

Abstract
Primary ciliary dyskinesia (PCD) is caused when defects of motile cilia lead to chronic airway infections, male infertility, and situs abnormalities. Multiple causative PCD mutations account for only 65% of cases, suggesting that many genes essential for cilia function remain to be discovered. By using zebrafish morpholino knockdown of PCD candidate genes as an in vivo screening platform, we identified c21orf59, ccdc65, and c15orf26 as critical for cilia motility. c21orf59 and c15orf26 knockdown in zebrafish and planaria blocked outer dynein arm assembly, and ccdc65 knockdown altered cilia beat pattern. Biochemical analysis in Chlamydomonas revealed that the C21orf59 ortholog FBB18 is a flagellar matrix protein that accumulates specifically when cilia motility is impaired. The Chlamydomonas ida6 mutant identifies CCDC65/FAP250 as an essential component of the nexin-dynein regulatory complex. Analysis of 295 individuals with PCD identified recessive truncating mutations of C21orf59 in four families and CCDC65 in two families. Similar to findings in zebrafish and planaria, mutations in C21orf59 caused loss of both outer and inner dynein arm components. Our results characterize two genes associated with PCD-causing mutations and elucidate two distinct mechanisms critical for motile cilia function: dynein arm assembly for C21orf59 and assembly of the nexin-dynein regulatory complex for CCDC65.
Introduction
Apical cilia are essential cellular organelles that regulate embryonic development and organ function.1 Cilia are microtubule-based structures projecting from the apical surface of nearly all cell types.2 Embryonic cells employ nonmotile cilia to anchor membrane receptors and process signals from morphogens and to generate and respond to mechanical signals in the form of fluid flow and shear force in confined spaces.1 Motile cilia and flagella propel germ cells through fluid environments and motile cilia on epithelial cells drive mucociliary clearance and create flow within fluid-filled lumens.3 A wide spectrum of human pathologies, collectively termed ciliopathies, is associated with mutations in genes required for cilia function and includes Meckel-Gruber syndrome (MKS [MIM 249000]), nephronophthisis (NPHP [MIM 256100]), Bardet-Biedl syndrome (BBS [MIM 209900]), Joubert syndrome (JS [MIM 213300]), Senior-Løken syndrome (SLSN [MIM 266900]), Leber congenital amaurosis (LCA [MIM 204000]), polycystic kidney disease (PKD [MIM 173900]), oral-facial-digital syndrome (OFD [MIM 311200]), and cranioectodermal dysplasia (CED [MIM 614068]).4–7 Clinical features of these syndromes include cystic kidney, organ laterality defects, nervous system development defects, and retinal degeneration, highlighting the ubiquity of cilia function.4 Mutations disrupting cilia motility in humans cause primary ciliary dyskinesia (PCD, CILD [MIM 244400]), which has an estimated incidence of 1 in 16,000 to 20,000 live births.8 Affected individuals present with sterility, organ laterality defects, hydrocephalus, chronic upper and lower respiratory tract infections, and otitis media. Twenty-one genes harboring PCD-causing mutations have been identified by a combination of traditional genetic mapping and selective screening of cilia motility genes identified in model systems such as Chlamydomonas reinhardtii and Danio rerio (Table S2).8
Motile cilia require outer dynein arms (ODAs) and inner dynein arms (IDAs) to achieve proper waveform. Mutations in genes encoding the structural elements of dynein arms or cytoplasmic components required for arm preassembly and transport result in loss of ODAs and IDAs with subsequent cilia paralysis.9–12 Cilia dysmotility can also arise from mutations in genes encoding one of two complexes required for the coordination of dynein arm activity: the N-DRC and the radial spoke complex (RS).13–16 The N-DRC generates resistance to microtubule sliding, and this resistance contributes to optimal alignment of microtubule doublets for productive flagellar motility.17 Recently, mutations in DRC1/CCDC164 (MIM 615288) and DRC4 were shown to impair N-DRC function,14,17 although the function of other N-DRC proteins remains to be determined.
More than one-third of PCD cases remain genetically uncharacterized and have yet to be linked to mutations in known genes.8 At the same time, informatic and proteomic analyses of cilia have identified more than 1,000 cilia-associated proteins and genes that might harbor mutations in affected individuals.18 To identify cilia proteome genes associated with yet-uncharacterized PCD mutations, we screened for ciliopathy phenotypes in zebrafish after morpholino-induced knockdown of genes encoding proteins present in multiple cilia proteomes and highly expressed in ciliated tissues. We focused further on three genes that when knocked down produced robust ciliopathy phenotypes resulting from defects in cilia motility. We show that two of these genes, C21orf59 and CCDC65 (MIM 611088), are mutated in human PCD, validating this strategy for identifying human disease mutations.
Material and Methods
Zebrafish Strains and Maintenance
TU-AB and Tg(ubiquitin:arl13b-GFP) zebrafish lines were maintained according to standard procedures.19 All embryos were raised in E3 egg water. Embryos analyzed at 24 hpf were treated with phenylthiourea (PTU) so that pigment would not develop. The Tg(ubiquitin:arl13b-GFP) fish line was generated by a standard Tol2-mediated transposition approach.20 The vector for transgenesis was generated by multisite Gateway recombination of the following vectors: pDONR221 containing the zebrafish arl13b coding sequence (from Zhaoxia Sun), p5E containing the ubiquitin promoter (from Leonard Zon), p3E-EGFPpA, and pDESTTol2CG2, both from the Tol2 kit.21
In Situ Hybridization
For some of the candidates assessed in the morpholino screen, expression data were available from a large-scale in situ hybridization screen.22 We generated digoxygenin-labeled antisense RNA probes for zebrafish c21orf59, ccdc65, c15orf26, ccdc63, and enkurin/c10orf63 by in vitro transcription from EST clones purchased from Open Biosystems of Thermo Scientific. Whole-mount in situ hybridization was performed as previously described.23 Stained embryos were cleared with dimethyl formamide and imaged on a Leica MZ12 stereomicroscope with a Spot Image digital camera.
Morpholino Injections
Foxj1a knockdown was achieved with previously described morpholinos.23 For the morpholino screen, the morpholinos listed in Table S1 (available online) were diluted to 0.25 mM and 0.5 mM concentrations in 1× injection solution containing 100 mM KCl, 10 mM HEPES, and 0.1% phenol red. TU-AB embryos were injected with 4.6 nl diluted morpholino injection mixes in the yolk at the 1–4 cell stage and allowed to develop at 28°C. At 24 hpf, injected clutches were screened for dead embryos and embryos displaying significant neural cell death. When these phenotypes were observed in >30% of the clutch, the morpholino dose was considered to be toxic, and embryos were not included in further analyses. Remaining embryos were incubated from 33 to 48 hpf in a low dose of pronase enzyme so that chorions would be removed. At 48 hpf, axis curvature, otolith number, and hydrocephalus phenotypes were assessed. The presence of pronephric cysts was scored at 60 hpf.
RT-PCR
To assess splicing defects caused by morpholino injection, we extracted RNA from pools of five embryos per condition by using TRI reagent (Molecular Research Center). cDNA was synthesized with gene-specific primers and Superscript II reverse transcriptase (Invitrogen). We then used primers binding in exons adjacent to the morpholino binding site (Table S2) in nested PCR to amplify wild-type and morphant products. PCR products were separated on agarose gels, and wild-type and morphant bands were extracted for sequencing. Products from PCR1 were used as templates for PCR2.
In Vivo Imaging of Zebrafish Cilia
Olfactory placode and pronephric duct cilia motility defects were assessed at 48 hpf by high-speed video microscopy. For imaging, embryos were anesthetized in 0.2 mg/ml tricaine and immobilized in 3% methylcellulose. Movies were acquired at 240 frames per second by a high-speed Dragonfly Express video camera (Point Gray Research) and converted to 15 frames per second for visualization of beat rate and amplitude. Kymograms were generated with the line scan function in ImageJ. For imaging of live Kupffer's vesicle (KV) cilia, Tg(ubiquitin:arl13b-GFP) fish were embedded in 3% low-melt agarose at the 7–12 somite stage. Z stacks of the entire vesicle were acquired on a Zeiss LSM5 Pascal confocal microscope. The percentage of motile cilia per KV was scored on the basis of cilia appearance (zig zags versus straight lines) in acquired z series.
Immunoblots
Immunoblotting on 24 hpf zebrafish embryo extracts was performed as previously described24 with mouse anti-human C21orf59 (1:500, Santa Cruz) or mouse anti-α-tubulin B512 (Sigma 1:5,000) and a peroxidase-conjugated goat anti-mouse secondary antibody (Jackson Immunoresearch, 1:10,000).
Immunofluorescence
For staining of zebrafish pronephric cilia, 2 dpf wild-type and morphant embryos were fixed in Dents fixative (80% MeOH, 20% DMSO) overnight at 4°C. Embryos were transferred to 100% MeOH and incubated for 1 hr at room temperature prior to rehydration through a MeOH gradient. Samples were blocked for 2 hr at room temperature in PBST + 10% normal goat serum (NGS) prior to incubation in primary antibodies at 4°C overnight. Primary antibodies were diluted in blocking buffer as follows: rabbit anti-IFT88 (1:400, gift from Brain Perkins), 6-11B-1 anti-acetylated α-tubulin (1:400, Sigma), and GTU88 anti-γ-tubulin (1:400, Sigma). After PBST washes, embryos were incubated overnight in a 1:800 dilution of Alexa 488- or 546-conjugated goat anti-rabbit or goat anti-mouse secondary antibodies (Invitrogen). For double staining by two mouse antibodies, a modified immunostaining protocol was performed as previously described.25 For imaging, stained embryos were equilibrated in clearing solution (53% benzyl alcohol, 45% glycerol, 2% N-propyl gallate), and two-color z series were acquired on a Zeiss LSM5 Pascal confocal microscope via sequential laser excitation.
Immunofluorescence of Human Respiratory Epithelium
Cells were treated with 4% paraformaldehyde, 0.2% Triton X-100, and 1% skim milk (all percentages are v/v) before incubation with primary (2–3 hr at room temperature or overnight at 4°C) and secondary (25 min at room temperature) antibodies. Appropriate controls were performed omitting the primary antibodies. Anti-acetylated tubulin was used at a 1:10,000 dilution, and rabbit polyclonal anti-a/b-tubulin was used at a 1:300 dilution (#2148; Cell Signaling). Mouse monoclonal anti-DNAH5 and rabbit polyclonal anti-DNALI1 antibodies were generated as reported.11,26 Highly cross-adsorbed secondary antibodies goat anti-mouse Alexa Fluor 488 (1:1,000; A11029) and goat anti-rabbit Alexa Fluor 546 (1:1,000; A11035) were from Molecular Probes (Invitrogen). DNA was stained with Hoechst33342 (1:1,000; 14533-100MG, Sigma). Immunofluorescence images were taken with a Zeiss Apotome Axiovert 200 microscope and processed with AxioVision 4.8 and Adobe Creative Suite 4.
Electron Microscopy
D. rerio, S. mediterranea, and human biopsy samples were fixed and prepared for transmission electron microscopy (TEM) as previously described.27–29 S. mediterranea was prepared for scanning electron microscopy (SEM) as previously described.29
Zebrafish mRNA Rescue
Full-length human C21orf59 and CCDC65 coding domains were cloned into the pDONR221 plasmid (Invitrogen) and subcloned into the pCSmCherryDest expression vector21 via Gateway technology (Invitrogen). For mutation analysis, the pCSmCherryDest-hsC21orf59 construct was mutagenized with a standard PCR-based site-directed mutagenesis approach.30 Capped RNAs were synthesized from linearized vectors with the SP6 mMessage mMachine kit (Invitrogen). RNA products were column purified with the RNeasy Mini Kit (QIAGEN) prior to injection at 25–400 pg doses into 1 cell stage zebrafish embryos. For rescue of morphant phenotypes, morpholinos were injected into 2–4 cell embryos that had previously been injected with RNA.
Schmidtea mediterranea
Planarians were acquired and maintained according to previously described methods.29 For generation of the RNAi vectors, part of the FBB18 coding sequence was cloned into the L4440 vector.31 RNAi knockdown and confirmatory RT-PCR were conducted according to standard protocols.32,33 For gliding assays, planarians were placed in petri dishes, and animal movement was recorded with a color CCD camera (DFK 31BU03; The Imaging Source). The distance traveled by each planarian was measured directly from the video. Movies were decompiled, cropped, combined, and labeled with Blaze Media Pro, VirtualDub, and ImageJ.
Chlamydomonas reinhardtii
All strains were obtained from the Chlamydomonas center and were raised according to standard protocols.34 For examination of flagellar protein content in Figure 2, cells were harvested by centrifugation and deflagellated by dibucaine treatment. Flagella were isolated by previously described methods. Proteins were separated on 10% SDS gels and either stained with Coomassie brilliant blue or blotted to nitrocellulose and probed with antibodies. Primary antibodies used were anti-D. rerio C21orf59/FBB18 (raised against multiple synthetic peptides distributed throughout the protein sequence; AbMart) and anti-D1blC.35
Figure 2.
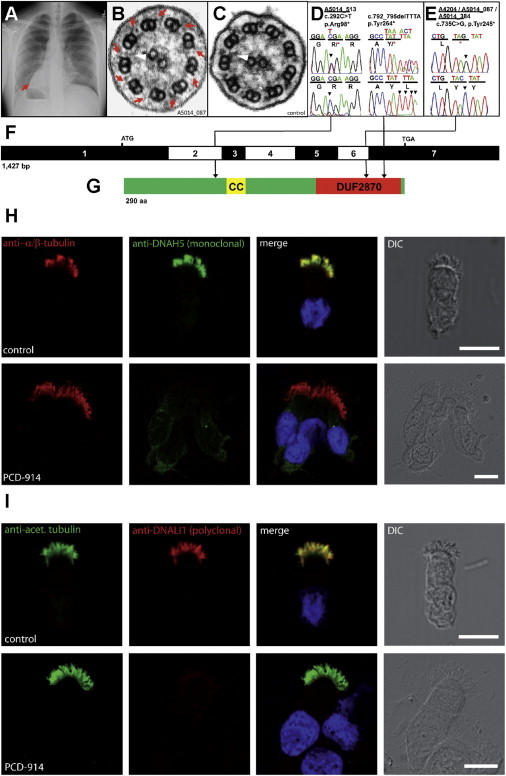
Identification of Recessive Mutations in C21orf59 in Four Families with Primary Ciliary Dyskinesia and Loss of Ciliary Dynein Arms in Individuals with PCD
(A) Chest X-ray of individual A5014_087-21, showing situs inversus. Red arrow indicates the apex of the heart on the right side.
(B and C) TEM of individual A5014_087-21 (B) showing shortened/missing outer (red arrows) and inner (white arrowheads) dynein arms compared to control cilia (C).
(D and E) Three different C21orf59 mutations detected in four independent families with PCD. Family number (underlined), mutation (arrowhead), and predicted translational changes are indicated. For the mutations detected, arrows indicate positions in relation to exons. Sequence trace is shown for mutation above normal controls.
(F) Exon structure of human C21orf59 cDNA. Positions of start codon (ATG) and stop codon (TGA) are indicated.
(G) C21orf59 protein putatively contains a coiled-coil (CC) and a domain of unknown function (DUF2870) at the C-terminal end.
(H and I) Immunolocalization of DNAH5 and DNALI1 in human respiratory epithelial cells from individual A5014_087-21 carrying the C21orf59 c.735C>G (p.Tyr245∗) mutation.
(H) Immunofluorescence analysis of human respiratory epithelial cells via specific antibodies directed against the outer dynein arm heavy chain DNAH5 (green). As a control, axoneme-specific antibodies against α/β-tubulin (red) were used. Nuclei were stained with Hoechst 33342 (blue). In respiratory epithelial cells from healthy probands, DNAH5 (green) localizes along the entire length of the axonemes. DNAH5 (green) absent from cilia in respiratory epithelial cells from individual A5014_087-21.
(I) Immunofluorescence analysis of human respiratory epithelial cells using specific antibodies directed against the inner dynein arm intermediate chain DNALI1 (red). As a control, axoneme-specific antibodies against acetylated α-tubulin (green) were used. Nuclei were stained with Hoechst 33342 (blue). DNALI1 is localized along the entire length of the axonemes of healthy probands. In contrast, in respiratory cells of individual A5014_087-21, DNALI1 is absent from cilia.
Scale bars in (H) and (I) represent 10 μm.
FAP250/ida6 Cloning
An ∼9.5 kb genomic fragment encoding FAP250 was subcloned from BAC 31n21 with DraI and SpeI. The FAP250 subclone was linearized with NotI and cotransformed into ida6 along with the plasmid pSI103 containing the selectable marker aphVIII.36 Transformants were selected on Tris-acetate-phosphate medium containing 10 μg/ml paromomycin and screened for improved motility by phase-contrast microscopy as described previously.17 The ida6 mutation was identified by the direct sequencing of PCR products obtained from wild-type and ida6 genomic DNA via primers spanning 500–1,000 bp regions across the FAP250 transcription unit. An RNA blot containing total wild-type RNA isolated from cells before (0 min) and 45 min after deflagellation was probed with an ∼1.3 kb RT-PCR product obtained from wild-type cDNA by the forward primer 5′-CCGCAAGGACGCCATTATG-3′ and the reverse primer 5′-TGTTCCGCTCCGTGAGTGTG-3′. For Figure 3, isolated axonemes were prepared and subjected to immunoblot analysis with antibodies against FAP250, RSP16, and tektin. Alternatively, axonemes were fixed for electron microscopy, and computer image averaging was performed as described.17,37
Figure 3.
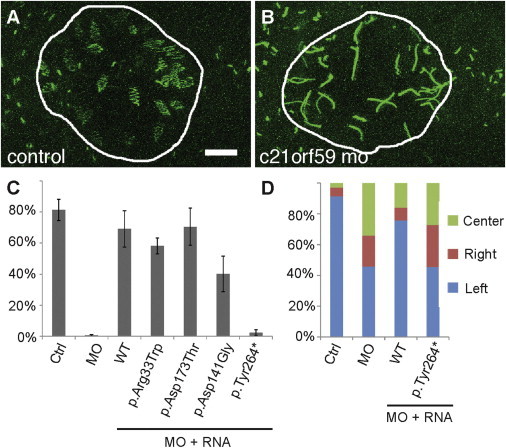
Functional Analysis of C21orf59 Alleles in Zebrafish
(A and B) Control (A) or c21orf59 (B) morpholinos were injected into the transgenic Tg(ubiquitin:arl13b-GFP) zebrafish line. Laser scanning confocal imaging reveals motile, GFP-positive Kupffer’s vesicle cilia as “zigzags” (A). Cilia in c21orf59 morphant KV’s (B) were paralyzed and appear as straight lines. Scale bar represents 5 μm.
(C) Quantification of the percentage of motile cilia within KV. Coinjection of c21orf59 morpholino with a full-length human C21orf59 mRNA or three C21orf59 missense alleles rescued the motility of KV cilia, whereas injection of human C21orf59 mRNA bearing the c.792_795delTTTA (p.Tyr264∗) mutation failed to rescue cilia motility. n = 3–6 embryos per sample, as indicated. Error bars indicate SEM.
(D) Injection of WT, but not c.792_795delTTTA (p.Tyr264∗) mutant, C21orf59 human mRNA rescued heart looping morphogenesis in zebrafish c21orf59 morphants.
Research Subjects
We obtained blood samples and pedigrees after receiving informed consent from individuals with PCD and their family members. Approval for research on human subjects was obtained from the institutional review boards of the universities of North Carolina, Michigan, Freiburg, and Münster, the Boston Children’s Hospital, and the other institutions involved. The diagnosis of PCD was based on published clinical criteria, including compatible clinical phenotype, hallmark diagnostic defects in ciliary ultrastructure, and low nasal nitric oxide (nNO).38 Genomic DNA was isolated by standard methods from blood samples or from lymphocyte cultures after Epstein-Barr virus transformation.
Mutation Analysis
Mutation analysis in candidate genes was performed with a bar-coded, high-throughput exon-sequencing technique that we recently developed.39 Segregation analysis was done by Sanger dideoxy-terminator resequencing for confirmation and segregation of potential disease-causing variants in the respective affected individuals, their affected siblings, and their parents. Sequencing was performed with BigDye Terminator v3.1 Cycle Sequencing Kit on an ABI 3730 XL sequencer (Applied Biosystems). Sequence traces were analyzed with Sequencher (v.4.8) software (Gene Codes Corporation). Polymerase chain reaction (PCR) was performed by a touchdown protocol described previously.40 At the University of North Carolina, we used Sanger sequencing with an ABI 3130XL and analysis with Sequencher. At Universitaetsklinik Muenster, Sanger sequencing was performed with an Applied Biosystems 3730xl DNA Analyzer, and data were analyzed with the CodonCode software provided by CodonCode Corporation.
Results
A Functional Screen of Cilia Proteome Genes in Zebrafish and Planaria
To characterize the function of genes not previously linked to PCD, we screened a set of uncharacterized genes encoding proteins present in multiple cilia proteomes41–50 by knockdown in zebrafish (Table S3). Knockdown of three genes, c21orf59/FBB18 (RefSeq accession number NM_200088.1), ccdc65/FAP250 (RefSeq NM_001002603.2), and c15orf26 (RefSeq NM_001017774.1) resulted in strong ciliopathy phenotypes, including those involving pronephric cysts, axis curvature, left-right asymmetry defects, and hydrocephalus (Figures 1A–1D and S1), suggesting that these genes play essential roles in cilia function. Immunostaining via antibodies against acetylated-tubulin (axonemes), γ-tubulin (basal bodies), and Ift88 (intraflagellar transport particles) did not detect structural cilia abnormalities (Figure S2), suggesting that ciliopathy phenotypes might be due to altered cilia motility. Consistent with other essential motile cilia genes,23,51 c21orf59, ccdc65, and c15orf26 mRNA expression was dependent on the transcriptional regulator Foxj1a (Figures 1E, 1F, and S3). Live imaging of olfactory placode and pronephric cilia by high-speed video microscopy revealed that compared to controls (Movies S1 and S2; Figures 1H and 1O), cilia in c21orf59, ccdc65, and c15orf26 morphants were either paralyzed or dyskinetic (Movies S3, S4, S5, and S6; Figures 1J, 1L, 1M, and 1P). Cilia ultrastructure analysis revealed that outer dynein arms were missing in c21orf59 and c15orf26 morphants but not in ccdc65 morphants, indicating that these genes control cilia motility by distinct mechanisms (Figures 1G, 1I, 1K, and 1N; insets).
Figure 1.
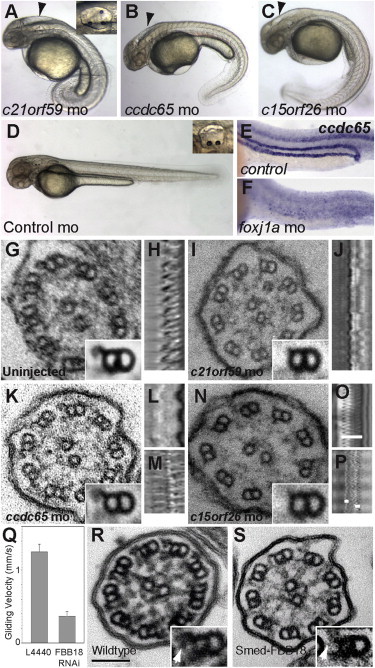
Zebrafish Knockdown Screen Identifies c21orf59, ccdc65, and c15orf26 as Essential for Cilia Motility
(A–D) Morpholino knockdown of c21orf59 (A), ccdc65 (B), and c15orf26 (C) produces ciliopathy phenotypes in zebrafish, including axis curvature, hydrocephalus (arrowheads), and supernumerary otoliths (insets in A and D). Phenotypes are not observed in control injected embryos (D).
(E and F) Expression of ccdc65 mRNA in ciliated tissues, visible in control (E), is eliminated by foxj1a morpholino knockdown (F).
(G) Ultrastructure of wild-type cilia shows the 9+2 arrangement of microtubules with inner and outer dynein arms projecting from the A subfibers of each doublet in cross sections and averaged doublets (inset; n = 75).
(H) Kymogram of wild-type olfactory cilia beat from high-speed microvideo (Movie S1) shows rhythmic cilia wave form at 31 Hz.
(I) c21orf59 morphant cilia ultrastructure showing loss of dynein outer arm motor domains in computer-averaged microtubule doublets (inset; n = 30 doublets).
(J) Kymogram of c21orf59 morphant olfactory cilia beat (Movie S3) shows near-complete cilia paralysis.
(K) ccdc65 morphant cilia show normal dynein arm ultrastructure in averaged doublets (inset; n = 27).
(L and M) Kymograms of ccdc65 morphant olfactory cilia beat shows either complete cilia paralysis (L; Movie S4) or dyskinetic, faster beat rate (48 Hz; M; Movie S5).
(N) Similar to c21orf59, outer dynein arms are lost in c15orf26 morphants (inset; n = 8).
(O) Kymogram of control pronephric cilia (Movie S2) shows a beat rate of 66 Hz and a wave amplitude of 8.6 μm (white bar in O).
(P) Kymogram of c15orf26 morphant pronephric cilia (Movie S6) shows severely reduced beat amplitude (white bars = 1.8 μm) or paralysis.
(Q–S) RNAi knockdown of the c21orf59 ortholog in planaria, FBB18.
(Q) Smed-FBB18 (RNAi) significantly reduces cilia-driven gliding locomotion of planaria (Movie S7).
(R and S) Ultrastructure of control (R) and Smed-FBB18 (RNAi) (S) cilia. Insets show defects in Smed-FBB18 (RNAi) ODA assembly (arrows). The scale bar represents 100 nm.
Ventral cilia in the planarian Schmidtea mediterranea generate a synchronized beating pattern used for locomotion.29 Consistent with previously published planarian motility mutants,29 complete knockdown of planaria c21orf59/fbb18 (mk4.000443.01.01)52 (Figure S4) significantly reduced cilia-driven gliding motion (Figure 1K; Movie S7). Ventral cilia were normal in number and length (Figure S4) but showed defects in assembly of outer dynein arms; these results are similar to those from zebrafish c21orf59 knockdown (Figures 1L and 1M), suggesting that the requirement for C21orf59 in cilia motility is well conserved.
Mutations in C21orf59 Cause Human Primary Ciliary Dyskinesia
To investigate whether C21orf59 mutations occur in humans with PCD, we examined a worldwide cohort of 295 individuals with PCD by targeted exon resequencing of all seven exons of C21orf59 (RefSeq accession number NM_021254.2) by a bar-coded, high-throughput exon-resequencing technique.39 All screened individuals suffered from recurrent bronchitis, sinusitis, and/or otitis media. Situs inversus (Figure 2A) and situs ambiguous were present in 112 and 26 individuals, respectively. Ultrastructural analysis of nasal brushings revealed that 85 affected individuals showed defective or missing outer dynein arms, 45 showed defective or missing outer and inner dynein arms, 53 showed defective inner dynein arms with microtubular disorganization, and nine showed central apparatus defects. In the remaining individuals, PCD was ascertained on the basis of clinical phenotype, the presence of situs abnormalities, and low levels of nasal nitric oxide. In total, we identified disease-causing C21orf59 mutations in four out of 295 families with PCD (A4204 [#46], A5014_087 [#143], A5014_384 [#508], 5014_513 [#652]) with 3 different truncating mutations: c.292C>T (p.Arg98∗), c.735C>G (p.Tyr245∗), and c.792_795delTTTA (p.Tyr264∗) (Table 1; Figures 2B–2E). One of the mutations (c.735C>G) was putatively homozygous in three unrelated families, two of which had known Ashkenazi Jewish ethnicity (Table 1; Figure 2E). Further analysis of all three affected individuals by the use of 20 SNP markers ∼2 Mb up and downstream of the c.735C>G mutation revealed that this mutation is located within a shared haplotype of ∼1 Mb (Figure S5). This result strongly indicates the presence of a founder effect involving a common distant ancestor for all three families. Because parental DNA for segregation analysis was not available in family A5014_087, we cannot rule out hemizygosity in this affected individual. However, if hemizygosity rather than homozygosity were present, that would not alter the assumption of the absence of a normal allele in the affected individual and thereby would not affect our conclusions. Given the parental segregation in the other two families, carrying the same mutation and its supposed founder effect, homozygosity is nevertheless most likely to be present in all three families. For exclusion of known genetic causes of PCD, 19 known genes previously linked to PCD were screened in all four families with C21orf59 mutations, but no explanatory mutations were detected (Table S4). In addition to truncating mutations, single heterozygous C21orf59 missense variants were found in three individuals without identification of a second mutated allele: c.97C>T (p.Arg33Trp), c.422A>G (p.Asp141Gly), and c.517G>T (p.Asp173Thr). Phenotypically, these individuals presented with low nasal nitric oxide, sinus disease, otitis media, and/or bronchiectasis but normal ciliary ultrastructure, suggesting that these mutations might be either hypomorphic or not causative of PCD.
Table 1.
Mutations of C21orf59 or CCDC65 in Six Families with Primary Ciliary Dyskinesia
| Family-Individual | Ethnic Origin (Sex) | Nucleotide Alteration (Segregation) | Deduced Protein Change | Exon/Intron (Zygosity) | nNO (nl/min) | Situs Inversus | TEM | Video Microscopy | Additional Clinical Presentation |
|---|---|---|---|---|---|---|---|---|---|
| C21orf59 | |||||||||
| A4204 (#46)-21 (#274) | Brazilian (male) | c.735C>Ga (P, het; M, het) | p.Tyr245∗ | 6 (hom) | 42.5 | yes | ODA+IDA defects | immotile cilia | NRD, SD, BE |
| A5014_087 (#143)-21 (#914) | Ashkenazi Jewish (male) | c.735C>Ga (P, ND; M, ND) | p.Tyr245∗ | 6 (hom/hem) | 8.9 | yes | ODA+IDA defects | ND | NRD, OM, SD |
| A5014_384 (#508)-21 (#1637) | Ashkenazi Jewish (female) | c.735C>Ga (P, ND; M, het) | p.Tyr245∗ | 6 (hom/hem) | 13.9 | yes | ODA+IDA defects | ND | NRD, OM, SD, BE |
| A5014_513 (#652)-21 (#2003) | European-American (female) | c.292C>Tb (P, WT; M, het), c.792_795delTTTA (P, het; M, WT) | p.Arg98∗, p.Tyr264∗ | 2 (het),7 (het) | 15.7 | no | partial ODA+IDA defects | ND | NRD, OM, SD |
| CCDC65 | |||||||||
| A5014_493 (#629)-21 (#1929) and -22 (#2005) | Ashkenazi Jewish (male and male) | c.877_878delAT (P, ND; M, ND) | p.Ile293Profs∗2 | 6 (hom and hem) | 33.2 (-21) and ND (-22) | no | normal DA | dyskinetic cilia | OM, SD, BE (-21) and OM, BE (-22) |
| A5014_656 (#798) -21 (#2272) | Ashkenazi Jewish (female) | c.877_878delAT (P, het; M, het) | p.Ile293Profs∗2 | 6 (hom) | 20 | no | normal DA+CA | dys-/ hyperkinetic cilia | NRD, OM, SD, BE |
Abbreviations are as follows: BE, bronchiectasis; CA, central apparatus/central pair; DA, dynein arms; hom, homozygous; het, heterozygous; hem, hemizygous; M, maternal; nNO, nasal nitric oxide, normal range: 376 ± 124 nl/min (mean ± SD);53 ODA, outer dynein arm; OM, otitis media; ND, no data; NRD, neonatal respiratory distress; P, paternal; SD, sinus disease or chronic sinusitis.
Mutation is in 1000 Genomes database: rs202094637 (GG = 0/GC = 1/CC = 661).
Mutation is in EVS (Exome variant server) database: rs143740376 (TT = 0/TC = 2/CC = 6501).
Human C21orf59 is a 290 aa protein that contains a coiled-coil (CC) and a domain of unknown function (DUF2870) at the C-terminal end, as does its zebrafish homolog (90% similarity, 73% identity) (Figures 2F, 2G, and S6). The identified human mutations would be predicted to encode truncated proteins that lack either the last 193 (p.Arg98∗), 45 (p.Tyr245∗), or 26 (p.Tyr264∗) amino acids (Figures 2D–2G). Alternatively, these mutant mRNAs might be subject to nonsense-mediated mRNA decay (see below). Cilia ultrastructure of all affected individuals carrying mutations in C21orf59 showed an absence of both outer- and inner-dynein-arm components (Figure 2B). To confirm the absence of both outer- and inner-dynein-arm components in C21orf59 mutant cilia, we obtained nasal brushing respiratory epithelial cells from individual A5014_087-21 carrying the c.735C>G (p.Tyr245∗) mutation (Table 1) and immunostained the cells with antibodies specific to DNAH5 (ODAs) and DNALI1 (IDAs). Both DNAH5 (Figure 2H) and DNALI1 (Figure 2I) were absent from C21orf59 mutant cilia, confirming the absence of both outer and DNALI1-related inner arms. Consistent with results for zebrafish and planaria c21orf59/fbb18-deficient cilia, live imaging of C21orf59 mutant cilia in human nasal epithelia revealed complete paralysis (Movie S8).
To test the pathogenicity of the identified mutations, we injected synthetic mRNA containing C21orf59 missense variants c.97C>T (p.Arg33Trp), c.422A>G (p.Asp141Gly), or c.517G>T (p.Asp173Thr) or the most C-terminal truncating mutation, c.792_795delTTTA (p.Tyr264∗) into zebrafish c21orf59 morphants and assayed KV cilia motility (Figure 3). C21orf59-deficient zebrafish have paralyzed KV cilia (Figures 3A–3C) and abnormal heart looping (Figure 3D). Injection of wild-type human C21orf59 mRNA rescued morphant KV cilia motility (Figure 3C) and heart looping (Figure 3D). In contrast, c.792_795delTTTA (p.Tyr264∗) mutant RNA injection failed to rescue cilia motility or heart looping (Figures 3C and 3D). Of the missense variants, only the c.422A>G (p.Asp141Gly) variant showed reduced rescue (Figure 3C), indicating that these mutations are likely to be benign polymorphisms or, for c.422A>G, potentially a hypomorphic mutation. The p.Tyr264∗ protein lacking the last 26 amino acids was not stable in zebrafish embryos, even when mRNA was injected at concentrations up to 10-fold higher than the wild-type (Figure S7), suggesting that this protein may be rapidly degraded or that the mRNA may be subject to nonsense-mediated mRNA decay.
The C. reinhardtii C21orf59 Ortholog FBB18 Is a Flagellar Matrix Protein Regulated by Cilia Motility
Failure of ODA assembly in C21orf59/FBB18-deficient cilia suggested that C21orf59/FBB18 may act as an ODA structural component or be involved in trafficking and assembly of cilia dynein arms. Using a cross-reacting C21orf59/FBB18 antibody (Figure S8), we found that C21orf59/FBB18 (RefSeq accession number XM_001699148.1) localized exclusively to the flagellar matrix in wild-type and ODA mutant C. reinhardtii axonemes (Figure 4A), similar to the IFT particle. C21orf59/FBB18 protein levels were low in wild-type axonemes but, surprisingly, were dramatically elevated in flagella of motility-impaired mutants, but not in a mutant with close to wild-type cilia motility (oda11; Figure 4B). Increased abundance of C21orf59/FBB18 protein was also observed in cell bodies of motility-impaired mutants, suggesting that C21orf59/FBB18 protein is induced or stabilized when cilia motility is impaired (Figure 4C). Although associated with the flagellar matrix, C21orf59/FBB18 did not comigrate with IFT particle proteins in Superose 6 filtration (Figure 4D). Consistent with its dynamic pattern of flagellar association in C. reinhardtii, C21orf59/FBB18 protein was predominantly cytoplasmic in rat trachea (Figures 5A, 5B, and S9). In rat trachea, C21orf59/FBB18 partially colocalized with SAS6 in cytoplasmic puncta, suggesting a centrosomal localization (Figure 5B).
Figure 4.
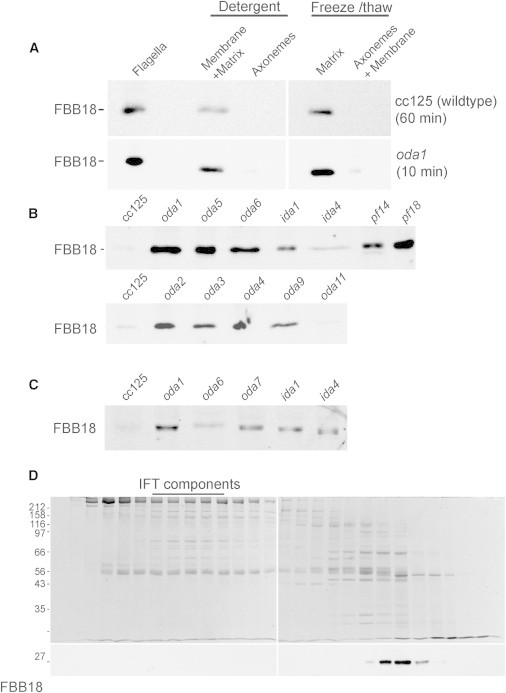
Analysis C21orf59/FBB18 in C. reinhardtii
(A) Flagella from wild-type and oda1 mutant Chlamydomonas strains were extracted by detergent or freeze-thaw methods. FBB18 is found exclusively within the flagellar matrix in both strains. Note that blot exposure time was significantly longer for the wild-type strain.
(B) Probing flagella extracts from wild-type (cc125) and Chlamydomonas mutant strains with an FBB18 antibody reveals that flagella protein levels are significantly enhanced in cilia mutants with severe motility defects but not in oda11, which retains most of the outer arm and exhibits close to wild-type ciliary motility.
(C) FBB18 immunoblot of wild-type (cc125) and cilia mutant cell bodies shows increased FBB18 protein abundance in mutants with impaired cilia motility.
(D) Fractionation of Chlamydomonas flagella matrix extract in a Superose 6 gel filtration column. FBB18 migrates in a single peak distinct from core IFT complex proteins.
Figure 5.
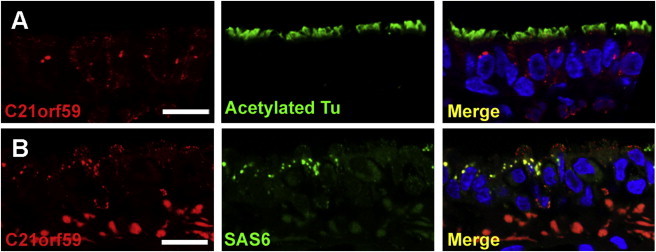
Cellular Localization of C21orf59 Protein
(A) C21orf59 immunostaining in rat trachea ciliated epithelial cells reveals predominant cytoplasmic localization in perinuclear puncta.
(B) C21orf59 colocalizes with SAS6 in cytoplasmic puncta in rat trachea ciliated epithelial cells.
Scale bars represent QA.
Mutations in CCDC65 Cause Human Primary Ciliary Dyskinesia
To screen for human CCDC65 (RefSeq NM_033124.4) mutations in individuals with PCD, we examined all eight CCDC65 coding exons and exon-intron boundaries in our cohort of 295 individuals with PCD by applying the above-described high-throughput technique (Figures 6A–6F). We identified truncating mutations in three individuals from two families (Figure 6A) whose affected members shared the same putatively homozygous frameshift mutation, c.877_878delAT (p.Ile293Profs∗2) (Table 1; Figures 6D–6F). Because parental DNA for family A5014_493 [#629] was not available, we cannot completely exclude hemizygosity in this case. Taken together with the segregation of family A5014_656 [#798], who shares not only the same mutation but also the same Ashkenazi Jewish ethnicity, a founder effect and thereby homozygosity of this mutation is very likely to be present in both families. In the affected individuals (A5014_493 [#629] -21, -22, and A5014_656 [#798]), cilia ultrastructure showed normal outer dynein arms, radial spokes, and central pairs but a reduction in inner dynein arms and nexin links (Figures 6B and 6C). Microtubule disorganization was also observed in 5%–15% of cilia cross sections (Figure 6B, inset), suggesting that the reduction in nexin links may lead to overall structural instability in a subset of PCD individual cilia. Live imaging of PCD individual nasal epithelial cilia revealed a stiff, dyskinetic cilia waveform (Movie S9), similar to that of zebrafish ccdc65 morphant cilia (Movie S5). The affected individuals suffered from recurrent bronchitis, sinusitis, and/or otitis media. Situs abnormalities were not present in affected individuals, and fertility status could not be ascertained. Biallelic mutations in any of 19 genes previously linked to PCD were excluded by the same mutation-analysis approach (Table S4). The human CCDC65 protein shows a 69% similarity and 41% identity to its zebrafish homolog (Figure S10). CCDC65 comprises 484 amino acids and contains two coiled-coil domains, one of which is truncated by the identified frameshift mutation (Figures 6E, 6F, and S10).
Figure 6.
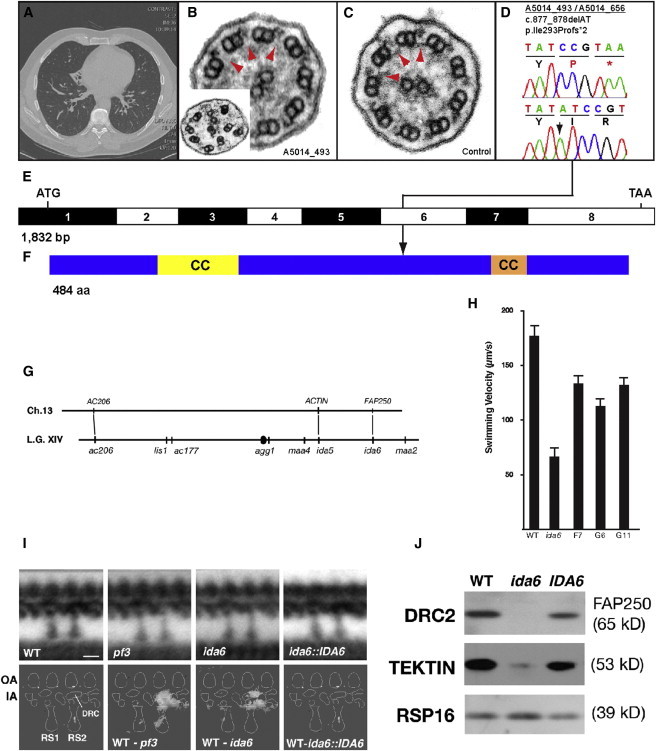
Identification of Recessive Mutations in CCDC65 in Two Families with Primary Ciliary Dyskinesia and Identification of FAP250/CCDC65 as the Causative Gene in the ida6 Mutant
(A) Chest CT scan of individual A5014_493-21 showing volume loss and bronchiectasis in the right middle lobe and bronchiectasis in both lower lobes.
(B) TEM of individual A5014_493-21 cilia showing normal dynein arms and central pairs. Note that upon close examination, N-DRC links are missing (red arrowheads). Microtubule disorganization (B, inset) was observed in 5%–15% of PCD individual cilia sections.
(C) Control cilia TEM showing normal dynein inner arms and nexin links (red arrowheads).
(D) A homozygous truncating CCDC65 mutation detected in two independent families with PCD. Family number (underlined), mutation (arrowhead), and predicted translational changes are indicated. Sequence trace is shown for mutation above normal control. For the mutations detected, arrows indicate positions in relation to exons.
(E) Exon structure of human CCDC65 cDNA. Positions of start codon (ATG) and of stop codon (TAA) are indicated.
(F) Domain structure of the CCDC65 protein indicating two coiled-coil (CC) domains and position of truncating stop codon mutation.
(G–J) The C. reinhardtii IDA6 gene encodes FAP250/DRC2/CCDC65.
(G) FAP250 maps near the ACTIN gene on the right arm of C. reinhardtii Linkage Group XIV (chromosome 13), in the vicinity of the ida6 mutation. AC206 and ACTIN have been linked to the ac206 and ida5 mutations on the left and right arms of Linkage Group XIV, respectively (bottom line). ida6 is an inner arm motility mutation located ∼6 cM from ida554 and the FAP250 gene is located ∼6.1 Mb away from the ACTIN gene.
(H) The ida6 motility mutant was transformed with an ∼9.5 kb genomic subclone encoding FAP250, and transformants were screened for improved motility by phase-contrast microscopy. The forward swimming velocities of three rescued strains are shown here relative to wild-type and ida6.
(I) Longitudinal views of the 96 nm repeat of axonemes from WT, pf3, ida6, and an IDA6 rescued strain (G11) were computer averaged and then compared to identify regions of statistically significant differences. The top row shows the average of the 96 nm repeat for each strain, whereas the bottom row shows the difference plots between the wild-type and each sample. A schematic diagram of the densities observed in each repeat is labeled on the bottom left. The crescent-shaped region associated with the dynein regulatory complex (DRC) is located above radial spoke 2 (RS2), between the outer arms (OA) and inner arms (IA). Both pf3 and ida6 are associated with significant defects in the assembly of the DRC and closely associated inner arm dyneins (see difference plots). These defects are not observed in the rescued ida6::IDA6 strain. The number of axonemes and 96 nm repeats analyzed for each strain are as follows: wild-type (9 axonemes, 62 repeats), pf3 (7 axonemes, 63 repeats), ida6 (4 axonemes, 35 repeats), and ida6::IDA6 (7 axonemes, 60 repeats).
(J) Axonemes from WT, ida6, and a rescued IDA6 strain were analyzed on an immunoblot probed with antibodies against several flagellar proteins. Both FAP250 (DRC2) and tektin are missing or reduced in ida6 and restored to near wild-type levels in the rescued IDA6 strain. The RSP16 subunit of the radial spokes serves as a loading control.
Scale bars in (A) and (B) represent 20 μm.
The CCDC65 C. reinhardtii Ortholog FAP250 Is an Essential Component of the Nexin-Dynein Regulatory Complex
Unlike C21orf59l/FBB18-deficient cilia, Ccdc65-deficient zebrafish and human cilia did not show ODA defects (Figures 1I and 6B), suggesting a different role for this protein in ciliary function. Recent studies in C. reinhardtii have identified the CCDC65 ortholog FAP250 (Cre13.g607750.t1.2)55 as a subunit of the nexin-dynein regulatory complex.17,56 The gene encoding FAP250 maps to the right arm of linkage group XIV (chromosome 13), in the vicinity of the ida6 mutation (Figure 6G). ida6 fails to assemble a single-headed, inner arm dynein and displays a slow swimming motility defect,54 but it is not linked to any of the genes encoding DHC subunits.57,58 To determine whether ida6 might be a mutation in an accessory protein required for dynein assembly and motility, we transformed ida6 with an ∼9.5 kb genomic subclone containing FAP250 and tested transformants for rescue of the slow-swimming phenotype. The observed restoration of forward swimming velocity (Figure 6H) and direct sequencing of FAP250 demonstrated that ida6 is a mutation that alters the stop codon of FAP250/CCDC65 and thus adds 106 amino acids to the C terminus of the protein (Figure S11). To assess whether ida6 also affects assembly of the N-DRC, we analyzed isolated axonemes by thin-section electron microscopy and computer image averaging.37 Longitudinal views of the 96 nm axoneme repeats of WT, pf3 (a previously characterized DRC mutant), ida6, and IDA6 rescued (G11) strains revealed defects in the assembly of both the inner dynein arms and the N-DRC in ida6 (Figure 6I). These defects were not observed in the rescued ida6::IDA6 strain (Figure 6I), demonstrating that the structural defect was specific to the FAP250 mutation. Immunoblotting revealed that both FAP250 and tektin were missing or reduced in isolated ida6 axonemes (Figure 6J), indicating that the additional amino acids in ida6 inhibited assembly of both FAP250 and tektin. As expected, both proteins were restored in axonemes from the rescued ida6::IDA6 strain (Figure 6J). These data, together with the predicted size of the FAP250 polypeptide, strongly suggest that FAP250 corresponds to the DRC2 subunit of N-DRC and that DRC2/FAP250/CCDC65 plays a critical role in the assembly of the N-DRC.
Discussion
Although the structure of cilia and flagella has been studied for decades, proteomic analysis has only recently revealed the complexity of these organelles and their component protein subunits.41,45,47,49 Our screen of cilia proteome candidate genes identified two genes, C21orf59/FBB18 and CCDC65/FAP250, that are mutated in PCD and that act via distinct mechanisms to control cilia motility.
We found that both c21orf59 zebrafish knockdown and C21orf59 mutations in individuals with PCD resulted in complete cilia paralysis and the loss of both outer and inner dynein arms. Failure of the C21orf59 nonsense mutant mRNA to rescue cilia motility in zebrafish was linked to a loss of stable protein levels, suggesting that human phenotypes might be due to the complete absence of C21orf59 protein, leading to failed ODA and DNALI1 related IDA assembly. Partial loss of rescuing activity by the c.422A>G (p.Asp141Gly) C21orf59 mutant mRNA suggests that this allele could represent a hypomorphic mutation. Although the affected individuals harboring this mutation were heterozygous at the C21orf59 locus, it remains possible that a hypomorphic mutation in C21orf59 inherited with mutation(s) in other genes associated with dynein function or assembly could contribute to digenic or oligogenic transmission of PCD. Evidence for digenic inheritance in short-rib polydactyly syndrome type Majewski, a ciliopathy linked to paired heterozygous mutations in NEK1 and DYNC2H1, has been reported.59
The stepwise sequence of cytoplasmic dynein arm assembly, transport, and axonemal docking has been well characterized in Chlamydomonas reinhardtii flagellar mutants. The mutated genes encode dynein motor subunits, axonemal docking complexes, and nonaxonemal dynein arm assembly and transport factors.11,60,61 The C21orf59/FBB18 accumulation we observed at the basal body suggests a role in cytoplasmic dynein assembly or transport into the axoneme. However, the behavior of C21orf59/FBB18 appears to be unique relative to known ODA assembly proteins in that C21orf59/FBB18 accumulates dramatically in the matrix fraction of mutant axonemes that exhibit reduced motility. C21orf59/FBB18 is not strongly associated with the IFT particle in fractionated axonemes, suggesting that it might act as an adaptor protein for ODA and IDA transport. An increased abundance of flagellar C21orf59/FBB18 in axonemes correlated with increased protein levels in cell bodies as well, indicating that C21orf59/FBB18 is either stabilized or induced in response to flagellar immotility. Elevated cell body and axonemal C21orf59/FBB18 in flagellar mutants suggests that cells can detect impaired cilia motility and initiate molecular responses to compensate for motility defects. Mammalian multiciliated cells are known to modulate dynein arm activity in response to increased fluid viscosity,62,63 indicating that cells monitor cilia movement and detect resistance to cilia beating. Intriguingly, a cellular abundance of Chlamydomonas Lis1 protein, a dynein regulatory factor that controls microtubule binding affinity, is enhanced in response to both genetic and environmental disruption of axonemal beating.64 It could be that the abundance of C21orf59/FBB18 in cilia and flagella is differentially regulated in response to flagellar motility and that this protein might enhance ODA stability in “high-load” environments.
Our functional analysis of CCDC65/FAP250/DRC2 revealed that this gene encodes a protein that is required for proper cilia motility. In contrast to C21orf59, CCDC65 is not required for ODA structure and instead plays an essential role in regulating dynein activity. CCDC65 is the vertebrate ortholog of Chlamydomonas FAP250, which encodes a protein recently identified as the DRC2 subunit of the N-DRC.17,56 The N-DRC is a 12 subunit protein complex positioned on axonemes adjacent to inner dynein arms.17,56 The N-DRC mediates both outer doublet alignment and the control of dynein activity in response to signals from radial spokes and the central pair apparatus.13,17,65,66 Structural analysis of the N-DRC suggests that CCDC65/FAP250/DRC2, together with DRC1, forms the “base plate” of N-DRC, which makes extensive connections with the outer doublet microtubules, radial spokes, and inner dynein arms.13,17 Interestingly, the loss of CCDC65/FAP250 in the Chlamydomonas ida6 mutant is also associated with a reduction of the protein Tektin in axonemes.67 Tektin forms rod-like structures that have been proposed to stabilize microtubules and participate in the longitudinal spacing of axonemal components such as inner dynein arms and radial spokes.68 The integrity of the 9+2 axoneme did not appear to be significantly affected in the Chlamydomonas ida6 mutant but we did observe axonemal microtubular disorganization (MTD) in a subset of cilia sections from affected individuals bearing a mutation in CCDC65. MTD has been reported in other N-DRC mutants and in radial spokehead mutants,69,70 suggesting that this defect might arise from loss of the putative radial spoke attachment site in CCDC65 mutant axonemes. No clear evidence for MTD was observed in the Chlamydomonas ida6 mutant, however. This and the low penetrance of MTD in CCDC65 mutant axonemes suggests that MTD might occur only after prolonged cilia beating and that it might represent a terminal breakdown of cilia structure.
The third gene we identified in our zebrafish screen, c15orf26, was not mutated in our PCD cohort. However, a locus containing C15orf26 (RefSeq accession number NM_173528.2) on chromosome 15 has previously been associated with PCD.71 Cilia immotility in these individuals was associated with a lack of outer dynein arms, similar to the c15orf26 knockdown phenotype in zebrafish. Although coding mutations in C15orf26 were not found, mutations affecting mRNA splicing or transcriptional regulation were not ruled out. Further analysis of C15orf26 expression in these individuals could therefore be informative.
Acknowledgments
We are grateful to all PCD subjects and family members for their participation and thank the US PCD Foundation and to the investigators and the coordinators of the “Genetic Disorders of Mucociliary Clearance Consortium.” We are also grateful to the funding agencies that supported this work; these include the National Institutes of Health, the Howard Hughes Medical Institute, the Deutsche Forschungsgemeinschaft, the IZKF Muenster, and the European Community’s Seventh Framework Programme. Detailed acknowledgements can be found in the Supplemental Data.
Contributor Information
Iain A. Drummond, Email: idrummond@partners.org.
Friedhelm Hildebrandt, Email: friedhelm.hildebrandt@childrens.harvard.edu.
Supplemental Data
Normal olfactory cilia beat rhythmically around the perimeter of the olfactory placode. This and all microvideo were acquired at 250 frames per second (fps) and slowed to 15 fps for viewing.
Pronephric multicilia beat in a coordinated helical pattern to drive fluid down the lumen of the pronephric tubule.
Cilia are largely paralyzed, consistent with loss of outer dynein arms.
Most ccdc65 morphants showed complete paralysis of olfactory cilia.
Some ccdc65 morphants showed faster, dyskinetic cilia beat patterns.
Cilia bundles showed greatly reduced beat amplitude or paralysis and bundle coordination was lost.
Time-lapse video of control (left) and Smed fbb18(RNAi) (right) -treated planaria. fbb18 knockdown planaria resort to a parastaltic mode of locomotion when cilia motility is disrupted.
Web Resources
The URLs for data presented herein are as follows:
Chlamydomonas Resource Center, http://chlamycollection.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
ZFIN, http://zfin.org
References
- 1.Veland I.R., Awan A., Pedersen L.B., Yoder B.K., Christensen S.T. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron, Physiol. 2009;111:39–53. doi: 10.1159/000208212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fisch C., Dupuis-Williams P. Ultrastructure of cilia and flagella - back to the future! Biol. Cell. 2011;103:249–270. doi: 10.1042/BC20100139. [DOI] [PubMed] [Google Scholar]
- 3.Ibañez-Tallon I., Heintz N., Omran H. To beat or not to beat: roles of cilia in development and disease. Hum. Mol. Genet. 2003;12(Spec No 1):R27–R35. doi: 10.1093/hmg/ddg061. [DOI] [PubMed] [Google Scholar]
- 4.Waters A.M., Beales P.L. Ciliopathies: an expanding disease spectrum. Pediatr. Nephrol. 2011;26:1039–1056. doi: 10.1007/s00467-010-1731-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arts H.H., Bongers E.M., Mans D.A., van Beersum S.E., Oud M.M., Bolat E., Spruijt L., Cornelissen E.A., Schuurs-Hoeijmakers J.H., de Leeuw N. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J. Med. Genet. 2011;48:390–395. doi: 10.1136/jmg.2011.088864. [DOI] [PubMed] [Google Scholar]
- 6.Walczak-Sztulpa J., Eggenschwiler J., Osborn D., Brown D.A., Emma F., Klingenberg C., Hennekam R.C., Torre G., Garshasbi M., Tzschach A. Cranioectodermal dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am. J. Hum. Genet. 2010;86:949–956. doi: 10.1016/j.ajhg.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bredrup C., Saunier S., Oud M.M., Fiskerstrand T., Hoischen A., Brackman D., Leh S.M., Midtbø M., Filhol E., Bole-Feysot C. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am. J. Hum. Genet. 2011;89:634–643. doi: 10.1016/j.ajhg.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zariwala M.A., Omran H., Ferkol T.W. The emerging genetics of primary ciliary dyskinesia. Proc. Am. Thorac. Soc. 2011;8:430–433. doi: 10.1513/pats.201103-023SD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guichard C., Harricane M.C., Lafitte J.J., Godard P., Zaegel M., Tack V., Lalau G., Bouvagnet P. Axonemal dynein intermediate-chain gene (DNAI1) mutations result in situs inversus and primary ciliary dyskinesia (Kartagener syndrome) Am. J. Hum. Genet. 2001;68:1030–1035. doi: 10.1086/319511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olbrich H., Häffner K., Kispert A., Völkel A., Volz A., Sasmaz G., Reinhardt R., Hennig S., Lehrach H., Konietzko N. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 2002;30:143–144. doi: 10.1038/ng817. [DOI] [PubMed] [Google Scholar]
- 11.Omran H., Kobayashi D., Olbrich H., Tsukahara T., Loges N.T., Hagiwara H., Zhang Q., Leblond G., O’Toole E., Hara C. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panizzi J.R., Becker-Heck A., Castleman V.H., Al-Mutairi D.A., Liu Y., Loges N.T., Pathak N., Austin-Tse C., Sheridan E., Schmidts M. CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat. Genet. 2012;44:714–719. doi: 10.1038/ng.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heuser T., Raytchev M., Krell J., Porter M.E., Nicastro D. The dynein regulatory complex is the nexin link and a major regulatory node in cilia and flagella. J. Cell Biol. 2009;187:921–933. doi: 10.1083/jcb.200908067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wirschell M., Olbrich H., Werner C., Tritschler D., Bower R., Sale W.S., Loges N.T., Pennekamp P., Lindberg S., Stenram U. The nexin-dynein regulatory complex subunit DRC1 is essential for motile cilia function in algae and humans. Nat. Genet. 2013;45:262–268. doi: 10.1038/ng.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams B.D., Velleca M.A., Curry A.M., Rosenbaum J.L. Molecular cloning and sequence analysis of the Chlamydomonas gene coding for radial spoke protein 3: flagellar mutation pf-14 is an ochre allele. J. Cell Biol. 1989;109:235–245. doi: 10.1083/jcb.109.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castleman V.H., Romio L., Chodhari R., Hirst R.A., de Castro S.C.P., Parker K.A., Ybot-Gonzalez P., Emes R.D., Wilson S.W., Wallis C. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am. J. Hum. Genet. 2009;84:197–209. doi: 10.1016/j.ajhg.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bower R., Tritschler D., Vanderwaal K., Perrone C.A., Mueller J., Fox L., Sale W.S., Porter M.E. The N-DRC forms a conserved biochemical complex that maintains outer doublet alignment and limits microtubule sliding in motile axonemes. Mol. Biol. Cell. 2013;24:1134–1152. doi: 10.1091/mbc.E12-11-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gherman A., Davis E.E., Katsanis N. The ciliary proteome database: an integrated community resource for the genetic and functional dissection of cilia. Nat. Genet. 2006;38:961–962. doi: 10.1038/ng0906-961. [DOI] [PubMed] [Google Scholar]
- 19.Westerfield M. University of Oregon Press; Eugene, OR: 2007. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio rerio) [Google Scholar]
- 20.Suster M.L., Kikuta H., Urasaki A., Asakawa K., Kawakami K. Transgenesis in zebrafish with the tol2 transposon system. Methods Mol. Biol. 2009;561:41–63. doi: 10.1007/978-1-60327-019-9_3. [DOI] [PubMed] [Google Scholar]
- 21.Kwan K.M., Fujimoto E., Grabher C., Mangum B.D., Hardy M.E., Campbell D.S., Parant J.M., Yost H.J., Kanki J.P., Chien C.B. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 2007;236:3088–3099. doi: 10.1002/dvdy.21343. [DOI] [PubMed] [Google Scholar]
- 22.Thisse B., Heyer V., Lux A., Alunni V., Degrave A., Seiliez I., Kirchner J., Parkhill J.P., Thisse C. Spatial and temporal expression of the zebrafish genome by large-scale in situ hybridization screening. Methods Cell Biol. 2004;77:505–519. doi: 10.1016/s0091-679x(04)77027-2. [DOI] [PubMed] [Google Scholar]
- 23.Hellman N.E., Liu Y., Merkel E., Austin C., Le Corre S., Beier D.R., Sun Z., Sharma N., Yoder B.K., Drummond I.A. The zebrafish foxj1a transcription factor regulates cilia function in response to injury and epithelial stretch. Proc. Natl. Acad. Sci. USA. 2010;107:18499–18504. doi: 10.1073/pnas.1005998107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panizzi J.R., Jessen J.R., Drummond I.A., Solnica-Krezel L. New functions for a vertebrate Rho guanine nucleotide exchange factor in ciliated epithelia. Development. 2007;134:921–931. doi: 10.1242/dev.02776. [DOI] [PubMed] [Google Scholar]
- 25.Pathak N., Austin C.A., Drummond I.A. Tubulin tyrosine ligase-like genes ttll3 and ttll6 maintain zebrafish cilia structure and motility. J. Biol. Chem. 2011;286:11685–11695. doi: 10.1074/jbc.M110.209817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rashid S., Breckle R., Hupe M., Geisler S., Doerwald N., Neesen J. The murine Dnali1 gene encodes a flagellar protein that interacts with the cytoplasmic dynein heavy chain 1. Mol. Reprod. Dev. 2006;73:784–794. doi: 10.1002/mrd.20475. [DOI] [PubMed] [Google Scholar]
- 27.Drummond I.A., Majumdar A., Hentschel H., Elger M., Solnica-Krezel L., Schier A.F., Neuhauss S.C., Stemple D.L., Zwartkruis F., Rangini Z. Early development of the zebrafish pronephros and analysis of mutations affecting pronephric function. Development. 1998;125:4655–4667. doi: 10.1242/dev.125.23.4655. [DOI] [PubMed] [Google Scholar]
- 28.Loges N.T., Olbrich H., Becker-Heck A., Häffner K., Heer A., Reinhard C., Schmidts M., Kispert A., Zariwala M.A., Leigh M.W. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009;85:883–889. doi: 10.1016/j.ajhg.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rompolas P., Patel-King R.S., King S.M. An outer arm Dynein conformational switch is required for metachronal synchrony of motile cilia in planaria. Mol. Biol. Cell. 2010;21:3669–3679. doi: 10.1091/mbc.E10-04-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Costa G.L., Bauer J.C., McGowan B., Angert M., Weiner M.P. Site-directed mutagenesis using a rapid PCR-based method. Methods Mol. Biol. 1996;57:239–248. doi: 10.1385/0-89603-332-5:239. [DOI] [PubMed] [Google Scholar]
- 31.Timmons L., Fire A. Specific interference by ingested dsRNA. Nature. 1998;395:854. doi: 10.1038/27579. [DOI] [PubMed] [Google Scholar]
- 32.Newmark P.A., Reddien P.W., Cebrià F., Sánchez Alvarado A. Ingestion of bacterially expressed double-stranded RNA inhibits gene expression in planarians. Proc. Natl. Acad. Sci. USA. 2003;100(Suppl 1):11861–11865. doi: 10.1073/pnas.1834205100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rompolas P., Patel-King R.S., King S.M. Schmidtea mediterranea: a model system for analysis of motile cilia. Methods Cell Biol. 2009;93:81–98. doi: 10.1016/S0091-679X(08)93004-1. [DOI] [PubMed] [Google Scholar]
- 34.Witman G.B., Carlson K., Berliner J., Rosenbaum J.L. Chlamydomonas flagella. I. Isolation and electrophoretic analysis of microtubules, matrix, membranes, and mastigonemes. J. Cell Biol. 1972;54:507–539. doi: 10.1083/jcb.54.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rompolas P., Pedersen L.B., Patel-King R.S., King S.M. Chlamydomonas FAP133 is a dynein intermediate chain associated with the retrograde intraflagellar transport motor. J. Cell Sci. 2007;120:3653–3665. doi: 10.1242/jcs.012773. [DOI] [PubMed] [Google Scholar]
- 36.Sizova I., Fuhrmann M., Hegemann P. A Streptomyces rimosus aphVIII gene coding for a new type phosphotransferase provides stable antibiotic resistance to Chlamydomonas reinhardtii. Gene. 2001;277:221–229. doi: 10.1016/s0378-1119(01)00616-3. [DOI] [PubMed] [Google Scholar]
- 37.O’Toole E., Mastronarde D., McIntosh J.R., Porter M.E. Computer-assisted analysis of flagellar structure. Methods Cell Biol. 1995;47:183–191. doi: 10.1016/s0091-679x(08)60808-0. [DOI] [PubMed] [Google Scholar]
- 38.Leigh M.W., Pittman J.E., Carson J.L., Ferkol T.W., Dell S.D., Davis S.D., Knowles M.R., Zariwala M.A. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet. Med. 2009;11:473–487. doi: 10.1097/GIM.0b013e3181a53562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Halbritter J., Diaz K., Chaki M., Porath J.D., Tarrier B., Fu C., Innis J.L., Allen S.J., Lyons R.H., Stefanidis C.J. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J. Med. Genet. 2012;49:756–767. doi: 10.1136/jmedgenet-2012-100973. [DOI] [PubMed] [Google Scholar]
- 40.Otto E.A., Helou J., Allen S.J., O’Toole J.F., Wise E.L., Ashraf S., Attanasio M., Zhou W., Wolf M.T.F., Hildebrandt F. Mutation analysis in nephronophthisis using a combined approach of homozygosity mapping, CEL I endonuclease cleavage, and direct sequencing. Hum. Mutat. 2008;29:418–426. doi: 10.1002/humu.20669. [DOI] [PubMed] [Google Scholar]
- 41.Avidor-Reiss T., Maer A.M., Koundakjian E., Polyanovsky A., Keil T., Subramaniam S., Zuker C.S. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004;117:527–539. doi: 10.1016/s0092-8674(04)00412-x. [DOI] [PubMed] [Google Scholar]
- 42.Broadhead R., Dawe H.R., Farr H., Griffiths S., Hart S.R., Portman N., Shaw M.K., Ginger M.L., Gaskell S.J., McKean P.G., Gull K. Flagellar motility is required for the viability of the bloodstream trypanosome. Nature. 2006;440:224–227. doi: 10.1038/nature04541. [DOI] [PubMed] [Google Scholar]
- 43.Stolc V., Samanta M.P., Tongprasit W., Marshall W.F. Genome-wide transcriptional analysis of flagellar regeneration in Chlamydomonas reinhardtii identifies orthologs of ciliary disease genes. Proc. Natl. Acad. Sci. USA. 2005;102:3703–3707. doi: 10.1073/pnas.0408358102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ostrowski L.E., Blackburn K., Radde K.M., Moyer M.B., Schlatzer D.M., Moseley A., Boucher R.C. A proteomic analysis of human cilia: identification of novel components. Mol. Cell. Proteomics. 2002;1:451–465. doi: 10.1074/mcp.m200037-mcp200. [DOI] [PubMed] [Google Scholar]
- 45.Efimenko E., Bubb K., Mak H.Y., Holzman T., Leroux M.R., Ruvkun G., Thomas J.H., Swoboda P. Analysis of xbx genes in C. elegans. Development. 2005;132:1923–1934. doi: 10.1242/dev.01775. [DOI] [PubMed] [Google Scholar]
- 46.Liu Q., Tan G., Levenkova N., Li T., Pugh E.N., Jr., Rux J.J., Speicher D.W., Pierce E.A. The proteome of the mouse photoreceptor sensory cilium complex. Mol. Cell. Proteomics. 2007;6:1299–1317. doi: 10.1074/mcp.M700054-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J.B., Gerdes J.M., Haycraft C.J., Fan Y., Teslovich T.M., May-Simera H., Li H., Blacque O.E., Li L., Leitch C.C. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117:541–552. doi: 10.1016/s0092-8674(04)00450-7. [DOI] [PubMed] [Google Scholar]
- 48.Andersen J.S., Wilkinson C.J., Mayor T., Mortensen P., Nigg E.A., Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426:570–574. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- 49.Pazour G.J., Agrin N., Leszyk J., Witman G.B. Proteomic analysis of a eukaryotic cilium. J. Cell Biol. 2005;170:103–113. doi: 10.1083/jcb.200504008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keller L.C., Romijn E.P., Zamora I., Yates J.R., 3rd, Marshall W.F. Proteomic analysis of isolated chlamydomonas centrioles reveals orthologs of ciliary-disease genes. Curr. Biol. 2005;15:1090–1098. doi: 10.1016/j.cub.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 51.Yu X., Ng C.P., Habacher H., Roy S. Foxj1 transcription factors are master regulators of the motile ciliogenic program. Nat. Genet. 2008;40:1445–1453. doi: 10.1038/ng.263. [DOI] [PubMed] [Google Scholar]
- 52.Robb S.M., Ross E., Sánchez Alvarado A. SmedGD: the Schmidtea mediterranea genome database. Nucleic Acids Res. 2008;36(Database issue):D599–D606. doi: 10.1093/nar/gkm684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Noone P.G., Leigh M.W., Sannuti A., Minnix S.L., Carson J.L., Hazucha M., Zariwala M.A., Knowles M.R. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am. J. Respir. Crit. Care Med. 2004;169:459–467. doi: 10.1164/rccm.200303-365OC. [DOI] [PubMed] [Google Scholar]
- 54.Kato T., Kagami O., Yagi T., Kamiya R. Isolation of two species of Chlamydomonas reinhardtii flagellar mutants, ida5 and ida6, that lack a newly identified heavy chain of the inner dynein arm. Cell Struct. Funct. 1993;18:371–377. doi: 10.1247/csf.18.371. [DOI] [PubMed] [Google Scholar]
- 55.Goodstein D.M., Shu S., Howson R., Neupane R., Hayes R.D., Fazo J., Mitros T., Dirks W., Hellsten U., Putnam N., Rokhsar D.S. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40(Database issue):D1178–D1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin J., Tritschler D., Song K., Barber C.F., Cobb J.S., Porter M.E., Nicastro D. Building blocks of the nexin-dynein regulatory complex in Chlamydomonas flagella. J. Biol. Chem. 2011;286:29175–29191. doi: 10.1074/jbc.M111.241760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Porter M.E., Knott J.A., Myster S.H., Farlow S.J. The dynein gene family in Chlamydomonas reinhardtii. Genetics. 1996;144:569–585. doi: 10.1093/genetics/144.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perrone C.A., Yang P., O’Toole E., Sale W.S., Porter M.E. The Chlamydomonas IDA7 locus encodes a 140-kDa dynein intermediate chain required to assemble the I1 inner arm complex. Mol. Biol. Cell. 1998;9:3351–3365. doi: 10.1091/mbc.9.12.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thiel C., Kessler K., Giessl A., Dimmler A., Shalev S.A., von der Haar S., Zenker M., Zahnleiter D., Stöss H., Beinder E. NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am. J. Hum. Genet. 2011;88:106–114. doi: 10.1016/j.ajhg.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ahmed N.T., Gao C., Lucker B.F., Cole D.G., Mitchell D.R. ODA16 aids axonemal outer row dynein assembly through an interaction with the intraflagellar transport machinery. J. Cell Biol. 2008;183:313–322. doi: 10.1083/jcb.200802025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fowkes M.E., Mitchell D.R. The role of preassembled cytoplasmic complexes in assembly of flagellar dynein subunits. Mol. Biol. Cell. 1998;9:2337–2347. doi: 10.1091/mbc.9.9.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson N.T., Villalón M., Royce F.H., Hard R., Verdugo P. Autoregulation of beat frequency in respiratory ciliated cells. Demonstration by viscous loading. Am. Rev. Respir. Dis. 1991;144:1091–1094. doi: 10.1164/ajrccm/144.5.1091. [DOI] [PubMed] [Google Scholar]
- 63.Andrade Y.N., Fernandes J., Vázquez E., Fernández-Fernández J.M., Arniges M., Sánchez T.M., Villalón M., Valverde M.A. TRPV4 channel is involved in the coupling of fluid viscosity changes to epithelial ciliary activity. J. Cell Biol. 2005;168:869–874. doi: 10.1083/jcb.200409070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rompolas P., Patel-King R.S., King S.M. Association of Lis1 with outer arm dynein is modulated in response to alterations in flagellar motility. Mol. Biol. Cell. 2012;23:3554–3565. doi: 10.1091/mbc.E12-04-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Piperno G., Mead K., Shestak W. The inner dynein arms I2 interact with a “dynein regulatory complex” in Chlamydomonas flagella. J. Cell Biol. 1992;118:1455–1463. doi: 10.1083/jcb.118.6.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Piperno G., Mead K., LeDizet M., Moscatelli A. Mutations in the “dynein regulatory complex” alter the ATP-insensitive binding sites for inner arm dyneins in Chlamydomonas axonemes. J. Cell Biol. 1994;125:1109–1117. doi: 10.1083/jcb.125.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yanagisawa H.A., Kamiya R. A tektin homologue is decreased in Chlamydomonas mutants lacking an axonemal inner-arm dynein. Mol. Biol. Cell. 2004;15:2105–2115. doi: 10.1091/mbc.E03-11-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Amos L.A. The tektin family of microtubule-stabilizing proteins. Genome Biol. 2008;9:229. doi: 10.1186/gb-2008-9-7-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Merveille A.C., Davis E.E., Becker-Heck A., Legendre M., Amirav I., Bataille G., Belmont J., Beydon N., Billen F., Clément A. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011;43:72–78. doi: 10.1038/ng.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ziętkiewicz E., Bukowy-Bieryłło Z., Voelkel K., Klimek B., Dmeńska H., Pogorzelski A., Sulikowska-Rowińska A., Rutkiewicz E., Witt M. Mutations in radial spoke head genes and ultrastructural cilia defects in East-European cohort of primary ciliary dyskinesia patients. PLoS ONE. 2012;7:e33667. doi: 10.1371/journal.pone.0033667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Geremek M., Schoenmaker F., Zietkiewicz E., Pogorzelski A., Diehl S., Wijmenga C., Witt M. Sequence analysis of 21 genes located in the Kartagener syndrome linkage region on chromosome 15q. Eur. J. Hum. Genet. 2008;16:688–695. doi: 10.1038/ejhg.2008.5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Normal olfactory cilia beat rhythmically around the perimeter of the olfactory placode. This and all microvideo were acquired at 250 frames per second (fps) and slowed to 15 fps for viewing.
Pronephric multicilia beat in a coordinated helical pattern to drive fluid down the lumen of the pronephric tubule.
Cilia are largely paralyzed, consistent with loss of outer dynein arms.
Most ccdc65 morphants showed complete paralysis of olfactory cilia.
Some ccdc65 morphants showed faster, dyskinetic cilia beat patterns.
Cilia bundles showed greatly reduced beat amplitude or paralysis and bundle coordination was lost.
Time-lapse video of control (left) and Smed fbb18(RNAi) (right) -treated planaria. fbb18 knockdown planaria resort to a parastaltic mode of locomotion when cilia motility is disrupted.