

Abstract
Sodium leak channel, nonselective (NALCN) is a voltage-independent and cation-nonselective channel that is mainly responsible for the leaky sodium transport across neuronal membranes and controls neuronal excitability. Although NALCN variants have been conflictingly reported to be in linkage disequilibrium with schizophrenia and bipolar disorder, to our knowledge, no mutations have been reported to date for any inherited disorders. Using linkage, SNP-based homozygosity mapping, targeted sequencing, and confirmatory exome sequencing, we identified two mutations, one missense and one nonsense, in NALCN in two unrelated families. The mutations cause an autosomal-recessive syndrome characterized by subtle facial dysmorphism, variable degrees of hypotonia, speech impairment, chronic constipation, and intellectual disability. Furthermore, one of the families pursued preimplantation genetic diagnosis on the basis of the results from this study, and the mother recently delivered healthy twins, a boy and a girl, with no symptoms of hypotonia, which was present in all the affected children at birth. Hence, the two families we describe here represent instances of loss of function in human NALCN.
Main Text
NALCN (MIM 611549), encoding sodium leak channel, nonselective (NALCN), also known as voltage-gated channel-like protein 1, extends on a large genomic region around 363 kb in 13q33.1 (Ensembl), and contains 44 exons.1,2 Human NALCN was cloned in 1999, but its critical role and function on nerve resting conductance, neonatal death, and neuronal rhythm in animals were not elucidated until recently.1–3 NALCN belongs to the “four homologous repeats of six transmembrane-spanning segments” (4×6TM) family and acts as a TTX-resistant leak sodium channel. Neuropeptides, substance P, and neurotensin are able to activate NALCN through the Src family tyrosine kinase (SFK)-dependent pathway rather than through G-protein-coupled receptors, implicating a unique modulation mechanism for the coupling.3,4 Moreover, NALCN is also activated similarly by M3 muscarinic receptors in a pancreatic β cell line through G-protein-independent and SFK-dependent mechanisms.5,6
Genetic studies have been performed in search of human diseases associated with NALCN variants. Two consecutive studies presented interesting results for the putative involvement of NALCN in schizophrenia and bipolar disorder.7,8 Although a genome-wide meta-analysis identified an association between these diseases and an intragenic SNP of NALCN,8 an earlier study reported a lack of evidence of a similar association for schizophrenia.7 A recent study examined 240 schizophrenic individuals and did not find any significant association between NALCN and treatment-resistant schizophrenia.9 A current report examined pancreatic cancer genomes by using exome sequencing and revealed mutations in five genes, including NALCN, indicating involvement of axon-guidance genes in pancreatic carcinogenesis.10 Lastly, a molecular-karyotyping study reported that a female person with congenital heart defects, facial anomalies, developmental delay, and intellectual disability had a large deletion extending from 13q33.1 to 13q34.11 The deleted region harbors several genes, including NALCN.
We have been actively searching for genes, mutations, and copy-number variations predisposing to autism spectrum disorders, intellectual disability, and dysmorphia. We have ascertained more than 200 familial and simplex cases under protocols approved by the King Faisal Specialist Hospital and Research Center institutional review board (IRB). A total of six individuals and their family members from two large consanguineous families were included in this study after IRB-approved consent forms were obtained (Figure 1A). Individuals IV-1, V-1, and V-2 presented with absence of speech development, severe hypotonia, chronic constipation, global developmental delay, and facial dysmorphia. Metabolic and genetic workups—including an acylcarnitine profile, a gas chromatography-mass spectrometry (GC-MS) profile of urine, an amino acid profile, a urine creatine-metabolism-disorder panel, molecular cytogenetic arrays, and mitochondrial genome analysis—were normal. Using Affymetrix’s axiom SNP calls, our initial linkage study (as well as screening of genome-wide loss-of-heterozygosity [LOH] blocks among the affected individuals) revealed an approximately 4.7 Mb (chr13: 98,184,670–102,934,400 Mb) genomic interval comprising 48 genes, including NALCN (NCBI Map Viewer Build 37.3) (Figures S1 and S2, available online). On the basis of its function, NALCN was among the first genes screened during our targeted sequencing study. This revealed on exon 13 a single base deletion (c.1489delT [p.Tyr497Thrfs∗21]) (Figure S3) resulting in a frameshift and consequently creating a stop codon (21 amino acids downstream) producing a severely truncated protein (RefSeq accession number NM_052867.2). Moreover, we screened more than 600 (n = 625) ethnically matched control samples. Complete segregation of the mutation in the tested family members (n = 17) and the absence of the deletion among the control cohort suggested that this alteration is not a polymorphism (Figure 1A). The findings were then reconfirmed by exome sequencing of individual V-1’s DNA sample.
Figure 1.
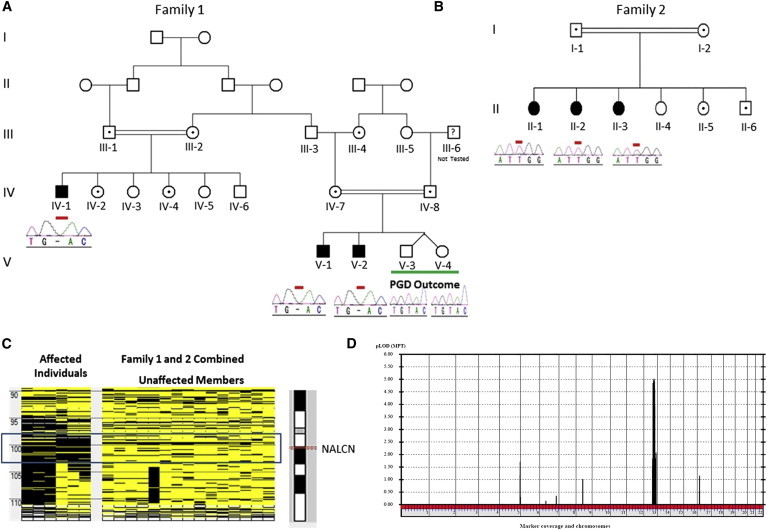
Genetic Analysis of the Autosomal-Recessive Disorder Caused by NALCN Mutations
(A and B) Pedigrees and selected chromatograms indicating mutations and carrier status of the tested members of families 1 (A) and 2 (B) are depicted. V-3 and V-4 are the outcome of preimplantation genetic diagnosis (PGD). PGD was performed on the basis of homozygosity mapping results, and neither individual has any NALCN mutations.
(C) Affymetrix GeneChip Human Mapping Axiom custom SNP arrays were used for genotyping, and the SNP calls were used as an input for the AutoSNPa tool. Homozygosity analysis indicated a shared run of homozygosity in both families. The block contains 48 genes, including NALCN.
(D) Parametric multipoint LOD score analysis was performed with the GeneHunter algorithm with Easy Linkage software. A recessive mode of inheritance, a disease allele frequency of 0.0001%, and no phenocopies were used in the analysis. Combined linkage results for both families indicated a single dominant peak (LOD score 5.01) on chromosome 13.
We then identified another pedigree with the same linkage interval (Figure 1B and Figures S4 and S5). Family 2 included three affected girls with delayed speech development, infantile hypotonia, easily controlled seizure disorder, hyperactivity, chronic constipation, and cognitive delay. Similar to in family 1, the metabolic and genetic workups were normal. Given the shared interval, we speculated that both families might have the same region of shared homozygosity, and we therefore decided to evaluate LOH blocks on chromosome 13. Using AutoSNPa, we aligned the LOH blocks from all the affected individuals from both families, as well as remaining family members. This analysis confirmed the shared overlapping LOH block between both families and resulted in a region that includes NALCN (Figure 1C). Combined linkage results for both families improved the LOD score to 5.01 (Figure 1D). Although milder, the phenotype of this family was similar to that in family 1. Hence, we decided to sequence NALCN in this family as a primary target and identified a missense mutation (c.3860G>T [p.Trp1287Leu]) in exon 34 (Figure 2A and Figure S6). The mutation site is highly conserved among different species (n = 66, based on PolyPhen prediction) and is seated in a region encoding the ion-transport domain and voltage-gated potassium-channel superfamily domain. The location of the mutation’s corresponding amino acid change is indicated on a predicted three-dimensional structure of NALCN (Figure S7).
Figure 2.
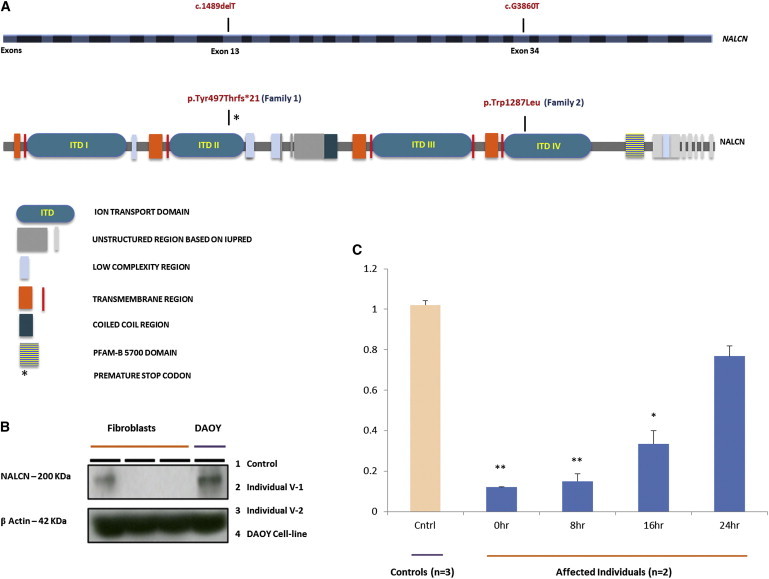
Location of NALCN Mutations and Immunoblotting and NMD Analyses
(A) Coding exons of NALCN are sequentially colored with dark gray and white (upper panel). Mutations are indicated in the relevant exons [13 and 34]). Pfam domains and low-complexity, transmembrane, coiled-coil, and unstructured regions are depicted on the basis of the Pfam source from the Howard Hughes Medical Institute (Web Resources). The protein structure is positioned according to the coding exons and amino acids (lower panel).
(B) Immunoblotting experiments performed on fibroblasts (from individuals V-1 and V-2) and DAOY cell lines indicated the absence of NALCN in the affected individuals but the presence of the protein in the control cell line. 100 μg/ml of the protein was used for immunoblotting, and β-actin was used as an internal control.
(C) qRT-PCR experiments were performed on RNA extracted from V-1’s and V-2’s fibroblasts treated with CHX (100 μg/ml) at different periods of time (0, 8, 16, and 24 hr). Control fibroblasts and samples from affected individuals at hour 0 were not treated with CHX. The data represent the mean fold change ± SE of three control samples and samples from two affected individuals for each time point (each sample was run in triplicate). Compared to controls, the affected individuals showed a significant decrease in expression. ∗p < 0.05, ∗∗p < 0.01 (Student’s t test).
Subsequently, we screened a large control cohort (n = 625) and did not find the mutation in the ethnically matched controls. Furthermore, the mutation was not reported in the Human Gene Mutation Database12 or the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server (Web Resources). The mutation was also not annotated in human variation databases such as HapMap,13 the 1000 Genomes Project,14 dbSNP, or the specialized databases for next-generation sequencing data, such as the Next Generation Sequencing Catalog15 (Web Resources). Furthermore, we performed an extensive in silico pathogenicity analysis of the missense mutation by using several publicly available tools and databases, and all predicted a damaging effect. Collectively, these results suggest that the above identified NALCN variants in these two families are responsible for the autosomal-recessive disorder causing a spectrum of severe to mild hypotonia, speech delay and impairment, and global developmental and cognitive delay (medical histories and clinical evaluations are summarized in Table 1 and are also extensively presented in Table S1).
Table 1.
Demographic, Clinical, and Laboratory Findings
|
Family 1 |
Family 2 |
|||||
|---|---|---|---|---|---|---|
| IV-1 | V-1 | V-2 | II-1 | II-2 | II-3 | |
| Age | 7 years, 3 months | 7 years, 1 month | 4 years, 4 months | 17 years, 6 months | 16 years | 9 years, 4 months |
| Gender | male | male | male | female | female | female |
| Age at presentation | birth | birth | birth | birth | birth | birth |
| Birth weight | 2.6 kg | 4 kg | 2.5 kg | 2.8 kg | 3 kg | 2.8 kg |
| Neonatal hypotonia | severe | severe | severe | moderate | moderate | mild |
| hypotonia | severe | severe | severe | mild | mild | mild |
| Feeding difficulties | severe | severe | severe | moderate | moderate | mild |
| Growth | normal | normala | normala | normal | normal | normal |
| Vision | squint | anisometropia | normal | normal | normal | squint |
| Hearing | normal | normal | normal | normal | normal | normal |
| Dysmorphism | yes | yes | yes | yes | yes | yes |
| Motor delay | severe | severe | severe | moderate to severe | moderate to severe | mild to moderate |
| Speech delay | severe | severe | severe | moderate to severe | moderate to severe | mild to moderate |
| Cognitive delay | very severe | very severe | very severe | severe | severe | severe |
| IQ | NA | NA | NA | 45/100 | 42/100 | 43/100 |
| Seizure | no | no | no | yesb | yesb | yesb |
| Hyperactivity | no | no | no | yes | yes | yes |
| Constipation | yes | yes | yes | yes | yes | yes |
| Karyotype | 46,XY | 46,XY | 46,XY | 46,XX | 46,XX | 46,XX |
| Metabolic screen | unremarkable | unremarkable | unremarkable | unremarkable | unremarkable | unremarkable |
| Brain MRI | normal | abnormal | normal | normal | normal | normal |
| EEG | - | normal | normal | normalc | normalc | normal |
| EMG and NC | - | normal | normal | normal | normal | normal |
The following abbreviations are used: EEG, electroencephalogram; EMG, electromyogram; NC, nerve conduction; and NA, not assessable.
Initial failure to thrive.
Infrequent and easily controlled by monotherapy.
Intermittent activity with slow background, but no epileptic activity.
To examine and better understand the truncated NALCN expression in human tissues, we initially sought an extensive in silico search by using publicly and commercially available tools and databases. Moreover, on the basis of previous work,1,2 it is known that Nalcn is expressed in all brain regions and throughout the nervous system, in addition to in the heart, skeletal muscle, pituitary glands, adrenal glands, and pancreatic islet cells.1,2,5,6 Although NALCN expression in mouse muscle was not detectable by in situ hybridization and RNA blotting,2 we speculated that this might be attributable to the detection power of the techniques or species-related tissue-specific expression of the gene. Therefore, we decided to perform RT-PCR experiments on readily available RNA from control samples and on affected individuals’ RNA from muscle and skin biopsies taken during diagnostic procedures. These experiments showed that NALCN mRNA was present in the muscle and the fibroblasts with moderate to low expression (data not shown). Given these results, we examined the NALCN levels in the fibroblasts of individuals V-1 and V-2 by using an immunoblotting technique, which revealed the absence of the protein (Figure 2B). It is known that the nonsense-mediated mRNA decay (NMD) pathway is likely to be activated if the premature termination codon is located more than 50–55 nt upstream of the 3′ exon-exon junction and does not reside within either the last exon or an exon followed by one or two exon-exon junctions that exist no more than 50–55 nt downstream.16 Our mutation is positioned 74 nt before the next exon-exon boundary, thus implicating involvement of NMD. Therefore, we decided to measure NALCN mRNA levels in cultured fibroblasts from individuals V-1 and V-2 by quantitative RT-PCR (qRT-PCR). The qRT-PCR showed a significant reduction of NALCN mRNA in the affected individuals (n = 2) compared to controls (n = 3), consistent with the NMD. We next performed similar qRT-PCR experiments on individuals’ fibroblasts treated with cycloheximide (CHX) and control fibroblasts without CHX treatments. The treatment recovered the reduced expression of NALCN in the affected individuals’ fibroblasts (Figure 2C).
Family 1 decided to go through preimplantation genetic diagnosis (PGD) at our hospital. PGD was solely done on the basis of the genetic analysis using the information from our study. The mother recently delivered healthy normal twins, a boy and girl, with no symptoms of hypotonia, which was present from birth in the affected boys in this family. Postnatal DNA analysis of NALCN from the twins confirmed their normal status (Figure 1A).
NALCN, unlike other members of the 4×6TM voltage-gated-channel family (comprising 21 members2,17), is noninactivating and voltage independent and has the capacity to be a cation-nonselective channel permeable to sodium, potassium, and calcium. These features have been attributed to the fewer positive charges in the voltage-sensing S4 segment, one of the six α-helical transmembrane segments (S1–S6) belonging to each of four homologous domains, which normally contain more positively charged amino acid residues.2–4,18 The channel becomes very important in that it not only is an elusive background leakage for Na+ currents at synapses but also has the ability to stimulate firing action potentials with the help of substance P and neurotensin.2–4 These unique structural and biochemical properties in animals are likely to be preserved in humans given the high interspecies conservation (99% homology between rat and human NALCN). In our individuals in family 1, the single-base deletion leads to a radically truncated protein lacking both three of four ion-transport domains and the same number of voltage-gated potassium-channel superfamily domains, which are highly conserved among different species. As a result, the abnormal protein is obviously incompatible with the typical function of NALCN. Therefore, it is conceivably safe to speculate that, just as in the knockout mouse that showed a severe phenotype and was unable to reach the second day of life, such a drastically truncated channel will have severe phenotypic consequences, as seen in our affected individuals, because of (1) a significantly reduced capacity of the permeability of the channel and (2) the consequent accumulation of cations (specifically Na+ ions); together, these will lead to an imbalance in the maintenance of nerve resting potentials and hence create a disturbance in neuronal conductance. However, the missense mutation is predicted to be sitting on a site probably close to the outer region of the channel (Figure S7) and is therefore likely to have lesser functional consequences; hence, it might be attributed to the milder phenotype seen in the affected individuals from family 2. Intriguingly, in addition to displaying mild to severe hypotonia, global developmental, and speech delay, all the affected individuals from both families presented with chronic constipation. It is known that NALCN is highly expressed in the murine small intestine19 and, together with substance P, is involved in induced depolarization of pacemaking activity of interstitial cells of Cajal in the gastrointestinal muscles.19 Therefore, disturbed pacemaking activity due to NALCN mutations might be related to such symptoms as seen in our affected individuals. Although more work needs to be done for better understanding the link between the NALCN mutations and the phenotypic consequences in these individuals, we believe NALCN’s involvement in an inherited human disease will open new venues for research and continue to be a subject of further studies.
Acknowledgments
The authors thank the families for their participation in this study. This work was supported by King Faisal Specialist Hospital and Research Centre seed research grants, and the authors are grateful for the financial support. We thank Fowzan S. Alkuraya, Yufei Shi, and Bedri Karakas for helpful discussions. We are grateful to our hospital’s purchasing department for facilitating all ordering and receiving inquiries, including reagents, assays, and chemicals, as well as members of our core facilities (Genotyping/Microarray and Sequencing) for their continuous technical support.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org/index.html
HHMI Pfam, http://pfam.janelia.org
NGS Catalog, http://bioinfo.mc.vanderbilt.edu/NGS/index.html
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Sequence Read Archive, http://www.ncbi.nlm.nih.gov/Traces/sra
References
- 1.Lee J.H., Cribbs L.L., Perez-Reyes E. Cloning of a novel four repeat protein related to voltage-gated sodium and calcium channels. FEBS Lett. 1999;445:231–236. doi: 10.1016/s0014-5793(99)00082-4. [DOI] [PubMed] [Google Scholar]
- 2.Lu B., Su Y., Das S., Liu J., Xia J., Ren D. The neuronal channel NALCN contributes resting sodium permeability and is required for normal respiratory rhythm. Cell. 2007;129:371–383. doi: 10.1016/j.cell.2007.02.041. [DOI] [PubMed] [Google Scholar]
- 3.Lu B., Zhang Q., Wang H., Wang Y., Nakayama M., Ren D. Extracellular calcium controls background current and neuronal excitability via an UNC79-UNC80-NALCN cation channel complex. Neuron. 2010;68:488–499. doi: 10.1016/j.neuron.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu B., Su Y., Das S., Wang H., Wang Y., Liu J., Ren D. Peptide neurotransmitters activate a cation channel complex of NALCN and UNC-80. Nature. 2009;457:741–744. doi: 10.1038/nature07579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swayne L.A., Mezghrani A., Varrault A., Chemin J., Bertrand G., Dalle S., Bourinet E., Lory P., Miller R.J., Nargeot J., Monteil A. The NALCN ion channel is activated by M3 muscarinic receptors in a pancreatic beta-cell line. EMBO Rep. 2009;10:873–880. doi: 10.1038/embor.2009.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swayne L.A., Mezghrani A., Lory P., Nargeot J., Monteil A. The NALCN ion channel is a new actor in pancreatic β-cell physiology. Islets. 2010;2:54–56. doi: 10.4161/isl.2.1.10522. [DOI] [PubMed] [Google Scholar]
- 7.Souza R.P., Rosa D.V., Romano-Silva M.A., Zhen M., Meltzer H.Y., Lieberman J.A., Remington G., Kennedy J.L., Wong A.H. Lack of association of NALCN genetic variants with schizophrenia. Psychiatry Res. 2011;185:450–452. doi: 10.1016/j.psychres.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 8.Wang K.S., Liu X.F., Aragam N. A genome-wide meta-analysis identifies novel loci associated with schizophrenia and bipolar disorder. Schizophr. Res. 2010;124:192–199. doi: 10.1016/j.schres.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 9.Teo C., Zai C., Borlido C., Tomasetti C., Strauss J., Shinkai T., Le Foll B., Wong A., Kennedy J.L., De Luca V. Analysis of treatment-resistant schizophrenia and 384 markers from candidate genes. Pharmacogenet. Genomics. 2012;22:807–811. doi: 10.1097/FPC.0b013e3283586c04. [DOI] [PubMed] [Google Scholar]
- 10.Biankin A.V., Waddell N., Kassahn K.S., Gingras M.C., Muthuswamy L.B., Johns A.L., Miller D.K., Wilson P.J., Patch A.M., Wu J., Australian Pancreatic Cancer Genome Initiative Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang C., Yang Y.F., Yin N., Chen J.L., Wang J., Zhang H., Tan Z.P. Congenital heart defect and mental retardation in a patient with a 13q33.1-34 deletion. Gene. 2012;498:308–310. doi: 10.1016/j.gene.2012.01.083. [DOI] [PubMed] [Google Scholar]
- 12.Stenson P.D., Ball E., Howells K., Phillips A., Mort M., Cooper D.N. Human Gene Mutation Database: towards a comprehensive central mutation database. J. Med. Genet. 2008;45:124–126. doi: 10.1136/jmg.2007.055210. [DOI] [PubMed] [Google Scholar]
- 13.Consortium I.H., International HapMap Consortium The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 14.Siva N. 1000 Genomes project. Nat. Biotechnol. 2008;26:256. doi: 10.1038/nbt0308-256b. [DOI] [PubMed] [Google Scholar]
- 15.Xia J., Wang Q., Jia P., Wang B., Pao W., Zhao Z. NGS catalog: A database of next generation sequencing studies in humans. Hum. Mutat. 2012;33:E2341–E2355. doi: 10.1002/humu.22096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maquat L.E. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 2004;5:89–99. doi: 10.1038/nrm1310. [DOI] [PubMed] [Google Scholar]
- 17.Catterall W.A. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 18.Yu F.H., Catterall W.A. Overview of the voltage-gated sodium channel family. Genome Biol. 2003;4:207. doi: 10.1186/gb-2003-4-3-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim B.J., Chang I.Y., Choi S., Jun J.Y., Jeon J.H., Xu W.X., Kwon Y.K., Ren D., So I. Involvement of Na(+)-leak channel in substance P-induced depolarization of pacemaking activity in interstitial cells of Cajal. Cell. Physiol. Biochem. 2012;29:501–510. doi: 10.1159/000338504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.