

Abstract
Facioscapulohumeral muscular dystrophy type 1 (FSHD1) is caused by contraction of the D4Z4 repeat array on chromosome 4 to a size of 1–10 units. The residual number of D4Z4 units inversely correlates with clinical severity, but significant clinical variability exists. Each unit contains a copy of the DUX4 retrogene. Repeat contractions are associated with changes in D4Z4 chromatin structure that increase the likelihood of DUX4 expression in skeletal muscle, but only when the repeat resides in a genetic background that contains a DUX4 polyadenylation signal. Mutations in the structural maintenance of chromosomes flexible hinge domain containing 1 (SMCHD1) gene, encoding a chromatin modifier of D4Z4, also result in the increased likelihood of DUX4 expression in individuals with a rare form of FSHD (FSHD2). Because SMCHD1 directly binds to D4Z4 and suppresses somatic expression of DUX4, we hypothesized that SMCHD1 may act as a genetic modifier in FSHD1. We describe three unrelated individuals with FSHD1 presenting an unusual high clinical severity based on their upper-sized FSHD1 repeat array of nine units. Each of these individuals also carries a mutation in the SMCHD1 gene. Familial carriers of the FSHD1 allele without the SMCHD1 mutation were only mildly affected, suggesting a modifier effect of the SMCHD1 mutation. Knocking down SMCHD1 in FSHD1 myotubes increased DUX4 expression, lending molecular support to a modifier role for SMCHD1 in FSHD1. We conclude that FSHD1 and FSHD2 share a common pathophysiological pathway in which the FSHD2 gene can act as modifier for disease severity in families affected by FSHD1.
Main Text
Facioscapulohumeral muscular dystrophy (FSHD [MIM 158900]) is one of the three most common muscular dystrophies in adults with an estimated prevalence of 1:20,000.1 Individuals with FSHD have facial, shoulder girdle, and upper extremity weakness that can spread with progression of the disease to abdominal, humeral, anterior lower leg muscles, and (in more severely affected individuals) to pelvic girdle muscles. The peculiar involvement of specific muscles is such a striking feature that it often distinguishes FSHD from other forms of muscular dystrophy.2 The disease usually becomes manifest in the second decade, but the progression and severity are highly variable with one-fifth of affected individuals becoming wheelchair dependent while an equal proportion of gene carriers remain asymptomatic throughout their lives.3
Autosomal-dominant FSHD1 represents the most common form, accounting for at least 95% of cases.4 It is caused by a contraction of the D4Z4 macrosatellite repeat array located in the subtelomeric region of chromosome 4q. The D4Z4 repeat is highly polymorphic in size, varying between 11 and 150 units in the general population, each unit being 3.3 kb in size.5–7 Individuals with FSHD1 have at least one allele of 1–10 units on chromosome 4.7,8
Despite the extensive interfamilial and intrafamilial variability in clinical severity and disease progression in FSHD1 families, there is a rough and inverse correlation between the residual size of the D4Z4 repeat, the age at onset, and the severity of muscular involvement. Indeed, small repeat arrays of 1–3 units tend to be associated with earlier onset and more rapid disease progression.9–12 Gender differences may also account for variability in clinical severity with males being more severely affected then females.1,13 The marked intrafamilial clinical variability further suggests the involvement of other genetic or environmental factors that modify the disease severity of a commonly inherited contraction size.
A minimum of one D4Z4 repeat is required to develop FSHD, suggesting that the repeat itself plays a critical role in the development of the disease.14 Indeed, each unit contains a copy of the DUX4 retrogene (MIM 606009) that becomes inappropriately derepressed in skeletal muscle of individuals with FSHD.15–18 DUX4 is a germline transcription factor that is normally repressed in somatic cells most probably by a mechanism of repeat-mediated heterochromatin formation. Its expression in skeletal muscle triggers germline and early stem cell programs, eventually causing muscle cell death.16,19–21 Consequent to the contraction, the repressive chromatin structure is compromised as evidenced by a loss of CpG methylation (hypomethylation) and repressive chromatin proteins and modifications22–24 and the concomitant gain in transcription-activating chromatin marks.25 Together, these changes in D4Z4 chromatin increase the likelihood of DUX4 expression in skeletal muscle, leading to a variegated pattern of DUX4-positive myonuclei.17,18,26
For FSHD to result, D4Z4 contraction needs to occur on FSHD-permissive chromosomal backgrounds.27,28 Internal copies of the DUX4 retrogene do not have a polyadenylation signal (PAS) but the distal copy of the D4Z4 repeat array can make use of a polymorphic PAS present on approximately one-half of the chromosome 4, commonly referred to as 4qA chromosomes (e.g., 4A161).15,29,30 In the absence of this DUX4 PAS, such as on 4qB chromosomes and on chromosome 10 where a highly homologous repeat array resides, transcriptional derepression of the FSHD locus does not normally lead to the production of stable DUX4 transcripts or to the appearance of FSHD clinical phenotype.29,31
A small group of individuals with FSHD show D4Z4 chromatin changes and DUX4 derepression in skeletal muscle in the absence of repeat array contraction. These individuals with FSHD2 (MIM 158901) carry at least one FSHD-permissive 4qA allele and are clinically identical to individuals with FSHD1.22,23,32 Unlike families with FSHD1 where the chromatin changes are mostly occurring on the contracted allele, in families with FSHD2, D4Z4 chromatin relaxation occurs on the D4Z4 repeat arrays of both chromosomes 4 and 10. Recently we showed that in the majority of families with FSHD2, the disease is caused by digenic inheritance of a DUX4 PAS-containing chromosome 4 and a mutation in the structural maintenance of chromosomes flexible hinge domain containing 1 (SMCHD1 [MIM 614982]) gene on chromosome 18.33 SMCHD1 is a chromatin modifier necessary for the establishment and maintenance of CpG methylation of the inactive X chromosome and specific classes of repeated elements.34–36 FSHD2 individuals have reduced SMCHD1 binding to the D4Z4 repeats on all four chromosomes, suggesting a haploinsufficiency mechanism. In support, knockdown of SMCHD1 in control myotube cultures with normal-sized D4Z4 repeat arrays on FSHD-permissive DUX4-PAS containing chromosomes leads to the transcriptional activation of DUX4.33
Because both FSHD1 and FSHD2 result from the somatic derepression of DUX4, we investigated whether SMCHD1 may act as a modifier for disease severity in families with FSHD1 and may have a role in the marked variability of clinical expression that is encountered in some families. Previous studies were only partially successful in explaining the variation in clinical severity in individuals with FSHD1. In addition to the inverse correlation with the residual repeat size,9,11,12,37 CpG methylation and histone modification studies of D4Z4 have likewise uncovered only rough correlations between residual methylation levels and clinical severity, with the most severely affected individuals with the smallest repeat sizes showing the largest reduction in repressive chromatin modifications.38,39
To identify modifiers of disease severity, of particular interest are those families with FSHD1 carrying upper-sized D4Z4 repeat arrays of 8–10 units, as shown by the fact that carriers of these alleles are more likely to have a partial or less severe form of FSHD or to be asymptomatic.40–42
To explore the possibility that mutations in SMCHD1 may modify the disease severity in families affected by FSHD1, we investigated the SMCHD1 locus in six unrelated individuals with FSHD1 from a cohort of 53 independent families with FSHD1 which carried a FSHD allele of 8–10 D4Z4 units. These individuals were selected based on D4Z4 methylation levels <25% indicative for FSHD2.33 We report that three of them have a mutation in SMCHD1 although in the remaining cases the cause for D4Z4 hypomethylation remains to be identified. These three cases have a repeat array of nine D4Z4 units on a FSHD-permissive DUX4 PAS-containing chromosome and show an unusually severe clinical presentation of the disease based on clinical evaluation that included manual muscle testing of 60 muscles and determination of the clinical severity score.39 The relevant biometric and genetic observations in these families are summarized in Table 1 and Table S1 available online. Pedigrees and genetic analyses of these three families are presented in Figure 1A. Signed and informed consent was obtained from all participants and family members according to protocols approved by the Institutional Ethics Review Boards of the University Hospital of Nice and collaborating institutes.
Table 1.
Clinical and Biometric Data of the Three Families
| Rf | Nr | Sex | AAE | Age at Onset | CSS | MMT Score | Diagnosis |
|---|---|---|---|---|---|---|---|
| Rf1021 | I-1 | M | 71 | unknowna | 3 | 261/300 | FSHD1 |
| I-2 | F | 67 | unknowna | 4 | 255/300 | FSHD2 | |
| II-1 | M | 48 | 12 | 10 | 65/300 | FSHD1+FSHD2 | |
| II-2 | F | 38 | – | 0 | 300/300 | unaffected | |
| III-1 | F | 15 | – | 0 | 300/300 | unaffected | |
| III-2 | M | 6 | 5 | 6 | 198/300 | FSHD1+FSHD2 | |
| Rf1110 | I-1 | M | 55 | 6 | 10 | 55/300 | FSHD1+FSHD2 |
| I-2 | F | 53 | – | 0 | 300/300 | unaffected | |
| II-1 | F | 26 | 24 | 5 | 280/300 | FSHD2 | |
| II-2 | M | 21 | unknowna | 2 | 269/300 | FSHD1 | |
| Rf1121 | II-1 | M | 67 | 15 | 10 | 75/300 | FSHD1+FSHD2 |
Abbreviations are as follows: CSS, clinical severity score; AAE, age at the last examination; MMT, manual muscle testing.
These patients do not report any symptoms.
Figure 1.
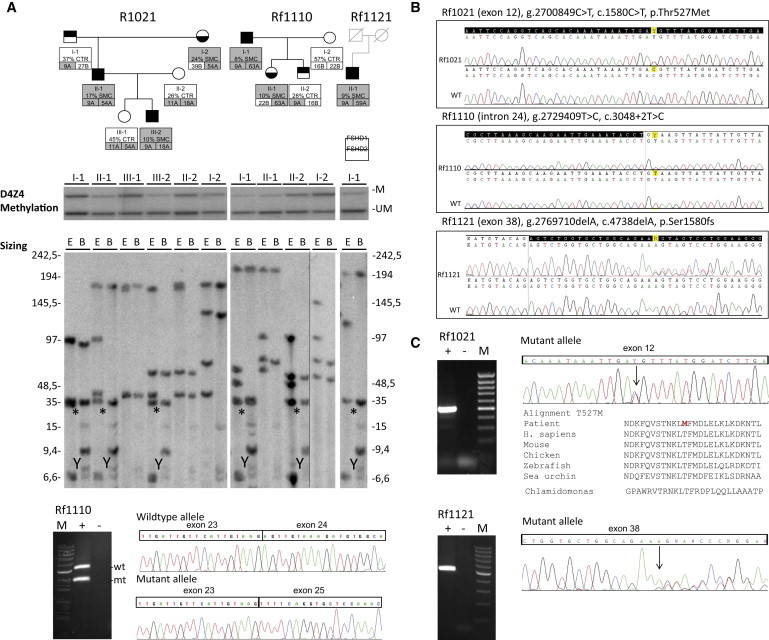
Genetic and Epigenetic Characterization of Three Families Affected by FSHD
(A) Pedigrees of the families with complete genetic data. Top: Pedigrees are shown of all three families, individuals in light gray were not available. Below each individual, information is given for the methylation level at the FseI site in the first units of the D4Z4 arrays (%) and the presence (SMC) or absence (CTR) of a SMCHD1 mutation. In addition, information is given for the size of the D4Z4 repeat arrays on chromosome 4 (in units) and the distal variation (A or B). Shaded boxes indicate FSHD-permissive genetic features. For example, individual I-1 of family Rf1110 has an FseI methylation level of 8% and is carrier of a SMCHD1 mutation. He carries one D4Z4 repeat array of 9 units on a 4A chromosome and one array of 63 units on a 4A chromosome. Middle: Methylation data of all three families with genomic DNA digested with restriction enzymes EcoRI, BglII, and methylation-sensitive FseI. Methylated (M) and unmethylated (UM) D4Z4 fragments are indicated. Bottom: D4Z4 repeat sizing data of samples in middle panel by double digestion of genomic DNA with EcoRI and HindIII (E) or with EcoRI and BlnI (B), followed by hybridization with probe p13E-11. Chromosome 4-derived D4Z4 arrays are BlnI resistant whereas chromosome 10-derived arrays are not. The disease-associated 9 units arrays (35 kb) are indicated with an asterisk. The cross-hybridizing DNA fragment on the Y chromosome is labeled with Y. Marker lanes are indicated on the right and left of the gel. Individual I-II of family Rf1110 was run on a separate gel as indicated with the vertical hairline.
(B) Sequence traces of the SMCHD1 mutations in all three families and in control samples. Exonic sequences are indicated in black and intronic sequences in white. The position of the heterozygous mutations are indicated above the sequence traces and are highlighted in yellow. The genomic position is based on UCSC Genome Browser hg19 chromosome 18, the transcript position on RefSeq accession number NM015295, and the protein position on NP056110.
(C) Overview of RNA analysis. Left: RT-PCR analysis via primers flanking the identified mutation on cDNA prepared from total RNA isolated from blood. The agarose gel images show RT-PCR fragments from the FSHD2 individuals (+). In addition, water control (−) and marker (M) lane are indicated. Right: RT-PCR sequence traces of the region covering the mutation in all three families. For the missense mutation in Rf1021, the evolutionary conservation is shown. For Rf1110, sequence traces are shown from wild-type (WT) and mutant allele (mt), the latter showing a skip of exon 24. In Rf1121 the 1 bp deletion indicated by the arrow causes mixed sequence traces.
In the first family (Rf1021), the proband (II-1) has been followed in our Center since the age of 35 when he became wheelchair dependent. He started to experience asymmetric scapular weakness at the age of 18, but he reported inability to blow into a flute at the age of 12. During the last examination, at age 48, he presented with severe and asymmetric facial weakness of orbicularis oculi and oris, marked shoulder girdle weakness associated with bilateral scapular winging, and humeral weakness. He displays marked hyperlordosis resulting from abdominal muscle weakness and weakness and atrophy of lower legs. Distal upper limb muscles are also becoming involved. He has a clinical severity score (CSS) of 10.11
Genetic analysis showed that this proband carries a nine D4Z4 unit 4A161 allele, confirming the diagnosis of FSHD1. His 6-year-old son (III-2) was referred to our Center because of difficulties in raising his arms and a history of frequent falls. At examination he presented with weakness of the orbicularis oculi muscles, mild shoulder girdle weakness with scapular winging, Gower’s sign, and asymmetric foot dorsiflexor weakness (CSS 6).
We also examined the proband’s daughter (III-1), 15 years old, and concluded that she was clinically unaffected. The father of the proband (I-1), at age 71, displayed mild facial and asymmetric shoulder girdle weakness (CSS 3) and the mother of the proband (I-2: 67 years old) showed asymmetric facial weakness and shoulder girdle involvement (CSS 4). Neither of them was complaining of any symptom except for arm fatigability. Extensive genotype, methylation, and SMCHD1 mutation analysis in this family showed that the proband’s father is a mildly affected individual with FSHD1 carrying the nine D4Z4 units on a 4A161 allele without a mutation in SMCHD1, consistent with the intermediate D4Z4 methylation levels. The proband’s mother, also mildly affected, is an individual with FSHD2 carrying a normal-sized 4A161 allele and a mutation in the SMCHD1 gene (c.1580C>T, Figure 1B). This mutation has not been reported previously in dbSNP, the 1000 Genomes Project, the ESP Exome Variant Server, or in-house databases and predicts a change of the amino acid threonine to methionine at protein position 527. The mutation was classified as damaging by the prediction programs PolyPhen-2, PMUT, and SNAP and confirmed in the RNA of the proband (Figure 1C). The proband and his affected son have inherited alleles for both FSHD1 (nine units 4A161 allele) and FSHD2 (marked hypomethylation of D4Z4 loci associated with c.1580C>T mutation in SMCHD1), suggesting a possible explanation for the severity of clinical phenotype in the proband and the unusual early onset in the son.
In the second family (Rf1110), the proband (I-1) was examined in our Center at the age of 56 showing, since the age of 6, a progressive and asymmetric weakness of facial muscles, shoulder girdle muscle weakness associated with scapular winging, and humeral muscle weakness. Abdominal weakness was also observed with marked hyperlordosis and pelvic girdle muscle weakness. Upon examination, the lower leg muscles were also affected (CSS 10). This person became wheelchair dependent by the age of 39. The diagnosis of FSHD1 was confirmed by the presence of a D4Z4 repeat array of nine units on a 4A161 allele.
His mother and father have no history of muscle disease and have not been examined. His son (II-2), 21 years old, was diagnosed as an asymptomatic FSHD1 carrier because he inherited the contracted 4A161 allele. At examination he showed very mild facial weakness and shoulder girdle involvement with mild right scapular winging (CSS 2). The daughter (II-1), 26 years old, did not inherit the contracted D4Z4 repeat array of nine units and was considered unaffected. On clinical examination, however, she had mild asymmetric facial weakness, scapular winging, and right anterior leg weakness. Functionally, this person’s only complaints are fatigability in right foot dorsiflexion and in arm raising above her head (CSS 5).
Methylation studies revealed slightly lower D4Z4 methylation levels in II-2, consistent with FSHD1 diagnosis and a marked hypomethylation in individuals I-1 and II-1, suggestive of FSHD2. These results prompted us to perform SMCHD1 mutation analysis resulting in the identification of a mutation in both individuals in a highly conserved nucleotide of the 5′ splice consensus site of exon 24, which has not been reported in any of the public databases (Figure 1B). RNA analysis in I-1 showed that this mutation leads to skipping of exon 24 (Figure 1C). In summary, in this family, the proband (I-1) carries the diagnosis of FSHD1 and FSHD2 and has a severe phenotype. The proband’s daughter (II-1), diagnosed with FSHD2, and his son (II-2), diagnosed with FSHD1, are only mildly affected.
In the last family (Rf1121), only individual II-1 was available for examination because his mother died at age 28 from delivery complications and his father died at age 35 from a work accident. He has no family history of muscle disease and no offspring. At age 15 he was noted to have asymmetric facial weakness of orbicularis oculi and oris muscles, shoulder girdle involvement with scapular winging, and abdominal muscle weakness. Anterior lower leg weakness appeared at the age of 30 and pelvic girdle muscles were subsequently involved. At the age of 33 he was walking with a cane. At the age of 40 he became wheelchair dependent and was unable to lift his arms. At his most recent examination (at age 65), his CSS was 10. Extensive genotyping of D4Z4 repeats on chromosomes 4 and 10 showed nine D4Z4 units on a 4A161 allele, confirming the diagnosis of FSHD1. Methylation studies revealed profound D4Z4 hypomethylation and SMCHD1 mutation analysis identified a SNP in exon 38 that is predicted to result in a frameshift in the SMCHD1 open reading frame, confirming the additional diagnosis of FSHD2 (Figure 1B). The frameshift mutation was confirmed in the RNA of individual II-1 (Figure 1C). In conclusion, these genetic and clinical data suggest that a mutation in SMCHD1 gene can act as a modifier of disease severity in individuals with FSHD1.
Previously, we showed that depletion of SMCHD1 by RNA interference in control myotubes carrying a normal-sized D4Z4 repeat leads to the transcriptional activation of DUX4.33 According to our clinical observations implying a synergistic effect of a D4Z4 contraction and a SMCHD1 mutation on transcriptional derepression of DUX4, we hypothesized that depletion of SMCHD1 in FSHD1 myotubes may result in transcriptional activation of DUX4 beyond that observed in FSHD1 myotubes, potentially resulting in a more severe phenotype. We tested this hypothesis by lentiviral transduction of SMCHD1 shRNAs into three independent FSHD1 myotube cultures and observed increased levels of DUX4 mRNA in myotubes with sufficient SMCHD1 knockdown (Figure 2). Consequent to the increase in DUX4 protein levels, we also detected transcriptional activation of the known DUX4 target genes ZSCAN4, RFLP2B, and TRIM43. Because two of the three myotube cultures are heterozygous for an FSHD-permissive 4qA chromosome with contracted D4Z4 repeat and a nonpermissive 4qB chromosome that cannot produce DUX4 in somatic cells (see Table S2), the increase in DUX4 levels upon SMCHD1 knockdown can be explained only by further chromatin relaxation of the contracted FSHD1 repeat.
Figure 2.
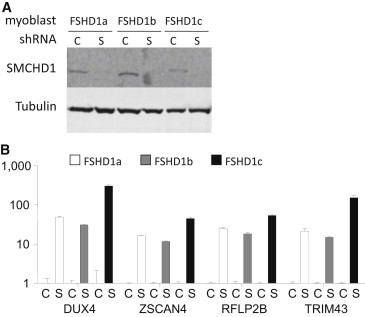
Depletion of SMCHD1 in FSHD1 Myoblast Augments DUX4 Expression
SMCHD1 depletion in FSHD1 primary myotubes leads to upregulation of DUX4 and DUX4 target transcripts RFLP2B, TRIM43, and ZSCAN4. Three FSHD1 myoblast cell lines (FSHD1a, FSHD1b, and FSHD1c) were transduced 48 hr prior to differentiation with lentiviral particles harboring scrambled shRNA constructs or SMCHD1 targeting constructs as described previously.33 After myotube formation, cells were harvested for protein and RNA isolation. cDNA was prepared and mRNA levels were measured by qRT-PCR as described.33
(A) Immunoblot analysis shows that SMCHD1 protein is depleted in myotubes transducted with shRNA constructs against SMCHD1 (S) and not with scrambled shRNA (C). Tubulin is used as a loading control.
(B) Relative expression levels of DUX4 and DUX4 target genes RFLP2B, TRIM43, and ZSCAN4 in SMCHD1-depleted FSHD1 myotubes. y axis depicts relative changes in expression levels corrected against housekeeping gene beta-glucuronidase after SMCHD1 depletion normalized to expression levels of samples without SMCHD1 depletion. Error bars represent standard error of the mean.
The marked variability in clinical severity, age at onset, and rate of progression, even between close relatives, is a clinical hallmark of FSHD. This feature, already reported in the first description of this disease in the late 1800s,43 was confirmed in later studies.1,12 Although residual repeat size D4Z4 methylation levels account for some of the variability, other (genetic) modifiers must contribute to the disease presentation, as was also concluded from population-based studies.27,44,45
In this context, the identification of three unrelated individuals with unusually severe clinical phenotypes associated with a borderline FSHD1 allele, and the presence of marked intrafamilial variability of disease severity in some familial carriers of the FSHD1 allele prompted us to consider the possibility that additional genetic defects may segregate in these families acting as disease modifiers. “Double trouble” in FSHD has been reported previously in isolated cases.46–48 In these cases, however, it was the result of concomitant mutations in other dystrophy genes and resulted in atypical FSHD phenotypes, suggesting synergistic effects of the two genetic lesions, rather than a genetic modifier acting on the D4Z4 locus itself. Nevertheless, we excluded mutations in VCP, FHL1, and CAPN3 by sequencing and (by histology, immunohistochemistry, and immunoblot studies on probands’ muscle biopsy) known muscular dystrophies and myopathies that could present with a clinical phenotype resembling FSHD49 (data not shown).
Our recent discovery that mutations in SMCHD1 cause FSHD2, the direct role for SMCHD1 in the chromatin structure of D4Z4, and the indistinguishable phenotypes between individuals with FSHD1 and those with FSHD2 led us to investigate SMCHD1 as a potential genetic modifier in FSHD1. We proposed that even though FSHD1 and FSHD2 do not have the same genetic defect, both probably have a common pathophysiological pathway converging on transcriptional derepression of DUX4 in skeletal muscle.50 Our observations in the present study, in addition to suggesting that SMCHD1 can act as a disease modifier, further support the hypothesis that FSHD1 and FSHD2 share a common pathophysiological pathway. Individuals with combined FSHD1 and FSHD2 genetic defects have a more severe clinical phenotype than expected based on the borderline repeat size of the FSHD1 allele, whereas individuals within the same family with only the FSHD1 borderline allele or the FSHD2 genetic defect were less severely affected. These findings suggest an additive effect of the SMCHD1 mutation on the pathophysiological pathway triggered by D4Z4 repeat array contraction by creating additional chromatin relaxation above the levels caused by D4Z4 contraction. This observation may explain why in occasional FSHD families the disease does not seem to segregate with a contracted 4qA allele.42,51 Considering the frequency of borderline D4Z4 repeat arrays in the population, we have to be alert for the possibility of an SMCHD1 mutation, or SMCHD1 variants that might influence its activity on D4Z4, when counseling families with FSHD. Based on this study, we would recommend SMCHD1 mutation testing in cases with upper FSHD-sized repeat arrays (8–10 units) with an unusual early onset or rapid disease progression. However, further studies are necessary in a larger cohort of individuals with FSHD1 in order to assess the effect of SMCHD1 as disease modifier.
In addition to SMCHD1, it is likely that other environmental or (epi)genetic factors are involved in modifying disease severity by facilitating or inhibiting the clinical expression. These might be trans-acting factors like other chromatin modifiers involved in repeat mediated gene silencing or cis-acting factors such as other SNPs in the distal end of the repeat that are unique to the FSHD-permissive alleles and that may affect D4Z4 chromatin structure by facilitating or inhibiting chromatin factor binding or processing of the D4Z4 transcripts. There have been only a few successful attempts at identifying modifier genes in Mendelian disorders.52 Recently, exome sequencing in an extreme phenotype study design facilitated the identification of variants in DCTN4 predisposing to specific parameters of P. aeruginosa airway infections in individuals with cystic fibrosis.53 To identify additional modifiers in FSHD, it is therefore imperative to study in detail FSHD-affected individuals with unusual phenotypes or genotypes as we have done here and to continue to study the complex regulation of D4Z4 during development and in different populations.
Acknowledgments
We thank all families for participating in this study. This work was supported by grants from the US National Institutes of Health (NIH) (National Institute of Neurological Disorders and Stroke [NINDS] P01NS069539 and National Institute of Arthritis and Musculoskeletal and Skin Diseases [NIAMS] R01AR045203), the Muscular Dystrophy Association (MDA; 217596), the Fields Center for FSHD Research, the Geraldi Norton and Eklund family foundation, the FSH Society, The Friends of FSH Research, the French Association Against Myopathies (AFM), European Union Framework Programme 7 2012-305121 (NEUROMICS), the Prinses Beatrix Spierfonds (W.OR-12-20), and the Stichting FSHD.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Mutalyzer, https://mutalyzer.nl/index
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
Richard Fields Center for FSHD Research, http://www.urmc.rochester.edu/fields-center/
UCSC Genome Browser, http://genome.ucsc.edu
Accession Numbers
Variant and phenotype data were submitted to the SMCHD1 gene variant database (http://www.LOVD.nl/SMCHD1/) with patient accession numbers 30789–30794 (see Table S1).
References
- 1.Padberg, G.W. (1982). Facioscapulohumeral disease. PhD thesis, Leiden University, Leiden.
- 2.Padberg G.W., Lunt P.W., Koch M., Fardeau M. Diagnostic criteria for facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 1991;1:231–234. doi: 10.1016/0960-8966(91)90094-9. [DOI] [PubMed] [Google Scholar]
- 3.Pandya S., King W.M., Tawil R. Facioscapulohumeral dystrophy. Phys. Ther. 2008;88:105–113. doi: 10.2522/ptj.20070104. [DOI] [PubMed] [Google Scholar]
- 4.Upadhyaya M., Maynard J., Rogers M.T., Lunt P.W., Jardine P., Ravine D., Harper P.S. Improved molecular diagnosis of facioscapulohumeral muscular dystrophy (FSHD): validation of the differential double digestion for FSHD. J. Med. Genet. 1997;34:476–479. doi: 10.1136/jmg.34.6.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hewitt J.E., Lyle R., Clark L.N., Valleley E.M., Wright T.J., Wijmenga C., van Deutekom J.C., Francis F., Sharpe P.T., Hofker M. Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 1994;3:1287–1295. doi: 10.1093/hmg/3.8.1287. [DOI] [PubMed] [Google Scholar]
- 6.van Deutekom J.C., Wijmenga C., van Tienhoven E.A., Gruter A.M., Hewitt J.E., Padberg G.W., van Ommen G.J., Hofker M.H., Frants R.R. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 1993;2:2037–2042. doi: 10.1093/hmg/2.12.2037. [DOI] [PubMed] [Google Scholar]
- 7.van Deutekom J.C., Bakker E., Lemmers R.J., van der Wielen M.J., Bik E., Hofker M.H., Padberg G.W., Frants R.R. Evidence for subtelomeric exchange of 3.3 kb tandemly repeated units between chromosomes 4q35 and 10q26: implications for genetic counselling and etiology of FSHD1. Hum. Mol. Genet. 1996;5:1997–2003. doi: 10.1093/hmg/5.12.1997. [DOI] [PubMed] [Google Scholar]
- 8.Wijmenga C., Hewitt J.E., Sandkuijl L.A., Clark L.N., Wright T.J., Dauwerse H.G., Gruter A.M., Hofker M.H., Moerer P., Williamson R. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat. Genet. 1992;2:26–30. doi: 10.1038/ng0992-26. [DOI] [PubMed] [Google Scholar]
- 9.Goto K., Lee J.H., Matsuda C., Hirabayashi K., Kojo T., Nakamura A., Mitsunaga Y., Furukawa T., Sahashi K., Arahata K. DNA rearrangements in Japanese facioscapulohumeral muscular dystrophy patients: clinical correlations. Neuromuscul. Disord. 1995;5:201–208. doi: 10.1016/0960-8966(94)00055-e. [DOI] [PubMed] [Google Scholar]
- 10.Lunt P.W., Jardine P.E., Koch M., Maynard J., Osborn M., Williams M., Harper P.S., Upadhyaya M. Phenotypic-genotypic correlation will assist genetic counseling in 4q35- facioscapulohumeral muscular dystrophy. Muscle Nerve. 1995;2:103–109. [PubMed] [Google Scholar]
- 11.Ricci E., Galluzzi G., Deidda G., Cacurri S., Colantoni L., Merico B., Piazzo N., Servidei S., Vigneti E., Pasceri V. Progress in the molecular diagnosis of facioscapulohumeral muscular dystrophy and correlation between the number of KpnI repeats at the 4q35 locus and clinical phenotype. Ann. Neurol. 1999;45:751–757. doi: 10.1002/1531-8249(199906)45:6<751::aid-ana9>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 12.Tawil R., Forrester J., Griggs R.C., Mendell J., Kissel J., McDermott M., King W., Weiffenbach B., Figlewicz D., The FSH-DY Group Evidence for anticipation and association of deletion size with severity in facioscapulohumeral muscular dystrophy. Ann. Neurol. 1996;39:744–748. doi: 10.1002/ana.410390610. [DOI] [PubMed] [Google Scholar]
- 13.Zatz M., Marie S.K., Cerqueira A., Vainzof M., Pavanello R.C., Passos-Bueno M.R. The facioscapulohumeral muscular dystrophy (FSHD1) gene affects males more severely and more frequently than females. Am. J. Med. Genet. 1998;77:155–161. [PubMed] [Google Scholar]
- 14.Tupler R., Berardinelli A., Barbierato L., Frants R., Hewitt J.E., Lanzi G., Maraschio P., Tiepolo L. Monosomy of distal 4q does not cause facioscapulohumeral muscular dystrophy. J. Med. Genet. 1996;33:366–370. doi: 10.1136/jmg.33.5.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dixit M., Ansseau E., Tassin A., Winokur S., Shi R., Qian H., Sauvage S., Mattéotti C., van Acker A.M., Leo O. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl. Acad. Sci. USA. 2007;104:18157–18162. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geng L.N., Yao Z., Snider L., Fong A.P., Cech J.N., Young J.M., van der Maarel S.M., Ruzzo W.L., Gentleman R.C., Tawil R., Tapscott S.J. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev. Cell. 2012;22:38–51. doi: 10.1016/j.devcel.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones T.I., Chen J.C., Rahimov F., Homma S., Arashiro P., Beermann M.L., King O.D., Miller J.B., Kunkel L.M., Emerson C.P., Jr. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis. Hum. Mol. Genet. 2012;21:4419–4430. doi: 10.1093/hmg/dds284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snider L., Geng L.N., Lemmers R.J.L.F., Kyba M., Ware C.B., Nelson A.M., Tawil R., Filippova G.N., van der Maarel S.M., Tapscott S.J., Miller D.G. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6:e1001181. doi: 10.1371/journal.pgen.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bosnakovski D., Xu Z., Gang E.J., Galindo C.L., Liu M., Simsek T., Garner H.R., Agha-Mohammadi S., Tassin A., Coppée F. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 2008;27:2766–2779. doi: 10.1038/emboj.2008.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitsuhashi H., Mitsuhashi S., Lynn-Jones T., Kawahara G., Kunkel L.M. Expression of DUX4 in zebrafish development recapitulates facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 2013;22:568–577. doi: 10.1093/hmg/dds467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wallace L.M., Liu J., Domire J.S., Garwick-Coppens S.E., Guckes S.M., Mendell J.R., Flanigan K.M., Harper S.Q. RNA interference inhibits DUX4-induced muscle toxicity in vivo: implications for a targeted FSHD therapy. Mol. Ther. 2012;20:1417–1423. doi: 10.1038/mt.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Greef J.C., Lemmers R.J., Camaño P., Day J.W., Sacconi S., Dunand M., van Engelen B.G., Kiuru-Enari S., Padberg G.W., Rosa A.L. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology. 2010;75:1548–1554. doi: 10.1212/WNL.0b013e3181f96175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Overveld P.G., Lemmers R.J., Sandkuijl L.A., Enthoven L., Winokur S.T., Bakels F., Padberg G.W., van Ommen G.J., Frants R.R., van der Maarel S.M. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet. 2003;35:315–317. doi: 10.1038/ng1262. [DOI] [PubMed] [Google Scholar]
- 24.Zeng W., de Greef J.C., Chen Y.Y., Chien R., Kong X., Gregson H.C., Winokur S.T., Pyle A., Robertson K.D., Schmiesing J.A. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD) PLoS Genet. 2009;5:e1000559. doi: 10.1371/journal.pgen.1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cabianca D.S., Casa V., Bodega B., Xynos A., Ginelli E., Tanaka Y., Gabellini D. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell. 2012;149:819–831. doi: 10.1016/j.cell.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tassin A., Laoudj-Chenivesse D., Vanderplanck C., Barro M., Charron S., Ansseau E., Chen Y.W., Mercier J., Coppée F., Belayew A. DUX4 expression in FSHD muscle cells: how could such a rare protein cause a myopathy? J. Cell. Mol. Med. 2013;17:76–89. doi: 10.1111/j.1582-4934.2012.01647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemmers R.J., Wohlgemuth M., van der Gaag K.J., van der Vliet P.J., van Teijlingen C.M., de Knijff P., Padberg G.W., Frants R.R., van der Maarel S.M. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2007;81:884–894. doi: 10.1086/521986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemmers R.J., van der Vliet P.J., van der Gaag K.J., Zuninga S., Frants R.R., de Knijff P., van der Maarel S.M. Worldwide population analysis of the 4q and 10q subtelomeres identifies only four discrete interchromosomal sequence transfers in human evolution. Am. J. Hum. Genet. 2010;86:364–377. doi: 10.1016/j.ajhg.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lemmers R.J., van der Vliet P.J., Klooster R., Sacconi S., Camaño P., Dauwerse J.G., Snider L., Straasheijm K.R., van Ommen G.J., Padberg G.W. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Snider L., Asawachaicharn A., Tyler A.E., Geng L.N., Petek L.M., Maves L., Miller D.G., Lemmers R.J., Winokur S.T., Tawil R. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum. Mol. Genet. 2009;18:2414–2430. doi: 10.1093/hmg/ddp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lemmers R.J., Wohlgemuth M., Frants R.R., Padberg G.W., Morava E., van der Maarel S.M. Contractions of D4Z4 on 4qB subtelomeres do not cause facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2004;75:1124–1130. doi: 10.1086/426035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Greef J.C., Lemmers R.J., van Engelen B.G., Sacconi S., Venance S.L., Frants R.R., Tawil R., van der Maarel S.M. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum. Mutat. 2009;30:1449–1459. doi: 10.1002/humu.21091. [DOI] [PubMed] [Google Scholar]
- 33.Lemmers R.J., Tawil R., Petek L.M., Balog J., Block G.J., Santen G.W., Amell A.M., van der Vliet P.J., Almomani R., Straasheijm K.R. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012;44:1370–1374. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blewitt M.E., Vickaryous N.K., Hemley S.J., Ashe A., Bruxner T.J., Preis J.I., Arkell R., Whitelaw E. An N-ethyl-N-nitrosourea screen for genes involved in variegation in the mouse. Proc. Natl. Acad. Sci. USA. 2005;102:7629–7634. doi: 10.1073/pnas.0409375102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blewitt M.E., Gendrel A.V., Pang Z., Sparrow D.B., Whitelaw N., Craig J.M., Apedaile A., Hilton D.J., Dunwoodie S.L., Brockdorff N. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat. Genet. 2008;40:663–669. doi: 10.1038/ng.142. [DOI] [PubMed] [Google Scholar]
- 36.Gendrel A.V., Apedaile A., Coker H., Termanis A., Zvetkova I., Godwin J., Tang Y.A., Huntley D., Montana G., Taylor S. Smchd1-dependent and -independent pathways determine developmental dynamics of CpG island methylation on the inactive X chromosome. Dev. Cell. 2012;23:265–279. doi: 10.1016/j.devcel.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lunt P.W., Jardine P.E., Koch M.C., Maynard J., Osborn M., Williams M., Harper P.S., Upadhyaya M. Correlation between fragment size at D4F104S1 and age at onset or at wheelchair use, with a possible generational effect, accounts for much phenotypic variation in 4q35-facioscapulohumeral muscular dystrophy (FSHD) Hum. Mol. Genet. 1995;4:951–958. doi: 10.1093/hmg/4.5.951. [DOI] [PubMed] [Google Scholar]
- 38.Balog J., Thijssen P.E., de Greef J.C., Shah B., van Engelen B.G., Yokomori K., Tapscott S.J., Tawil R., van der Maarel S.M. Correlation analysis of clinical parameters with epigenetic modifications in the DUX4 promoter in FSHD. Epigenetics. 2012;7:579–584. doi: 10.4161/epi.20001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Overveld P.G., Enthoven L., Ricci E., Rossi M., Felicetti L., Jeanpierre M., Winokur S.T., Frants R.R., Padberg G.W., van der Maarel S.M. Variable hypomethylation of D4Z4 in facioscapulohumeral muscular dystrophy. Ann. Neurol. 2005;58:569–576. doi: 10.1002/ana.20625. [DOI] [PubMed] [Google Scholar]
- 40.Butz M., Koch M.C., Müller-Felber W., Lemmers R.J., van der Maarel S.M., Schreiber H. Facioscapulohumeral muscular dystrophy. Phenotype-genotype correlation in patients with borderline D4Z4 repeat numbers. J. Neurol. 2003;250:932–937. doi: 10.1007/s00415-003-1116-y. [DOI] [PubMed] [Google Scholar]
- 41.Felice K.J., Whitaker C.H. The clinical features of facioscapulohumeral muscular dystrophy associated with borderline (>/=35 kb) 4q35 EcoRI fragments. J. Clin. Neuromuscul. Dis. 2005;6:119–126. doi: 10.1097/01.cnd.0000158302.79014.c4. [DOI] [PubMed] [Google Scholar]
- 42.Scionti I., Fabbri G., Fiorillo C., Ricci G., Greco F., D’Amico R., Termanini A., Vercelli L., Tomelleri G., Cao M. Facioscapulohumeral muscular dystrophy: new insights from compound heterozygotes and implication for prenatal genetic counselling. J. Med. Genet. 2012;49:171–178. doi: 10.1136/jmedgenet-2011-100454. [DOI] [PubMed] [Google Scholar]
- 43.Landouzy L., Dejerine J. De la myopathy atrophique progressive. Rev. Med. 1885;5:81–117. [Google Scholar]
- 44.Scionti I., Greco F., Ricci G., Govi M., Arashiro P., Vercelli L., Berardinelli A., Angelini C., Antonini G., Cao M. Large-scale population analysis challenges the current criteria for the molecular diagnosis of fascioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2012;90:628–635. doi: 10.1016/j.ajhg.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y., Forner J., Fournet S., Jeanpierre M. Improved characterization of FSHD mutations. Ann. Genet. 2001;44:105–110. doi: 10.1016/s0003-3995(01)01075-9. [DOI] [PubMed] [Google Scholar]
- 46.Ricci G., Scionti I., Alì G., Volpi L., Zampa V., Fanin M., Angelini C., Politano L., Tupler R., Siciliano G. Rippling muscle disease and facioscapulohumeral dystrophy-like phenotype in a patient carrying a heterozygous CAV3 T78M mutation and a D4Z4 partial deletion: Further evidence for “double trouble” overlapping syndromes. Neuromuscul. Disord. 2012;22:534–540. doi: 10.1016/j.nmd.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rudnik-Schöneborn S., Weis J., Kress W., Häusler M., Zerres K. Becker’s muscular dystrophy aggravating facioscapulohumeral muscular dystrophy—double trouble as an explanation for an atypical phenotype. Neuromuscul. Disord. 2008;18:881–885. doi: 10.1016/j.nmd.2008.06.387. [DOI] [PubMed] [Google Scholar]
- 48.Sacconi S., Salviati L., Bourget I., Figarella D., Péréon Y., Lemmers R., van der Maarel S., Desnuelle C. Diagnostic challenges in facioscapulohumeral muscular dystrophy. Neurology. 2006;67:1464–1466. doi: 10.1212/01.wnl.0000240071.62540.6f. [DOI] [PubMed] [Google Scholar]
- 49.Sacconi S., Camaño P., de Greef J.C., Lemmers R.J., Salviati L., Boileau P., Lopez de Munain Arregui A., van der Maarel S.M., Desnuelle C. Patients with a phenotype consistent with facioscapulohumeral muscular dystrophy display genetic and epigenetic heterogeneity. J. Med. Genet. 2012;49:41–46. doi: 10.1136/jmedgenet-2011-100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van der Maarel S.M., Miller D.G., Tawil R., Filippova G.N., Tapscott S.J. Facioscapulohumeral muscular dystrophy: consequences of chromatin relaxation. Curr. Opin. Neurol. 2012;25:614–620. doi: 10.1097/WCO.0b013e328357f22d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tonini M.M., Passos-Bueno M.R., Cerqueira A., Matioli S.R., Pavanello R., Zatz M. Asymptomatic carriers and gender differences in facioscapulohumeral muscular dystrophy (FSHD) Neuromuscul. Disord. 2004;14:33–38. doi: 10.1016/j.nmd.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 52.Hamilton B.A., Yu B.D. Modifier genes and the plasticity of genetic networks in mice. PLoS Genet. 2012;8:e1002644. doi: 10.1371/journal.pgen.1002644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Emond M.J., Louie T., Emerson J., Zhao W., Mathias R.A., Knowles M.R., Wright F.A., Rieder M.J., Tabor H.K., Nickerson D.A., National Heart, Lung, and Blood Institute (NHLBI) GO Exome Sequencing Project. Lung GO Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat. Genet. 2012;44:886–889. doi: 10.1038/ng.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.