

Abstract
We searched for disruptive, genic rare copy-number variants (CNVs) among 411 families affected by sporadic autism spectrum disorder (ASD) from the Simons Simplex Collection by using available exome sequence data and CoNIFER (Copy Number Inference from Exome Reads). Compared to high-density SNP microarrays, our approach yielded ∼2× more smaller genic rare CNVs. We found that affected probands inherited more CNVs than did their siblings (453 versus 394, p = 0.004; odds ratio [OR] = 1.19) and that the probands’ CNVs affected more genes (921 versus 726, p = 0.02; OR = 1.30). These smaller CNVs (median size 18 kb) were transmitted preferentially from the mother (136 maternal versus 100 paternal, p = 0.02), although this bias occurred irrespective of affected status. The excess burden of inherited CNVs among probands was driven primarily by sibling pairs with discordant social-behavior phenotypes (p < 0.0002, measured by Social Responsiveness Scale [SRS] score), which contrasts with families where the phenotypes were more closely matched or less extreme (p > 0.5). Finally, we found enrichment of brain-expressed genes unique to probands, especially in the SRS-discordant group (p = 0.0035). In a combined model, our inherited CNVs, de novo CNVs, and de novo single-nucleotide variants all independently contributed to the risk of autism (p < 0.05). Taken together, these results suggest that small transmitted rare CNVs play a role in the etiology of simplex autism. Importantly, the small size of these variants aids in the identification of specific genes as additional risk factors associated with ASD.
Introduction
Discovering the mutations and the genes responsible for autism spectrum disorder (ASD) requires an assessment of the full spectrum of genetic variation, including both de novo and inherited events, within families. There is compelling evidence that a diverse range of de novo mutations, including copy-number variants (CNVs),1–5 single-nucleotide variants (SNVs), and insertions and deletions (indels),6–10 play an important role. However, all together, de novo variation does not fully explain the genetic etiology of ASD: only ∼8% of probands carry a de novo CNV, and ∼10%–20% carry a pathogenic de novo SNV or indel. Many of these mutations most likely play a pathogenic role in the development of ASD, especially in the context of sporadic (or “simplex”) ASD. However, the heritability of ASD is estimated to be between 50% and 90%11–13—much higher than the explained fraction of disease to date—suggesting that additional genetic factors contribute to the etiology of ASD.
The prevalence of rare CNVs smaller than 50 kb has been underestimated in previous surveys using oligonucleotide microarrays,1,2 and their role in ASD has not been extensively explored (but see Prasad et al.14 for an analysis of small CNVs in a case-control ASD cohort). Such pathogenic events could in principle provide as much specificity as exonic de novo mutations with respect to genes and informative protein networks. Several recent methods based on exome sequencing read-depth data, such as CoNIFER (Copy Number Inference from Exome Reads), employed in this work, have enabled the discovery of small genic CNVs previously missed by microarray.15 In this study, we tested the hypothesis that small genic inherited CNVs also contribute to the genetic etiology of sporadic autism. Several lines of evidence—including increased prevalence of the broader autism phenotype (BAP) in parents of affected children,16,17 trends for higher burden of extremely rare singly transmitted CNVs in simplex families,1 and enrichment of large CNVs in cases versus unrelated controls5—are potentially supportive of this hypothesis. In contrast, other previous studies that examined inherited CNVs in ASD found no significant excess of inherited burden in probands with ASD, although these studies were mainly designed to detect de novo CNVs.2
We investigated families in which both affected and unaffected siblings had been exome sequenced, and here we present evidence of transmission disequilibrium for smaller CNVs (median size ∼18 kb). By leveraging normalized read depth from whole-exome sequence data, we have added nearly 2-fold inherited, small, genic CNVs to the body of known variants in these samples. Finally, we present a model that integrates both rare SNVs and CNVs to more fully explain the genetic architecture of ASD.
Material and Methods
CNV Detection from Exome Sequence Data
We analyzed exome sequence data from families ascertained as part of the Simons Simplex Collection (SSC).18 Underlying FASTQ sequence data were obtained from 391 published ASD quads (consisting of a proband, an unaffected sibling, a mother, and a father),6–8 and we generated sequence data for an additional 19 unaffected siblings from published trios.10 The data set included sequence data (median coverage > 50×) from 411 families in which a quad had been sequenced for a total of 1,644 samples (see Tables S1 and S2, available online, for details). Sequence reads were split into 36-mers and were mapped with the mrsFAST alignment program19 to the Nimblegen EZ-SeqCap v.2 targets (including 300 bp around each target and allowing two mismatches per 36-mer). We used CoNIFER15 to calculate exon-level coverage and removed systematic bias between samples and targets. Using a custom pipeline (Figure S1), we (1) segmented our CoNIFER SVD-ZRPKM values by using the DNACopy algorithm,20 (2) minimized false negatives by a quad-based genotyping method, (3) clustered CNVs into overlapping CNV regions (CNVRs), and (4) removed CNVs found in duplicated or repetitive genomic space. We limited our final call set to inherited CNVs (i.e., transmitted CNVs) that were present in ten or fewer families (or approximately 1% population frequency), and we excluded CNVs that primarily fell within duplicated or highly polymorphic regions of the genome. We considered a CNV “rare” if it occurred in ten or fewer families and a CNV “private” if it was observed only in one family. Lastly, we did not include CNVs on the X chromosome in any analysis, and all de novo CNVs were excluded from burden analyses except where noted. Throughout this paper, we define “CNV burden” as the number of rare CNVs per individual. This study was approved by the institutional review board of the University of Washington.
Array Comparative Genomic Hybridization
We designed a customized comparative genomic hybridization (CGH) microarray (Agilent SurePrint G3 4x180K CGH microarray; probe density ranged from 1/125 bp to 1/5 kb depending on the size of the event to be validated) and selected 161 CNVs from a subset of 80 samples, stratified by affected status (36 probands and 44 siblings) and by data set (26 from Iossifov et al.,6 22 from O’Roak et al.,7 32 from Sanders et al.8 [Tables S1 and S5]). The minimum deletion and duplication thresholds for validation were determined by receiver operating characteristic (ROC) curve analysis of known positive- and negative-control CNVs (Figure S3).
Phenotypic Measures and Models
The Social Responsiveness Scale (SRS) was used as a quantitative measure of social deficits.21 We had complete phenotype information based on data from the SSC (SRS scores for both probands and siblings and full-scale IQs [FSIQs] for probands) for 389 families in this study (Table S7). The probands in this study had a median SRS t-score of 82, significantly higher (i.e., indicating a more severe phenotype) than the median SRS score of our unaffected siblings (45; p < 0.00001, two-tailed paired t test). We defined mild, moderate, and severely affected individuals on the basis of published thresholds.21
Expression Analysis
Gene-expression data were from the Human U133A/GNF1H Gene Atlas (Gene Expression Omnibus accession number GSE1133), comprising 79 human tissues, including 18 nervous system tissues.22 Expression values were averaged across multiple probes when available. We defined a gene to be expressed in a given tissue if it ranked in the top 5% of all genes for that tissue. To measure enrichment, we compared the fraction of genes unique to either siblings or probands expressed in each tissue and calculated empirical p values by shuffling proband and sibling labels 20,000 times and recomputing tissue-level expression enrichment. We used the false discovery rate (FDR) method to correct for 79 tests (i.e., for tissues) and assessed statistical significance at q < 0.05.
Combined Mutation Model
We generated a list of truncating de novo SNVs (nonsense, frameshift, or splice mutations) discovered in our 411 quads from published lists.6–8,10 Both de novo and inherited CNV burden was derived from this work (Tables S3 and S4). We used a logistic regression model, which transforms the binary outcome (i.e., affected versus unaffected) such that linear predictors can be used. The model shown in Figure 5 is summarized as follows:
Figure 5.

A Combined Model of Inherited and De Novo Mutations Reveals Independent Risk for Both
A logistic regression model estimates the OR for each inherited CNV (blue), de novo CNV (red), or disruptive de novo SNV (gray; nonsense and splice mutations and indels only) in probands and siblings. ORs and burden (proband-sibling ratio) given in the accompanying table reveal independent risk for each type of mutation. The line width for each type of mutation in the figure indicates whether a bias was observed for new mutations arising on the maternal or paternal haplotypes (see also O’Roak et al.7 for SNVs and Hehir-Kwa et al.,34 for CNVs).
Results
Samples and CNV Discovery
We discovered a total of 847 transmitted, exonic, rare, autosomal CNVs (Table 1), including 453 transmitted to probands and 394 transmitted to unaffected siblings. Overall, the median estimated CNV size was 18.1 kb (range = 150 bp to 5.18 Mb or 2–320 exons). The median size of inherited CNVs was slightly larger in probands (19.4 kb) than in unaffected siblings (16.6 kb), but this difference was not statistically significant. As expected, duplications outnumbered deletions (519 versus 328, p < 1 × 10−10, binomial two-tailed test), and duplications were significantly larger than deletions (two-sided Mann-Whitney U test, p < 1 × 10−16). The excess of duplications depended upon the size of the event. For example, rare CNVs involving 20 or more exons were overwhelmingly duplications (139 duplications versus 25 deletions), whereas small events were not significantly different (73 duplications and 93 deletions for 2-exon CNVs). This difference was observed irrespective of disease status (Figure S4).
Table 1.
Summary of Transmitted CNVs in 411 ASD Quads
| Category | CNVs | Dups | Dels | Median Size (Estimate) | Percentage of Samples | CNVs > 500 kb | CNVRs |
|---|---|---|---|---|---|---|---|
| All proband CNVs | 453 | 277 | 176 | 19.4 kb | 64% | 21 | 390 |
| All sibling CNVs | 394 | 242 | 152 | 16.6 kb | 60% | 16 | 345 |
| Father → both | 199 | 130 | 69 | 16.7 kb | 41% | 7 | 94 |
| Father → proband only | 100 | 67 | 33 | 25.0 kb | 19% | 7 | 93 |
| Father → sibling only | 82 | 52 | 30 | 15.4 kb | 18% | 2 | 80 |
| Mother → both | 233 | 127 | 106 | 15.0 kb | 48% | 10 | 118 |
| Mother → proband only | 136 | 82 | 54 | 24.9 kb | 26% | 5 | 128 |
| Mother → sibling only | 97 | 61 | 36 | 21.7 kb | 21% | 6 | 94 |
| Either parent → proband only | 236 | 149 | 87 | 25.0 kb | 39% | 12 | 211 |
| Either parent → sibling only | 179 | 113 | 66 | 19.3 kb | 36% | 8 | 168 |
| Mother → either offspring | 466 | 270 | 196 | 17.8 kb | 86% | 21 | 313 |
| Father → either offspring | 381 | 249 | 132 | 18.6 kb | 72% | 16 | 252 |
| Totals | 847 | 519 | 328 | 18.1 kb | 62% | 37 | 525 |
The following abbreviations are used: dups, duplications; and dels, deletions.
Validation Using SNP Microarray and Targeted Array CGH
We assessed the specificity of our call set by comparing our larger calls to Illlumina 1M/Duo SNP microarrary data and then selecting a subset of 80 samples for validation of smaller CNVs by array CGH validation. These 80 samples carried a total of 161 exome-based CNV calls, of which 69 (43%) were confirmed by SNP microarray (Figure 1A). Using a customized microarray design (Material and Methods), we were able to test 86 of the 92 remaining calls and confirmed an additional 65 events (nearly a 2-fold increased yield of CNVs) (Table S5). Of the 27 events not validated by array CGH, 14 (or 9% of all 161 calls) were found to be specifically part of processed pseudogenes (i.e., retrotranscribed mRNA), which masquerade as duplications in exome-based discovery of CNVs, indicating that these events—although not genomic CNVs—were in fact true duplications of these genes or exons. Thus, we estimated an overall false-positive rate (FPR) of 4%–8% (7/155 tested or 13/161 in total; Figure 1A), depending on the number of probes (or exons) in each call: for calls with fewer than ten exons, the FPR was ∼7% (6/104), whereas only one call with ten or more exons did not validate (1/51 [2%]). There was no difference in the FPR between probands and siblings (3/68 [4.2%] for probands versus 4/80 [4.5%] for siblings; Table S6).
Figure 1.
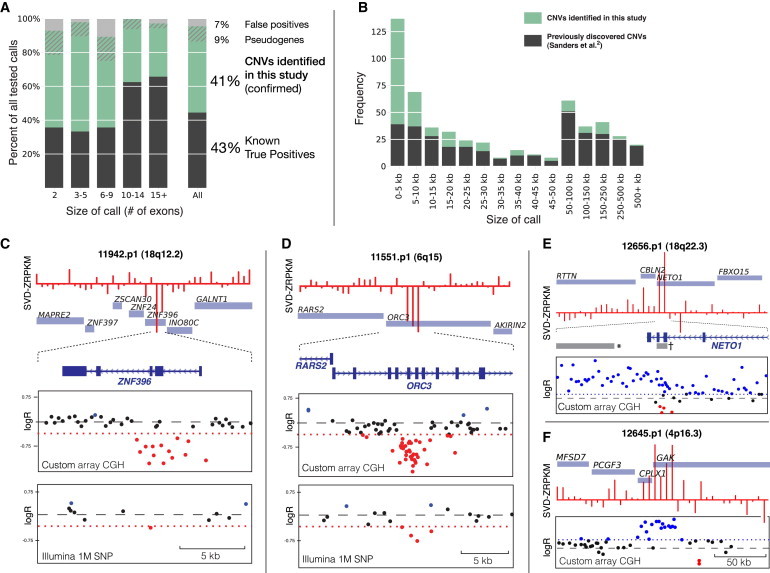
Discovery and Validation of CNVs with the Use of Exomes
(A) Fraction of CNVs previously identified via Illumina 1M SNP microarray (gray, “known true positives”), the fraction of CNVs identified and confirmed by targeted array CGH in this study (green, “CNVs identified in this study”), confirmed processed pseudogenes (hatched green), and the overall FPR for unconfirmed CNVs (gray).
(B) The majority (73% [152/207]) of all calls (green) identified in this study with the use of exomes were smaller than 20 kb.
(C–D and F) Three examples of CNVs in this study. Top: CoNIFER output and normalized coverage at each exon. Middle: targeted array CGH at CNV locus; the threshold for deletion or duplication (dotted red line) was determined by ROC-curve analysis of known CNVs (Supplemental Data). Bottom: Illumina 1M SNP microarray data for locus shows poor probe coverage (C and D only).
(E) Exome-based CNV discovery affords high exon-level specificity, as indicated by duplication of NETO1 exons (†, CoNIFER call). Previous work (Sanders et al.2) discovered this CNV (∗), but the (incorrect) breakpoints did not extend into NETO1.
We also assessed the sensitivity (or false-negative rate [FNR]) of our calls versus the previously identified CNVs from SNP microarray data. We found that our pipeline identified 72% (FNR of 0.28) of all known CNVs intersecting at least two exons and supported by ten SNP microarray probes. False-negative CNVs corresponded to samples with reduced mapped sequence coverage (Figure S2). For example, the Iossivof data set6 had an approximately 2-fold higher FNR, most likely due to the lower overall sequence coverage in these exomes (a known factor in exome-based CNV discovery14,15), and the FNR for calls affecting only two exons was significantly higher than for those with three or more exons (Table S6). We found no differences in the mapped coverage, estimated FPRs, or estimated FNRs among siblings and probands (p > 0.3, Fisher’s two-sided exact test and Table S6).
Increased Inherited CNV Burden among Autism Probands
We compared the burden of inherited CNVs in the 411 probands and their siblings in terms of the total number of CNVs and the total number of genes “hit.” We found that probands inherited more CNVs than did their siblings (453 versus 394; Figure 2A) and that the probands’ CNVs harbored more genes (921 versus 726; Figure 2B). These comparisons were significant when a paired t test was used for proband-sibling pairs (p = 0.02 for genes and p = 0.004 for CNVs, two-tailed paired t test) and when the summed values for probands and siblings were compared in aggregate (p < 1 × 10−6 for genes and p = 0.046 for CNVs, binomial two-tailed test). In order to ensure that these results were not driven by a few outlier families, we bootstrapped our data and calculated the confidence intervals (CIs) for the proband-to-sibling burden (Figure S5). We found a median burden increase of 1.19 (95% CI = 1.09–1.29) for CNVs and a burden increase of 1.30 (95% CI = 1.10–1.52) for genes across 10,000 bootstrap replicates, thereby rejecting the null hypothesis that the inherited CNV burden in probands is not higher than that in their siblings (Figure S5). Proband CNV burden was elevated over that of siblings across all size ranges, although individual quintile bins did not independently achieve statistical significance because of their smaller size (Figure 2C). We found no significant enrichment of burden in either the smallest or the largest CNVs (by chi-square test: χ2 = 1.18, p = 0.95, 5 degrees of freedom [df]), suggesting that the burden was not exclusively the result of either small or large CNVs.
Figure 2.
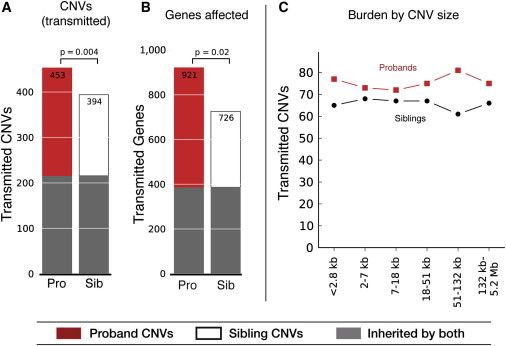
Increased Inherited CNV Burden in ASD Probands for Large and Small CNVs
(A) Total number of rare (observed in fewer than ten families) inherited CNVs (at least two exons) for 411 ASD probands (Pro) and their unaffected siblings (Sib).
(B) Total number of affected genes in rare inherited CNVs. p values refer to two-tailed paired t tests between proband and sibling counts.
(C) Burden of inherited CNVs across six size categories.
Previous work has indicated that private or ultra-rare CNVs might be more likely to be pathogenic than simply “rare” (e.g., <1% frequency) CNVs.1 We therefore examined whether the inherited burden in probands was due to private CNVs in a small subset of the 411 families. We examined 271 private CNVs in probands and 245 private CNVs in siblings but found no enrichment of private burden when compared to rare CNVs (p = 0.74, Fisher’s exact test; Figure S6A), nor did we find enrichment of the number of affected genes (p = 0.46, fisher’s exact test; Figure S6B). (Note: the burden was in fact slightly increased when all rare events were considered.) We searched for additional factors that could underlie the proband-sibling burden differential. We found no significant differences in CNV burden dependent on the sex of the proband or sibling, the concordance of their sexes, or the birth order of the proband and sibling (p > 0.5, Fisher’s exact test; Table S8). However, we note that the highest overall CNV burden was found in families with one affected proband and at least three unaffected siblings. In fact, there was a linear increase in burden between probands and siblings across increasing family size, culminating in a 1.38-fold higher burden of CNVs in probands with three or more unaffected siblings.
Finally, we analyzed our data set for parent-of-origin effects and found a greater number of maternally transmitted CNVs (136 maternal versus 100 paternal, binomial two-tailed p value = 0.02), but this effect was not significantly enriched in probands versus siblings (Fisher’s exact test odds ratio [OR] = 1.14, two-tailed p = 0.49). Nonetheless, when we considered a null hypothesis in which a given transmitted CNV was equally likely to be transmitted to the proband only, the sibling only, or both (each with 1/3 probability), we found strong evidence that CNVs were not transmitted in equal fashion (Table 1; chi-square test with equal expected proportions: χ2 = 16.4, p = 0.0058, 5 df) and that CNVs transmitted from the mother to the proband only were significantly more common than other transmissions.
Correlation between CNV Burden and the ASD Phenotype
We used phenotype data from the SSC to assess whether the increased inherited CNV burden would segregate with markers of ASD phenotypic severity. First, we utilized the SRS, a standardized parent- or teacher-completed questionnaire that measures the severity of autism symptoms in social settings (but is not a diagnostic indicator of ASD and was not used in the ascertainment of the SSC). We partitioned our 411 families into two groups on the basis of the SRS t-score: we defined (1) “SRS-discordant quads” as those where the proband was severely affected (SRS t > 75) and the sibling was mildly affected (SRS t < 60) and (2) “SRS-concordant quads” as all others (Figure S7). The concordant group encompassed a range of moderately affected probands and some moderately affected siblings (Figure S7). There were a total of 276 SRS-discordant proband-sibling pairs and 115 concordant pairs according to this definition. We found a striking split between the discordant and concordant proband-sibling pairs: the increased CNV and gene burden was almost completely driven by the discordant pairs (p < 0.0002 for CNVs, p < 0.02 for genes, two-tailed paired t test [Figure 3A]), and there was virtually no difference at a group or family level for SRS-concordant pairs overall (1.04×, p > 0.5). Moreover, the burden ratio between probands and siblings was increased in the discordant group (1.27× for CNVs and 1.41× for genes) over the ratio for the full set of 411 quads. Finally, we found that offspring (probands and siblings) with SRS scores ≥ 60 (“moderate” and “severe” range) had higher CNV burden than did all offspring with SRS scores < 60 (361 CNVs in 390 mildly affected offspring [1.12] versus 436 CNVs in 388 moderately or severely affected offspring [0.92]; two-tailed independent t test p < 0.0094). There was no statistically significant difference in burden between probands and siblings within each group (i.e., SRS < 60 or ≥ 60); however, the relatively low number of “affected” siblings and “unaffected” probands hampered these comparisons.
Figure 3.
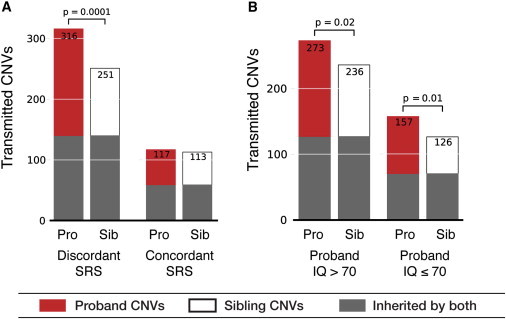
Inherited CNV Burden Correlates with SRS Phenotype
(A) The SRS measures autism features in social settings via parent report on 65 items. We classified proband-sibling pairs with severely affected probands but mildly or unaffected siblings as SRS-discordant quads (276 quads) and all other quads as SRS-concordant quads (115 quads). Strikingly, the SRS-discordant quads fully recapitulated the inherited CNV transmission bias, whereas the SRS-concordant quads did not show a differential burden.
(B) CNV burden was independent of FSIQ, and probands with either low FSIQ (≤70) or high FSIQ had more CNVs than did their siblings. p values refer to two-tailed paired t tests between probands and siblings.
Next, we considered whether the FSIQ of the probands was affected by inherited CNVs. Because FSIQ scores were only available for probands (Table S7), we grouped quads into three groups: FSIQ ≤ 70 (“low,” consistent with a diagnosis of intellectual disability), between 71 and 85 (“intermediate”), or greater than 85 (“high”). The CNV burden was significantly greater for probands in the “low” and “intermediate” proband FSIQ bins (1.25×–1.27× burden, Table 2). Probands with “high” FSIQ did not show statistically significant enrichment over siblings, although a trend was still apparent (1.11×, Table 2). When we examined the effect of SRS and FSIQ together (Table 2, Table S8, and Figure S8), we found that the burden differential was strongest for the most severely affected probands (those with FSIQ ≤ 85 and part of SRS-discordant quads); it reached 1.32×–1.40× for CNVs (p = 0.004). However, there was no significant burden between probands and siblings in SRS-concordant quads, even with “low” FSIQ probands (0.8×–1.09×, p > 0.5; Table 2), indicating that the inherited burden might be most closely aligned with the SRS score rather than FSIQ (however, we caution that there were only 22 quads total in this group).
Table 2.
Summary of IQ and SRS Burden
| Proband FSIQ | Proband CNVs | Sibling CNVs | Ratio | Two-Tailed t Test: Probands versus Siblings |
|---|---|---|---|---|
| All Quads | ||||
| ≤70 | 157 | 126 | 1.25 | p = 0.014 |
| 71–85 | 89 | 70 | 1.27 | p = 0.029 |
| ≥86 | 184 | 166 | 1.11 | NS |
| SRS-Discordant Quads (Proband SRS < 60 and Sibling SRS > 75) | ||||
| ≤70 | 138 | 104 | 1.32 | p = 0.004 |
| 71–85 | 62 | 44 | 1.40 | p = 0.012 |
| ≥86 | 113 | 101 | 1.12 | NS |
| SRS-Concordant Quads | ||||
| ≤70 | 19 | 22 | 0.86 | NS |
| 71–85 | 27 | 26 | 1.04 | NS |
| ≥86 | 71 | 65 | 1.09 | NS |
p values represent two-tailed paired t tests between probands and siblings in each group. The following abbreviation is used: NS, nonsignificant.
Enrichment of Brain-Expressed Genes in Inherited CNVs
We observed a trend in which more of the proband-only genes were highly expressed in brain-related tissues (19/317 [6%] for proband only versus 6/224 [2.7%] for sibling only; Table S8; see Material and Methods). The effect became most pronounced when we considered SRS-discordant quads (15/256 [5.9%] genes in probands and 2/170 [1.2%] genes in siblings, p = 0.007) (Figure 4). When we considered all genes highly expressed in at least one brain-related tissue, we found that significantly more probands than siblings had a CNV (57/411 [13.9%] for probands versus 33/411 [8.0%] for siblings; OR = 1.85, p = 0.009, Fisher’s exact test). These results suggest that a fraction of proband-specific genes were expressed in the nervous system tissues and that this fraction was higher in proband-only genes than in sibling-only genes. Although we caution that expression does not definitively imply pathogenicity, many of these genes and their biological pathways might be of interest for further study, both for these particular individuals and for ASD genetics in general.
Figure 4.
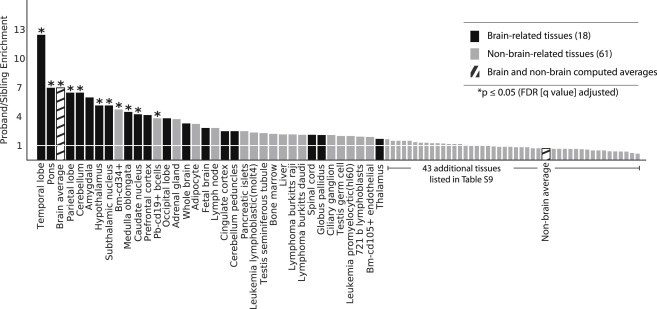
Genes in Proband-Only CNVs from SRS-Discordant Quads Are More Likely Brain-Expressed Genes
We defined a gene to be expressed in a tissue if it ranked in the top 5% of all genes in that tissue and calculated the fold enrichment of proband and sibling genes expressed in each tissue. The tissues with the strongest proband enrichment were part of brain structures (black bars), as was the computed average of expression across 18 brain regions (“brain average”). However, the particular brain tissues with the strongest apparent enrichment should not be considered as independently enriched, given that expression values for individual genes between brain regions are highly correlated. Asterisks indicate a FDR-corrected p value < 0.05. See Figure S9 for results from all 411 quads.
We compared the genes detected in the CNVs in this study to a set of 1,560 genes that were previously observed in ASD, intellectual disability, or schizophrenia (Table S10 and Figure S10). Among SRS-discordant quads, we found significant enrichment of autism genes among proband CNVs compared to those of unaffected siblings (66 versus 35 genes, p = 0.006 two tailed paired t test; Table S8). In contrast, there was no enrichment among SRS-concordant proband-sibling pairs for previously observed genes (in fact, siblings had more genes: 17 versus 24, p = 0.069). Overall, 16% (44/276) of probands in the SRS-discordant group had a CNV in a previously observed gene, whereas only 10% (12/115) of probands in the concordant group had such an event. Intersecting the brain-expressed genes and previously observed disease genes, we found that 13 genes, corresponding to 1.7% of all proband genes and only two genes (0.3%) in siblings, matched both criteria (Figure S10). The 13 convergent proband-only genes were found exclusively in SRS-discordant families, indicating that these genes might be associated with more severe phenotypes (Table S11).
Discussion
In this study, we found an enrichment of inherited CNVs in sporadic cases versus their unaffected siblings, indicating a significant CNV transmission bias for autism. The targeted nature of exome sequencing enabled us to explore a smaller CNV landscape largely inaccessible by high-density SNP microarray data.1,2,5 We estimate that the use of exome data increased our power to detect gene-disruptive CNVs smaller than 20 kb by ∼2.25-fold. These CNVs provide potential insight into the pathophysiology of inherited CNVs in sporadic autism. We found that the CNV burden was more strongly correlated with measures of ASD phenotypes (such as the SRS score) than with IQ; for proband-sibling pairs with concordant SRS scores, IQ was not dependent on the probands’ CNV burden. Genes already associated with autism and/or highly expressed in the brain were more likely to be disrupted. Private CNVs (seen once) were no more likely than simply rare variants (seen fewer than ten times in our families) to be found in probands. Burden was consistent across all sizes of CNVs, and we did not find any enrichment of either small or large events. Mothers were significantly more likely than fathers to be carriers of transmitted CNVs, irrespective of the disease status of the child, consistent with our recent finding that “secondary” CNVs are transmitted from mothers to children with developmental delay and multiple CNVs.23 We also noted that the transmission bias became more significant in probands from ASD-affected families with many siblings than in ASD-affected families with fewer individuals. Although this observation is inconsistent with the assumption that probands in larger families with many unaffected siblings are more likely to have an underlying sporadic genetic etiology, it most likely reflects an ascertainment in selecting the “least affected” sibling in a large family as the “designated sibling” for the purposes of forming a quad.8 This suggests that a significant fraction of the underlying genetic etiology in the SSC might be inherited, a notion that has been examined previously.17
Our study benefited from the quad-based design of the SSC,18 which provided a robust genetic control for each ASD proband, as well as the detailed phenotypic information available, which sharpened the contrasts between severely affected and less affected probands and their siblings, some of which showed subtle signs of the BAP.17 Most of our observations were strengthened or restricted to SRS-discordant quads, where the proband was severely affected in terms of the SRS scale but the sibling was unaffected. Approximately 67% (276/411) of the quads in this study were categorized as SRS discordant, and these quads explained virtually the entire CNV burden, encompassed the majority of brain-expressed genes, and strengthened the association with previously implicated disease genes. This effect might have been driven by inherent ambiguity in the simplex and multiplex classification scheme—a scheme that is not truly binary but rather a continuous probability based on the number of unaffected siblings in the family (the more unaffected siblings, the greater the likelihood that the family is simplex). In essence, by only concentrating on the SRS-discordant quads, we have focused our study on a more severe proband phenotype but also a truly “simplex” genetic etiology (as opposed to an environmental and/or stochastic one), thus enhancing the observed transmission disequilibrium of CNVs.
Our results should be viewed carefully in the context of previous studies. Notably, two recent studies1,2 failed to find statistically significant enrichment of inherited CNVs in sporadic autism probands compared to their siblings. These studies, which also analyzed families from the SSC, used high-density microarray platforms to discover CNVs in a genome-wide fashion. It is possible that the increased sensitivity of our exome-based method for genic events— which are most strongly implicated by studies of both de novo CNVs and de novo SNVs—revealed the difference in burden between probands and siblings. Additionally, our study found that the differential burden was dependent on the SRS score and not IQ, a factor which has not been previously examined in the context of ASD and CNVs. In contrast, our results are in good agreement with those of the case-control study by Pinto and colleagues,5 who found an overall case-control ratio of 1.19 for genic CNVs and no enrichment for “ultra-rare” CNVs seen only once in their cases (although this study was largely limited to CNVs larger than 50 kb).
In contrast to previous studies focused on large CNVs, which typically encompass dozens of genes, our study’s sensitivity for smaller CNVs provides increased specificity to define individual genes. The deletion or duplication of a subset of exons can have, in principle, the same impact on gene function as disruptive point mutations. Accordingly, several genes in our brain-expressed and SRS-discordant set of CNVs have been previously identified as part of severe neurological disorders. Among these was a CNV affecting DDHD2, which encodes an intracellular phospholipase that plays an essential role in synaptic function and which has recently been implicated in a recessive form of complex hereditary spastic paraplegia (MIM 615033), a syndrome characterized by early-onset intellectual disability and spastic paraplegia.24 We did not observe any CNVs in this gene in 2,972 control exomes. Similarly, another proband (and 0/2,972 controls) carried an inherited CNV affecting only PACS2 (MIM 610423), one of six genes in a critical region of 14q32 deletion syndrome, characterized by intellectual disability and mild facial dysmorphology.25 Lastly, in two families, we identified a small (∼5 kb), 2-exon deletion in ZNF396 (Figure 1C), which was identified as a candidate gene for alopecia with mental retardation syndrome (MIM 613930) by microsatellite linkage analysis26 (in fact, ZNF396 was the closest gene to the linkage peak). The frequency of this deletion in our control set was 3/2,972 (0.1%). Although these identified genes and CNVs might play an important role in the pathogenesis of ASD on the basis of their previously identified roles in Mendelian disorders, we would like to emphasize that their individual rarity and overall small effects prevent them from being conclusively identified as having Mendelian effects.
Other genes disrupted by CNVs have functional roles in neural function, brain development, or neurobehavioral phenotypes in model organisms (Table 3). For example, we identified two independent disruptions of ORC3 (MIM 604972; one shown in Figure 1D), encoding a protein in the origin recognition complex. The complex regulates dendritic spines and dendrite arborization in postmitotic neurons and has been implicated in olfactory learning and memory in Drosophila.27 Notable also was a CNV affecting CPLX1 (MIM 605032; Figure 1F), specific to the SNARE neuronal vesicle exocytosis pathway in neurons, as well as CNVs affecting neural receptors such as HTR3E (MIM 610123; a subunit of the ionotropic seratonin receptor) and NETO1 (MIM 607973; Figure 1E), a key component of the NMDA-receptor complex and critical for synaptic plasticity and learning in mice28 (however, this CNV was transmitted to both probands and siblings). Previous work has implicated the ubiquitin-processing pathway,4 and we found a rare CNV in UCHL1 (MIM 191342), which encodes a ubiquitin-adduct-processing enzyme and has strong and specific brain expression; UCHL10-knockout mice show specific neurodegenerative phenotypes,29 and recent work has shown this gene to regulate the NCAM1 neural cell adhesion molecule (MIM 116930; Wobst et al.30). Finally, we found several interesting mutations on the basis of brain expression pattern, including (1) an inherited deletion of IQSEC1 (MIM 610166), which is strongly expressed in the prefrontal cortex and involved in clathrin-mediated endocytosis of AMPA receptors critical to long-term potentiation in mice,31 (2) a duplication at 8q24.3 of ZNF251 and ZNF517, which have tissue-specific expression highest in the fetal brain and cerebellum,32 and (3) a duplication of AQP4 (MIM 600308), which encodes the primary water transporter in brain glial cells, especially in the amygdala and prefrontal cortex, and has been implicated in epilepsy.33
Table 3.
Selected Inherited CNVs
| Sample | Chromosomal Position (UCSC Genome Browser hg19) | Size (kb) | State | No. of Exons | Frequency in 411 Quads | Genes in Transmitted CNVs (MIM Numbers) | Summary of De Novo Mutations in Proband | SRS (Proband, Sibling, Delta) | Proband IQ |
|---|---|---|---|---|---|---|---|---|---|
| 12647.p1 | chr1: 32,084,793–32,110,465 | 25.7 | dup | 12 | 2 | HCRTR1a (602392), PEF1 (610033) | - | 88, 40, 48 | 72 |
| 11872.p1 | chr1: 65,730,593–65,831,879 | 101.3 | dup | 4 | 1 | DNAJC6a (608375) | KATNAL2 (SS) | 88, 49, 39 | 62 |
| 12719.p1 | chr1: 146,715,494–146,767,190 | 51.7 | del | 23 | 1 | CHD1Lb (613039) | - | 90, 48, 42 | 46 |
| 12997.p1 | chr2: 230,632,269–230,724,290 | 92 | dup | 39 | 1 | TRIP12c (604506) | - | 81, 40, 41 | 97 |
| 12394.p1 | chr2: 241,538,067–241,709,123 | 171.1 | dup | 42 | 3 | KIF1Aa,b (601255), GPR35 (602646), AQP12B, AQP12A (609789), CAPN10 (605286) | - | 77, 36, 41 | 88 |
| 12534.p1 | chr3: 12,940,888–12,978,197 | 37.3 | del | 13 | 2 | IQSEC1a (610166) | - | 90, 42, 48 | 81 |
| 13099.p1 | chr3: 97,486,951–97,634,880 | 147.9 | del | 19 | 1 | ARL6b (608845) | - | 86, 38, 48 | 92 |
| 12645.p1 | chr4: 818,279–845,762 | 27.5 | dup | 5 | 1 | CPLX1a,b (605032), GAKb (602052) | ANK2 (NS) | 90, 36, 54 | 86 |
| 11773.p1 | chr4: 2,641,461–2,835,561 | 194.1 | dup | 34 | 1 | TNIP2b (610669), FAM193A, SH3BP2b (602104) | - | 90, 42, 48 | 43 |
| 11066.p1 | chr4: 41,258,993–41,259,143 | 150 bp | dup | 2 | 3 | UCHL1a,b (191342) | - | 82, 37, 45 | 88 |
| 13385.p1 | chr4: 169,083,678–169,086,477 | 2.8 | del | 3 | 1 | ANXA10a (608008) | - | 90, 52, 38 | 16 |
| 13293.p1 | chr5: 619,104–644,540 | 25.4 | dup | 9 | 2 | CEP72a | - | 90, 41, 49 | 83 |
| 12758.p1 | chr6: 24,454,242–24,523,153 | 68.9 | dup | 20 | 1 | ALDH5A1a,b (610045), GPLD1b (602515) | - | 80, 40, 40 | 74 |
| 11551.p1 | chr6: 88,315,634–88,318,947 | 3.3 | del | 3 | 1 | ORC3a (604972) | - | 90, 39, 51 | 98 |
| 11459.p1 | chr6: 88,317,390–88,366,700 | 49.3 | del | 10 | 1 | ORC3a (604972) | - | 90, 42, 48 | 80 |
| 13412.p1 | chr7: 33,102,179–33,185,976 | 83.8 | dup | 7 | 3 | RP9 (607331), BBS9b (607968), NT5C3 (606224) | - | 90, 36, 54 | 33 |
| 11722.p1 | chr7: 48,308,576–48,416,169 | 107.6 | del | 20 | 1 | ABCA13b (607807) | - | 81, 42, 39 | 97 |
| 11716.p1 | chr8: 38,090,512–38,117,639 | 27.1 | del | 16 | 1 | DDHD2a (615003) | - | 90, 42, 48 | 48 |
| 13412.p1 | chr8: 86,351,940–86,575,726 | 223.8 | dup | 14 | 1 | CA3 (114750), CA2a,b (611492), REXO1L1 | - | 90, 36, 54 | 33 |
| 12534.p1 | chr8: 145,947,028–146,033,780 | 86.8 | dup | 18 | 1 | ZNF251, ZNF34 (14526), ZNF517,aRPL8 (604177) | - | 90, 42, 48 | 81 |
| 11356.p1 | chr9: 139,634,401–139,651,044 | 16.6 | dup | 16 | 1 | LCN6a (609379), LCN10c (612904), LCN8a (612902) | NAPRT1 (SS) | 90, 42, 48 | 72 |
| 13162.p1 | chr10: 5,203,384–5,260,723 | 57.3 | del | 12 | 1 | AKR1C4a (600451), AKR1CL1 | RIMS1 (FS) | 90, 36, 54 | 74 |
| 13843.p1 | chr11: 43,772,460–43,775,671 | 3.2 | del | 2 | 1 | HSD17B12a (609574) | - | 90, 42, 48 | 66 |
| 11241.p1 | chr12: 120,875,929–120,884,632 | 8.7 | dup | 7 | 2 | GATC, COX6A1a (602072), TRIAP1 (614943) | - | 90, 38, 52 | 76 |
| 12396.p1 | chr14: 105,836,177–105,861,009 | 24.8 | dup | 17 | 1 | PACS2a (610423) | - | 90, 51, 39 | 99 |
| 11479.p1 | chr15: 43,696,610–43,701,294 | 4.7 | dup | 5 | 1 | TP53BP1a (605230), TUBGCP4c (609610) | - | 79, 46, 33 | 133 |
| 13843.p1 | chr15: 55,475,512–55,497,903 | 22.4 | dup | 6 | 2 | RAB27Ab (613262), RSL24D1 | - | 90, 42, 48 | 66 |
| 12837.p1 | chr15: 57,730,197–57,754,090 | 23.9 | dup | 7 | 2 | CGNL1c (607856) | - | 86, 57, 29 | 89 |
| 13543.p1 | chr15: 91,488,121–91,520,001 | 31.9 | del | 25 | 2 | RCCD1, PRC1c (603484), UNC45A (611219) | - | 82, 40, 42 | 42 |
| 13215.p1 | chr16: 15,596,178–15,609,285 | 13.1 | del | 6 | 1 | C16orf45a | - | 90, 51, 39 | 74 |
| 14201.p1 | chr16: 68,710,287–68,713,877 | 3.6 | dup | 5 | 2 | CDH3c (114021) | - | 90, 45, 45 | 43 |
| 12100.p1 | chr16: 70,714,696–70,714,928 | 232 bp | dup | 2 | 3 | MTSS1La | chr16: 29,675,049–30,199,897 (CNV) | 90, 38, 52 | 71 |
| 12373.p1 | chr16: 81,314,461–81,396,216 | 81.8 | dup | 10 | 1 | GANa,b (605379), BCMO1 (605748) | - | 90, 52, 38 | NA |
| 12697.p1 | chr18: 24,436,174–24,628,467 | 192.3 | dup | 10 | 1 | CHST9 (610191), AQP4a (600308), CHST9-AS1 | - | 80, 46, 34 | 85 |
| 12869.p1 | chr18: 72,229,281–72,251,798 | 22.5 | dup | 8 | 1 | CNDP1a (609064) | - | 90, 36, 54 | 31 |
| 11356.p1 | chr18: 77,470,345–77,891,075 | 420.7 | dup | 28 | 2 | KCNG2a (605696), RBFA, CTDP1b (604927), ADNP2, TXNL4A (611595), PQLC1 | NAPRT1 (SS) | 90, 42, 48 | 72 |
| 13296.p1 | chr19: 6,681,951–6,686,913 | 5 | dup | 8 | 1 | C3a (120700) | - | 87, 40, 47 | 30 |
| 11298.p1 | chr19: 18,704,375–18,704,917 | 542 bp | dup | 2 | 1 | CRLF1a (604237) | - | 90, 37, 53 | 141 |
| 13815.p1 | chr19: 57,835,049–57,932,849 | 97.8 | del | 15 | 1 | ZNF547, ZNF304 (613840), ZNF17,aZNF548, ZNF543 | CNTNAP4 (CNV) | 82, 44, 38 | 51 |
| 13396.p1 | chr21: 19,628,825–19,632,603 | 3.8 | del | 3 | 1 | CHODLa (607247) | - | 83, 43, 40 | 103 |
| 13327.p1 | chr21: 35,742,777–35,899,047 | 156.3 | dup | 8 | 2 | KCNE2a (603796), RCAN1 (602917), KCNE1 (176261), FAM165B | - | 90, 45, 45 | 103 |
The following abbreviations are used: dup, duplication; del, deletion; SS, splice site; NS, nonsense; and FS, frameshift.
Brain expression.
Previously associated with disease.
Target of previously discovered disruptive de novo SNV.
Because the affected individuals and families had been analyzed for both de novo CNVs and SNVs, we were able to develop a model to assess the relative contribution of each class of genetic variant to autism. First, we confirmed that inherited CNVs were enriched in the set of probands without other known de novo CNVs or SNVs (368 inherited CNVs in probands versus 327 CNVs in siblings of 336 quads; p < 0.03, two-tailed paired t test). Second, we developed a logistic regression model, in which the binary outcome of either probands or siblings was predicted by the count of disruptive de novo SNVs, de novo CNVs, and our rare transmitted CNVs. We performed regressions on both the set of all 411 quads and the set of 276 proband-sibling pairs with SRS-discordant scores. The results (Figure 5 and Table S12) revealed a strong effect for disruptive (nonsense, splice, and frameshift) de novo SNVs (OR = 4.30, p < 0.001) and CNVs (OR = 6.65, p < 0.02) and also confirmed a statistically independent effect for transmitted CNVs (OR = 1.16, p < 0.04); again, in this model, the effect was primarily driven by SRS-discordant quads. Although the strength of de novo SNVs strongly outweighed the pathogenic effect of inherited CNVs, our model predicted that the inherited CNV would contribute significantly to sporadic disease, especially in the case of SRS-discordant pairs (where the OR increased to 1.26, p < 0.015). We did not find any significant interactions between our predictors, reflecting the relative infrequency of co-occurring CNVs and de novo SNVs but also the limited sample size. It is also possible that careful consideration of rare and disruptive inherited SNVs could statistically interact with other classes of mutation, but we did not take these into account in building our model. Our model suggests that disruptive de novo SNVs and both inherited and de novo CNVs contribute independently to the risk of autism. We believe that future studies of ASD and other complex neurological disorders will contribute significantly to the understanding of the genetic underpinnings of disease, especially if an integrated approach considering all disruptive mutations—inherited and de novo, CNV and SNV, small and large—is applied.
Acknowledgments
We thank the National Institutes of Health (NIH) National Heart, Lung, and Blood Institute Grand Opportunity (GO) Exome Sequencing Project and its ongoing studies, which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the Women’s Health Initiative Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010). We thank all the families at the participating Simons Simplex Collection (SSC) sites, as well as the principal investigators (A. Beaudet, R.B., J. Constantino, E. Cook, E. Fombonne, D. Geschwind, E. Hanson, D. Grice, A. Klin, R. Kochel, D. Ledbetter, C. Lord, C. Martin, D. Martin, R. Maxim, J. Miles, O. Ousley, K. Pelphrey, B. Peterson, J. Piggot, C. Saulnier, M. State, W. Stone, J. Sutcliffe, C. Walsh, Z. Warren, and E. Wijsman). We also acknowledge M. State and the SSC Genetics Consortium for providing Illumina genotyping data and T. Lehner and the Autism Sequencing Consortium for data exchange among the participating groups. We are grateful for helpful discussion and manuscript preparation from T. Brown, P. Sudmant, and all members of the Eichler lab. This work was supported by the Simons Foundation Autism Research Initiative (SFARI 137578 and 191889 to E.E.E., J.S., and R.B.) and NIH HD065285 (E.E.E. and J.S.). E.B. is an Alfred P. Sloan Research Fellow. E.E.E. is an Investigator of the Howard Hughes Medical Institute and is on the scientific advisory boards for Pacific Biosciences, Inc., SynapDx Corp., and DNAnexus, Inc.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://omim.org
National Database of Autism Research (NDAR), http://ndar.nih.gov
Accession Numbers
The CoNIFER output files for 1,644 samples were deposited in the National Database of Autism Research (NDAR) under NDAR Collection ID 1878 and the title of this article.
References
- 1.Levy D., Ronemus M., Yamrom B., Lee Y.-H., Leotta A., Kendall J., Marks S., Lakshmi B., Pai D., Ye K. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 2.Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glessner J.T., Wang K., Cai G., Korvatska O., Kim C.E., Wood S., Zhang H., Estes A., Brune C.W., Bradfield J.P. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J., Yamrom B., Lee Y.-H., Narzisi G., Leotta A. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neale B.M., Kou Y., Liu L., Ma’ayan A., Samocha K.E., Sabo A., Lin C.-F., Stevens C., Wang L.-S., Makarov V. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Roak B.J., Deriziotis P., Lee C., Vives L., Schwartz J.J., Girirajan S., Karakoc E., Mackenzie A.P., Ng S.B., Baker C. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bailey A., Le Couteur A., Gottesman I., Bolton P., Simonoff E., Yuzda E., Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol. Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 12.Constantino J.N., Todorov A., Hilton C., Law P., Zhang Y., Molloy E., Fitzgerald R., Geschwind D. Autism recurrence in half siblings: strong support for genetic mechanisms of transmission in ASD. Mol. Psychiatry. 2013;18:137–138. doi: 10.1038/mp.2012.9. [DOI] [PubMed] [Google Scholar]
- 13.Hallmayer J., Cleveland S., Torres A., Phillips J., Cohen B., Torigoe T., Miller J., Fedele A., Collins J., Smith K. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry. 2011;68:1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prasad A., Merico D., Thiruvahindrapuram B., Wei J., Lionel A.C., Sato D., Rickaby J., Lu C., Szatmari P., Roberts W. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3 (Bethesda) 2012;2:1665–1685. doi: 10.1534/g3.112.004689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krumm N., Sudmant P.H., Ko A., O’Roak B.J., Malig M., Coe B.P., Quinlan A.R., Nickerson D.A., Eichler E.E., NHLBI Exome Sequencing Project Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22:1525–1532. doi: 10.1101/gr.138115.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Losh M., Childress D., Lam K., Piven J. Defining key features of the broad autism phenotype: a comparison across parents of multiple- and single-incidence autism families. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2008;147B:424–433. doi: 10.1002/ajmg.b.30612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davidson J., Goin-Kochel R.P., Green-Snyder L.A., Hundley R.J., Warren Z., Peters S.U. Expression of the Broad Autism Phenotype in Simplex Autism Families from the Simons Simplex Collection. J. Autism Dev. Disord. 2012 doi: 10.1007/s10803-012-1492-1. Published online March 1, 2012. [DOI] [PubMed] [Google Scholar]
- 18.Fischbach G.D., Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010;68:192–195. doi: 10.1016/j.neuron.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 19.Hach F., Hormozdiari F., Alkan C., Hormozdiari F., Birol I., Eichler E.E., Sahinalp S.C. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat. Methods. 2010;7:576–577. doi: 10.1038/nmeth0810-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venkatraman E.S., Olshen A.B. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23:657–663. doi: 10.1093/bioinformatics/btl646. [DOI] [PubMed] [Google Scholar]
- 21.Constantino J.N., Gruber C.P. Western Psychological Services; Los Angeles: 2005. Social Responsiveness Scale (SRS) Manual. [Google Scholar]
- 22.Su A.I., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Girirajan S., Rosenfeld J.A., Coe B.P., Parikh S., Friedman N., Goldstein A., Filipink R.A., McConnell J.S., Angle B., Meschino W.S. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N. Engl. J. Med. 2012;367:1321–1331. doi: 10.1056/NEJMoa1200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schuurs-Hoeijmakers J.H.M., Geraghty M.T., Kamsteeg E.-J., Ben-Salem S., de Bot S.T., Nijhof B., van de Vondervoort I.I.G.M., van der Graaf M., Nobau A.C., Otte-Höller I., FORGE Canada Consortium Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am. J. Hum. Genet. 2012;91:1073–1081. doi: 10.1016/j.ajhg.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holder J.L., Jr., Lotze T.E., Bacino C., Cheung S.W. A child with an inherited 0.31 Mb microdeletion of chromosome 14q32.33: further delineation of a critical region for the 14q32 deletion syndrome. Am. J. Med. Genet. A. 2012;158A:1962–1966. doi: 10.1002/ajmg.a.35289. [DOI] [PubMed] [Google Scholar]
- 26.Wali A., Ali G., John P., Lee K., Chishti M.S., Leal S.M., Ahmad W. Mapping of a gene for alopecia with mental retardation syndrome (APMR3) on chromosome 18q11.2-q12.2. Ann. Hum. Genet. 2007;71:570–577. doi: 10.1111/j.1469-1809.2007.00362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Z., Zang K., Reichardt L.F. The origin recognition core complex regulates dendrite and spine development in postmitotic neurons. J. Cell Biol. 2005;170:527–535. doi: 10.1083/jcb.200505075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng D., Pitcher G.M., Szilard R.K., Sertié A., Kanisek M., Clapcote S.J., Lipina T., Kalia L.V., Joo D., McKerlie C. Neto1 is a novel CUB-domain NMDA receptor-interacting protein required for synaptic plasticity and learning. PLoS Biol. 2009;7:e41. doi: 10.1371/journal.pbio.1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saigoh K., Wang Y.L., Suh J.G., Yamanishi T., Sakai Y., Kiyosawa H., Harada T., Ichihara N., Wakana S., Kikuchi T., Wada K. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat. Genet. 1999;23:47–51. doi: 10.1038/12647. [DOI] [PubMed] [Google Scholar]
- 30.Wobst H., Förster S., Laurini C., Sekulla A., Dreiseidler M., Höhfeld J., Schmitz B., Diestel S. UCHL1 regulates ubiquitination and recycling of the neural cell adhesion molecule NCAM. FEBS J. 2012;279:4398–4409. doi: 10.1111/febs.12029. [DOI] [PubMed] [Google Scholar]
- 31.Scholz R., Berberich S., Rathgeber L., Kolleker A., Köhr G., Kornau H.-C. AMPA receptor signaling through BRAG2 and Arf6 critical for long-term synaptic depression. Neuron. 2010;66:768–780. doi: 10.1016/j.neuron.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 32.Lorenz P., Dietmann S., Wilhelm T., Koczan D., Autran S., Gad S., Wen G., Ding G., Li Y., Rousseau-Merck M.F., Thiesen H.J. The ancient mammalian KRAB zinc finger gene cluster on human chromosome 8q24.3 illustrates principles of C2H2 zinc finger evolution associated with unique expression profiles in human tissues. BMC Genomics. 2010;11:206. doi: 10.1186/1471-2164-11-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Binder D.K., Nagelhus E.A., Ottersen O.P. Aquaporin-4 and epilepsy. Glia. 2012;60:1203–1214. doi: 10.1002/glia.22317. [DOI] [PubMed] [Google Scholar]
- 34.Hehir-Kwa J.Y., Rodríguez-Santiago B., Vissers L.E., de Leeuw N., Pfundt R., Buitelaar J.K., Pérez-Jurado L.A., Veltman J.A. De novo copy number variants associated with intellectual disability have a paternal origin and age bias. J. Med. Genet. 2011;48:776–778. doi: 10.1136/jmedgenet-2011-100147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.