ribosylation-like factor 6 interacting protein 2 | 64225 | NM_022374 | AF449176-AF449187 |
| BLOCK 22 | Unknown | AF447702-AF447707 | ||
| BLOCK 23 | 97% homology to ribosomal phosphoprotein, acidic, P0 | AY064377 | ||
| BLOCK 24 | Unknown, similar to human heterogeneous nuclear ribonuclear protein L | 92906 | AK000155 | |
| BLOCK 25 | Unknown, 88% homology with aldose-1-epimerase from Sus scrofa | BC014916 | AY064378-AY064385 | |
| SFRS7 | Splicing factor, arginine/serine-rich 7 | 6432 | NM_006276 | AY062913-AY062919 |
| BLOCK 26 | Unknown | |||
| BLOCK 27 | Unknown | AF449188-AF449191 | ||
| BLOCK 28 | Unknown | AF454826-AF454826 | ||
| SOS1 | Son of sevenless (D. melanogaster) homolog 1 | 6654 | NM_005633 | AF441466-AF441485 |
| MAP4K3 | Mitogen-activated protein kinase kinase kinase kinase 3 | 8491 | NM_003618 | AF445385-AF445413 |
| BLOCK 35 | Unknown, SAM-dependent methyltransferase | 80745 | AF380566-AF380576 | |
| SLC8A1 | Solute carrier family 8 (sodium/calcium exchanger), member 1 | 6546 | NM_021097 |
Note.— Genes were identified as indicated in the “Subjects and Methods” section. No gene without an EST and/or cDNA sequence was considered.
Linkage Analyses
Results of linkage analysis for 26 STRP markers spanning the candidate interval are given in table 3 (two-point analyses) and figure 3 (multipoint analysis). On the basis of haplotype analysis, we were able to refine the candidate interval to ∼2.3 Mb, within a genetic interval flanked by the novel marker loci 92F3-01 and D2S2238. The maximum two-point LOD score was for the marker D2S2220, Zmax=14.26 at recombination fraction (θ) 0.00. The marker locus 173C1-01 is located within intron 7 of the SOS1 gene (Zmax=12.08 at θ=0.00). The maximum multipoint location score was Zmax=20.18 for D2S2220. Using the criteria of ZMAX –1.0 LOD, to determine the 95% CI, we placed the HGF1 locus within an ∼1.6-Mb interval flanked by 92F3-01 and 288L6-01 (Conneally et al. 1985).
Table 3.
Two-Point Linkage Analysis: LOD Scores of 26 Markers Spanning 7.17 cM on Chromosome 2
| LOD Score at θ = |
||||||||
| Marker | Distancea(kb) | .0 | .01 | .05 | .10 | .20 | .30 | .40 |
| D2S2230 | 0 | 1.82 | 5.87 | 6.12 | 5.76 | 4.54 | 3.09 | 1.56 |
| 501007 | 140 | 3.19 | 3.14 | 2.92 | 2.64 | 2.02 | 1.35 | .64 |
| D2S2186 | 430 | −.07 | 3.23 | 3.78 | 3.75 | 3.14 | 2.20 | 1.10 |
| 531C11-02 | 680 | 6.51 | 9.40 | 8.87 | 8.08 | 6.28 | 4.27 | 2.10 |
| 423P10-02 | 780 | 10.00 | 9.93 | 9.50 | 8.78 | 7.00 | 4.90 | 2.55 |
| 429J10-01 | 1,050 | 1.28 | 5.76 | 5.96 | 5.60 | 4.42 | 2.99 | 1.47 |
| 278G12-03 | 1,130 | 7.85 | 8.11 | 8.06 | 7.47 | 5.78 | 3.71 | 1.52 |
| 278G12-02 | 1,140 | 2.23 | 2.20 | 2.08 | 1.89 | 1.40 | .85 | .34 |
| D2S2163 | 1,220 | .67 | 6.53 | 7.30 | 6.99 | 5.54 | 3.64 | 1.60 |
| D2S177 | 1,330 | 11.29 | 12.97 | 12.70 | 11.70 | 9.18 | 6.27 | 3.07 |
| 92F3-01 | 1,400 | −6.78 | −3.12 | −.53 | .39 | .94 | .88 | .54 |
| 92F3-02 | 1,520 | 7.54 | 7.42 | 6.94 | 6.30 | 4.86 | 3.24 | 1.44 |
| 081H20-01 | 1,565 | 7.63 | 7.50 | 6.95 | 6.26 | 4.80 | 3.19 | 1.43 |
| D2S1346 | 1,570 | 14.25 | 14.00 | 12.96 | 11.62 | 8.82 | 5.83 | 2.72 |
| GATA190D04 | 1,730 | 6.93 | 6.81 | 6.30 | 5.63 | 4.22 | 2.72 | 1.20 |
| D2S2220 | 1,800 | 14.26 | 14.02 | 13.04 | 11.75 | 9.01 | 6.05 | 2.92 |
| 173C1-01 | 2,570 | 12.08 | 11.91 | 11.19 | 10.21 | 8.03 | 5.61 | 2.92 |
| 288L6-01 | 3,210 | 7.27 | 7.18 | 6.78 | 6.21 | 4.90 | 3.40 | 1.75 |
| D2S2238 | 3,470 | 11.33 | 11.66 | 11.54 | 10.73 | 8.40 | 5.51 | 2.32 |
| 509C20-01 | 3,580 | 6.96 | 7.06 | 7.10 | 6.81 | 5.67 | 4.08 | 2.13 |
| 3F16-01 | 3,710 | 5.32 | 10.61 | 10.55 | 9.82 | 7.79 | 5.37 | 2.65 |
| D2S2272 | 4,190 | −.16 | 7.85 | 7.83 | 7.21 | 5.54 | 3.60 | 1.56 |
| D2S2172 | 5,270 | −.09 | 7.37 | 7.40 | 6.82 | 5.25 | 3.43 | 1.55 |
| D2S1356 | 5,670 | 6.47 | 10.15 | 10.56 | 10.02 | 8.07 | 5.56 | 2.71 |
| D2S2259 | 6,570 | −2.36 | 6.71 | 7.45 | 7.27 | 6.05 | 4.29 | 2.19 |
| D2S119 | 7,170 | 9.43 | 9.61 | 9.57 | 9.02 | 7.31 | 5.17 | 2.67 |
Marker distances relative to D2S2230, as reported from an integrated physical and genetic map of the candidate interval (M.C.G. and T.C.H., unpublished data). D2S2230 is 56.15 cM from the 2p telomere (Marshfield genetic map; Broman et al. 1998).
Figure 3.

Multipoint LOD analysis of the family in the 7.5-cM interval flanking the refined HGF1 candidate interval. LOD scores are given on the X-axis, and genetic distances are indicated on the Y-axis from the telomeric end of chromosome 2p, on the left. The position of each of the 26 STRP markers tested are indicated by Roman numerals in the figure. The name corresponding to each number is indicated in the key below the figure.
Genomic Structure of the SOS1 Gene
The alignment of available ESTs (batch download from Unigene Hs.326392) and mRNA for SOS1 with genomic sequences from two BACs (RP11-173C1 and R911-603F24) permitted identification and localization of 24 exons of the SOS1 gene. One splice variant involving exon 22 was identified (Rojas et al. 1996). The genomic organization of the SOS1 gene is shown in figure 4. The SOS1 gene spans 136 kb and consists of 24 exons. The intronic sizes range from 30 bp to 53 kb. The open reading frame contains 4,002 nucleotides and encodes 1,333 amino acids (hSOS1 mRNA; GenBank accession number 306777). The translation initiation codon is located at nucleotides 45–47 of exon 2, whereas the open reading frame is terminated at nucleotide 492 in exon 24.
Figure 4.

Structure of the human SOS1 gene. A, Genomic organization of the SOS1 gene. The exons and introns are represented as vertical and horizontal lines, respectively. The sizes of each intron are shown in kb. B, cDNA structure of the SOS1 gene. The location of each exons are shown with the nucleotide numbers (bp). Since we did not determine the exact termini of the SOS1 cDNA, the N- and C-terminals are marked with dashed lines. The translation start and stop codon are labeled as ATG and TGA, respectively. The asterisk (*) marks the single-nucleotide (C) insertion between nucleotides 3248 and 3249. The 5′ and 3′ UTRs are shaded. C, Structure of the wild-type SOS1 protein. The location of each domain is marked by the amino acid numbers, with the initiation Met as amino acid 1. RhoGEF = guanine nucleotide-exchange factor for the Rho-Rac/cdc42-like GTPases region. PH = Pleckstrin homology domain. RasGEFN = guanine nucleotide-exchange factor for Ras-like GTPases; N-terminal motif. RasGEF = guanine nucleotide-exchange factor for Ras-like GTPases. PRGB = proline-rich Grbs-binding domain. In the PRGB domain, the location of the four proline-rich SH3 binding sites and the five phosphorylated serine residues are indicated by P and S, respectively. Codon 1083 (proline), where the single-nucleotide insertion occurs in HGF1, is indicated as P1083. D, Structure of the mutant SOS1 protein identified in patients with HGF. The 22–amino acid missense addition at the C-terminal end are marked with a black-diamond pattern.
Mutational Analysis
A total of 16 genes were sequenced from the 2.3-Mb candidate interval. A number of exonic and intronic polymorphisms were identified but were found not to segregate with the HGF phenotype (data not shown). A complete list of these genes is presented in table 2. Direct DNA sequencing of the SOS1 gene identified 12 intronic SNPs spanning the SOS1 gene (data not shown). None of these SNPs appeared to affect splice-site junctions, and none segregated with the HGF disease phenotype in this extended kindred. One exonic sequence alteration differed from the wild-type SOS1 mRNA structure reported elsewhere. This single-nucleotide (cytosine) insertion (c.3248-3249insC) in exon 21 was identified in all individuals affected with HGF1 (fig. 5). This alteration segregated with the HGF phenotype and was not identified in any unaffected family members nor in any of the control chromosomes tested (104 from white subjects and 104 from Chinese subjects). The insertion of a single C nucleotide into codon 1083 does not change the coded amino acid (proline), but it does introduce a frameshift at codon 1084 of the SOS1 gene. This frameshift mutation is predicted to alter the subsequent 22 amino acids (1084–1105) and to introduce a premature stop codon at codon 1106 (fig. 5). The predicted protein chimera does not contain the proline-rich Grb2 binding domains nor the five MAP kinase phosphorylation sites that are normally present in the wild-type SOS1 protein. The presence of both the normal and mutant SOS1 transcripts was demonstrated in RT-PCR from mRNA isolated from both white blood cells and from gingival tissue of affected individuals, verifying that the mutant SOS1 allele is transcribed.
Figure 5.

Mutation in SOS1, revealed by sequencing of exon 21. The ATG translation initiation codon, encoded by exon 2, is taken as nucleotide 1. A, Sequences of (a) wild type, (b) genomic DNA from an affected individual, and (c) cloned affected cDNA allele amplification from an affected individual. The insertion of a C nucleotide in genomic sequences between nucleotides 126,142 and 126,143 (b) and in cDNA between nucleotides 3248 and 3249 (c) are shown by arrows. p.R1084fsX1105 indicates that this mutation produces a frameshift with Arginine1084 as the first amino acid affected and that the new reading frame is open for 22 amino acids. B, Amino acid sequences of codons 1083–1106 of the wild type and predicted amino acid sequences of the mutant SOS1 proteins. The premature termination codon is indicated by “Stop.”
Western Blot Analysis
Western blot analysis with a mouse monoclonal antibody directed against the N-terminus of the SOS1 protein revealed the expected ∼170 kDa wild-type band in all samples, as well as an ∼150-kDa band that is consistent with a cross-reactive band for the smaller-sized Son of sevenless–2 protein (SOS2) (data not shown). An additional, smaller band of ∼143 kDa, consistent with the truncated SOS1 protein, was present in western blots for HGF gingival-tissue extracts. Western blot analysis with a polyclonal antibody directed against amino acids 1241–1260 of the mouse Sos1 protein revealed a predominant band of 170 kDa in all samples tested, corresponding to the wild-type protein. In addition to the wild-type protein, we consistently observed a doublet band at a high molecular weight (>200 kDa) in gingival tissue from an affected individual. The stoichiometry of the two bands in the doublet differed for the two different antibodies. When the C-terminal SOS1-specific antibody was used, both bands in the doublet were of equal intensity. The intensity of the lower–molecular-weight band in the doublet was significantly enhanced, with a ratio of ∼1:2 (upper:lower bands) when the N-terminus–specific antibody was used. These findings are consistent with the upper–molecular-weight band containing the wild-type SOS1 protein and the lower–molecular-weight band containing both the wild-type and the mutant SOS1 protein.
Discussion
The findings of this study indicate that alteration of the carboxyl-terminal domain of the SOS1 protein is responsible for HGF1 in an extended kindred. Genetic linkage studies localized a gene for HGF1 to a genetic interval containing the SOS1 gene locus. Sequence analysis of genomic DNA and mRNA from individuals affected with HGF1 identified a single-cytosine insertion in exon 21 of the SOS1 gene. On the basis of nomenclature guidelines (Antonarakis 1988), we have designated the insertion between nucleotides 126,142 and 126,143 in the genomic sequence (g.126,142-126,143insC) and between nucleotides 3248 and 3249 in the SOS1 cDNA sequence (c.3248-3249insC). The codon for amino acid 1083 is changed from CCA to CCC. Both CCA and CCC code for proline, but the additional nucleotide produces a frameshift and an early termination of the protein (fig. 5). This mutation yields a chimeric 1,105–amino acid protein that consists of 1,083 SOS1 N-terminal amino acids followed by 22 replaced amino acids and a premature stop codon at codon 1106 (p.K1084fsX1105).
We believe that this mutation is the cause of HGF1 on the basis of several findings. First, there is complete cosegregation between the g.126,142-126,143insC insertion and the HGF phenotype in this kindred. This nucleotide change is not present in 208 control chromosomes from unrelated healthy individuals. Second, SOS1 is expressed in gingival tissues, the site that is clinically affected in HGF. Both the wild-type and mutant transcripts are expressed in gingival tissues of affected individuals. Western blot experiments are consistent with expression of both wild type and mutant SOS1 protein in gingival tissue from individuals affected with HGF1. Third, the highly conserved 1,333–amino acid wild-type human SOS1 protein is constitutively maintained in a down-regulated state. The carboxyl-terminal domain exerts negative allosteric control on the interaction of the SOS1 catalytic domain with Ras (Corbalan-Garcia et al. 1998). The p.K1084fsX1105 mutation deletes ∼20% of the SOS1 protein, including functional domains that maintain SOS1 in a down-regulated state. The truncated SOS1 protein is therefore predicted to have enhanced activity. Studies in several species—including yeast, Drosophila melanogaster, mouse, and human—indicate that truncation of the carboxyl-terminal portion of SOS1 is associated with a gain of function. Finally, a transgene construct with an SOS1 carboxyl-terminal deletion induces skin-tumor development in mice. On the basis of these findings, we hypothesize that the g.126,142-126,143insC mutation produces the truncated protein chimera p.K1084fsX1105 that causes HGF1 through a gain-of-function mechanism.
The guanine nucleotide-exchange factor SOS1 mediates the coupling of receptor tyrosine kinases to Ras activation (Corbalan-Garcia et al. 1996). The SOS1 protein has been studied extensively and the functional domains and crystal structure of the SOS1 protein are known (Soisson et al. 1998; Chen et al. 1999; Wasiak et al. 2001). The human SOS1 protein contains six well-characterized functional domains, two of which are encoded by nucleotides 3′ to codon 1083 (fig. 4). These include four proline-rich SH3-binding sites and five mitogen-activated protein (MAP) kinase phosphorylation sites. Although the wild-type SOS1 protein is constitutively maintained in a down-regulated state, human SOS1 truncation mutants that lack either the amino terminus or the carboxyl-terminal domain display guanine nucleotide-exchange activity that is significantly greater than that in the full-length protein (Corbalan-Garcia et al. 1998).
Similar to the situation in humans (Aronheim et al. 1994), deletion of the proline-rich region in SOS1 is also associated with enhanced SOS1 activity in D. melanogaster (McCollam et al. 1995) and yeast. Additionally, a transgenic mouse model of a similar SOS1 mutation (called “SOS-F”) is associated with a dominant skin phenotype (Sibilia et al. 2000). In the SOS-F transgene construct, the C-terminal region containing the Grb2 binding site has been deleted. When the SOS-F construct is expressed in basal keratinocytes by use of a keratin 5′ promoter, multiple papillomas and skin hypertrophy were observed. The skin-tumor phenotype is 100% penetrant in mice with the SOS-F mutation. These data suggest that a C-terminal truncation of the SOS1 protein is functional and could produce the gingival overgrowth clinically observed in human HGF1 with the SOS1 mutation. Why the HGF phenotype appears localized to the gingiva is unknown. The keratinized masticatory mucosa is recognized to be developmentally unique (Gao and Mackenzie 1992), and different tissue-specific signaling pathways in this unique tissue may be responsible for the limited tissue distribution of the gingival fibromatosis phenotype. Although the site-specific localization of this phenotype is remarkable, the gingiva is also the only site affected in drug-induced and most syndromic forms of gingival fibromatosis.
Interpretation of western blot analyses of extracts from human gingiva with SOS1 antibodies are complicated by the cross-reactivity of SOS1 antibodies with the related SOS2 protein (∼150 kDa), and by the presence of SOS1 as part of high–molecular-weight complexes (>200 kDa) in gingiva. Nonetheless, protein extracts from HGF-affected tissues were consistent with the presence of a truncated SOS1 protein band (143 kDa). However, it appears that the majority of the truncated (mutant) protein is associated in the high molecular-weight complex, thus supporting a functional role for the truncated protein. Although the components of the high molecular-weight species are not known, SOS1 has been reported to form a complex with Grb2 (Li et al. 1993), src-type tyrosine kinase (Park et al. 1998), and phospholipase C-γ1 (Kim et al. 2000). A complex is also observed between Grb2/SOS1 and the epidermal growth factor (EGF) receptor after EGF stimulation (Hu and Bowtell 1996). The ability of mutant SOS1 protein to form complexes may lead to constitutive activation of its downstream targets.
The genetic basis for gingival fibromatosis in humans is heterogeneous (Witkop 1971; Gorlin 1990; Hart et al. 2000). Loci for autosomal dominant HGF have been localized to an overlapping interval on chromosome 2p21-p22 (Hart et al. 1998; Shashi et al. 1999; Xiao et al. 2000), suggesting that a gene locus within the 3.8-cM overlap may be causal for HGF1. Evaluation of linkage data presented here, by use of a recently developed 6-Mb integrated physical and genetic map of the HGF1 candidate interval, indicates that the two chromosome 2p21-p22 HGF linkage intervals are distinct (fig. 2). The SOS1 gene locus is ∼1.3 Mb centromeric to the linkage interval reported in Chinese families affected with HGF (Xiao et al 2000). Whether there are two distinct HGF loci in close proximity on chromosome 2p21-p22 is unknown. Mutational analysis of the SOS1 gene in families demonstrating linkage to chromosome 2p21-p22 may clarify genetic heterogeneity of HGF.
The SOS1 mutation in individuals with HGF described in this report represents the first genetic mutation identified for isolated HGF. Whether the increase in gingival tissue observed in the HGF1 phenotype is a result of increased cellular proliferation, increased cell survival, and/or inhibition of cellular differentiation is not known. Identification of the specific genetic basis in HGF1 should help to elucidate the pathogenic mechanism by which gingival overgrowth occurs. Understanding the mechanism by which gingival fibromatosis develops may have implications for understanding how certain drugs induce gingival overgrowth and may have applications for the nonsurgical treatment of gingival insufficiency.
Acknowledgments
This investigation was supported in part by United States Public Health Service research grant DE10990 from the National Institute of Dental and Craniofacial Research, National Institutes of Health.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- LocusLink, http://www.ncbi.nlm.nih.gov/LocusLink/ (for loci shown in )
- Marshfield Center for Medical Genetics, http://research.marshfieldclinic.org/genetics/ (for position of marker D2S2230)
- NCBI dbEST database, http://www.ncbi.nlm.nih.gov/blast/Blast.cgi
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for fibrous enlargement of maxillary and mandibular keratinized gingiva [MIM 135300], anomalies of chromosome 4q [MIM 252500], anomalies of chromosome 8 [MIM 266270], anomalies of chromosome 19p [MIM 246200], and chromosome 19q [MIM 248500])
References
- Antonarakis SE (1998) Recommendations for a nomenclature system for human gene mutations. Hum Mutat 11:1–3 [DOI] [PubMed] [Google Scholar]
- Aronheim A, Engelberg D, Li N, Al Alawi N, Schlessinger J, Karin M (1994) Membrane targeting of the nucleotide exchange factor Sos is sufficient for activating the Ras signaling pathway. Cell 78:949–961 [DOI] [PubMed] [Google Scholar]
- Broman KW, Murray JC, Sheffield VC, White RL, Weber JL (1998) Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am J Hum Genet 63:861–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JM, Friedman FK, Hyde MJ, Monaco R, Pincus MR (1999) Molecular dynamics analysis of the structures of ras-guanine nucleotide exchange protein (SOS) bound to wild-type and oncogenic ras-p21: identification of effector domains of SOS. J Protein Chem 18:867–874 [DOI] [PubMed] [Google Scholar]
- Conneally PM, Edwards JH, Kidd KK, Lalouel JM, Morton NE, Ott J, White R (1985) Report of the committee on methods of linkage analysis and reporting. Cytogenet Cell Genet 40:356–359 [DOI] [PubMed] [Google Scholar]
- Corbalan-Garcia S, Margarit SM, Galron D, Yang SS, Bar-Sagi D (1998) Regulation of SOS activity by intramolecular interactions. Mol Cell Biol 18:880–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbalan-Garcia S, Yang SS, Degenhardt KR, Bar-Sagi D (1996) Identification of the mitogen-activated protein kinase phosphorylation sites on human Sos1 that regulate interaction with Grb2. Mol Cell Biol 16:5674–5682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottingham RW Jr, Idury RM, Schäffer AA (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53:252–263 [PMC free article] [PubMed] [Google Scholar]
- Elston RC, Stewart J (1971) A general model for the genetic analysis of pedigree data. Hum Hered 21:523–542 [DOI] [PubMed] [Google Scholar]
- Fryns JP (1996) Gingival fibromatosis and partial duplication of the short arm of chromosome 2 (dup(2)(p13→p21)). Ann Genet 39:54–55 [PubMed] [Google Scholar]
- Gao Z, Mackenzie IC (1992) Patterns of phenotypic expression of human junctional, gingival and reduced enamel epithelia in vivo and in vitro. Epithelial Cell Biology 1:156–167 [PubMed] [Google Scholar]
- Gorlin RJ, Cohen MM, Levi LS (1990) Syndromes of the head and neck. In: Gorlin RJ, Cohen MM Jr, Levin LS (eds) Syndromes of the head and neck. Oxford University Press, New York, pp 847–857 [Google Scholar]
- Hart TC, Pallos D, Bowden DW, Bolyard J, Pettenati MJ, Cortelli JR (1998) Genetic linkage of hereditary gingival fibromatosis to chromosome 2p21. Am J Hum Genet 62:876–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart TC, Pallos D, Bozzo L, Almeida OP, Marazita ML, O'Connell JR, Cortelli JR (2000) Evidence of genetic heterogeneity for hereditary gingival fibromatosis. J Dent Res 79:1758–1764 [DOI] [PubMed] [Google Scholar]
- Hassell TM, Hefti AF (1991) Drug-induced gingival overgrowth: old problem, new problem. Crit Rev Oral Biol Med 2:103–137 [DOI] [PubMed] [Google Scholar]
- Hu Y, Bowtell DD (1996) Sos1 rapidly associates with Grb2 and is hypophosphorylated when complexed with the EGF receptor after EGF stimulation. Oncogene 12:1865–1872 [PubMed] [Google Scholar]
- Jorgenson RJ, Cocker ME (1974) Variation in the inheritance and expression of gingival fibromatosis. J Periodontol 45:472–477 [DOI] [PubMed] [Google Scholar]
- Kim MJ, Chang JS, Park SK, Hwang JI, Ryu SH, Suh PG (2000) Direct interaction of SOS1 Ras exchange protein with the SH3 domain of phospholipase C-gamma1. Biochemistry 39:8674–8682 [DOI] [PubMed] [Google Scholar]
- Lathrop GM, Lalouel JM (1984) Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet 36:460–465 [PMC free article] [PubMed] [Google Scholar]
- Li N, Batzer A, Daly R, Yajnik V, Skolnik E, Chardin P, Bar-Sagi D, Margolis B, Schlessinger J (1993) Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature 363:85–88 [DOI] [PubMed] [Google Scholar]
- Macias-Flores MA, Garcia-Cruz D, Rivera H, Escobar-Lujan M, Melendrez-Vega A, Rivas-Campos D, Rodriguez-Collazo F, Moreno-Arellano I, Cantu JM (1984) A new form of hypertrichosis inherited as an X-linked dominant trait. Hum Genet 66:66–70 [DOI] [PubMed] [Google Scholar]
- McCollam LL, Bonfini CA, Karlovich BR, Conway LM, Kozma Banerjee U, Czech MP (1995) Functional roles for the pleckstrin and Dbl homology regions in the Ras exchange factor Son-of sevenless. J Biol Chem 270:15954–15957 [DOI] [PubMed] [Google Scholar]
- O'Connell JR, Weeks DE (1995) The VITESSE algorithm for rapid exact multilocus linkage analysis via genotype set-recoding and fuzzy inheritance. Nat Genet 11:402–408 [DOI] [PubMed] [Google Scholar]
- Park C, Choi Y, Yun Y (1998) Son of sevenless binds to the SH3 domain of src-type tyrosine kinase. Mol Cells 8:518–523 [PubMed] [Google Scholar]
- Raeste AM, Collan Y, Kilpinen E (1978) Hereditary fibrous hyperplasia of the gingiva with varying penetrance and expressivity. Scand J Dent Res 86:357–365 [DOI] [PubMed] [Google Scholar]
- Rivera HM, Ramirez-Duenas L, Figuera LE, Gonzalez-Montes RM, Vasquez AI (1992) Opposite imbalances of distal 14q in two unrelated patients. Ann Genet 35:97–100 [PubMed] [Google Scholar]
- Rojas JM, Coque JJ, Guerrer C, Aroca P, deMora JF, de la Cruz X, Lorenzi MV, Esteban LM, Santos E (1996) A 15 amino acid stretch close to the Grb2-binding domain defines two differentially expressed hSos1 isoforms with markedly different Grb2 binding affinity and biological activity. Oncogene 12:2291–2300 [PubMed] [Google Scholar]
- Scannapieco FA (1998) Position paper of The American Academy of Periodontology: periodontal disease as a potential risk factor for systemic diseases. J Periodontol 69:841–850 [PubMed] [Google Scholar]
- Seymour RA, Thomason JM, Ellis JS (1996) The pathogenesis of drug-induced gingival overgrowth. J Clin Periodontol 23:165–175 [DOI] [PubMed] [Google Scholar]
- Shashi V, Pallos D, Pettenati MJ, Cortelli JR, Fryns JP, Kap-Herr C, Hart TC (1999) Genetic heterogeneity of gingival fibromatosis on chromosome 2p. J Med Genet 36:683–686 [PMC free article] [PubMed] [Google Scholar]
- Sibilia MA, Fleischmann A, Behrens A, Stingl L, Carroll J, Watt FM, Schlessinger J, Wagner EF (2000) The EGF receptor provides an essential survival signal for SOS-dependent skin tumor development. Cell 102:211–220 [DOI] [PubMed] [Google Scholar]
- Smith RG (1997) Gingival recession: reappraisal of an enigmatic condition and a new index for monitoring. J Clin Periodontol 24:201–205 [DOI] [PubMed] [Google Scholar]
- Sobel E, Lange K (1996) Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 58:1323–1337 [PMC free article] [PubMed] [Google Scholar]
- Soisson SM, Nimnual AS, Uy M, Bar-Sagi D, Kuriyan J (1998) Crystal structure of the Dbl and pleckstrin homology domains from the human Son of sevenless protein. Cell 95:259–268 [DOI] [PubMed] [Google Scholar]
- Terwilliger JD, Ott J (1994) Handbook of human genetic linkage. Johns Hopkins University Press, Baltimore [Google Scholar]
- Wasiak S, Quinn C, Ritter B, de Heuvel E, Baranes D, Plomann M, McPherson PS (2001) The Ras/Rac guanine nucleotide exchange factor mammalian Son-of-sevenless interacts with PACSIN 1/syndapin I, a regulator of endocytosis and the actin cytoskeleton. J Biol Chem 276:26622–26628 [DOI] [PubMed] [Google Scholar]
- Witkop CJ Jr (1971) Heterogeneity in gingival fibromatosis. Birth Defects Orig Artic Ser 7:210–221 [PubMed] [Google Scholar]
- Xiao S, Bu L, Zhu L, Zheng G, Yang M, Qian M, Hu L, Liu J, Zhao G, Kong X (2001) A new locus for hereditary gingival fibromatosis (GINGF2) maps to 5q13-q22. Genomics 74:180–185 [DOI] [PubMed] [Google Scholar]
- Xiao S, Wang X, Qu B, Yang M, Liu G, Bu L, Wang Y, Zhu L, Lei H, Hu L, Zhang X, Liu J, Zhao G, Kong X (2000) Refinement of the locus for autosomal dominant hereditary gingival fibromatosis (GINGF) to a 3.8-cM region on 2p21. Genomics 68:247–252 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gorry MC, Hart PS, Pettenati MJ, Marks MJ, Lu X, Hart TC. Localization, genomic organization, and alternative transcription of a novel human SAM-dependent methyltransferase gene on chromosome 2p21-p22. Cytogenet Genome Res (in press) [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lundgren T, Renvert S, Tatakis DN, Firatli E, Uygur C, Hart PS, Gorry MC, Marks JJ, Hart TC (2001) Evidence of a founder effect for four cathepsin C gene mutations in Papillon-Lefevre syndrome patients. J Med Genet 38:96–101 [DOI] [PMC free article] [PubMed] [Google Scholar]