

Abstract
Primary ciliary dyskinesia (PCD) is a genetically heterogeneous, autosomal-recessive disorder, characterized by oto-sino-pulmonary disease and situs abnormalities. PCD-causing mutations have been identified in 20 genes, but collectively they account for only ∼65% of all PCDs. To identify mutations in additional genes that cause PCD, we performed exome sequencing on three unrelated probands with ciliary outer and inner dynein arm (ODA+IDA) defects. Mutations in SPAG1 were identified in one family with three affected siblings. Further screening of SPAG1 in 98 unrelated affected individuals (62 with ODA+IDA defects, 35 with ODA defects, 1 without available ciliary ultrastructure) revealed biallelic loss-of-function mutations in 11 additional individuals (including one sib-pair). All 14 affected individuals with SPAG1 mutations had a characteristic PCD phenotype, including 8 with situs abnormalities. Additionally, all individuals with mutations who had defined ciliary ultrastructure had ODA+IDA defects. SPAG1 was present in human airway epithelial cell lysates but was not present in isolated axonemes, and immunofluorescence staining showed an absence of ODA and IDA proteins in cilia from an affected individual, thus indicating that SPAG1 probably plays a role in the cytoplasmic assembly and/or trafficking of the axonemal dynein arms. Zebrafish morpholino studies of spag1 produced cilia-related phenotypes previously reported for PCD-causing mutations in genes encoding cytoplasmic proteins. Together, these results demonstrate that mutations in SPAG1 cause PCD with ciliary ODA+IDA defects and that exome sequencing is useful to identify genetic causes of heterogeneous recessive disorders.
Main Text
Primary ciliary dyskinesia (PCD) is a rare autosomal-recessive disorder (MIM 244400) that results from genetic abnormalities of motile cilia.1–3 Disease manifestations include neonatal respiratory distress, chronic sino-pulmonary disease, otitis media, male infertility, and laterality defects in ∼50% of affected individuals. PCD is highly genetically heterogeneous, and mutations in 20 genes account for ∼65% of cases.2–16 Most of the genes implicated in PCD encode components of the ciliary axonemal structure, including the outer and inner dynein arms (ODAs and IDAs, respectively), which are multiprotein complexes essential for ciliary movement.2–16
For PCD cases with absence of both the ODA and IDA, mutations have been described in five genes involved in cytoplasmic assembly of dynein arms: DNAAF1 (previously LRRC50 [MIM 613190]), DNAAF2 (previously KTU [MIM 612517]), DNAAF3 (previously PF22 [MIM 614566]), HEATR2 (MIM 614864), and LRRC6 (MIM 614930).11–16 The absence of both ODA and IDA occurs in as many as 30% of the ciliary ultrastructural defects in PCD, but only a minority are caused by mutations in the five genes involved in cytoplasmic assembly of dynein arms.11–16 Given this heterogeneity, we sought to identify additional mutations that cause absence of both ODA and IDA by exome sequencing in three unrelated individuals with PCD with ODA+IDA defects (Table S1 available online, demographic and clinical phenotype). Study participants were recruited at the UNC and collaborating sites of Genetic Disorders of Mucociliary Clearance Consortium and signed informed consents were obtained. The study was approved by the institutional review board for the Protection of the Rights of Human subjects at the UNC and collaborating institutions.
Exome sequencing was performed as previously described.9,17 In brief, sheared genomic DNA was ligated to sequencing adaptors bearing an 8 bp barcode, followed by PCR amplification and hybridization-based, solution-phase capture with EZSeqCap V1 (corresponding to CCDS2008 gene set) probes (Roche Nimblegen). Postenrichment libraries were amplified, pooled, and sequenced on an Illumina GAIIx (PE76 + barcode read). After barcode-based sample deconvolution, sequence reads were mapped with BWA to the reference human genome (UCSC Genome Browser hg18).18 On average, we achieved 76× mean fold coverage of the targeted exome (26.4 megabases [Mb]), with 98% covered at ≥8× (details in Table S2). SAMtools were used for sequence variant calling after removal of potential PCR duplicates, with a minimum quality threshold of 30 for SNPs and 200 for indels.19 Average calls per individuals were 17,553 base substitutions and 504 insertions and/or deletions (Table S2), and SeattleSeq server was used for genotype annotation (Table S3). After removal of common variants as previously described,9 only genes with biallelic variants consistent with an autosomal-recessive model were considered further (Tables S2 and S3). Variants were prioritized for validation by Sanger sequencing (capillary electrophoresis; Life Technologies) based on manual investigation of their pathogenic potential, their expression in ciliated airway epithelia (RNA-seq data set, unpublished), their potential relevance to PCD through published literature, and analysis of large-scale coexpression data sets.20,21 The results of validations and the primer sequences are presented in Tables S3 and S4. Segregation analysis of the confirmed variants in the proband was carried out on the available DNA from family members.
We identified an apparently homozygous missense variant of the start codon (c.2T>G [p.Met1?]) in exon 2 of SPAG1 (sperm-associated antigen 1, RefSeq accession number NM_172218.2 [MIM 603395]) in an individual with PCD (#711 [III-2]) and ODA+IDA defects in ciliary ultrastructure (Table 1). This variant in the translation start site is predicted to abrogate SPAG1 protein translation. Sanger sequencing confirmed the mutation in a heterozygous state in the unaffected mother (#713 [II-2]) but failed for the unaffected father (#713 [II-1]), which prompted us to consider the possibility of a large deletion in this region of SPAG1 encompassing the start codon. Intragenic high-frequency SNPs were genotyped in this propositus with PCD (#711 [III-2]) and his unaffected father (#713 [II-1]) by means of 17 primer pairs (Table S4) across SPAG1, plus two adjacent genes, FBXO43 (MIM 609110) and POLR2K (MIM 606033) (Figure 1A). The genotyping revealed loss of heterozygosity encompassing the C terminus of POLR2K and the N terminus of SPAG1 as well as intergenic sequences between these genes (Figure 1A and Table S5). Amplification by long-range PCR coupled with Sanger sequencing identified the deletion breakpoint leading to an 11,971 bp deletion (c.61+201_POLR2K:c.140+1169_SPAG1del) in the affected propositus (#711 [III-2]) and his unaffected father (#713 [II-1]) (Figures 1A, 1B, and S1A). This deletion encompassed the stop codon and 3′ UTR of POLR2K, the 5′ UTR and start codon of SPAG1, and intergenic sequences and is predicted to abrogate protein translation for SPAG1. Subsequently, a PCR-based assay was developed to detect this large deletion (Figure S1B). This deletion analysis, coupled to Sanger sequencing, confirmed recessive inheritance of the start codon variant (c.2T>G [p.Met1?]) and deletion in the propositus (#711 [III-2]) and two affected siblings (#710 [III-1] and #712 [III-3]) (Table 1 and Figure 1B). Because the large deletion on one allele led to a loss-of-function mutation in POLR2K, we performed mutation analysis in the propositus (#711 [III-2]); however, no other mutations were identified in POLR2K, suggesting that mutations in SPAG1 are indeed responsible for the PCD phenotype. The concept that mutations in SPAG1 (and not POLR2K) cause PCD is because (1) only one mutant allele in POLR2K does not explain the autosomal-recessive inheritance of PCD, (2) functional data from zebrafish (see below) show that SPAG1 is important for cilia function, and (3) PCD occurred in seven unrelated individuals who had biallelic mutations in SPAG1 but did not have the POLR2K deletion. Additionally, SPAG1 is a strong candidate because analysis of the gene expression data set (UGET) showed coordinate regulation between SPAG1 and multiple other genes known to harbor PCD-causing mutations;20,21 plus SPAG1 has never been implicated with any other inherited disorders.
Table 1.
Demographic, Clinical Phenotype, and SPAG1 Mutations in 14 PCD-Affected Subjects from 11 Unrelated Families
| Subject # | Family # | Sex | Age in Years | Situs Status | Ciliary Ultrastructural Defect | Ethnicity | nNO nl/mina | Neo RDS | Bxsis | Sinusitis | Otitis Media |
Allele 1 |
Allele 2 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exon # | Base Change | Amino Acid Change | Segregationb | Exon # | Base Change | Amino Acid Change | Segregationb | ||||||||||||
| Homozygous Mutations | |||||||||||||||||||
| OP-94 (II-2) | OP94 | F | 17 | SS | ODA+IDA | white | NA | NA | NA | yes | yes | 7 | c.679G>T | p.Glu227∗ | paternal | 7 | c.679G>T | p.Glu227∗ | maternal |
| #1854 (II-1) | UNC-608 | F | 4 mo | SI | ODA+IDA | white | 9.4 | yes | no | yes | yes | 9 | c.902_906delAAGTA | p.Lys301Thrfs∗4 | paternal | 9 | c.902_906delAAGTA | p.Lys301Thrfs∗4 | maternal |
| OP-16 (II-1)c,d | OP16 | M | 35 | SI | ODA+IDA | white | NA | yes | yes | yes | no | 16 | c.2014C>T | p.Gln672∗ | paternal | 16 | c.2014C>T | p.Gln672∗ | maternal |
| OP-46 (II-1) | OP46 | M | 14 | SI | ODA+IDA | NA | NA | NA | yes | yes | yes | 16 | c.2014C>T | p.Gln672∗ | NA | 16 | c.2014C>T | p.Gln672∗ | NA |
| #1391 (III-1) | UNC-372 | M | 5 | SI | ODA+IDA | white | 11.3 | yes | NA | yes | yes | 16 | c.2014C>T | p.Gln672∗ | paternal | 16 | c.2014C>T | p.Gln672∗ | maternal |
| #1802 (II-1)c | UNC-587e | M | 14 | SI | ODA+IDA | South Asian | 107.2 | yes | no | no | no | 18 | c.2542delG | p.Asp848Ilefs∗10 | NA | 18 | c.2542delG | p.Asp848Ilefs∗10 | NA |
| #1803 (II-2)c | UNC-587e | M | 2 | SI | ODA+IDA | South Asian | 8.8 | no | no | no | yes | 18 | c.2542delG | p.Asp848Ilefs∗10 | NA | 18 | c.2542delG | p.Asp848Ilefs∗10 | NA |
| Compound Heterozygous Mutations | |||||||||||||||||||
| #710 (III-1) | UNC-121e | M | 12 | SS | ODA+IDA | white | 41.1 | yes | yes | yes | yes | multif | large deletionf | untranslated | paternal | 2 | c.2T>G | p.Met1?g | maternal |
| #711 (III-2) | UNC-121e | M | 10 | SI | ODA+IDA | white | 13.1 | yes | no | yes | yes | multif | large deletionf | untranslated | paternal | 2 | c.2T>G | p.Met1?g | maternal |
| #712 (III-3) | UNC-121e | F | 8 | SS | ODA+IDA | white | 14.6 | yes | no | yes | yes | multif | large deletionf | untranslated | paternal | 2 | c.2T>G | p.Met1?g | maternal |
| OP-13 (II-2) | OP13 | M | 19 | SS | ODA+IDA | white | NA | no | yes | yes | NA | multif | large deletionf | untranslated | maternal | 16 | c.2014C>T | p.Gln672∗ | paternal |
| OP-90 (II-2) | OP90 | M | 22 | SI | ODA+IDA | white | NA | yes | yes | yes | yes | multif | large deletionf | untranslated | paternal | 16 | c.2014C>T | p.Gln672∗ | maternal |
| #26 (II-1) | UNC-4 | F | 16 | SI | NA | white | NA | yes | yes | yes | yes | multif | large deletionf | untranslated | maternal | 16 | c.2089C>T | p.Arg697∗ | paternal |
| #2103 (II-1) | UNC-712 | F | 12 | SS | ODA+IDA | white | 1.0 | yes | NA | yes | yes | 9 | c.902_906delAAGTA | p.Lys301Thrfs∗4 | NA | 16 | c.1993_1996delCTCT | p.Leu665Valfs∗3 | NA |
Abbreviations are as follows: M, male; F, female; SI, situs inversus; SS, situs solitus; NA, not available; ODA+IDA, outer + inner dynein arms; Neo RDS, neonatal respiratory distress in full-term neonates; Bxsis, bronchiectasis; nNO, nasal nitric oxide.
Normal nNO levels are 376 ± 124 nl/min (mean ± SD), calculated from 27 healthy subjects.5
Mutant allele shown to segregate with either the paternal or maternal side of the family (pedigrees in Figures 1 and S2).
Parental consanguinity was noted.
Family was linked to SPAG1 locus.
Affected siblings are listed under same family numbers.
Large 11,971 bp deletion (c.61+201_POLR2K:c.140+1169_SPAG1del) comprised of multiple introns, exons, and untranslated regions from 3′ of POLR2K and 5′ of SPAG1. This deletion includes translation start codon and is predicted to abrogate SPAG1 translation.
Base substitution leading to apparent missense mutation in the translation start codon is predicted to abrogate SPAG1 translation.
Figure 1.
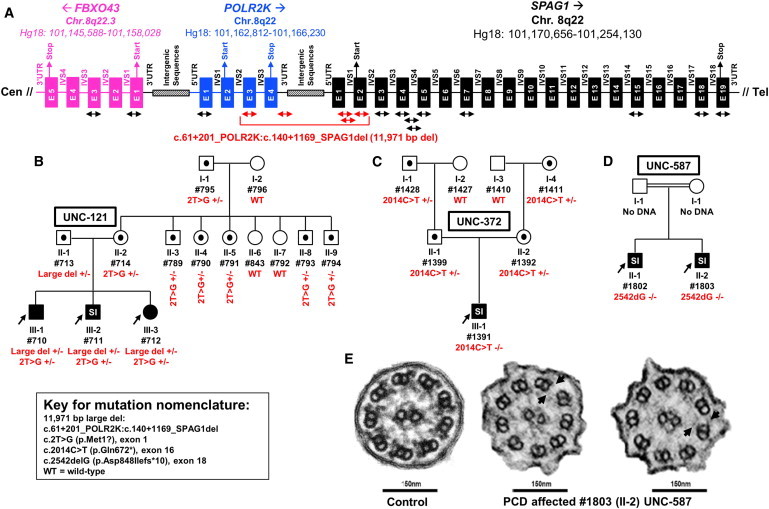
Deletion Analysis in SPAG1, Segregation of Mutations in Pedigrees, and Ciliary Ultrastructural Defects
(A) A total of 17 primer pairs (see the arrows; listed in Table S4 as SPAG1 exons 1, 2, 3, 5, 7, 15, 18, 19, introns 1-1, 1-2, 4-1, 4-2, POLR2K exons 1, 3, 3′ UTR, FBXO43 exons 1 and 3) were designed for SNP genotyping to ascertain loss of heterozygosity in affected individual (#711 [III-2] from family UNC-121). Genomic structure and coordinates for FBXO43, POLR2K, and SPAG1 are shown. Filled boxes designate exons (E), horizontal lines designate introns (IVS), and location of 5′ UTR, 3′ UTR, translation start and stop, and intergenic sequences are shown. Introns and exons are not drawn to scale. Absence of paternal alleles from affected individual at certain loci is shown with the red arrows for primers. The exact genotype is presented in Table S5.
(B–D) Pedigrees with mutations segregating in trans, consistent with autosomal-recessive mode of inheritance (see additional pedigrees in Figure S2). Males and females are designated by squares and circles, respectively. Filled symbols indicate affected individuals with PCD and symbols with a dot within indicate carrier status. Proband is designated by an arrow, presence of parental consanguinity is designated by double horizontal lines between the parents, and SI refers to situs inversus. Shown are families UNC-121 (B), UNC-372 (C), and UNC-587 (D).
(E) Normal ciliary ultrastructure from healthy subject (left) and individual (#1803 [II-2] of family UNC-587) with biallelic mutations in SPAG1 with defective outer+inner dynein arms (middle) and/or stubs (right) (see arrows).
We sought to identify additional cases of PCD with biallelic mutations in SPAG1 by testing 62 affected individuals with similar ultrastructural findings as our SPAG1 discovery case (ODA+IDA defects), another cohort of 35 affected individuals with isolated ODA defects, and a singleton case with unknown ciliary ultrastructure. A combination of methods were used to test SPAG1 in these 98 individuals, including screening for the large deletion (Figure S1B) in all cases, Sanger-based high-throughput sequencing through the Genotyping and Resequencing Services in Seattle (RC2-HL-102923) or at UNC via capillary electrophoresis ([Life Technologies], primer sequences shown in Table S4) (89 unrelated cases), and additional whole-exome sequencing and targeted screening for SPAG1 (nine unrelated cases) (Table S6).9,22 Full details of the exomes for six individuals with ODA defects is described in Knowles et al.9 (subjects 144, 359, 453, 483, 961, and 980) and two individuals with ODA+IDA defects are described in Zariwala et al.22 (subjects A4207 and A4218), and one individual (#1802 [II-1]) with ODA+IDA defects harboring biallelic mutations in SPAG1 is listed in Table 1.
All variants were confirmed by Sanger sequencing at UNC. We identified biallelic mutations in an additional 11 individuals (10 unrelated families) (Table 1); thus, biallelic mutations in SPAG1 were found in a total of 11 families (14 affected individuals), and segregation analysis confirmed that the mutations were inherited in trans in the 8 families that could be tested, consistent with recessive inheritance (Figures 1B–1D and S2). All 13 subjects with defined ciliary ultrastructure had ODA+IDA defects (Figures 1E). The clinical phenotypes of all individuals with biallelic mutations (Table 1) were indistinguishable from individuals with “classic” PCD with ODA defects and mutations in DNAI1 (MIM 604366) or DNAH5 (MIM 603335), including situs inversus and low levels of nasal nitric oxide (nNO).23–26 Videomicroscopy was carried out to explore the functional consequences of biallelic mutations in SPAG1 on respiratory cilia obtained from two affected individuals (#712 [III-3] UNC-121 and #1391 [II-1] UNC-372). Consistent with ODA+IDA defects, cilia exhibited near complete ciliary immotility (Movies S1, S2, and S3).
The genomic organization of SPAG1 and location of mutations is shown in Figure 2. SPAG1 consists of 18 coding exons and encodes a large protein with 926 amino acid residues (Figures 2 and S3). SPAG1 contains nine tetratricopeptide repeat motifs (TPRs) that form scaffolds to mediate protein-protein interactions and are often involved in the assembly of multiprotein complexes (Figure S3).27 In total, there were eight different mutant alleles (three frameshift, three nonsense, one large deletion, and one missense or null at the start codon), and all mutant alleles are predicted to lead to loss of function of full-length SPAG1 (Table 1 and Figure 2). Five of the eight mutant alleles were private; however, three alleles were seen to occur in more than one unrelated families. The three recurrent mutant alleles (c.2014C>T [p.Gln672∗]; a large 11,971 bp deletion; and c.902_906delAAGTA [p.Lys301Thrfs∗4]) were seen in five, four, and two unrelated families, respectively. For subjects harboring the three recurrent mutant alleles, we constructed haplotypes for all variants and polymorphisms detected during Sanger sequencing of SPAG1. We found that all affected individuals carried an identical haplotype in cis with the c.2014C>T (p.Gln672∗) mutation, an identical haplotype in cis with the large deletion, and an identical haplotype in cis with the c.902_906delAAGTA (p.Lys301Thrfs∗4) mutation. These results suggest that these three variants are founder mutations (Figure S4). Mutation screening identified a large number of benign variants in SPAG1 that are listed in Table S7. To further investigate the role of SPAG1 in ciliary structure and function and in PCD, we examined its expression in a panel of human tissues. At least two different SPAG1 proteins are predicted (104 and 48 kDa);28,29 therefore, we designed primers to amplify both a full-length (long) transcript and a shorter transcript containing only the first ten exons (Ensembl; Table S4, Figure S3). Figure 3A demonstrates that both SPAG1 transcripts were present in most tissues, including lung, with the strongest expression in brain, colon, kidney, and testis, in agreement with the EST data set. To examine SPAG1 expression in human airway epithelial cells, RNA was isolated from air-liquid interface cultures of airway epithelia at different stages of differentiation and examined by reverse transcriptase-polymerase chain reaction (RT-PCR).30–32 Although the shorter SPAG1 transcript was expressed in human airway epithelial cells at all time points throughout the culture period, the full-length, longer transcript was strongly induced at later stages of culture, when ciliated cell differentiation was occurring (Figure 3B). This induction closely paralleled the expression pattern for DNAI1, a known component of the ciliary outer dynein arm, suggesting that the longer transcript of SPAG1 plays a specific role in ciliated cells. Interestingly, four of the eight mutant alleles would not be expected to alter the protein predicted from the short transcript, again suggesting that it is the longer, full-length SPAG1 that is important to ciliated cell function. Spag1 is induced during differentiation of mouse spermatogenic cells,28 and SPAG1 is expressed in human lung and testis (EST Profile Viewer in EST database), consistent with a potential function in ciliated cells.
Figure 2.
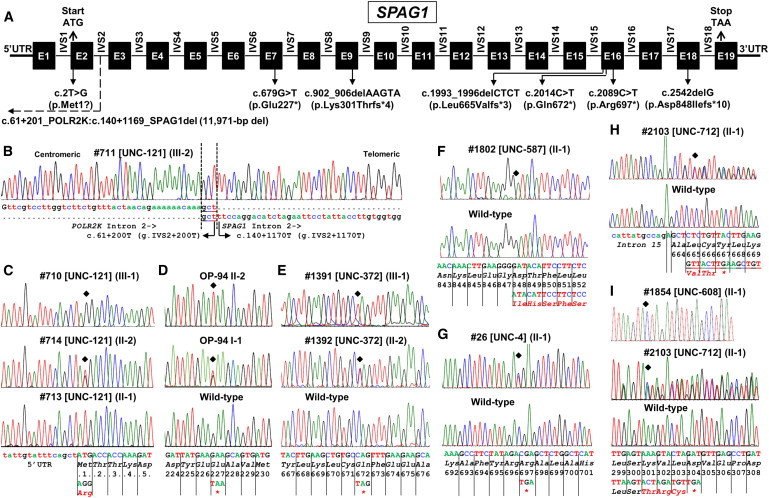
Genomic Organization of SPAG1 and Location of PCD-Causing Mutations
(A) The transcript of SPAG1 consists of 3,760 bp (RefSeq NM_172218.2). Filled boxes designate exons (E), horizontal lines designate introns (IVS), and locations of 5′ UTR, 3′ UTR, start and stop codons, and noncoding exon 1 are shown. Introns and exons are not drawn to scale. The positions of all eight identified SPAG1 putative mutations are indicated.
(B) Large 11,971 bp deletion involves deletion of part of intron 2 and upstream regions including start codon of SPAG1 as well as 3′ region of the proximal gene POLR2K that include stop codon. Precise breakpoints (dashed vertical lines) and the nucleotide sequence of the junction point, including microhomology (underlined) at the breakpoint, are shown. For the mutation nomenclature, the microhomology region is included in POLR2K. Deletion breakpoint sequence in an affected individual is shown with the vertical dotted line.
(C) Hemizygous mutation in affected (top), heterozygous carrier (middle), and hemizygous wild-type (bottom) for start codon c.2T>G (p.Met1?) mutation sequences.
(D and E) Homozygous mutation in affected (top) and heterozygous carrier (middle) individuals and corresponding wild-type sequences in control (bottom) individual for c.679G>T (p.Glu227∗) (D) and c.2014C>T (p.Gln672∗) (E) mutations.
(F) Homozygous mutation in affected (top) and corresponding wild-type sequences in control (bottom) individuals for c.2542delG (p.Asp848Ilefs∗10) mutation.
(G and H) Heterozygous mutation in affected (top) and corresponding wild-type sequence (bottom) in controls for c.2089C>T (p.Arg697∗) (G) and c.1993_1996delCTCT (p.Leu665Valfs∗3) (H) mutations.
(I) Homozygous mutation in affected (top) and heterozygous mutation in another affected (middle) and corresponding wild-type sequences in control (bottom) individuals for c.902_906delAAGTA (p.Lys301Thrfs∗4) mutation. Additionally, a polymorphism c.897G>A (p.Leu299Leu) was noted that is listed in Table S7.
Base sequence, amino acid sequence, and codon numbers are shown. Location of the mutation is shown by filled diamond and mutated amino acids are represented by red fonts. Pedigrees and segregation analysis are shown in Figures 1 and S2.
Figure 3.
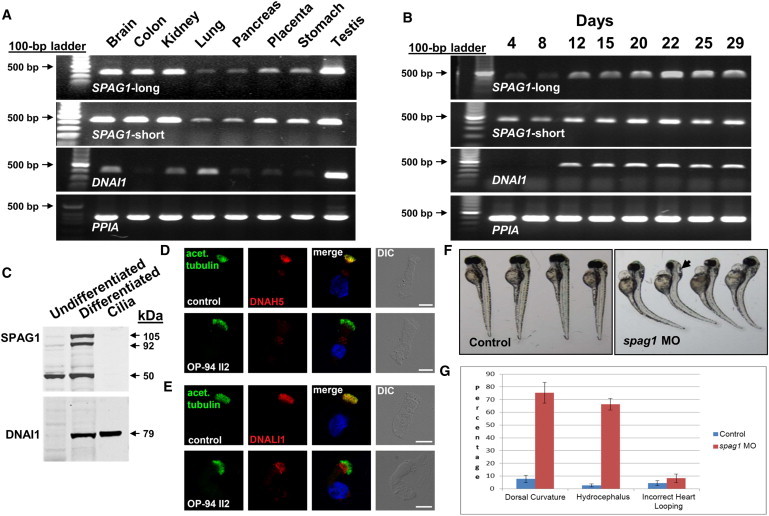
Expression of SPAG1 and Morpholino Knockdown in Zebrafish
(A) Expression pattern of two SPAG1 transcripts in a panel of normal human tissues. Specific primers to the full-length (long) and truncated transcripts (Table S4) were used for RT-PCR on total RNA from different tissues (Ambion, Life Technologies).
(B) Expression pattern of long and short forms of SPAG1 (top and second from top) during ciliogenesis in human airway cells. RNA was isolated from human airway epithelial cells at different times after plating on collagen-coated culture inserts at an air-liquid interface. Under these conditions the cells initially grow as a single mono-layer of undifferentiated “basal-like” cells. After 10–14 days, ciliated cells begin to appear, increasing over the next 2–3 weeks before plateauing at approximately 60%–80% of the culture.30,31 For both expression analyses, DNAI1 (third from top) was used as a known ciliary differentiation gene control and PPIA (cyclophilin A) (bottom) as a positive RNA control. The full-length SPAG1 transcript is induced coordinately with ciliated cell differentiation.
(C) Immunoblot showing expression of SPAG1 (top) and DNAI1 (bottom) in undifferentiated and differentiated human airway epithelia cells and isolated cilia. Total cell lysates and ciliary axonemes were isolated as previously described.33 DNAI1 is present in the ciliary axoneme; SPAG1 is not.
(D and E) High-resolution immunofluorescence analysis of the localization of DNAH5 (D) and DNALI1 (E) in respiratory cells from a control (top) and affected individual OP-94 (II-2) (bottom) carrying biallelic mutations in SPAG1. In control cells (top), axoneme-specific acetylated alpha tubulin (green), DNAH5 (red), and DNALI1 (red) colocalizes to the entire length of the axonemes. The yellow immunofluorescence in the ciliary axonemes indicates colocalization of both proteins in cilia of control. In contrast, in respiratory epithelial cells from an affected individual, OP-94 (II-2) (bottom), DNAH5 and DNALI1 are completely missing from the ciliary axoneme. Nuclei were stained with Hoechst 33342 (blue). Scale bars represent 10 μm.
(F) Knockdown of zebrafish ortholog of SPAG1, using splice blocking morpholino targeting splice acceptor site at the intron 4-exon 5 junction of zebrafish (5′-TCTGGCTGTGGAGAAAGTAGATACA-3′; QA accession # ZDB-GENE-030131-9443). Standard control morpholino (5′-CCTCTTACCTCAGTTACAATTTATA-3′; Gene Tools LLC) was injected to zebrafish, as control. Unlike controls (left), spag1 morpholino-injected zebrafish show tail curls (right) 72 hr after injection. Hydrocephalus is indicated by an arrow (right).
(G) Quantification of the zebrafish phenotype data shows increased incidence of dorsal curvature (p = 0.00017 by t test) and hydrocephalus (p = 0.00018 by t test); however, increase in incorrect heart looping (p = 0.17 by t test) was not significant (actual values shown in Table S8).
Error bars indicate ±1 SD from the mean of three replicates.
A monoclonal antibody targeting the COOH region of SPAG1 (kind gift of T. Crnogorac-Jurcevic29) (Figure S3) reacted strongly with two proteins in immunoblots of total cell lysates from fully ciliated cultures (Figure 3C) that were not detected in lysates from undifferentiated cells, again paralleling the expression of DNAI1. These proteins of ∼92 and 105 kDa are close to the expected size predicted for full-length SPAG1 (104 kDa) and may be the result of an alternative splicing event or posttranslational modification. In addition, the antibody detected a smaller protein of ∼50 kDa in immunoblots of total cell lysates from both the undifferentiated and ciliated airway epithelia; however, it is not clear whether this band represents an additional form of SPAG1 or whether this signal is due to cross-reactivity of the antibody. Interestingly, little or no SPAG1 was detected in isolated ciliary axonemes (Figure 3C), in contrast to the strong signal detected for DNAI1.33 This result agrees with our earlier proteomic analysis that did not identify SPAG1 as a component of isolated ciliary axonemes,34 indicating that SPAG1 is probably not an integral axonemal protein but plays a role in the cytoplasmic assembly of the ciliary dynein arms. To better understand the effects of SPAG1 mutations at the protein level, we performed immunofluorescence (IF) analysis in respiratory epithelia from the PCD individual (OP-94 [II-2]) with biallelic mutations in SPAG1. The IF studies were carried out with antibodies targeting the ODA heavy chain, DNAH5, and the IDA light chain component, DNALI1, as previously described.35,36 Both DNAH5 and DNALI1 were completely absent from the ciliary axonemes of this individual with PCD (Figures 3D and 3E), which is consistent with the ciliary ultrastructural analyses. We have previously shown that respiratory cilia have two distinct ODA types: (1) a proximally located ODA type 1 containing DNAH5 and probably DNAH11 (DNAH9 is absent) and (2) distally located ODA type 2 containing DNAH5 and DNAH9.35 In SPAG1 mutant cilia, the IF analysis revealed absence of both ODA types (Figure 3D), which is similar to findings reported for cytoplasmic proteins involved in preassembly of cilia (DNAAF1/LRRC50;13 DNAAF3;14 LRRC6 and ZMYND10 mutant cilia22). Thus, SPAG1 is probably involved in similar cytoplasmic preassembly processes as these four proteins. These findings differ from another cytoplasmic preassembly protein DNAAF2 (KTU) mutant cilia, where only the distal type 1 ODA is defective.15 The concept that SPAG1 is involved in preassembly of axonemal components is further supported by the absence of the IDA component DNALI1 in the axoneme of SPAG1 mutant cilia (Figure 3E). SPAG1 mutant cilia are different when compared to cilia with mutations in DNAH5, DNAI1, DNAI2 (ODA components), and ARMC4 (targeting ODA complexes), because these gene mutations cause isolated ODA defects without affecting the localization of IDA protein DNALI1.7,37
The zebrafish (D. rerio) ortholog spag1 (ZFIN database accession number ZDB-GENE-030131-9443) has 45% identity to SPAG1 at the protein level. To directly test the function of spag1 in zebrafish, antisense mopholinos, targeting the splice acceptor site at the junction of intron 4 and exon 5, were injected into embryos. Knockdown efficiency was confirmed by RT-PCR (data not shown). Mutant zebrafish exhibited dorsal body axis curvature (75% versus 8% in control) and hydrocephalus (66% versus 3% in control); however, heart looping defects showed only a modest increase (8% versus 4% in control) that was not significant (Figures 3F and 3G, Table S8). It is possible that the use of a different morpholino, or a translation-blocking morpholino, may show a significant increase in incorrect heart looping, but that was not possible to design because of the incomplete annotation of zebrafish spag1. These cilia-related phenotypes have previously been noted in several zebrafish models of PCD-causing mutations in genes that encode cytoplasmic proteins, including DNAAF3,14 LRRC50,38 and LRRC6.39,40
In the current study, three unrelated individuals underwent exome sequencing as the initial cohort, which led to the identification of mutations in SPAG1 in one individual with PCD. For the remaining two individuals with PCD and ODA+IDA defects who were exome sequenced, one individual (#SL0018) harbored a homozygous missense variant (c.2384C>T [p.Leu795Pro]) in HEATR2 (RefSeq NM_017802.3) (Tables S1 and S2). This individual belongs to a large Amish Mennonite cohort with multiple PCD-affected individuals, and the genetic and biological analysis of this missense mutation in HEATR2 as a cause of PCD have been published.11 In another individual (#158 [III-1]), contemporaneous studies by the candidate gene approach identified biallelic mutations in DYX1C1 (MIM 608706) (c.783+1G>T and 3,549 bp deletion), and targeted sequencing reveals the same genetic defects in an affected sibling (#159 [III-3]) as well.41 Further review of our exome genotype calls for DYX1C1 revealed that indeed the splice mutation was annotated; however, a second mutant allele (large deletion) was missed because of its size. Hence, our exome filtration analysis that assumes a recessive trait for PCD failed to identify DYX1C1 as a potential candidate gene in this pedigree. Interestingly, in addition to PCD, the proband (#158 [III-1]) also had an apparently autosomal-dominant retinitis pigmentosa (AD-RP [MIM 268000]), and exome data analysis identified a heterozygous nonsense mutation (c.2515G>T [p.Glu839∗]) in TOPORS (RefSeq NM_005802.4 [MIM 609507]) that has previously been implicated in AD-RP.42 This TOPORS mutation was not present in the PCD-affected sibling (#159 [III-3]) who did not have AD-RP, which revealed that AD-RP and PCD segregate independently (Figure S5). Previously, cosegregation of PCD and X-linked RP has been associated with the hemizygous mutations in RPGR (MIM 312610).43–45 However, our genetic analysis confirms that PCD and AD-RP in this family are caused by mutations in different genes (Figure S5). In conclusion, exome sequencing identified biallelic mutations in SPAG1 that cause PCD with ODA+IDA defects associated with severe ciliary dysmotility and laterality defects. SPAG1 was not observed in isolated ciliary axonemes and is probably involved in the cytoplasmic assembly or transport of the dynein arm complexes. Of the eight different mutant alleles identified in SPAG1, three were seen in at least two unrelated families, and haplotype analysis suggests that these are founder mutations in individuals with Northern European ancestry. Additionally, we identified a nonsense mutation in TOPORS as a cause of AD-RP. The identification of genetic causes of PCD in two families and AD-RP in one family via exome sequencing further demonstrates the power of this approach for identifying the molecular etiology in genetically heterogeneous disorders.
Acknowledgments
We thank individuals with PCD and family members, Michele Manion (founder of the US PCD Foundation), US PCD foundation, and German patient support group Kartagener Syndrome und Primaere Ciliaere Dyskinesie e.V. We thank investigators and the coordinators of the Genetic Disorders of Mucociliary Clearance Consortium that is part of the Rare Disease Clinical Research Network; Xueliang Guo, Kimberly Burns, Rhonda Pace, and Andrew Cutting from UNC; C. Westermann, D. Nergenau, M. Herting, and S. Weiser for technical assistance; Elizabeth Godwin for administrative support; and Syanne Olson for manuscript transcription. We thank T. Crnogorac-Jurcevic for providing the SPAG1 antibody, Scott Randell for access to unpublished RNA-seq data, and Hong Dang for bioinformatics assistance. For full listing, please see Supplemental Data.
Funding support for research was provided to M.R.K., M.W.L., J.L.C., M.J.H., S.D. Dell, S.D. Davis, T.W.F., S.D.S., K.N.O., and M.A.Z. by US NIH/ORDR/NHLBI grant 5U54HL096458-06; to M.R.K., L.E.O., and M.A.Z. by NIH-NHLBI grant 5R01HL071798; to J.S. and D.A.N. by NIH-NHLBI grant 5R01HL094976; to M.J.B. by NIH-NHLBI grant RC2 HL-102923; and to J.S. by NIH-NHGRI grant 5R21HG004749. K.N.O. is supported by the Intramural Research Program of NIH-NIAID. Resequencing was provided through RS&G by NIH-NHLBI contract # HHSN268201100037C. The work was supported by NIH-NCATS grants UL1 TR000083 to UNC-CH and UL1 TR000154 to Colorado CTSI and CF foundation grant CFF R026-CR07. H. Omran is supported by funding from DFG Om6/4, IZKF Muenster Om2/009/12, and BESTCILIA and SYSCILIA from the European Community. F.H. is an investigator of the Howard Hughes Medical Institute.
Contributor Information
Michael R. Knowles, Email: knowles@med.unc.edu.
Maimoona A. Zariwala, Email: zariwala@med.unc.edu.
Supplemental Data
Cilia show nearly complete immotility and stiffness consistent with PCD.
Cilia show nearly complete immotility and stiffness consistent with PCD.
Cilia show normal motility and waveform.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Ensembl Genome Browser, http://www.ensembl.org/index.html
GeneReviews, Zariwala, M., Knowles, M., and Leigh, M.W. (2012). Primary ciliary dyskinesia, http://www.ncbi.nlm.nih.gov/books/NBK1122
Genetic Disorders of Mucociliary Clearance Consortium (GDMCC), http://rarediseasesnetwork.epi.usf.edu/gdmcc/index.htm
MutationTaster, http://www.mutationtaster.org/
NCBI Expressed Sequence Tags database (dbEST), http://www.ncbi.nlm.nih.gov/dbEST/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
NHLBI Resequencing and Genotyping Service (RS&G), http://rsng.nhlbi.nih.gov/scripts/index.cfm
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
SeattleSeq Annotation 137, http://snp.gs.washington.edu/SeattleSeqAnnotation137/
UCLA Gene Expression Tool (UGET), http://genome.ucla.edu/∼jdong/GeneCorr.html
UCSC Genome Browser, http://genome.ucsc.edu
ZFIN, http://zfin.org
References
- 1.Fliegauf M., Benzing T., Omran H. When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 2.Zariwala M.A., Knowles M.R., Omran H. Genetic defects in ciliary structure and function. Annu. Rev. Physiol. 2007;69:423–450. doi: 10.1146/annurev.physiol.69.040705.141301. [DOI] [PubMed] [Google Scholar]
- 3.Leigh M.W., Pittman J.E., Carson J.L., Ferkol T.W., Dell S.D., Davis S.D., Knowles M.R., Zariwala M.A. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet. Med. 2009;11:473–487. doi: 10.1097/GIM.0b013e3181a53562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noone P.G., Leigh M.W., Sannuti A., Minnix S.L., Carson J.L., Hazucha M., Zariwala M.A., Knowles M.R. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am. J. Respir. Crit. Care Med. 2004;169:459–467. doi: 10.1164/rccm.200303-365OC. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy M.P., Omran H., Leigh M.W., Dell S., Morgan L., Molina P.L., Robinson B.V., Minnix S.L., Olbrich H., Severin T. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation. 2007;115:2814–2821. doi: 10.1161/CIRCULATIONAHA.106.649038. [DOI] [PubMed] [Google Scholar]
- 6.Bush A., Chodhari R., Collins N., Copeland F., Hall P., Harcourt J., Hariri M., Hogg C., Lucas J., Mitchison H.M. Primary ciliary dyskinesia: current state of the art. Arch. Dis. Child. 2007;92:1136–1140. doi: 10.1136/adc.2006.096958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hjeij R., Lindstrand A., Francis R., Zariwala M.A., Liu X., Li Y., Damerla R., Dougherty G.W., Abouhamed M., Olbrich H. ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am. J. Hum. Genet. 2013;93:357–367. doi: 10.1016/j.ajhg.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wirschell M., Olbrich H., Werner C., Tritschler D., Bower R., Sale W.S., Loges N.T., Pennekamp P., Lindberg S., Stenram U. The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nat. Genet. 2013;45:262–268. doi: 10.1038/ng.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knowles M.R., Leigh M.W., Ostrowski L.E., Huang L., Carson J.L., Hazucha M.J., Yin W., Berg J.S., Davis S.D., Dell S.D., Genetic Disorders of Mucociliary Clearance Consortium Exome sequencing identifies mutations in CCDC114 as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2013;92:99–106. doi: 10.1016/j.ajhg.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Onoufriadis A., Paff T., Antony D., Shoemark A., Micha D., Kuyt B., Schmidts M., Petridi S., Dankert-Roelse J.E., Haarman E.G., UK10K Splice-site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2013;92:88–98. doi: 10.1016/j.ajhg.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horani A., Druley T.E., Zariwala M.A., Patel A.C., Levinson B.T., Van Arendonk L.G., Thornton K.C., Giacalone J.C., Albee A.J., Wilson K.S. Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2012;91:685–693. doi: 10.1016/j.ajhg.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kott E., Duquesnoy P., Copin B., Legendre M., Dastot-Le Moal F., Montantin G., Jeanson L., Tamalet A., Papon J.F., Siffroi J.P. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2012;91:958–964. doi: 10.1016/j.ajhg.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loges N.T., Olbrich H., Becker-Heck A., Häffner K., Heer A., Reinhard C., Schmidts M., Kispert A., Zariwala M.A., Leigh M.W. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009;85:883–889. doi: 10.1016/j.ajhg.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchison H.M., Schmidts M., Loges N.T., Freshour J., Dritsoula A., Hirst R.A., O’Callaghan C., Blau H., Al Dabbagh M., Olbrich H. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 2012;44:381–389. doi: 10.1038/ng.1106. S1–S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Omran H., Kobayashi D., Olbrich H., Tsukahara T., Loges N.T., Hagiwara H., Zhang Q., Leblond G., O’Toole E., Hara C. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duquesnoy P., Escudier E., Vincensini L., Freshour J., Bridoux A.M., Coste A., Deschildre A., de Blic J., Legendre M., Montantin G. Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2009;85:890–896. doi: 10.1016/j.ajhg.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Roak B.J., Deriziotis P., Lee C., Vives L., Schwartz J.J., Girirajan S., Karakoc E., Mackenzie A.P., Ng S.B., Baker C. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Day A., Carlson M.R., Dong J., O’Connor B.D., Nelson S.F. Celsius: a community resource for Affymetrix microarray data. Genome Biol. 2007;8:R112. doi: 10.1186/gb-2007-8-6-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Day A., Dong J., Funari V.A., Harry B., Strom S.P., Cohn D.H., Nelson S.F. Disease gene characterization through large-scale co-expression analysis. PLoS ONE. 2009;4:e8491. doi: 10.1371/journal.pone.0008491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zariwala M.A., Gee H.Y., Kurkowiak M., Al-Mutairi D.A., Leigh M.W., Hurd T.W., Hjeij R., Dell S.D., Chaki M., Dougherty G.W. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. Am. J. Hum. Genet. 2013;93:336–345. doi: 10.1016/j.ajhg.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hornef N., Olbrich H., Horvath J., Zariwala M.A., Fliegauf M., Loges N.T., Wildhaber J., Noone P.G., Kennedy M., Antonarakis S.E. DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am. J. Respir. Crit. Care Med. 2006;174:120–126. doi: 10.1164/rccm.200601-084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olbrich H., Häffner K., Kispert A., Völkel A., Volz A., Sasmaz G., Reinhardt R., Hennig S., Lehrach H., Konietzko N. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 2002;30:143–144. doi: 10.1038/ng817. [DOI] [PubMed] [Google Scholar]
- 25.Zariwala M.A., Leigh M.W., Ceppa F., Kennedy M.P., Noone P.G., Carson J.L., Hazucha M.J., Lori A., Horvath J., Olbrich H. Mutations of DNAI1 in primary ciliary dyskinesia: evidence of founder effect in a common mutation. Am. J. Respir. Crit. Care Med. 2006;174:858–866. doi: 10.1164/rccm.200603-370OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zariwala M., Noone P.G., Sannuti A., Minnix S., Zhou Z., Leigh M.W., Hazucha M., Carson J.L., Knowles M.R. Germline mutations in an intermediate chain dynein cause primary ciliary dyskinesia. Am. J. Respir. Cell Mol. Biol. 2001;25:577–583. doi: 10.1165/ajrcmb.25.5.4619. [DOI] [PubMed] [Google Scholar]
- 27.Blatch G.L., Lässle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays. 1999;21:932–939. doi: 10.1002/(SICI)1521-1878(199911)21:11<932::AID-BIES5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 28.Takaishi M., Huh Nh. A tetratricopeptide repeat-containing protein gene, tpis, whose expression is induced with differentiation of spermatogenic cells. Biochem. Biophys. Res. Commun. 1999;264:81–85. doi: 10.1006/bbrc.1999.1477. [DOI] [PubMed] [Google Scholar]
- 29.Neesse A., Gangeswaran R., Luettges J., Feakins R., Weeks M.E., Lemoine N.R., Crnogorac-Jurcevic T. Sperm-associated antigen 1 is expressed early in pancreatic tumorigenesis and promotes motility of cancer cells. Oncogene. 2007;26:1533–1545. doi: 10.1038/sj.onc.1209961. [DOI] [PubMed] [Google Scholar]
- 30.Fulcher M.L., Gabriel S., Burns K.A., Yankaskas J.R., Randell S.H. Well-differentiated human airway epithelial cell cultures. Methods Mol. Med. 2005;107:183–206. doi: 10.1385/1-59259-861-7:183. [DOI] [PubMed] [Google Scholar]
- 31.Gray T.E., Guzman K., Davis C.W., Abdullah L.H., Nettesheim P. Mucociliary differentiation of serially passaged normal human tracheobronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1996;14:104–112. doi: 10.1165/ajrcmb.14.1.8534481. [DOI] [PubMed] [Google Scholar]
- 32.Kultgen P.L., Byrd S.K., Ostrowski L.E., Milgram S.L. Characterization of an A-kinase anchoring protein in human ciliary axonemes. Mol. Biol. Cell. 2002;13:4156–4166. doi: 10.1091/mbc.E02-07-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ostrowski L.E., Yin W., Rogers T.D., Busalacchi K.B., Chua M., O’Neal W.K., Grubb B.R. Conditional deletion of dnaic1 in a murine model of primary ciliary dyskinesia causes chronic rhinosinusitis. Am. J. Respir. Cell Mol. Biol. 2010;43:55–63. doi: 10.1165/rcmb.2009-0118OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ostrowski L.E., Blackburn K., Radde K.M., Moyer M.B., Schlatzer D.M., Moseley A., Boucher R.C. A proteomic analysis of human cilia: identification of novel components. Mol. Cell. Proteomics. 2002;1:451–465. doi: 10.1074/mcp.m200037-mcp200. [DOI] [PubMed] [Google Scholar]
- 35.Fliegauf M., Olbrich H., Horvath J., Wildhaber J.H., Zariwala M.A., Kennedy M., Knowles M.R., Omran H. Mislocalization of DNAH5 and DNAH9 in respiratory cells from patients with primary ciliary dyskinesia. Am. J. Respir. Crit. Care Med. 2005;171:1343–1349. doi: 10.1164/rccm.200411-1583OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rashid S., Breckle R., Hupe M., Geisler S., Doerwald N., Neesen J. The murine Dnali1 gene encodes a flagellar protein that interacts with the cytoplasmic dynein heavy chain 1. Mol. Reprod. Dev. 2006;73:784–794. doi: 10.1002/mrd.20475. [DOI] [PubMed] [Google Scholar]
- 37.Loges N.T., Olbrich H., Fenske L., Mussaffi H., Horvath J., Fliegauf M., Kuhl H., Baktai G., Peterffy E., Chodhari R. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am. J. Hum. Genet. 2008;83:547–558. doi: 10.1016/j.ajhg.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Rooijen E., Giles R.H., Voest E.E., van Rooijen C., Schulte-Merker S., van Eeden F.J. LRRC50, a conserved ciliary protein implicated in polycystic kidney disease. J. Am. Soc. Nephrol. 2008;19:1128–1138. doi: 10.1681/ASN.2007080917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun Z., Amsterdam A., Pazour G.J., Cole D.G., Miller M.S., Hopkins N. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development. 2004;131:4085–4093. doi: 10.1242/dev.01240. [DOI] [PubMed] [Google Scholar]
- 40.Kishimoto N., Cao Y., Park A., Sun Z. Cystic kidney gene seahorse regulates cilia-mediated processes and Wnt pathways. Dev. Cell. 2008;14:954–961. doi: 10.1016/j.devcel.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 41.Tarkar A., Loges N.T., Slagle C.E., Francis R., Dougherty G.W., Tamayo J.V., Shook B., Cantino M., Schwartz D., Jahnke C., UK10K DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat. Genet. 2013 doi: 10.1038/ng.2707. Published online July 21, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chakarova C.F., Papaioannou M.G., Khanna H., Lopez I., Waseem N., Shah A., Theis T., Friedman J., Maubaret C., Bujakowska K. Mutations in TOPORS cause autosomal dominant retinitis pigmentosa with perivascular retinal pigment epithelium atrophy. Am. J. Hum. Genet. 2007;81:1098–1103. doi: 10.1086/521953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moore A., Escudier E., Roger G., Tamalet A., Pelosse B., Marlin S., Clément A., Geremek M., Delaisi B., Bridoux A.M. RPGR is mutated in patients with a complex X linked phenotype combining primary ciliary dyskinesia and retinitis pigmentosa. J. Med. Genet. 2006;43:326–333. doi: 10.1136/jmg.2005.034868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bukowy-Bieryłło Z., Ziętkiewicz E., Loges N.T., Wittmer M., Geremek M., Olbrich H., Fliegauf M., Voelkel K., Rutkiewicz E., Rutland J. RPGR mutations might cause reduced orientation of respiratory cilia. Pediatr. Pulmonol. 2013;48:352–363. doi: 10.1002/ppul.22632. [DOI] [PubMed] [Google Scholar]
- 45.Zito I., Downes S.M., Patel R.J., Cheetham M.E., Ebenezer N.D., Jenkins S.A., Bhattacharya S.S., Webster A.R., Holder G.E., Bird A.C. RPGR mutation associated with retinitis pigmentosa, impaired hearing, and sinorespiratory infections. J. Med. Genet. 2003;40:609–615. doi: 10.1136/jmg.40.8.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cilia show nearly complete immotility and stiffness consistent with PCD.
Cilia show nearly complete immotility and stiffness consistent with PCD.
Cilia show normal motility and waveform.