

Abstract
Anophthalmia and/or microphthalmia, pulmonary hypoplasia, diaphragmatic hernia, and cardiac defects are the main features of PDAC syndrome. Recessive mutations in STRA6, encoding a membrane receptor for the retinol-binding protein, have been identified in some cases with PDAC syndrome, although many cases have remained unexplained. Using whole-exome sequencing, we found that two PDAC-syndrome-affected siblings, but not their unaffected sibling, were compound heterozygous for nonsense (c.355C>T [p.Arg119∗]) and frameshift (c.1201_1202insCT [p.Ile403Serfs∗15]) mutations in retinoic acid receptor beta (RARB). Transfection studies showed that p.Arg119∗ and p.Ile403Serfs∗15 altered RARB had no transcriptional activity in response to ligands, confirming that the mutations induced a loss of function. We then sequenced RARB in 15 subjects with anophthalmia and/or microphthalmia and at least one other feature of PDAC syndrome. Surprisingly, three unrelated subjects with microphthalmia and diaphragmatic hernia showed de novo missense mutations affecting the same codon; two of the subjects had the c.1159C>T (Arg387Cys) mutation, whereas the other one carried the c.1159C>A (p.Arg387Ser) mutation. We found that compared to the wild-type receptor, p.Arg387Ser and p.Arg387Cys altered RARB induced a 2- to 3-fold increase in transcriptional activity in response to retinoic acid ligands, suggesting a gain-of-function mechanism. Our study thus suggests that both recessive and dominant mutations in RARB cause anophthalmia and/or microphthalmia and diaphragmatic hernia, providing further evidence of the crucial role of the retinoic acid pathway during eye development and organogenesis.
Main Text
Anophthalmia or microphthalmia (A/M) refers to the absence or reduced size of the axial diameter of the globe in the ocular orbit, respectively. In over 50% of cases, A/M is associated with other congenital abnormalities.1,2 The combination of pulmonary hypoplasia or agenesis, diaphragmatic hernia or eventration, A/M, and cardiac defects is characteristic of PDAC syndrome, which is also known as Matthew-Wood syndrome or Spear syndrome (MIM 601186).3 In some individuals, PDAC syndrome is caused by autosomal-recessive mutations in STRA6 (MIM 610745), encoding a membrane receptor for the retinol-binding protein.4,5 Many cases of PDAC syndrome, however, remain unexplained.
In this study, we performed whole-exome sequencing in a nonconsanguineous family affected by PDAC syndrome to uncover the genetic cause (Figure 1A). The parents have two healthy daughters and four affected children, of whom two have all the features of PDAC syndrome and two have at least two features of PDAC syndrome (individuals II-1, II-4, II-5, and II-6 in family A, see Table 1 for clinical details). This family was described previously (cases 1A [II-4], 2A [II-5], and 3A [II-6] from family A in Chitayat et al.3). STRA6 was previously sequenced in one affected individual of this family, and no mutation was identified.
Figure 1.
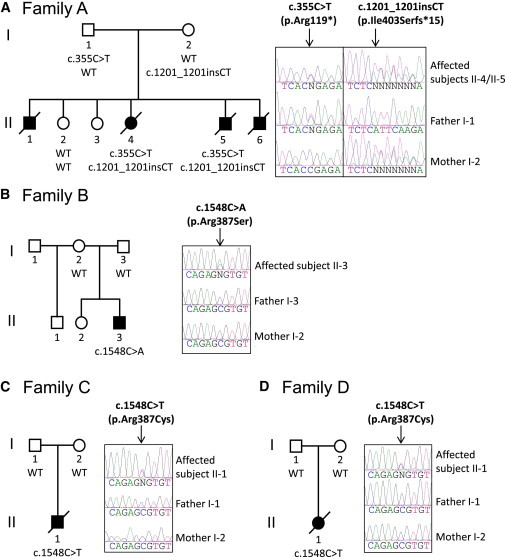
Mutations in RARB in Individuals with PDAC Syndrome
Sanger sequencing confirmed segregation of the recessive mutations in RARB in family A (A) and revealed that the mutations were de novo in families B (B), C (C), and D (D).
Table 1.
Clinical Characteristics of Subjects with Mutations in RARB
|
Family A |
Family B |
Family C |
Family D |
||||
|---|---|---|---|---|---|---|---|
| II-1 | II-4 | II-5 | II-6 | II-3 | II-1 | II-1 | |
| Ethnicity | French Canadian and English | French | African (Angola [father] and Congo [mother]) | French | |||
| Consanguinity | − | − | − | − | − | − | − |
| Mutation(s) | NA | c.355C>T (p.Arg119∗) and c.1201_1202insCT (p.Ile403Serfs∗15) | c.355C>T (p. Arg119∗) and c.1201_1202insCT (p.Ile403Serfs∗15) | NA | c.1159 > A (p.Arg387Ser)a | c.1159C>T (p.Arg387Cys)a | c.1159C>T (p.Arg387Cys)a |
| Gender | male | female | male | male | male | male | female |
| Age (age at death) | (few hours; 34 weeks of gestation)b | (few hours; 38 weeks of gestation) | (21-week-old fetus)c | (few hours; term) | 16 years | (34-week-old fetus)c | (few hours; 39 weeks of gestation) |
| Bilateral A/M | + | + | + | + | + | unilateral (left) microphthalmia; normal right eye | bilateral microphthalmia |
| Pulmonary hypoplasia | NA | + | + | NA | − | left lung with one hypoplastic lobe; normal right lung | bilateral (predominant on the left side) |
| Diaphragmatic hernia | NA | + | + | + | + | + (left) | + (left) |
| Cardiac abnormality | NA | + | − | NA | − | − | − |
| Intellectual disability | NA | NA | NA | NA | + | NA | NA |
| Other | − | cleft palate, dysmorphisms, small spleen, bicornate and small uterus | dysmorphisms, unfixed malrotated bowel | dysmorphisms | − | malrotated bowel, right cryptorchydy, mild IUGR (weight and length), and OFC at the fifth percentile | bicornate uterus |
Abbreviations are as follows: NA, not available; IUGR, intrauterine growth restriction; and OFC, occipitofrontal circumference.
De novo mutation.
Died shortly after birth at 34 weeks as a result of a presumed tangled cord, although a diagnosis of PDAC was strongly suspected.
Terminated pregnancy.
This study was approved by our institutional ethics committee, and informed consent was obtained from each participant or legal guardian. Blood genomic DNA from affected individuals II-4 and II-5 and the unaffected sister (II-2) from family A was captured with the Agilent SureSelect Human All Exon Capture V4 Kit and sequenced (two paired-end 100 bp reads, three exomes per lane) with Illumina HiSeq2000 at the McGill University Genome Quebec Innovation Center (Montreal). Sequence processing, alignment (with a Burrows-Wheeler algorithm), and variant calling were done according to the Broad Institute Genome Analysis Toolkit (GATK v.4) best practices, and variant annotation was done with ANNOVAR.6 The average exome coverage of the target bases was 111–114×, and 95% of the target bases were covered by at least 20 reads. Only the variants whose positions were covered ≥8× and supported by at least three variant reads constituting at least 20% of the total reads for each called position were retained. To identify potentially pathogenic variants, we filtered out (1) synonymous variants or intronic variants other than those affecting the consensus splice sites, (2) variants seen in more than 2% of our in-house exomes (n = 1,000) from unrelated projects, and (3) variants with a minor allele frequency greater than 0.5% in either the 1000 Genomes Project or the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server (EVS). The affected individuals did not share any rare coding or splicing variants in STRA6 or ALDH1A3 (MIM 600463), another gene involved in A/M and retinoic acid (RA) signaling.7
Given that transmission of the phenotype in this family was consistent with autosomal-recessive inheritance, we searched the whole-exome data sets for genes harboring homozygous or multiple rare variants in both affected probands, but not in their unaffected sibling. There were no such genes with rare homozygous variants. Only three genes containing multiple rare variants in both affected individuals were not shared by the unaffected sister: PRPF39 (MIM 614907), RARB (MIM 180220), and GAPVD1 (MIM 611714). Sanger sequencing in the parents and the siblings revealed that the variants in PRPF39 were inherited in cis, thus excluding this gene as a candidate. The affected probands, but not the unaffected sister, were compound heterozygous and the parents were singly heterozygous for the variants in RARB and GAPVD1 (Figure 1A). RARB (RefSeq accession number NM_000965.3), which encodes RA receptor beta (RARB), was found to harbor two protein-truncating mutations: nonsense c.355C>T (p.Arg119∗) and frameshift c.1201_1202insCT (p.Ile403Serfs∗15). These variants are absent from all public SNP databases (1000 Genomes, EVS, and dbSNP138) and from our in-house exomes (n > 1,000). RARB has two major isoforms noted in the UCSC Genome Browser and three additional isoforms noted in the Ensembl Genome Browser. The major RefSeq isoform (RefSeq NM_000965.3) has eight exons and encodes a 448 aa protein. Both identified mutations affect all known RARB isoforms (Figure 2A). GAPVD1 (RefSeq NM_015635), which encodes GTPase-activating protein and VPS9 domain 1, was found to harbor two rare missense variants, c.2809C>T (p.Arg937Trp) and c.3266G>T (p.Gly1089Val), in the probands. The p.Arg937Trp substitution is predicted to be damaging by both SIFT (score 0.0) and PolyPhen-2 (score 1.0), but the p.Gly1089Val substitution is predicted to be benign by both SIFT (score 0.2) and PolyPhen-2 (score 0.145). GAPVD1 is involved in endocytosis,8 phagosome maturation,9 and regulation of the epidermal growth factor receptor10 but is not known to have a role in eye development or embryogenesis. Because of the importance of the RA pathway for eye and diaphragm development (discussed below), mutations in RARB were deemed more likely to be pathogenic than those in GAPVD1.
Figure 2.
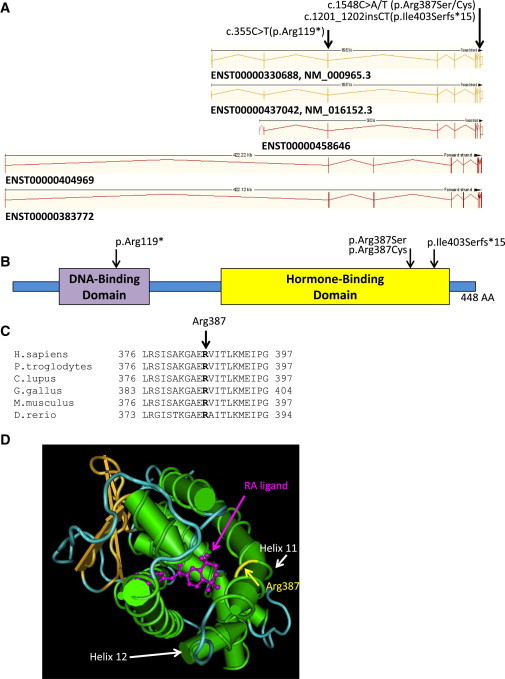
Localization and Impact of the Mutations in RARB
(A) Shown are the positions of the mutations with respect to the different RARB Ensembl-annotated transcripts that are predicted to produce proteins. Numbering on top is based on the cDNA positions of Ensembl ENST00000330688 (identical to RefSeq NM_000965.3).
(B) A schematic of RARB shows the DNA-binding and hormone-binding domains. Arrowheads above the protein show the positions of the variants.
(C) The HomoloGene-generated amino acid alignment of human RARB and its predicted orthologs shows the conservation of the p.Arg387 residue.
(D) The three-dimensional structure of RARB (Protein Data Bank ID 4DM8) in the presence of the RA ligand (purple) shows the proximity of the Arg387 residue in helix 11 to the RA ligand.
RA receptors bind to DNA motifs known as RA response elements (RAREs) to modulate transcription of target genes by interacting with transcriptional corepressors and coactivators. Upon binding to RA, the corepressor docking site becomes hindered by helix 12 positioning, resulting in the recruitment of coactivators and an increase in transcription of target genes.11 The c.355C>T (p.Arg119∗) nonsense mutation is predicted to result in an inactive truncated receptor lacking the second zinc finger of the DNA-binding domain and the entire ligand-binding domain (Figure 2B). Moreover, this mutation has the potential to activate the nonsense-mediated mRNA decay pathway, resulting in the degradation of the corresponding transcript. The c.1201_1202insCT (p.Ile403Serfs∗15) mutation results in the substitution of a hydrophobic isoleucine with serine, a polar residue, and the replacement of the last 52 amino acids with an aberrant extension of 15 amino acids (Figure 2B). As part of helix 12, residue Ile403 is thus predicted to play a key role in the recruitment of transcriptional cofactors and response to ligand. As such, in vitro studies have shown that Ile403 substitution with serine in RARB confers an increased binding to corepressor SMRT in the absence of ligand and thus results in reduced transcriptional activity.12 Both mutations found in family A are thus predicted to disrupt RARB function.
We tested the impact of these truncating mutations on RARB activity by using a cellular one-hybrid luciferase-reporter transcriptional assay as previously described.13 Expression plasmids encoding transcription factor Gal4 DNA-binding domain fusions to wild-type RARB or truncated variants p.Arg119∗ and p.Ile403Serfs∗15 were generated and used for transfecting human embryonic kidney 293 (HEK293) cells in the presence of a luciferase reporter gene construct under the control of a Gal4-binding DNA upstream-activating sequence (UAStkLuc). This assay allowed transcriptional activity and response to ligand to be directly compared between variants and the wild-type receptor, avoiding any background effect of endogenously expressed RARB in cells. Transfected HEK293 cells were treated with the natural ligands all-trans RA (atRA) and its stereoisomer, 9-cis RA, which both act as pan-RAR agonists. As expected, compared to that of wild-type RARB, the transcriptional response of the p.Arg119∗ variant to the two RA ligands was completely abolished, correlating with its lack of ligand-binding domain (Figure 3). Similarly, compared to wild-type RARB, the p.Ile403Serfs∗15 variant showed an impaired transcriptional response to retinoids. In the p.Ile403Serfs∗15 variant, the disruption of helix 12 and its replacement with an aberrant extension most likely interfered with its ability to occlude the corepressor docking site. All together, these results strongly suggest that these truncating variants confer a loss of RARB function and explain the occurrence of PDAC syndrome in family A.
Figure 3.
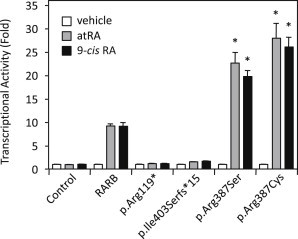
Transcriptional Response of Human RARB Variants to RA Ligands
HEK293 cells were seeded in 24-well plates and transfected with 100 ng per well of expression plasmid encoding either Gal4 fusion of wild-type human RARB or p.Arg119∗, p.Ile403Serfs∗15, p.Arg387Ser, or p.Arg387Cys RARB variants in the presence of 500 ng of UAStkLuc reporter-gene construct. All variant constructs, including p.Ile403Serfs∗15 (carrying the additional out-of-frame amino acid extension), were generated by site-directed mutagenesis. Cells were treated with 1 μM atRA, 1 μM 9-cis RA, or vehicle (DMSO; 1/1,000, v/v) for 16 hr. Luciferase values were normalized to β-galactosidase activity and expressed as a fold response compared to those of empty Gal4-transfected control cells. Data were derived from three independent experiments performed in triplicate. ∗p < 0.005 versus wild-type RARB response to the respective RA ligand. Values represent means, and error bars represent SEs.
We next sequenced all the coding exons and intron-exon boundaries of RARB and the alternate exon 1 (Ensembl accession number ENST00000404969) in 15 additional individuals who had bilateral or unilateral A/M and at least one additional feature of PDAC (diaphragmatic hernia, cardiac defect, or lung hypoplasia) and who had been previously screened for mutations in STRA6.14 In three unrelated subjects, who were all simplex cases, we identified single heterozygous RARB missense mutations affecting the same nucleotide. Of these, two subjects (II-1 from family C and II-1 from family D) harbored missense mutation c.1159C>T (p.Arg387Cys) and one individual (II-3 from family B) harbored missense mutation c.1159C>A (p.Arg387Ser) (Figures 1B and 1C). These mutations were absent from the genomic DNA of the parents, indicating that they occurred de novo. Using six informative unlinked microsatellite markers (D3S1754, D4S3351, D8S1179, D15S659, D14S587, and D19S215), we confirmed the paternity and maternity in these families, as previously described.15 Both c.1159C>T (p.Arg387Cys) and c.1159C>A (p.Arg387Ser) are absent from public SNP databases (EVS, 1000 Genomes, and dbSNP138) and from our entire collection of in-house exomes (n >1,000). They are both predicted to be damaging by SIFT (score 0.0) and PolyPhen-2 (score 1.0) and affect a highly conserved amino acid in helix 11 of the ligand-binding domain (Figure 2C). Subject II-1 from family C was a fetus for whom pregnancy was terminated because of unilateral microphthalmia and left diaphragmatic hernia on prenatal ultrasound (Table 1). Autopsy also showed hypoplasia of a pulmonary lobe on the left side. Subject II-1 from family D was a newborn who passed away within a few hours after birth because of a left diaphragmatic hernia. This subject also showed bilateral microphthalmia and pulmonary hypoplasia. Subject II-3 from family B is currently a 14-year-old male with bilateral microphthalmia, corrected diaphragmatic hernia, and abnormal cognitive development with spasticity (for clinical details, see case 6 in Chitayat et al.3). We also sequenced RARB in 11 cases with isolated bilateral A/M, but we did not find any mutation in this gene.
The fact that the de novo mutations involved the same residue (Arg387) suggests that they confer a specific property to the protein. These mutations could induce a dominant-negative effect or act through a gain-of-function mechanism. In order to distinguish between these possibilities, we sought to study the impact of these mutations by using our one-hybrid functional assay. The p.Arg387Ser and p.Arg387Cys altered RARB exhibited a significant increase in their transcriptional response to atRA; they reached 23- and 28-fold induction, respectively, compared to 9-fold induction for wild-type RARB (Figure 3). Similar activation levels were also obtained with the 9-cis RA ligand. These results suggest that the two variants at Arg387 provide increased transcriptional potential to respond to retinoid ligands through a gain-of-function mechanism. Explaining such an increase in activity will require a more detailed mechanistic analysis.
We have identified compound-heterozygous truncating mutations and de novo mutations affecting the same RARB nucleotide in individuals with PDAC syndrome. The occurrence of such mutations in individuals with a similar and rare phenotype strongly suggests that they cause PDAC syndrome. Indeed, several observations indicate that the RA pathway plays a major role in the development of the eyes, diaphragm, and lungs. RA is a metabolite of retinol, a derivative of vitamin A. The importance of the RA pathway in embryogenesis has been recognized for decades, given that rats deficient in vitamin A give birth to progeny with multiple congenital malformations, including ocular abnormalities and diaphragmatic hernia.16 Circulating retinol is bound to retinol-binding protein 4 (RBP4). The transmembrane protein STRA6 (stimulated by RA) facilitates the intracellular uptake of the retinol-RBP complex.4 Mutations in STRA6 have been identified in at least 24 individuals with A/M.5,14,17–19 Most of these subjects showed other features of PDAC syndrome, but some of them had isolated A/M. Once transported into the cell, retinol is successively oxidized to retinaldehyde and RA. Mutations in ALDH1A3, which encodes a retinaldehyde dehydrogenase that is responsible for the oxidation of retinaldehyde into RA, have been found to be responsible for A/M with variable neurodevelopmental and cardiac features.7
In target cells, RA acts as a ligand for nuclear RA receptors. Several observations in mice suggest that these receptors play a major role in eye, diaphragm, and lung development. Mice lacking all Rarb isoforms display microphthalmia.20,21 RA is generated in the epithelial ocular compartment and diffuses in the neural-crest-cell-derived periocular mesenchyme to activate RARB and RARG. In turn, these receptors regulate the remodeling of the periocular mesenchyme, the growth of the ventral retina, and the expression of Foxc1 and Pitx2, which play central roles in the development of the anterior eye segment.22 Studies of mutant mice lacking both Rara and Rarb subtypes have also demonstrated the presence of diaphragmatic hernias in a subset of offspring.23 Moreover, administration of nitrofen to pregnant rodents is thought to cause diaphragmatic hernias, in part through downregulation of RA receptor signaling (reviewed in Greer et al.24). Recent studies have indicated that Rarb functions along a pathway that directs development of the central tendon of the diaphragm.25 Finally, Rarb−/− mice have smaller and more numerous alveoli in their lungs.26 Rarb has been shown to have a critical role in lung morphogenesis by inducing Fgf10 expression in bud fields.27 Overall, these studies support our conclusion that loss of RARB function causes PDAC syndrome in family A.
Our transfection experiments suggest that the de novo mutations affecting Arg387 result in enhanced activity of RARB in response to RA ligands, suggesting a gain-of-function mechanism. Whether these two substitutions induce a conformational change that enhances protein stability, favors coactivator recruitment, and/or increases RA binding affinity remains to be determined. Consistent with this latter possibility, crystallographic studies have established that the Arg387 residue is facing inward relative to the ligand-binding pocket, in close proximity to the retinoid ligand (Figure 2D). Our model of gain-of-function mutations thus suggests that an increase in RARB response to retinoids might represent a primary cause of PDAC syndrome. Indeed, excess of vitamin A or RA during development in mice causes various malformations, including microphthalmia and diaphragmatic hernia.28–32 Moreover, expression of a constitutively active RAR transgene in the developing eye results in animals that exhibit microphthalmia.33 Similarly, zebrafish embryos exposed to 9-cis-RA develop multiple developmental abnormalities, including microphthalmia.34 RA is also teratogenic in humans.35 Interestingly, microphthalmia and diaphragmatic hernia have been reported in some babies from women exposed to RA during pregnancy.35,36
All together, our findings and the previous work described above therefore suggest that both decreased and increased RARB activity can result in PDAC syndrome. A recent study showed that RA exposure during embryonic development in mice was followed by decreased levels of Raldh transcripts encoding RA-synthesizing enzymes and increased levels of Cyp26a1 and Cyp26b1, mRNAs encoding enzymes that catabolize RA.32 Overall, these changes resulted in a decrease in RA levels. Restoration of RA levels by maternal supplementation with low doses of RA after the teratogenic insult rescued several developmental defects. Paradoxically, increased RARB signaling could thus result in a secondary state of RA deficiency, which could have an impact on this pathway at specific stages of development. Alternatively, it is possible that some developmental processes require a tight regulation of RARB targets, given that too little or too much signaling has the same consequence on these pathways.
In summary, we found that both recessive and dominant RARB mutations affect RARB function in the context of PDAC syndrome, opening a new window on the structural and mechanistic basis of RA receptor activity as it relates to human disease.
Acknowledgments
J.L.M. is a National Scientist of the Fonds de Recherche du Québec - Santé. M.S. holds a clinician-scientist award from the Canadian Institute for Health Research. We wish to thank the members of the bioinformatic analysis team of Réseau de Médecine Génétique Appliquée du Québec (Alexandre Dionne-Laporte, Dan Spiegelman, Edouard Henrion, and Ousmane Diallo) for the analysis of the exome sequencing data. This research was funded by a grant from the Canadian Institutes of Health Research (MOP 106499) to J.L.M. and by grants from the Clinical Research Hospital Program from the French Ministry of Health (PHRC 09 109 01) and Retina France to N.C.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://browser.1000genomes.org/index.html
Ensembl Genome Browser, http://www.ensembl.org/index.html
GATK Best Practices, http://www.broadinstitute.org/gatk/guide/topic?name=best-practices
HomoloGene (NCBI), http://www.ncbi.nlm.nih.gov/homologene
NHLBI Exome Sequencing Project (ESP) Exome Variant Server (EVS), http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Protein Data Bank, http://www.rcsb.org/pdb
SIFT, http://sift.jcvi.org/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Shaw G.M., Carmichael S.L., Yang W., Harris J.A., Finnell R.H., Lammer E.J. Epidemiologic characteristics of anophthalmia and bilateral microphthalmia among 2.5 million births in California, 1989-1997. Am. J. Med. Genet. A. 2005;137:36–40. doi: 10.1002/ajmg.a.30840. [DOI] [PubMed] [Google Scholar]
- 2.Källén B., Robert E., Harris J. The descriptive epidemiology of anophthalmia and microphthalmia. Int. J. Epidemiol. 1996;25:1009–1016. doi: 10.1093/ije/25.5.1009. [DOI] [PubMed] [Google Scholar]
- 3.Chitayat D., Sroka H., Keating S., Colby R.S., Ryan G., Toi A., Blaser S., Viero S., Devisme L., Boute-Bénéjean O. The PDAC syndrome (pulmonary hypoplasia/agenesis, diaphragmatic hernia/eventration, anophthalmia/microphthalmia, and cardiac defect) (Spear syndrome, Matthew-Wood syndrome): report of eight cases including a living child and further evidence for autosomal recessive inheritance. Am. J. Med. Genet. A. 2007;143A:1268–1281. doi: 10.1002/ajmg.a.31788. [DOI] [PubMed] [Google Scholar]
- 4.Kawaguchi R., Yu J., Honda J., Hu J., Whitelegge J., Ping P., Wiita P., Bok D., Sun H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science. 2007;315:820–825. doi: 10.1126/science.1136244. [DOI] [PubMed] [Google Scholar]
- 5.Pasutto F., Sticht H., Hammersen G., Gillessen-Kaesbach G., Fitzpatrick D.R., Nürnberg G., Brasch F., Schirmer-Zimmermann H., Tolmie J.L., Chitayat D. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am. J. Hum. Genet. 2007;80:550–560. doi: 10.1086/512203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fares-Taie L., Gerber S., Chassaing N., Clayton-Smith J., Hanein S., Silva E., Serey M., Serre V., Gérard X., Baumann C. ALDH1A3 mutations cause recessive anophthalmia and microphthalmia. Am. J. Hum. Genet. 2013;92:265–270. doi: 10.1016/j.ajhg.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hunker C.M., Galvis A., Kruk I., Giambini H., Veisaga M.L., Barbieri M.A. Rab5-activating protein 6, a novel endosomal protein with a role in endocytosis. Biochem. Biophys. Res. Commun. 2006;340:967–975. doi: 10.1016/j.bbrc.2005.12.099. [DOI] [PubMed] [Google Scholar]
- 9.Kitano M., Nakaya M., Nakamura T., Nagata S., Matsuda M. Imaging of Rab5 activity identifies essential regulators for phagosome maturation. Nature. 2008;453:241–245. doi: 10.1038/nature06857. [DOI] [PubMed] [Google Scholar]
- 10.Su X., Kong C., Stahl P.D. GAPex-5 mediates ubiquitination, trafficking, and degradation of epidermal growth factor receptor. J. Biol. Chem. 2007;282:21278–21284. doi: 10.1074/jbc.M703725200. [DOI] [PubMed] [Google Scholar]
- 11.Glass C.K., Rosenfeld M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- 12.Farboud B., Privalsky M.L. Retinoic acid receptor-alpha is stabilized in a repressive state by its C-terminal, isotype-specific F domain. Mol. Endocrinol. 2004;18:2839–2853. doi: 10.1210/me.2004-0236. [DOI] [PubMed] [Google Scholar]
- 13.Demers A., Caron V., Rodrigue-Way A., Wahli W., Ong H., Tremblay A. A concerted kinase interplay identifies PPARgamma as a molecular target of ghrelin signaling in macrophages. PLoS ONE. 2009;4:e7728. doi: 10.1371/journal.pone.0007728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chassaing N., Ragge N., Kariminejad A., Buffet A., Ghaderi-Sohi S., Martinovic J., Calvas P. Mutation analysis of the STRA6 gene in isolated and non-isolated anophthalmia/microphthalmia. Clin. Genet. 2013;83:244–250. doi: 10.1111/j.1399-0004.2012.01904.x. [DOI] [PubMed] [Google Scholar]
- 15.Berryer M.H., Hamdan F.F., Klitten L.L., Møller R.S., Carmant L., Schwartzentruber J., Patry L., Dobrzeniecka S., Rochefort D., Neugnot-Cerioli M. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum. Mutat. 2013;34:385–394. doi: 10.1002/humu.22248. [DOI] [PubMed] [Google Scholar]
- 16.Wilson J.G., Roth C.B., Warkany J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am. J. Anat. 1953;92:189–217. doi: 10.1002/aja.1000920202. [DOI] [PubMed] [Google Scholar]
- 17.Chassaing N., Golzio C., Odent S., Lequeux L., Vigouroux A., Martinovic-Bouriel J., Tiziano F.D., Masini L., Piro F., Maragliano G. Phenotypic spectrum of STRA6 mutations: from Matthew-Wood syndrome to non-lethal anophthalmia. Hum. Mutat. 2009;30:E673–E681. doi: 10.1002/humu.21023. [DOI] [PubMed] [Google Scholar]
- 18.Casey J., Kawaguchi R., Morrissey M., Sun H., McGettigan P., Nielsen J.E., Conroy J., Regan R., Kenny E., Cormican P. First implication of STRA6 mutations in isolated anophthalmia, microphthalmia, and coloboma: a new dimension to the STRA6 phenotype. Hum. Mutat. 2011;32:1417–1426. doi: 10.1002/humu.21590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White T., Lu T., Metlapally R., Katowitz J., Kherani F., Wang T.Y., Tran-Viet K.N., Young T.L. Identification of STRA6 and SKI sequence variants in patients with anophthalmia/microphthalmia. Mol. Vis. 2008;14:2458–2465. [PMC free article] [PubMed] [Google Scholar]
- 20.Ghyselinck N.B., Dupé V., Dierich A., Messaddeq N., Garnier J.M., Rochette-Egly C., Chambon P., Mark M. Role of the retinoic acid receptor beta (RARbeta) during mouse development. Int. J. Dev. Biol. 1997;41:425–447. [PubMed] [Google Scholar]
- 21.Zhou G., Strom R.C., Giguere V., Williams R.W. Modulation of retinal cell populations and eye size in retinoic acid receptor knockout mice. Mol. Vis. 2001;7:253–260. [PubMed] [Google Scholar]
- 22.Matt N., Dupé V., Garnier J.M., Dennefeld C., Chambon P., Mark M., Ghyselinck N.B. Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development. 2005;132:4789–4800. doi: 10.1242/dev.02031. [DOI] [PubMed] [Google Scholar]
- 23.Mendelsohn C., Lohnes D., Décimo D., Lufkin T., LeMeur M., Chambon P., Mark M. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development. 1994;120:2749–2771. doi: 10.1242/dev.120.10.2749. [DOI] [PubMed] [Google Scholar]
- 24.Greer J.J., Babiuk R.P., Thebaud B. Etiology of congenital diaphragmatic hernia: the retinoid hypothesis. Pediatr. Res. 2003;53:726–730. doi: 10.1203/01.PDR.0000062660.12769.E6. [DOI] [PubMed] [Google Scholar]
- 25.Coles G.L., Ackerman K.G. Kif7 is required for the patterning and differentiation of the diaphragm in a model of syndromic congenital diaphragmatic hernia. Proc. Natl. Acad. Sci. USA. 2013;110:E1898–E1905. doi: 10.1073/pnas.1222797110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Massaro G.D., Massaro D., Chan W.Y., Clerch L.B., Ghyselinck N., Chambon P., Chandraratna R.A. Retinoic acid receptor-beta: an endogenous inhibitor of the perinatal formation of pulmonary alveoli. Physiol. Genomics. 2000;4:51–57. doi: 10.1152/physiolgenomics.2000.4.1.51. [DOI] [PubMed] [Google Scholar]
- 27.Desai T.J., Chen F., Lü J., Qian J., Niederreither K., Dollé P., Chambon P., Cardoso W.V. Distinct roles for retinoic acid receptors alpha and beta in early lung morphogenesis. Dev. Biol. 2006;291:12–24. doi: 10.1016/j.ydbio.2005.10.045. [DOI] [PubMed] [Google Scholar]
- 28.Padmanabhan R., Singh G., Singh S. Malformations of the eye resulting from maternal hypervitaminosis A during gestation in the rat. Acta Anat. (Basel) 1981;110:291–298. doi: 10.1159/000145439. [DOI] [PubMed] [Google Scholar]
- 29.Sulik K.K., Dehart D.B., Rogers J.M., Chernoff N. Teratogenicity of low doses of all-trans retinoic acid in presomite mouse embryos. Teratology. 1995;51:398–403. doi: 10.1002/tera.1420510605. [DOI] [PubMed] [Google Scholar]
- 30.Ozeki H., Shirai S. Developmental eye abnormalities in mouse fetuses induced by retinoic acid. Jpn. J. Ophthalmol. 1998;42:162–167. doi: 10.1016/s0021-5155(97)00134-2. [DOI] [PubMed] [Google Scholar]
- 31.Ozeki H., Shirai S., Ikeda K., Ogura Y. Critical period for retinoic acid-induced developmental abnormalities of the vitreous in mouse fetuses. Exp. Eye Res. 1999;68:223–228. doi: 10.1006/exer.1998.0591. [DOI] [PubMed] [Google Scholar]
- 32.Lee L.M., Leung C.Y., Tang W.W., Choi H.L., Leung Y.C., McCaffery P.J., Wang C.C., Woolf A.S., Shum A.S. A paradoxical teratogenic mechanism for retinoic acid. Proc. Natl. Acad. Sci. USA. 2012;109:13668–13673. doi: 10.1073/pnas.1200872109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balkan W., Klintworth G.K., Bock C.B., Linney E. Transgenic mice expressing a constitutively active retinoic acid receptor in the lens exhibit ocular defects. Dev. Biol. 1992;151:622–625. doi: 10.1016/0012-1606(92)90200-z. [DOI] [PubMed] [Google Scholar]
- 34.Shi H., Zhu P., Sun Z., Yang B., Zheng L. Divergent teratogenicity of agonists of retinoid X receptors in embryos of zebrafish (Danio rerio) Ecotoxicology. 2012;21:1465–1475. doi: 10.1007/s10646-012-0900-9. [DOI] [PubMed] [Google Scholar]
- 35.Benke P.J. The isotretinoin teratogen syndrome. JAMA. 1984;251:3267–3269. [PubMed] [Google Scholar]
- 36.Loureiro K.D., Kao K.K., Jones K.L., Alvarado S., Chavez C., Dick L., Felix R., Johnson D., Chambers C.D. Minor malformations characteristic of the retinoic acid embryopathy and other birth outcomes in children of women exposed to topical tretinoin during early pregnancy. Am. J. Med. Genet. A. 2005;136:117–121. doi: 10.1002/ajmg.a.30744. [DOI] [PubMed] [Google Scholar]