

Abstract
Identifying genomic variation is a crucial step for unraveling the relationship between genotype and phenotype and can yield important insights into human diseases. Prevailing methods rely on cost-intensive whole-genome sequencing (WGS) or whole-exome sequencing (WES) approaches while the identification of genomic variants from often existing RNA sequencing (RNA-seq) data remains a challenge because of the intrinsic complexity in the transcriptome. Here, we present a highly accurate approach termed SNPiR to identify SNPs in RNA-seq data. We applied SNPiR to RNA-seq data of samples for which WGS and WES data are also available and achieved high specificity and sensitivity. Of the SNPs called from the RNA-seq data, >98% were also identified by WGS or WES. Over 70% of all expressed coding variants were identified from RNA-seq, and comparable numbers of exonic variants were identified in RNA-seq and WES. Despite our method’s limitation in detecting variants in expressed regions only, our results demonstrate that SNPiR outperforms current state-of-the-art approaches for variant detection from RNA-seq data and offers a cost-effective and reliable alternative for SNP discovery.
Introduction
Our ability to decipher the relationship between genotype and phenotype relies on the effective identification of genomic variation. The advent of next-generation sequencing has greatly facilitated this endeavor. Whole-genome sequencing (WGS) or whole-exome sequencing (WES) has been a common practice in many large-scale projects, such as the 1000 Genomes and The Cancer Genome Atlas projects, in which its main uses comprise the identification of genomic variants,1–3 many of which improve our understanding of human diseases.4–6 RNA sequencing (RNA-seq) is arguably a more popular application because it costs less than genome sequencing and has the ability to address a multitude of different questions, such as the quantification of gene expression levels, detection of alternative splicing, allele-specific expression, gene fusions,7–10 or RNA editing.11–13
Employing RNA-seq data for identifying genomic variants, however, remains a challenge because of the transcriptome’s intrinsic complexity (e.g., splicing), which leads to the technical difficulty of the computational analysis. What are the benefits of calling variants from RNA-seq data? First, a large number of samples with available RNA-seq data do not come with matched WGS or WES data. Calling variants in them is “free,” an additional deliverable of the existing RNA-seq data. Second, a large number of disease samples might have both RNA-seq and WGS or WES data. Calling SNPs from the WGS or WES data can be challenging because of the heterogeneity of the disease samples (e.g., tumors). De novo variant calling in RNA-seq data provides an efficient option to validate the findings from the WGS or WES data.
Recent developments in computational approaches to identifying SNPs in cancer14 and accurate mapping of RNA-seq reads15 have resulted in the identification of potentially disease-associated variations in RNA-seq data.16–18 These studies either imposed strong variant filtering criteria and thus limited their analysis to several candidate sites16 or required data from multiple individuals and the aid of additional WES for the accurate identification of variants.19 This handful of studies also demonstrates the utility of detecting genetic variants and somatic mutations with the use of RNA-seq data, underscores the considerable effort underlying these investigations, and highlights the need for automated, high-accuracy determination of RNA variants. Correct mapping of RNA-seq reads to the reference genome is crucial for avoiding mismatches that are incorrectly interpreted as SNPs. The assignment of reads to their original genomic location is mostly hampered by (1) highly similar regions in the genome,20 (2) artifacts in library construction,21 and (3) the inability of many computational pipelines to map reads in a splice-aware manner (this last hindrance is possibly the greatest challenge to the accurate detection of SNPs). The vast majority (>90%) of the transcripts in the human genome are spliced version of genes.22 In addition, recent studies have revealed that alternative splicing occurs in over 90% of genes.23 Given the average length of human exons (∼150 bp) and the read lengths of current sequencing technologies (two paired-end 100 bp reads), sequencing of these transcripts often results in sequencing reads that span splice junctions. Current methods24–26 might achieve satisfactory mapping performance for RNA-seq expression studies and the identification of alternative splicing. For the purpose of variant calling from RNA-seq data, however, they still suffer from an unacceptably high rate of wrongly mapped reads. In addition, they fail to account for other RNA-seq-study specifics that could hamper the accurate identification of genomic variants. Recently, several methods for the discovery of RNA-editing sites from transcriptome data have been described.11–13,27–29 Although many of them take some of the above concerns into consideration, there exists no account that describes a fully integrated approach for the detection of single-nucleotide variants from RNA-seq experiments.
Here, we present a simple yet highly accurate method termed SNPiR to identify SNPs in RNA-seq data. SNPiR consists of (1) a modified RNA-seq read-mapping procedure that allows alignment of reads to the reference in a splice-aware manner, (2) variant calling using the Genome Analysis Toolkit (GATK),30 and (3) vigorous filtering of false-positive calls. The steps in our computational pipeline are inspired by common practice for mapping, variant calling, and variant filtering in WGS and WES experiments but were modified to account for the specific characteristics of RNA-seq experiments, including errors introduced during RNA-seq library preparation, sequencing, and read-mapping difficulties due to highly similar genomic regions. The application of our method to two well-characterized samples allows a systematic assessment of sensitivity and specificity and highlights the immense importance of variant filtering for avoiding false-positive calls.
Material and Methods
SNPiR: A Pipeline for the Detection of SNPs in RNA-Seq Data
We have developed a highly efficient procedure (SNPiR) to reliably identify SNPs in RNA-seq data (Figure 1). First, RNA-seq reads are mapped to the reference genome and all known splice junctions. The presence of sequences that surround splice junctions allows the short read mapper to correctly assign spliced reads to their genomic location given that its originating junction is present in the reference. Uniquely mapped reads are then used for calling the initial set of candidate variants with the use of GATK,30 which takes the number of original and alternative alleles and their quality into account for variant calling. Subsequently, these candidates are subjected to several filtering criteria for ensuring the removal of technical artifacts that might have been introduced during RNA-seq library preparation, sequencing, or computational analysis.21 These filters include removal of false calls in duplicated regions, in homopolymeric regions, or close to splice junctions. The resulting set of RNA-seq variants is further compared to the catalog of known RNA-editing sites12,13 for the separation of genomic SNPs from RNA-editing sites.
Figure 1.

A Computational Framework for the Identification of SNPs from Transcriptome Data
Shown are RNA-seq reads mapped to the human reference genome (blue lines) and all regions spanning known splice junctions (yellow lines separated by dashes). Subsequent variant calling used GATK and filtering to remove spurious sites, generating a high-confidence set of SNVs.
RNA-Seq Mapping
We obtained poly(A)+ RNA-seq data for (1) whole GM12878 lymphoblastoid cells from the ENCODE project (Gene Expression Omnibus [GEO] accession number GSM758559) and (2) peripheral-blood mononuclear cells (PBMCs) from one healthy individual31 (GEO GSE33029). The strand-specific RNA-seq libraries were made as described previously.32 Both samples were deeply sequenced on the Illumina HiSeq platform. For GM12878 cells, the transcriptome was sequenced in two biological replicates, resulting in 235.8 and 263.7 million paired-end 76 bp sequencing reads, respectively (Table S1, available online). The PBMC data were obtained from samples of a 20-point time series, which resulted in a total of 3,232 million paired-end 101 bp reads. We chose the Burrows-Wheeler Aligner (BWA)33 as the mapper for RNA-seq reads because of its demonstrated high accuracy of alignment34 (although untested in our study, other gapped aligners with high mapping specificity35,36 might give similar results). We mapped each of the paired-end reads separately by using the commands “bwa aln fastqfile” and “bwa samse -n4.” In contrast to previous approaches, we mapped RNA-seq reads not only to the reference genome11,29 or the transcriptome27,37 but to a combination of the hg19 reference genome (UCSC Genome Browser) plus exonic sequences surrounding all currently known splice junctions from gene models available in annotations from GENCODE, RefSeq, Ensembl, and the UCSC Genome Browser. These short pseudochromosomes allowed us to capture reads derived from transcript regions that span splice junctions and to assign them to the correct genomic location. We chose the length of these splice-junction regions to be slightly shorter than the RNA-seq reads to avoid simultaneous hits to the reference genome and the splice junctions (for 76 bp reads, a 75 bp region upstream and downstream was chosen; for 101 bp reads, a 95 bp region upstream and downstream was chosen). When the adjacent exons upstream and/or downstream of a splice junction were shorter than the required length, we extended the regions across multiple exons. Although this strategy can avoid the mismapping of most split reads, some others might still be wrongly placed onto the genome or split incorrectly. SNPiR avoids such potential false-positive variant calls through an additional BLAT step, as described in the next paragraph. We only considered uniquely mapped reads with mapping quality q > 10 and used SAMtools rmdup38 to remove identical reads (PCR duplicates) that mapped to the same location. Of these identical reads, only the read with the highest mapping quality was retained for further analysis.
RNA-Seq Variant Calling and Filtering
Mapped reads were subject to local realignment, base-score recalibration, and candidate-variant calling with the IndelRealigner, TableRecalibration, and UnifiedGenotyper tools from GATK.30 In contrast to common-practice variant calling, we called variants with very loose criteria by using the UnifiedGenotyper tool with options stand_call_conf of 0 and stand_emit_conf of 0 and output mode EMIT_ALL_CONFIDENT_SITES, which allowed a high sensitivity of SNPiR. This set of candidate variants was subject to several filtering steps that increased the precision of SNPiR (Figure S1). More specifically, we required a variant call quality Q > 20, discarded variants if they occurred in the first six bases of a read, and removed variants in repetitive regions according to RepeatMasker annotation provided through the UCSC Genome Browser. Furthermore, we removed intronic variants if they were within 4 bp of splice junctions and filtered variants in homopolymer runs ≥ 5 bp. Both of these filter settings proved effective in the removal of false-positive variant calls in previous sequence analyses.12,13 Moreover, we ensured that reads supporting a variant were uniquely mapped to the genome. For that purpose, we used BLAT39 to remap all reads supporting a variant to the genome. For each read, we required that (1) the best hit overlap with the variant site and (2) the second best hit have a score < 95% of the best hit. We only retained variant sites if the majority of supporting reads fulfilled these criteria. Finally, we removed all currently known RNA-editing sites that were found by recent high-throughput studies.12,13 (This final step can be omitted if the user chooses to identify not only genomic variants but also RNA-editing sites.) We used ANNOVAR40 to annotate variants on the basis of gene models from GENCODE, RefSeq, Ensembl, and the UCSC Genome Browser. We defined all RNA-seq variants that can also be identified from WGS data or are present in dbSNP (version 135) as “known” variants. Conversely, all RNA-seq variants that cannot be found in WGS or in dbSNP were denoted as “novel.” The precision of SNPiR was calculated as the number of all known RNA-seq variants divided by the total number of known and novel RNA-seq variants. To allow a fair comparison between RNA-seq and WGS variants, we determined the sensitivity of SNPiR as the fraction of coding exonic variants identified from WGS.
WGS and WES Mapping and Variant Calling
WGS and WES data for the GM12878 cell line were provided in mapped form by the 1000 Genomes Project (see Web Resources). The genome was sequenced at 44× coverage.1 WGS and WES reads for the PBMCs were available from the Sequence Read Archive under accessions SRP008054.4 and SRA040093, respectively. We mapped the PBMC data by using the BWA in paired-end mode with commands “bwa aln fastqfile1,” “bwa aln fastqfile2,” and “bwa sampe.” Realignment, recalibration, and variant calling were performed with GATK. For variant calling and filtering in WGS and WES data, we applied the same parameter set as done by the 1000 Genomes Project Consortium. More precisely, we used the UnifiedGenotyper with options stand_call_conf of 30 and stand_emit_conf of 10 and filtering criteria as described by the 1000 Genomes Project Consortium.1 We chose to use these widely accepted guidelines for variant calling to obtain a high-confidence variant set that could be used as the gold standard in our analysis.
Expression Analysis
The expression of known genes (i.e., expected fragments per kilobase of transcript per million fragments mapped [FPKM]) was quantified with cufflinks41 (parameter -G) on the basis of Tophat242 mappings. Gene models were obtained from the UCSC Genome Browser for reference genes. If a variant overlapped with several gene models, the average FPKM for all overlapping genes was calculated.
Results
High Precision of SNP Detection in RNA-Seq Data
We applied SNPiR to data from the GM12878 human lymphoblastoid cell line1 and PBMCs from another healthy individual31 (Table S1). These resources have three major advantages. First, the transcriptome, exome, and whole genome of these samples have been deeply sequenced and allow accurate identification of variants from RNA and DNA of the same individual. Second, the matched RNA and DNA samples enable verification of RNA SNP calls because they can be compared to variation present in the DNA. Third, the GM12878 cell line has been extensively studied, and SNPs detected in its genome have been continuously deposited into dbSNP, making it a good candidate set for evaluating the precision and sensitivity of SNPiR.
Using the approach described in Figure 1, we identified SNPs in the transcriptomes of GM12878 cells and PBMCs. At the same time, we used common-practice variant calling as performed by the 1000 Genomes Project1 to catalog variants in the WGS and WES data of the same samples. In total, we were able to detect 172,982 variants in the GM12878 RNA-seq data and 299,153 variants in the PBMC RNA-seq data (Table S1). The larger number of PBMC variants can be attributed to the larger size of the RNA-seq data set and thus higher coverage and confidence in variant calls. We found that SNPiR detected genomic SNPs with high precision, given that 99.1% of the GM12878 variants and 96.6% of the PBMC variants that were discovered from RNA-seq were also called through WGS data (Figure 2) and that 99.6% (172,322) of the GM12878 variants and 97.7% (292,224) of the PBMC variants were supported by evidence from WGS or dbSNP v.135 (Figures S2A, S2C, S2D, and S2F). For both GM12878 cells and PBMCs, these known sites exhibited a transition-to-transversion (ts/tv) ratio of 2.25, which is similar to the overall ts/tv ratio of 2.0–2.1 for the entire human genome1,43–45 and estimates of ∼3 for exonic regions46 and thus is a good reflection of the genomic variation in transcribed regions.1 Also, the mutational profile of known variants matched well with the expectations for genomic regions given that similar profiles of variants were observed in WGS data (Figure S3). For the remaining (novel) sites (600 in GM12878 data and 6,929 in PBMC data), we observed higher ts/tv ratios than for the known sites (2.49 in GM12878 data and 3.58 in PBMC data). We found that ∼27% of our novel variants in GM12878 data and ∼7% in PBMC data were supported by variant reads in WGS data (Figure S4A). The remaining novel sites showed a clear enrichment of A>G and T>C variation (70.1% for GM12878 data and 71.1% for PBMC data), indicative of the dominant A-to-I RNA editing47 (Figure S4B). The fraction of A>G and T>C variants was even higher (92.1%) for the 64 novel sites shared between the two data sets. Given the fact that the genomes of these two individuals were deeply sequenced, most of the genomic SNPs had already been identified. It was therefore expected that novel SNPs identified in the RNA-seq data would be enriched with RNA-editing sites. Although our computational pipeline includes the removal of all currently known RNA-editing variants identified from high-throughput studies12,13 (1,369,030 sites in total), this catalog is still far from being complete, and thus some variants in our analysis might have remained unidentified as RNA editing. However, our results show that RNA-editing events are rare compared to the number of SNPs that can be found in a human genome. Moreover, the rapid growth of the RNA-editing catalog in humans will allow us to filter known RNA-editing sites and thus increase the precision of SNPiR to find genomic variants only.
Figure 2.
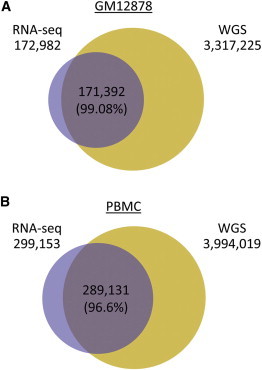
Comparison of SNPs Identified via RNA-Seq and WGS of GM12878 Cells and PBMCs
SNPiR achieved high precision for both GM12878 (A) and PBMC (B) data sets, given that most of the RNA-seq variants were also identified by WGS of the same subject. Numbers in parentheses give the percentage of RNA-seq variants found in WGS.
Enrichment of Variants in Functional Categories
For expressed genes, the use of RNA-seq data for SNP calling can be advantageous compared to WGS because it enriches for expressed genic regions and thus increases the power to detect functionally important SNPs. Using RNA-seq data rather than WGS or WES allowed us to enrich for variants in coding exons, UTRs, and introns (Figure 3A). The SNPs discovered by SNPiR were highly abundant in these three categories. Only a small fraction fell into intergenic regions. The large number of intronic variants in our analysis can be explained by the facts that (1) poly(A)+ RNA-capturing protocols can also capture a small fraction of pre-mRNAs (that still contain introns), (2) introns compose a much larger fraction of the human genome than do exonic regions, and (3) much more variation exists in introns than in exonic regions because of the higher selective pressures on the exonic portion of the genome to correctly encode proteins. Given the very high sequencing coverage of the GM12878 sample, many intronic regions were covered with low sequencing depth. The high sensitivity of our method allowed us to detect many intronic variants from these regions of low sequence coverage. Although exome-capturing techniques are commonly used to enrich genic regions, we have found that WES variant calls for GM12878 and PBMC data only overlap with variants discovered in the transcriptome to a surprisingly small extent (Figures S2B and S2E). This situation occurs because exome-capturing kits are mostly designed to capture the protein-coding portion of the genome, whereas transcriptome sequencing also provides information about UTRs and intronic regions. Nevertheless, the agreement between RNA-seq and WES is substantially higher in coding regions than in any other category (in Figure 3A, the fraction of coding sites is markedly higher in the overlap between RNA-seq and WES than in the single techniques). When focusing on coding sites only, we found that 33.4% of SNPs identified by WES in GM12878 cells were also identified by SNPiR using the RNA-seq data. As a result of no or very low coverage, 81.5% of the remaining 66.6% of SNPs were not identified. We have demonstrated that our method can achieve highly confident variant calls. For that reason, the small overlap between WES and RNA-seq variants suggests that RNA-seq has the power to uncover variants that are in UTRs and introns and that might have important regulatory functions but are missed in WES screens. Therefore, our results suggest that transcriptome variant discovery could serve as a complementary approach to WES for the detection of nucleotide variation.
Figure 3.
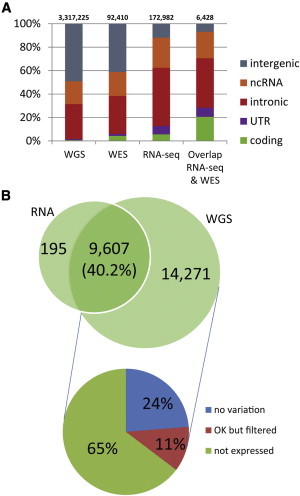
Characteristics of SNPs Identified from RNA-Seq Data of GM12878 Cells
(A) The composition of genomic regions for variants in WGS, WES, and RNA-seq suggests a high enrichment of RNA-seq variants in functionally important regions. Sites present in RNA-seq and WES occurred substantially more often in coding exons.
(B) Overlap in coding variants detected from RNA-seq and WGS. Of all coding variants, 40.2% were found by RNA-seq. The majority of the remaining sites were not detected as a result of the lack of expression. “No variation” indicates that the position was homozygous in RNA, “OK but filtered” indicates that the position was heterozygous but was removed by one of our filtering steps, and “not expressed” indicates that the position was not covered by RNA-seq reads.
High Sensitivity in Coding Regions
To calculate the sensitivity of SNPiR, we focused on variants in coding regions only. The comparison of all RNA variants to all whole-genome variants would not have been fair because of the limited representation of the human genome by RNA transcripts (Figure 3A). Nevertheless, we found that RNA-seq data alone enabled the discovery of 40.2% and 47.7% of all coding variants identified by WGS in GM12878 cells and PBMCs, respectively. At the same time, RNA-seq only required a fraction of the sequencing effort (e.g., 499 million [for RNA-seq] versus 2,976 million [for WGS] sequencing reads for GM12878 data). Naturally, SNPiR is restricted to the detection of variants in genic regions, specifically in genes that are being expressed in the cell under the sampling conditions. Therefore, the SNPs that had known function in coding exons but that were not detected by analysis of the transcriptome were mainly missed because of the lack of transcription of these genes (Figure 3B). When we compared the RNA-seq variants only to WGS variants in expressed genes (characterized by FPKM > 0.2), the sensitivity of SNPiR increased from 40%–50% to >70% (Figure 4A). This agrees with the fact that a large fraction of genes are expressed at very low levels (Figure 4B). Therefore, our initial results show that SNPiR achieves high sensitivity and precision for variant calling in expressed genes.
Figure 4.
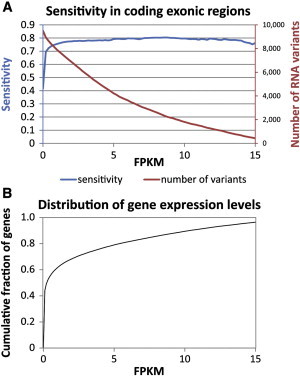
High Sensitivity of SNPiR Variant Calling in Coding Regions of Expressed Genes of GM12878 Cells
(A) Sensitivity and number of detected variants called from RNA-seq data in dependence of the minimum gene expression (in FPKM).
(B) Cumulative distribution of expression levels (in FPKM) for all reference genes.
Precision and Sensitivity for Low-Depth RNA-Seq Data
Many of the currently available RNA-seq data sets vary in read lengths and read numbers. Our results are based on very deeply sequenced RNA-seq libraries (499 million reads for GM12878 data and 3,232 million reads for PBMC data). However, in many other experimental settings, the sequencing depth is often lower. To test the performance of SNPiR for smaller RNA-seq data sets, we carried out three random samplings of 5, 10, 20, 50, and 100 million reads from the GM12878 RNA-seq data set. Our results were highly reproducible for all sample sizes (Table S2). In general, fewer sites were detected for smaller subsets (Figure 5A). Nevertheless, we detected more than half of the coding variants of the complete data set (499 million reads) by using a subsample of only 20 million reads (Figure 5B). In addition, we found an enrichment of coding variants for smaller sample sizes (Figure 5C) as a result of the overall higher coverage of coding regions with RNA-seq reads (higher coverage allows reliable variant calls despite the lower total read number). The precision of SNPiR remained very high (0.980 - 0.997) for all sampling sizes, given that nearly all detected variants are known (Figure 5A), and the mutational profile was similar to that generated from WGS data (Figure 5D).
Figure 5.
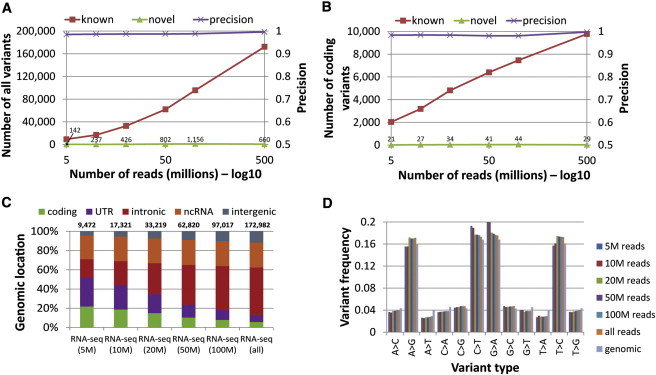
Subsampling of RNA-Seq Reads
Subsamplings of 5, 10, 20, 50, and 100 million reads were generated from the total set of 499 million GM12878 RNA-seq reads. We compared (A) the number of discovered variants, (B) the number of variants in coding regions, and (C) the genomic location of variants between the random samplings and the complete set RNA-seq reads, as well as (D) the mutational profile of known RNA-seq variants and genomic variants. In (A) and (B), “known” variants denote all variant sites that were discovered from RNA-seq and were either confirmed through WGS or present in dbSNP. Conversely, “novel” denotes all variants that were previously not found from WGS or dbSNP. The total amounts of novel variants per sample size are shown as small numbers above the data series.
Comparison of Sensitivity and Precision between RNA-Seq and WES Experiments
To evaluate the performance of SNPiR, we compared its sensitivity and precision to those of WES in (1) regions that are annotated as protein coding in the Consensus Coding Sequence (CCDS) database and are commonly targeted in WES and (2) exonic regions (coding exons and UTRs). For that purpose, we used the PBMC WES library of 94.1 million mapped reads and matched its size by subsampling the same number of reads from the larger PBMC RNA-seq data. Sensitivity was calculated as the number of correctly identified variants divided by the total number of variants identified from high-coverage WGS in the same regions. Precision was calculated as the number of correct variant calls divided by the number of correct and false variant calls.
In the CCDS regions, we identified 22,052 variants through WGS and were able to recover 17,922 (81.3%) and 9,892 (44.9%) of them through WES and RNA-seq, respectively (Figure 6A). The majority of the variants discovered by SNPiR overlapped with sites found in WES. Although the number of variants discovered by SNPiR decreased with coverage (Figures S5A and S5B), its sensitivity increased rapidly with larger numbers of covering reads (Figure S5C). SNPiR’s precision was remarkably high for all coverage levels and could compete with that of WES (Figure S5D). The smaller total number of variants detected by SNPiR compared to WES can be explained by the lower coverage of the CCDS regions by RNA-seq reads and the nature of our method to scale with the number of mapped reads. Only 35.1 million (37.3%) of the sampled 94.1 million RNA-seq reads covered CCDS annotations, whereas 60.2 million (64%) of the WES reads were located in the same regions and thus led to a larger number of detected variants in WES. The remaining 62.7% of RNA-seq reads that did not cover CCDS regions enabled us to discover variation in genomic regions not commonly covered by WES. When we targeted all 62,028 genomic variants in exonic regions (coding and UTR exons), the numbers of variants discovered through WES and RNA-seq were very close to each other: we were able to recover 23,693 (38.2%) WGS variants by using WES and 24,987 (40.3%) variants by using RNA-seq (Figure 6B and Figures S5A and S5B). This highlights the utility of SNPiR for the detection of genomic variants that have potential regulatory function but that are commonly not targeted by WES and emphasizes its importance as a complementary approach to variant discovery from WES.
Figure 6.
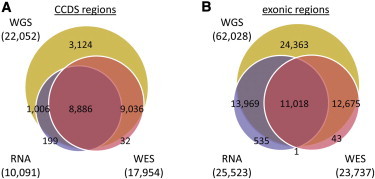
Comparison of Genomic Variants Identified in CCDS and Exonic Regions by WGS, WES, or RNA-Seq
An equal number of reads (94.1 million) of RNA-seq and WES data was used for fair comparison of variants identified in CCDS regions (A) and exonic regions (B).≥
SNPiR Achieves Higher Precision and Sensitivity than RNASEQR
To further evaluate the performance of SNPiR, we compared it to RNASEQR, which is the current most accurate method for RNA-seq mapping given that compared to other RNA-seq mappers, it yields the smallest number of false-positive RNA-seq variant calls.15 RNASEQR uses a three-step approach in which it maps (1) reads to the transcriptome, (2) unmapped reads to the reference genome in order to detect novel exons, and (3) unmapped reads in a split fashion to the reference genome and transcriptome to discover novel splice junctions. In contrast to SNPiR, which uses the BWA33 as a mapping algorithm, RNASEQR employs Bowtie (v.0.12.7),48 which we previously demonstrated to have inferior performance for the detection of transcriptomic variants because it does not support gapped alignment.21 We applied RNASEQR to the same GM12878 and PBMC data that were used in our pipeline. We called variants by using the same parameters as previously done with RNASEQR to identify SNPs in RNA-seq data15 (Table S3). SNPiR detected a slightly smaller number of variants (172,982 sites) in the GM12878 sample than did RNASEQR (200,318 sites) (Figure 7A), mainly because of an unexpected, large number of novel variants (18,840) identified by RNASEQR (SNPiR only identified 660) (Figure 7B). The ts/tv ratio for known variants identified by SNPiR appeared to be in the normal range, whereas novel variants showed the expected excess of A>G and T>C (see above). On the other hand, the low ts/tv ratio (1.23) for the novel SNPs identified by RNASEQR suggests a higher false-positive rate (Figure 7B and Figure S6A). In fact, a larger portion of the novel RNASEQR variants in the GM12878 sample did not show any support in WGS (compare Figures S4A and S6B) and did not show any enrichment of A>G and T>C types (compare Figures S4B and S6C).
Figure 7.
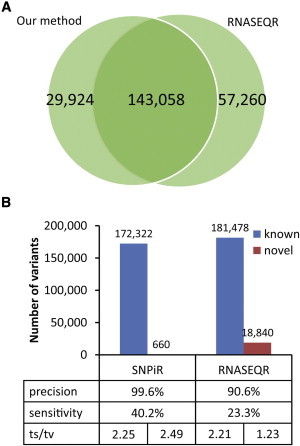
Comparison of SNPiR with RNASEQR
Overlap between the sites detected by SNPiR and RNASEQR on the same RNA-seq data set for GM12878 cells (A) and the number of known and novel variants discovered by SNPiR and RNASEQR, the precision and sensitivity of variant calling, and the ts/tv ratio for each category (B). Precision was calculated as the fraction of RNA-seq variants either supported by WGS or present in dbSNP. Sensitivity was determined as the fraction of WGS variants both found in coding regions and discovered in the RNA-seq data.
We examined the 18,840 novel sites identified by RNASEQR and found that the majority of them (>13,000) were false calls. First, we found that despite the efforts by RNASEQR to report uniquely mapped reads, 10,531 sites (55.9%) were supported by nonunique mappings only and were removed by the BLAT filter in SNPiR. Reads that support such variants have the same or even higher BLAT mapping scores in other genomic locations at which the alternative nucleotide matches the reference genome (Figure S7A). If mapped incorrectly, such reads result in mismatches from the reference genome, which in turn are wrongly identified as single-nucleotide variants. Second, our BLAT filter was also able to identify 1,273 false variants close to exon-intron junctions in the RNASEQR mappings. By correctly mapping spliced reads across junctions, it prevented them from extending into intronic regions, where they could have caused mismatches (Figure S7B). Third, variation at 5′ read ends, most of which was shown to be due to technical artifacts,21,49,50 accounted for 1,629 sites. Fourth, we found 53 sites located in homopolymers. All together, we identified 71.6% (13,486/18,840) of sites in the novel RNASEQR variant calls as potential false positives. When comparing the novel RNASEQR variants between GM12878 data and PBMC data, we found 3,173 shared sites between two individuals. Of the 3,173 sites, 2,882 (90.8%) were among the filtered sites, whereas only 1,266 (39.9%) were A>G and T>C variants (potential RNA-editing sites). These results suggest that systematic errors in the RNASEQR mapping can occur in multiple individuals.
Although SNPiR called fewer SNPs and achieved higher precision than did RNASEQR, its sensitivity was also higher in coding regions. Of the 23,878 coding SNPs identified from WGS, SNPiR identified 9,607 (40.2%) and RNASEQR identified 5,571 (23.3%) (Figure 7B). Considering all 54,891 coding and UTR SNPs that were identified from WGS, SNPiR was able to detect 21,608 (39.4%) and RNASEQR detected 13,562 (24.7%). This demonstrates the capability of our splice-aware mapping procedure to avoid the incorrect mapping of entire reads. It also highlights the importance of our filtering process, which specifically removes false-positive variants.
Discussion
In this work, we have devised SNPiR, a computational approach that allows the accurate identification of genomic variants from transcriptome sequencing through the combination of a splice-aware RNA-seq read-mapping procedure and subsequent variant filtering that takes the specifics of RNA-seq experiments into account. We applied SNPiR to the RNA-seq data from two individuals whose genomes and exomes had been deeply sequenced. On both data sets, SNPiR was able to detect genomic variants at high precision by removing false-positive calls. The usage of RNA-seq data allowed us to enrich for variants in functionally important regions and to achieve high sensitivity in variant calling in expressed exonic regions. This high precision and sensitivity were also maintained for low-coverage sequencing data.
Of paramount importance to us was to achieve the highest possible accuracy of SNP calling from RNA-seq experiments. For that purpose, we adapted a read-mapping strategy that allows us to reduce the number of falsely mapped reads. Reads are simultaneously mapped to the reference genome and to short pseudochromosomes created from sequences around all currently known splice junctions. Although this restricts our mapping to known isoforms and, unlike other RNA-seq mappers,15,24–26 lacks the ability to discover novel splice junctions, it avoids the incorrect placement of reads from highly similar locations. RNA-seq mappers that initially map reads to the transcriptome can be restricted by the incompleteness of the transcriptome and force reads from unannotated regions to be mapped into transcripts. Similarly, initial mapping to the reference genome alone can force split reads to be mapped in a continuous fashion to a suboptimal location. Both scenarios result in falsely mapped reads and thus false SNP calls. SNPiR is able to avoid both cases by using genome and transcriptome information simultaneously. Similarly to SNPiR, the most recent version of TopHat (TopHat2)42 can take genome and transcriptome information simultaneously into account during mapping. We tested the performance of TopHat2 as a replacement for our mapping strategy by calling variants and applying all SNPiR filtering steps to the complete set of GM12878 RNA-seq reads that were mapped with TopHat2. We found that TopHat2 mappings allowed the identification of more total variants. However, these were less precise and lacked the sensitivity of SNPiR mappings in coding regions (Figure S8). Moreover, our simple mapping procedure, based on the BWA as the mapper, is at least four times faster on the same RNA-seq sequencing library.
Calling of genomic variants from RNA-seq data can have manifold applications. It enables researchers to use their readily available RNA-seq data to profile samples for known variants or allows confirmation of variants that were detected by genome sequencing (e.g., validation of somatic mutations related to cancer). Furthermore, it permits the detection of previously unknown variants that might carry important functional implications. For instance, we observed that proportionally more novel variants were found in the previously unstudied PBMC data than in the GM12878 data (the novel/known ratio was 0.024 in PBMC data and 0.004 in GM12878 data), in which nucleotide variation is well characterized through the HapMap and 1000 Genomes projects.1,2 This confirms our initial hypothesis that more novel RNA variants would be found in previously unstudied data sets, promising a substantial yield of novel variants from other data sets. Although previous screens were able to uncover common genomic variants, the power of our analysis lies in the diverse origin of RNA-seq samples and individuals, empowering us to detect rare SNPs that were not observed before51 at minimal additional cost. Many of the rare and low-frequency variants are thought to be functionally important and responsible for the heritability of complex diseases.52 Given the relatively small overlap between the regions targeted by RNA-seq and WES, our method might also find its application in the discovery of variants associated with rare Mendelian diseases—especially in genomic regions that are not captured by WES experiments. For nonhuman species without large-scale genome sequencing efforts such as the 1000 Genomes Project, SNPiR has the potential to identify a lot more novel SNPs in the RNA-seq data, which can be obtained at a lower cost. For nonmodel species without available reference genomes, sequencing the RNA and calling RNA variants might be a very efficient approach to identifying genetic markers that allow genetic mapping of traits of interest.53 Further development of SNPiR will be needed for meeting the challenges of accurate read mapping without a well-assembled genome and a well-annotated transcriptome. Finally, in tumor sequencing projects such as the Cancer Genome Atlas (TCGA), often the genome and/or exome and RNA are both sequenced. Calling variants in the tumor samples with the genome and/or exome sequencing data is even more challenging because of the complexity of the tumors (such as the heterogeneous nature). The ability to independently call variants in the RNA-seq data will serve as an efficient means to validate a large number of somatic mutations identified in the genome and/or exome data.
SNPiR shows high performance in variant calling and opens the door to many applications by using RNA-seq data. At the same time, its abilities are limited by the nature of RNA-seq experiments. SNPiR is predicated on the discovery of functionally relevant variants, which, in most cases, requires the expression of the transcript harboring the variant. This becomes evident through the relatively small overlap between variants detected from WES and RNA-seq. WES experiments are specifically designed to target predefined regions of interest (predominantly the protein-coding portion of the genome). As such, WES can identify variation in these regions with high sensitivity, whereas comprehensive coverage and variant detection in the same portions of the genome are not guaranteed by RNA-seq because of the potential lack of expression. Furthermore, tissue-specific gene expression might hamper the discovery of genomic variants given that the collection of tissues related to the phenotype can be challenging and easily accessible tissues might not express the genes with disease-related variants. Furthermore, nonsense variants might be missed by our method as a result of nonsense-mediated decay. Nevertheless, SNPiR allows the detection of variants even for lowly expressed genes (Figure 4). In some cases, this translates to as few as two to three reads per genomic locus. For obtaining higher confidence in the called variants, pooling of multiple data sets from the same individual (e.g., RNA-seq from different tissues) can help to increase the coverage and to facilitate variant discovery in regions of interest that would otherwise lack sufficient coverage. This study, as well as our previous work,13 demonstrates that variant calling from RNA-seq experiments can tremendously profit from an increased number of reads as the coverage of genomic regions increases. Nevertheless, our subsampling of the RNA-seq data shows that small sample sizes also allow reliable calling of variants and enrich for variants in exonic regions (Figure 5) as a result of the overall higher coverage of exons compared to UTRs, introns, and intergenic positions. On the other hand, the simultaneous usage of multiple data sets from different individuals can help to avoid systematic errors in variant detection. Although the filtering steps in SNPiR effectively remove false positives from single data sets, systematic errors might still persist across data sets. In general, most common variants have already been discovered by previous sequencing projects and appear in dbSNP. Therefore, genuine novel variation is most likely to be restricted to few individuals. Variants present in many samples are likely to be either RNA-editing events, as exemplified by the shared novel variants between the two data sets in the SNPiR analysis, or systematic errors, as shown in the case of the shared variants in the RNASEQR mappings. These recurring variants can be identified via cross-comparison of variant calls between different RNA-seq data sets. We also anticipate that the rapidly growing atlas of known RNA-editing sites will permit the removal of such positions with increasing efficiency in the future. Alternatively, if highly confident variant calls are essential, all A>G variants may be removed.
In addition to failing to call genomic variants in genes that are not expressed, SNPiR might encounter difficulty in calling variants in expressed genes as a result of monoallelic expression (in which only one parental allele is expressed). When only the reference allele is expressed, the SNP will remain undetected. When only the nonreference allele is expressed, the SNP will be miscalled as a homozygous rather than a heterozygous variant. However, previous work suggests that only 5%–10% of human genes are subject to monoallelic expression,54 which is also reflected in our results. In the total set of genes with FPKM > 5, we detected >80% of all coding variants (Figure 4), suggesting that less than 20% of all coding variation will escape detection and only part of it might be attributable to monoallelic expression.
Despite the limitations of calling genomic variants from RNA-seq data, our work demonstrates the feasibility of SNP calling from RNA-seq data with high precision and sensitivity. The framework described in this work will not replace WGS or WES approaches but rather presents a viable alternative to these two approaches in cases where neither of them is available nor cost effective. Our approach might complement whole-exome variant calling and be used to validate SNPs that were discovered by either WGS or WES. Therefore, it presents a powerful tool that will empower the exploration of SNPs at the genomic level from RNA-seq data alone.
Acknowledgments
R.P. was supported by a fellowship from the German Academic Exchange Service. G.R. was supported by the Stanford Genome Training Program, funded by the National Institutes of Health (NIH) and a Stanford Graduate Fellowship. This work was supported by the Stanford University Department of Genetics, NIH, and Ellison Medical Foundation (to J.B.L.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org
Burrows-Wheeler Aligner (BWA), http://bio-bwa.sourceforge.net/
Consensus Coding Sequence (CCDS), http://www.ncbi.nlm.nih.gov/CCDS/
Gene Expression Omnibus (GEO), http://www.ncbi.nlm.nih.gov/geo/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altshuler D.M., Gibbs R.A., Peltonen L., Altshuler D.M., Gibbs R.A., Peltonen L., Dermitzakis E., Schaffner S.F., Yu F., Peltonen L., International HapMap 3 Consortium Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacArthur D.G., Balasubramanian S., Frankish A., Huang N., Morris J., Walter K., Jostins L., Habegger L., Pickrell J.K., Montgomery S.B., 1000 Genomes Project Consortium A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altshuler D., Hirschhorn J.N., Klannemark M., Lindgren C.M., Vohl M.C., Nemesh J., Lane C.R., Schaffner S.F., Bolk S., Brewer C. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat. Genet. 2000;26:76–80. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- 5.Chapman M.A., Lawrence M.S., Keats J.J., Cibulskis K., Sougnez C., Schinzel A.C., Harview C.L., Brunet J.P., Ahmann G.J., Adli M. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stransky N., Egloff A.M., Tward A.D., Kostic A.D., Cibulskis K., Sivachenko A., Kryukov G.V., Lawrence M.S., Sougnez C., McKenna A. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pickrell J.K., Marioni J.C., Pai A.A., Degner J.F., Engelhardt B.E., Nkadori E., Veyrieras J.B., Stephens M., Gilad Y., Pritchard J.K. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rozowsky J., Abyzov A., Wang J., Alves P., Raha D., Harmanci A., Leng J., Bjornson R., Kong Y., Kitabayashi N. AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol. Syst. Biol. 2011;7:522. doi: 10.1038/msb.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Montgomery S.B., Sammeth M., Gutierrez-Arcelus M., Lach R.P., Ingle C., Nisbett J., Guigo R., Dermitzakis E.T. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J., Lee W., Jiang Z., Chen Z., Jhunjhunwala S., Haverty P.M., Gnad F., Guan Y., Gilbert H.N., Stinson J. Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome Res. 2012;22:2315–2327. doi: 10.1101/gr.140988.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bahn J.H., Lee J.H., Li G., Greer C., Peng G., Xiao X. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res. 2012;22:142–150. doi: 10.1101/gr.124107.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramaswami G., Zhang R., Piskol R., Keegan L.P., Deng P., O’Connell M.A., Li J.B. Identifying RNA editing sites using RNA sequencing data alone. Nat. Methods. 2013;10:128–132. doi: 10.1038/nmeth.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramaswami G., Lin W., Piskol R., Tan M.H., Davis C., Li J.B. Accurate identification of human Alu and non-Alu RNA editing sites. Nat. Methods. 2012;9:579–581. doi: 10.1038/nmeth.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goya R., Sun M.G., Morin R.D., Leung G., Ha G., Wiegand K.C., Senz J., Crisan A., Marra M.A., Hirst M. SNVMix: predicting single nucleotide variants from next-generation sequencing of tumors. Bioinformatics. 2010;26:730–736. doi: 10.1093/bioinformatics/btq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L.Y., Wei K.C., Huang A.C., Wang K., Huang C.Y., Yi D., Tang C.Y., Galas D.J., Hood L.E. RNASEQR—a streamlined and accurate RNA-seq sequence analysis program. Nucleic Acids Res. 2012;40:e42. doi: 10.1093/nar/gkr1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah S.P., Morin R.D., Khattra J., Prentice L., Pugh T., Burleigh A., Delaney A., Gelmon K., Guliany R., Senz J. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 17.Kridel R., Meissner B., Rogic S., Boyle M., Telenius A., Woolcock B., Gunawardana J., Jenkins C., Cochrane C., Ben-Neriah S. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood. 2012;119:1963–1971. doi: 10.1182/blood-2011-11-391474. [DOI] [PubMed] [Google Scholar]
- 18.Shah S.P., Roth A., Goya R., Oloumi A., Ha G., Zhao Y., Turashvili G., Ding J., Tse K., Haffari G. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seo J.S., Ju Y.S., Lee W.C., Shin J.Y., Lee J.K., Bleazard T., Lee J., Jung Y.J., Kim J.O., Shin J.Y. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22:2109–2119. doi: 10.1101/gr.145144.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piskol R., Peng Z., Wang J., Li J.B. Lack of evidence for existence of noncanonical RNA editing. Nat. Biotechnol. 2013;31:19–20. doi: 10.1038/nbt.2472. [DOI] [PubMed] [Google Scholar]
- 21.Lin W., Piskol R., Tan M.H., Li J.B. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science. 2012;335:1302. doi: 10.1126/science.1210419. author reply 1302. [DOI] [PubMed] [Google Scholar]
- 22.Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 24.Wang K., Singh D., Zeng Z., Coleman S.J., Huang Y., Savich G.L., He X., Mieczkowski P., Grimm S.A., Perou C.M. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Au K.F., Jiang H., Lin L., Xing Y., Wong W.H. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 2010;38:4570–4578. doi: 10.1093/nar/gkq211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ju Y.S., Kim J.I., Kim S., Hong D., Park H., Shin J.Y., Lee S., Lee W.C., Kim S., Yu S.B. Extensive genomic and transcriptional diversity identified through massively parallel DNA and RNA sequencing of eighteen Korean individuals. Nat. Genet. 2011;43:745–752. doi: 10.1038/ng.872. [DOI] [PubMed] [Google Scholar]
- 28.Park E., Williams B., Wold B.J., Mortazavi A. RNA editing in the human ENCODE RNA-seq data. Genome Res. 2012;22:1626–1633. doi: 10.1101/gr.134957.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng Z., Cheng Y., Tan B.C., Kang L., Tian Z., Zhu Y., Zhang W., Liang Y., Hu X., Tan X. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol. 2012;30:253–260. doi: 10.1038/nbt.2122. [DOI] [PubMed] [Google Scholar]
- 30.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen R., Mias G.I., Li-Pook-Than J., Jiang L., Lam H.Y., Chen R., Miriami E., Karczewski K.J., Hariharan M., Dewey F.E. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell. 2012;148:1293–1307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parkhomchuk D., Borodina T., Amstislavskiy V., Banaru M., Hallen L., Krobitsch S., Lehrach H., Soldatov A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37:e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H., Homer N. A survey of sequence alignment algorithms for next-generation sequencing. Brief. Bioinform. 2010;11:473–483. doi: 10.1093/bib/bbq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marco-Sola S., Sammeth M., Guigó R., Ribeca P. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat. Methods. 2012;9:1185–1188. doi: 10.1038/nmeth.2221. [DOI] [PubMed] [Google Scholar]
- 37.Li M., Wang I.X., Li Y., Bruzel A., Richards A.L., Toung J.M., Cheung V.G. Widespread RNA and DNA sequence differences in the human transcriptome. Science. 2011;333:53–58. doi: 10.1126/science.1207018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kent W.J. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levy S., Sutton G., Ng P.C., Feuk L., Halpern A.L., Walenz B.P., Axelrod N., Huang J., Kirkness E.F., Denisov G. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bentley D.R., Balasubramanian S., Swerdlow H.P., Smith G.P., Milton J., Brown C.G., Hall K.P., Evers D.J., Barnes C.L., Bignell H.R. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pelak K., Shianna K.V., Ge D., Maia J.M., Zhu M., Smith J.P., Cirulli E.T., Fellay J., Dickson S.P., Gumbs C.E. The characterization of twenty sequenced human genomes. PLoS Genet. 2010;6:e1001111. doi: 10.1371/journal.pgen.1001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marth G.T., Yu F., Indap A.R., Garimella K., Gravel S., Leong W.F., Tyler-Smith C., Bainbridge M., Blackwell T., Zheng-Bradley X., 1000 Genomes Project The functional spectrum of low-frequency coding variation. Genome Biol. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kleinman C.L., Majewski J. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science. 2012;335:1302. doi: 10.1126/science.1209658. author reply 1302. [DOI] [PubMed] [Google Scholar]
- 50.Pickrell J.K., Gilad Y., Pritchard J.K. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science. 2012;335:1302. doi: 10.1126/science.1210484. author reply 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Broad GO. Seattle GO. NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McClellan J., King M.C. Genetic heterogeneity in human disease. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 53.Seeb J.E., Carvalho G., Hauser L., Naish K., Roberts S., Seeb L.W. Single-nucleotide polymorphism (SNP) discovery and applications of SNP genotyping in nonmodel organisms. Mol Ecol Resour. 2011;11(Suppl 1):1–8. doi: 10.1111/j.1755-0998.2010.02979.x. [DOI] [PubMed] [Google Scholar]
- 54.Chess A. Mechanisms and consequences of widespread random monoallelic expression. Nat. Rev. Genet. 2012;13:421–428. doi: 10.1038/nrg3239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.